

Abstract
In spite of significant progress in the field of targeted anticancer therapy, the FDA has approved only five ADC-based drugs. Hence the search for new targeted anticancer agents is an unfulfilled necessity. Here, we present novel types of protein–drug conjugates (PDCs) that exhibit superior anticancer activities. Instead of a monoclonal antibody, we used fibroblast growth factor 2 (FGF2) as a targeting molecule. FGF2 is a natural ligand of fibroblast growth factor receptor 1 (FGFR1), a transmembrane receptor overproduced in various types of cancers. We synthesized site-specific and stoichiometric-controlled conjugates of FGF2 with a highly potent, hydrophilic derivative of auristatin called auristatin Y. To increase the hydrophilicity and hydrodynamic radius of conjugates, we employed PEG4 and PEG27 molecules as a spacer between the targeting molecule and the cytotoxic payload. All conjugates were selective to FGFR1-positive cell lines, effectively internalized via the FGFR1-dependent pathway, and exhibited a highly cytotoxic effect only on FGFR1-positive cancer cell lines.
Keywords: FGF2, auristatin Y, hydrophilic drug, PEGylation, conjugate, cancer
Introduction
Conventional low molecular weight anticancer drugs do not effectively accumulate at the tumor site, leading to the high toxicity of the healthy tissues and, in consequence, exhibiting a narrow therapeutic window.1,2 On the other hand, antibody–drug conjugates (ADCs), thanks to the monoclonal antibody as a targeting molecule, ensure the specific delivery of drugs to cancer cells. However, their large size limits the ability to penetrate solid tumors.3 Therefore, small-format drug conjugates (SFDCs)4 or small molecule–drug conjugates (SMDCs)5 have been recently proposed as alternatives to ADCs. These conjugates utilize specific targeting molecules, usually much smaller than the full-length antibody, such as antibody fragments,6−9 designed ankyrin repeat proteins (DARPins),10,11 knottins,12,13 affibodies,14,15 bicyclic peptides,16,17 or small organic ligands.5,18 SFDCs or SMDCs retain the ability of ADCs to localize selectively in cancerous tissues, exhibiting at the same time favorable pharmacokinetics and accessibility.4,5 Thus, many of them are currently entering clinical trials.19
However, for effective delivery of the drug not only the size of the conjugate matters.20−23 An import issue is the hydrophobicity of the cytotoxic payload and of the linker that connects the payload to the targeting molecule. Reduced hydrophobic properties of the toxic cargo increase a therapeutic window due to the better exposure and solubility,20,24,25 but it has to be kept in mind that the potency of the conjugate increases with the higher drug loading (an increased drug-to-targeting-protein ratio, DPR (called DAR for ADCs)), leading to enhanced hydrophobicity and in consequence an unfavorable pharmacokinetic profile. Thus, there is a strong need in the ADC field for the reduction of off-target cytotoxicity, and the optimization of linkers and payloads.21,26,27 The most common approach used to improve the hydrophilicity of cargo is the attachment of polyethylene glycol moieties (PEGylation).28−32 Other strategies utilize glucuronidation of the cytotoxic agent or preparation of its hydrophilic derivatives.20,22,33,34
One of the most exploited cytotoxins in ADCs, currently in clinics and in many clinical trials, is hydrophobic monomethyl auristatin E (MMAE).21,35 Recently we showed that human fibroblast growth factor 2 (FGF2), in a similar manner as an antibody, can be used as a targeting protein to deliver MMAE to cancer cells overproducing fibroblast growth factor receptor 1 (FGFR1). FGF2 loaded with three MMAE molecules effectively killed U2OS stably transfected with FGFR1 (U2OS-R1) cells compared with nontransfected cells.36 Unfortunately, FGF2 loaded with three MMAE drugs showed a tendency to aggregate after overnight storage at 4 °C and could be used in micromolar concentrations. FGF2, due to a small molecular weight (17 kDa), undergoes renal filtration.37 In this study, we decided to use a hydrophilic derivative of auristatin (auristatin Y, AY). To increase the hydrodynamic size of AY-based conjugates and overcome the kidney filtration problem, we applied different PEGs of increasing size.
Experimental Section
Materials
Reagents
All chemical reagents were from commercial suppliers and used without further purification. Reagents used for the solid-phase peptide synthesis were as follows: amino acids Fmoc-l-Ser(tBu)–OH, Fmoc-l-Arg(Pbf)–OH, Fmoc-l-Cys(StBu)–OH; PEGs mal-dPEG(4)-NHS, mal-PEG(27)-NHS; COMU (1-[1-(cyano-2-ethoxy-2-oxoethylideneaminooxy)-dimethylamino-morpholino]-uronium hexafluorophosphate), piperidine, TIS (triisopropylsilane), DIPEA (N,N-diisopropylethylamine), DMF (N,N-dimethylformamide), DCM (dichloromethane), and TFA (trifluoroacetic acid) were purchased from Iris Biotech GmbH (Marktredwitz, Germany). HPLC-pure acetonitrile and Et2O (diethyl ether) were from Avantor (Gliwice, Poland). TentaGel S RAM resin (particle size: 90 μm, loading 0.21 mmol/g) was from Rapp Polymere GmbH (Tübingen, Germany). The cytotoxic agents, auristatin Y (AY) and maleimidocaproyl-threonine-valine-auristatin Y (tvAY), were from custom synthesis carried out by ChiroBlock GmbH (Wolfen, Germany).
The following dyes were used: CellTracker Red CMTPX, DyLight 488 NHS Ester, DyLight 550 NHS Ester, DAPI, CellLight Early Endosomes-GFP BacMam 2.0, CellLight Lysosomes-GFP BacMam 2.0, and alamarBlue were from Thermo Fisher Scientific (Waltham, MA). The chromatographic column HiTrap Desalting with Sephadex G-25 resin, Superdex 75 10/300 GL, HiTrap CM Sepharose FF, and HiTrap Heparin HP were from GE Healthcare (U.K.). Zeba Spin Desalting columns were from Thermo Fisher Scientific, and the Synergi 4 μm Fusion-RP 80 Å 250 × 10 mm LC column was from Phenomenex Inc. (Torrance, CA). All other reagents were obtained from Sigma-Aldrich (Saint Louis, MO) or BioShop Canada Inc. (Burlington, ON).
Cells
DMS114 (human small cell lung cancer, ATCC CRL-2066) cells were cultured in Waymouth’s MB 752/1 from Gibco (Waltham, MA). U2OS (human osteosarcoma, HTB-96) and U2OS stably transfected with FGFR1 (U2OS-R1) were cultured in DMEM High Glucose with stable glutamine and sodium pyruvate from Biowest (France). NCI-H520 (human squamous cell carcinoma, ATCC HTB-182) and HCC15 (human squamous cell lung carcinoma) and were cultured in RPMI-1640 medium from Gibco (Waltham, MA). All media were supplemented with 10% fetal bovine serum from Thermo Fisher Scientific, and 1% penicillin/streptomycin mix was from Biowest (France). Additionally, the U2OS-R1 cell medium contained 50 μg/mL gentamicin sulfate from Thermo Fisher Scientific. All cell lines were cultured in a humidified incubator at 37 °C in a 5% CO2 atmosphere. The DMS114, NCI-H520, and U2OS cell lines were obtained from American Type Culture Collection (Manassas, VA). HCC15 cells were supplied by the Leibniz Institute DSMZ, German Collection of Microorganisms and Cell Cultures. The U2OS cells stably expressing FGFR1 (U2OS-R1) were a kind gift from Dr. Ellen M. Haugsten from The Norwegian Radium Hospital.38 The E. coli expression strain Rosetta 2(DE3)pLysS was from Novagen-EMD Biosciences (Madison, WI).
Methods
Synthesis of PEGylated tvAY Moieties
Step 1: Synthesis and Purification of l-Cys-tvAY
l-Cysteine (184 mg, 1.52 mmol, 20 equiv), maleimidocaproyl-threonine-valine-auristatin Y (100 mg, 0.08 mmol), and DIPEA (26.5 μL, 0.16 mmol, 2 equiv) were added to 1 mL of DMAc. The reaction was conducted at 30 °C for 12 h. Next, the solvent was removed under a vacuum. The crude product was dissolved in 500 μL of 30% acetonitrile/water with 0.1% TFA, then the NH2–Cys-tvAY was separated from an excess of Cys by RP-HPLC, and the solvent was removed by lyophilization. The identity of the product was confirmed by MALDI-MS.
Step 2a: Synthesis and Purification of Maleimide-PEG4-tvAY
mal-dPEG(4)-NHS (89.9 mg, 0.175 mmol, 5 equiv) and DIPEA (12.4 μL, 0.075 mmol, 2 equiv) were added to a solution of NH2–Cys-tvAY (50 mg, 0.035 mmol) in 500 μL of DMAc. The reaction mixture was incubated at 30 °C for 12 h. The crude product was dissolved in 500 μL of 30% acetonitrile/water with 0.1% TFA, and then the desired product was separated by reverse-phase HPLC, lyophilized, and evaluated by MALDI-MS.
Step 2b: Synthesis and Purification of Maleimide-PEG27-tvAY
mal-PEG(27)-NHS (275 mg, 0.175 mmol, 5 equiv) and DIPEA (12.4 μL, 0.075 mmol, 2 equiv) were added to a solution of NH2–Cys-tvAY (50 mg, 0.035 mmol) in 500 μL of DMAc. The reaction mixture was incubated at 30 °C for 12 h. The crude product was dissolved in 500 μL of 30% acetonitrile/water with 0.1% TFA, and then the desired product was separated by RP-HPLC and lyophilized. The identity of the product was confirmed by MALDI-MS.
Synthesis of Maleimide-PEG27-(tvAY)3
In the first step, the all-l Fmoc-S(tBu)R(Pbf)C(StBu)R(Pbf)C(StBu)R(Pbf)C(StBu) peptide scaffold was synthesized on the solid support (TentaGel S RAM resin; 0.1 g, capacity 0.21 mmol/g) using COMU as coupling reagent (3 equiv of each reagent). In the next step, the orthogonal StBu-protecting group was removed from Cys residues using PBu3 in MeOH (three cycles of 5 min, 100 equiv), and then the maleimidocaproyl-threonine-valine-auristatin Y (155 mg, 0.13 mmol, 2 equiv) was coupled to the sulfhydryl group of Cys. After Fmoc group deprotection (20% piperidine in DMF, two cycles: 10, 5 min), mal-PEG(27)-NHS (100 mg, 0.06 mmol, 3 equiv) was attached to the N-terminal amino group. Finally, the maleimide-PEG27-(tvAY)3 was cut off from the resin with a mixture of TFA/TIS/DCM (% v/v/v, 96:2:2), triply precipitated in cold Et2O, purified by reverse-phase HPLC, and lyophilized. The identity of the product was confirmed by MALDI-MS.
Protein Production and Purification
The plasmids encoding fibroblast growth factor 2 with the N-terminal KCKSGG linker and FGF2 with the N-terminal KCKSGG linker and the two-point mutations C78S and C96S were used.36 The plasmids were transformed into the E. coli Rosetta 2(DE3)pLysS expression strain. Protein production was carried out in the Biostat C fermentor system (B. Braun Biotech International, Germany). Bacteria were grown in a TB medium with 100 μg/mL ampicillin at 37 °C, pO235–50% range, and 250 rpm stirring to OD600 = 8. Then, the temperature was decreased to 20 °C, and protein production was induced by the addition of IPTG to a final concentration of 0.5 mM and conducted for 12 h. After that, bacteria were harvested by centrifugation at 6500g, resuspended in lysis buffer (50 mM monosodium phosphate, 0.15 M NaCl, 1 mM DTT, 1 mM EDTA, 0.1% Triton X-100, 1 mM PMSF, pH 7.2) supplemented with 500 U/L of Pierce Universal Nuclease, and homogenized using a French press. The cell debris was separated by ultracentrifugation at 50 000g at 4 °C for 1 h. The clarified cell lysate was diluted in 50 mM monosodium phosphate, 0.7 M NaCl, 10 mM (NH4)2SO4, 1 mM DTT, and 1 mM EDTA, pH 7.2, and loaded on a HiTrap Heparin HP column. The column was washed with a washing buffer (50 mM monosodium phosphate, 1.0 M NaCl, 10 mM (NH4)2SO4, 1 mM DTT, 1 mM EDTA, pH 7.2), and the protein was eluted with a linear 1.0–2.0 M gradient of NaCl in the same buffer.
Synthesis of the Conjugates
Maleimide-tvAY, maleimide-PEG4-tvAY, maleimide-PEG27-tvAY, and maleimide-PEG27-(tvAY)3 were dissolved in DMAc at a concentration of 50 mg/mL. The attachment of the cytotoxic payload containing a maleimide moiety to the sulfhydryl group of protein was performed in the reaction buffer (25 mM monosodium phosphate, 10 mM Na2SO4, 10 mM methionine, 1 mM EDTA, pH 7.0) at a 0.5 mg/mL protein concentration and 5-fold molar excess of payload per sulfhydryl group. The reaction was incubated at 20 °C for 1 h. Next, the reaction mixture was loaded onto the HiTrap CM Sepharose column, the unreacted payload was washed out using 25 mM monosodium phosphate pH 7.0 with 10 mM Na2SO4, and finally, the conjugate was eluted with the same buffer containing 0.5 M NaCl.
Stability in Human Serum
FGF2 and FGF2 conjugates (1 μg/mL) were incubated at 37 °C for different time periods (0, 48, 72, 96, and 120 h) in human serum (H4522, Sigma-Aldrich) in the absence of heparin. Then samples were analyzed by immunoblotting with mouse anti-FGF2 (sc-74412) and donkey antimouse IgG-HRP (sc-2318) antibodies from Santa Cruz Biotechnology (Dallas, TX).
Fluorescence Labeling of Proteins and Conjugates
FGF2 and the conjugates were labeled with DyLight 488 or 550 coupled with NHS ester.
The 10 mM stock solutions of the fluorescence probes were prepared in N,N-dimethylacetamide (DMAc). Then, 100 μL of the protein samples at a concentration of 1 mg/mL in 25 mM HEPES pH 8.0 with 10 mM (NH4)2SO4 was mixed with 5 μL of fluorescence probes. The reactions were conducted at room temperature for 1 h in the dark. Labeled proteins or conjugates were purified using Zeba Spin Desalting columns using PBS supplemented with 10 mM (NH4)2SO4 as an elution buffer.
Mass Spectrometry (MS)
Molecular masses were determined by MALDI-TOF MS (Applied Biosystems AB 4800+) using α-cyano-4-hydroxycinnamic acid as a matrix.
Spectrofluorimetry
To validate the folded state of protein and conjugates, spectrofluorimetry measurements were performed. The fluorescence spectra were acquired using an FP-8500 spectrofluorimeter (Jasco, Japan) with excitation at 280 nm and emission in the 300–450 nm range, at 20 °C using a 4 × 10–6 M sample concentration in PBS.
Thermal Unfolding and Aggregation
The thermal denaturation and aggregation were analyzed using nanoDSF and light back-reflection, respectively. The measurements were conducted using the NanoTemper Technologies instrument Prometheus NT.48 fitted with back-reflection optics at the concentration of the proteins of 3 μM in PBS. The samples were loaded into Prometheus NT.48 Series nanoDSF grade high sensitivity capillaries and then heated at a defined, constant ramp, 1 °C/min, over a defined temperature range from 20 to 95 °C. Fluorescence was excited at 280 nm, and emission was recorded at 350 nm. The aggregation was measured in parallel, as light intensity loss due to scattering. Data recorded by back-reflection optics were expressed on the chart in milli-attenuation units.
Size-Exclusion Chromatography (SEC)
Size-exclusion chromatography runs were performed to estimate the hydrodynamic-radius-based molecular mass of FGF2 and conjugates. The analysis was performed at 20 °C using an ÄKTA explorer FPLC System with a Superdex 75 HR 10/30 column. Then, 2.5 mg/mL samples were loaded onto the column by a full 50 μL loop injection. The mobile phase (25 mM monosodium phosphate pH = 7.4, 10 mM Na2SO4) was pumped at a flow rate of 1 mL/min, and absorption at 280 nm was recorded. Molecular weight standards containing BPTI, cytochrome C, carbonic anhydrases, human serum albumin, α-lactoglobulin, chymotrypsinogen A, ovalbumin, and albumin were used to generate a standard curve from which the effective size of the PEGylated conjugates was estimated.
Biolayer Interferometry (BLI)
The kinetic rate constants were measured on ForteBio Octet K2 (Pall ForteBio, CA) and high-precision streptavidin biosensors (SAX) (Pall ForteBio, CA) were used. Biotinylated extracellular domains of FGFR1c fused to the Fc fragment were immobilized, and then binding of FGF2 or the conjugates was performed similarly as we reported previously.36 Association of the samples at a concentration of 50 nM was conducted for 400 s, and then the dissociation was monitored for 400 s. Kinetic parameters were calculated using a 1:1 model with Octet Data Analysis software 11.0.
Confocal Microscopy
Specific Internalization of FGF2 and Conjugates into Cells Expressing FGFR1
U2OS cells were stained with CellTracker Red CMTPX, according to the manufacturer’s protocol, then seeded on coverslips with an equal number of nonstained U2OS-R1 cells, and grown together to 70% confluence. Next, the cells were starved in a serum-free medium for 2 h. Subsequently, the cells were incubated with 500 ng/mL of FGF2 or conjugates labeled with DyLight 488 in serum-free medium supplemented with 1% BSA and 10 U/mL heparin at 4 °C for 40 min. Then, the cells were incubated at 37 °C for 35 min. Next, the cells were washed with PBS, fixed with 4% formaldehyde in PBS, and blocked with 1% BSA, and 0.1 M glycine in PBS, and DNA was stained with DAPI. The coverslips were mounted with the ProLong Gold antifade mountant and viewed under a ZEISS LSM 880 microscope using a Plan-Apochromat 63×/1.4 Oil DIC M27 objective and GaAsP PMT combined with 2 multialkali PMT spectral detectors (Zeiss, Germany). Images were processed with Zeiss ZEN 2.6 software (Zeiss, Germany).
Endocytosis of FGF2 and Conjugates
U2OS-R1 cells were seeded on coverslips and grown to 70% confluence. Subsequently, CellLight Early Endosomes-GFP or CellLight Lysosomes-GFP were added to the cultures in a final concentration of 30 particles per cell. After 16 h, the medium was removed; then cells were washed with PBS and starved in a serum-free medium for 2 h. Next, the cells were incubated with 500 ng/mL of FGF2 or conjugates labeled with DyLight 550 in serum-free medium supplemented with 1% BSA and 10 U/mL heparin at 4 °C for 40 min. Then, the cells were incubated at 37 °C for 35 min in the case of transfection with CellLight Early Endosomes-GFP and for 50 min in the case of transfection with CellLight Lysosomes-GFP. Subsequently, the cells were washed with PBS, fixed with 4% formaldehyde in PBS, blocked with 1% BSA, and 0.1 M glycine in PBS, and stained with DAPI. The coverslips were mounted with the ProLong Gold antifade mountant, and images were collected as described above. Raw data images were analyzed using Zeiss ZEN 2.6 software (Zeiss, Germany).
Flow Cytometry Analysis of Internalization
U2OS and U2OS-R1 cells were seeded onto 12-well plates (0.15 × 106 cells per well) in full medium and left to attach overnight. Then the medium was removed, and the cells were washed with PBS and starved with serum-free medium for 2 h. Next, plates were cooled on ice, and FGF2 or conjugates (500 ng/mL) labeled with DyLight 488 were added to the cells. After 40 min of incubation on ice, the cells were moved to 37 °C for 35 min. Then the medium was removed; the cells were washed with PBS (three times, 5 min) and subsequently detached by 10 mM EDTA in PBS, pH = 8.0. The cells were harvested by centrifugation, resuspended in PBS with 1% BSA, and analyzed by flow cytometry using a NovoCyte 2060R Flow Cytometer and NovoExpress software (ACEA Biosciences, San Diego, CA).
Cell Viability Assay
The cells in the appropriate full medium were seeded on 96-well plates and left overnight to attach. Then, the medium was removed, replaced with a fresh one, supplemented with 10 U/mL heparin, and cells were treated with six different concentrations of FGF2, conjugates, or free AY for 96 h. Next, the medium was removed, and the cells were incubated for 4 h in fresh, full medium with 10% alamarBlue. The fluorescence signal (excitation at 560 nm and emission at 590 nm) was measured by a TECAN Infinite M1000 Pro microplate reader (TECAN Group Ltd., Switzerland). The data were fitted to the Hill equation to calculate EC50 values using OriginPro 8 software (Northampton, MA). Statistical analyses were performed using SigmaPlot software for three independent experiments using the t test. The statistical significance of differences in cytotoxicity between the FGF21xC-PEG27-(tvAY)3 conjugate and other conjugates was assessed.
Results
Design Schemes of Novel FGF2 Conjugates
We applied a hydrophilic derivative of auristatin, auristatin Y (AY), as a cytotoxic agent. This compound contains a dimethylamine group at the N-terminus and l-norleucine at the C-terminus of the auristatin backbone (Figure 1A). The linker, which allows the conjugation of FGF2 to the
Figure 1.

Schematic representation of cytotoxic payloads and FGF2 conjugates: (A) chemical structure of cytotoxic agent–auristatin Y with dipeptide (l-Thr-d-Val) linker and maleimide (maleimide-tvAY); (B) PEGylated derivatives of auristatin Y; (C) conjugates utilized in this study vs previously published construct (in the frame).36
The C-terminus of auristatin Y contains hydrolyzable l-Thr-d-Val dipeptide (tv) and a maleimide moiety (Figure 1A).20,39
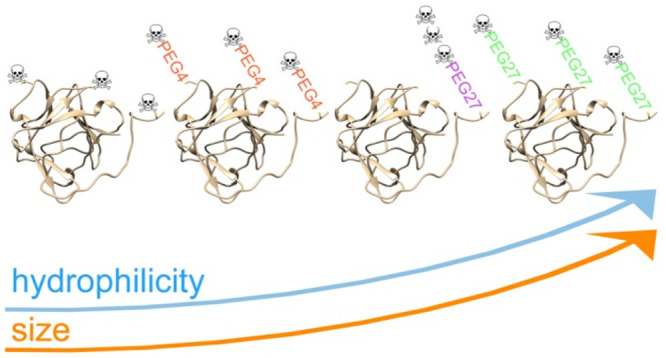
In order to further increase the hydrophilicity of the cytotoxic payload and to elevate the hydrodynamic radius of conjugates, we used a PEG molecule (either 4 or 27 mer) as a spacer between tvAY and FGF2. We applied two different schemes of FGF2-based conjugate synthesis. In both schemes, always three tvAY molecules were attached to a single FGF2 molecule to enable direct comparisons.
We used two previously described FGF2 variants with one or three cysteines exposed to a solvent.36 The first one, FGF23xC, contained three exposed Cys residues, two naturally occurring (C78, C96) and one introduced at the N-terminus (in the KCK extension) to facilitate the thiol-maleimide reaction. Previously we reported that cysteine surrounded by basic residues Lys or Arg, as in the KCK sequence, shows significantly higher reactivity.40 The second FGF2 variant (FGF21xC) contained a single solvent-exposed Cys residue, located in the N-terminal KCK extension, and C78 and C96 were mutated to serines (C78S/C96S) (Figure 1C).
In the first conjugation scheme, tvAYs were attached to three Cys in FGF23xC. tvAYs were attached either directly or via PEG4 or PEG27 linkers. We coupled maleimide-tvAY to the sulfhydryl group of free Cys, and then we used heterobifunctional PEG moieties (NHS-PEG4-mal or NHS-PEG27-mal) to obtain two PEGylated tvAY derivatives, maleimide-PEG4-tvAY and maleimide-PEG27-tvAY, respectively (Figure 1B). Three molecules of maleimide-tvAY, maleimide-PEG4-tvAY, or maleimide-PEG27-tvAY were conjugated via thiol-maleimide chemistry to three cysteine residues of FGF23xC (Figure 1C), providing three conjugates with cytotoxic payloads spread on the protein surface.
In the second scheme, three tvAY molecules were first attached to a short synthetic peptide scaffold (SRCRCRC), again containing highly reactive cysteines, and the peptide conjugate was attached to the N-terminal KCK extension of FGF21xC via PEG27. First, we synthesized a peptide scaffold comprising three Cys residues (H2N-SRCRCRC–CONH2) and attached three tvAY molecules to peptide sulfhydryl groups. Next, we coupled the NHS-PEG27-mal moiety to the α-amine group of the peptide scaffold via an NHS-primary amine reaction. We obtained a PEGylated, triply substituted with tvAY peptide scaffold (maleimide-PEG27-(tvAY)3) ready for conjugation with cysteine residues of proteins. (Figure 1B). Finally, we coupled one molecule of the peptide scaffold (maleimide-PEG27-(tvAY)3) with one cysteine residue of FGF21xC (Figure 1C).
As a consequence, in both schemes, the drug-to-protein ratio (DPR) was three, but the payloads were located differently on the FGF2 molecule. Unless otherwise stated, in all further experiments, we compared obtained conjugates with the wild-type FGF2 denoted as FGF2. Basic properties of FGF21xC and FGF23xC were examined previously36 and were similar to FGF2.
Conjugation of tvAY and Their PEGylated Derivatives to FGF2
We attached maleimide-tvAY, maleimide-PEG4-tvAY, or maleimide-PEG27-tvAY to the cysteine residues of the FGF23xC and maleimide-PEG27-(tvAY)3 to FGF21xC via maleimide–thiol reaction, as described previously.36 The homogeneity and purity of the products were verified by SDS-PAGE (Figure 2A, lanes 2, 3, 4, 5), and the identity of the conjugates was confirmed by MALDI-MS (Figure 2B).
Figure 2.

Conjugation of FGF2 variants to auristatin Y. (A) SDS-PAGE analysis of purified products of the conjugation reaction performed at 25 °C for 1 h. (B) Mass spectra of FGF2 variants and their conjugates.
Biophysical Properties of Proteins and Conjugates
The Native State of the FGF2 Proteins and Their Conjugates
The conformation of FGF23xC and FGF21xC before and after conjugation was evaluated by the fluorescence measurement of Trp123 emission. In the native state, the fluorescence signal of single Trp (at 353 nm) is quenched, and the spectrum is dominated by the emission of several Tyr residues (at 303 nm). Upon unfolding, the Trp residue recovers strong emission at 353 nm. The fluorescence spectra of all tested samples showed the highest emission at 303 nm and a very low signal at 353 nm (Figure 3A), in clear contrast to the spectrum of FGF2 chemically unfolded in
Figure 3.

Biophysical analysis of FGF2 conjugates. (A) Fluorescence emission spectra (300–450 nm) of FGF2 conjugates at a concentration of 4 μM upon excitation at 280 nm. The dashed line represents FGF2 unfolded in 6 M GdmCl. (B) Thermal denaturation of FGF2 conjugates at a concentration of 3 μM, monitored by the change in the fluorescence at 350 nm. (C) Analysis of the aggregation of FGF2 conjugates evaluated by changes in light back-reflection during thermal denaturation. (D) Retention volume of FGF2 conjugates determined by size-exclusion chromatography. Estimated molecular weights are given in Table 1. (E) Stability of FGF2 and FGF2 conjugates in human serum. The FGF2 and the conjugates were incubated at a concentration of 1 μg/mL without heparin in the human serum at 37 °C for indicated times. The samples were analyzed by SDS-PAGE and immunoblotting using the anti-FGF2 antibody. Experiments were performed in triplicate, and representative results are shown.
Six M GdmCl (Figure 3A dashes line). These results confirmed the native conformation of FGF2 variants before and after conjugation.
Stability and Susceptibility to the Aggregation of the Conjugates
The thermal stability of FGF2 and all tvAY conjugates was studied by the nanoDSF technique. In all cases, protein samples showed cooperative unfolding determined by the change of the intrinsic fluorescence emission at 350 nm in the temperature range from 20 to 95 °C (Figure 3B). Denaturation temperature of all conjugates differed only slightly from the Tden of FGF2 wild type, indicating that cytotoxic payloads do not substantially influence the stability of FGF2 (Figure 3B, Table 1). The most stable conjugate was FGF23xC-(PEG4-tvAY)3 – Tden was 3 °C higher than that observed for the wild type (TdenFGF2 = 54.5 °C). FGF21xC with three tvAY molecules located at the peptide scaffold linked by the PEG27 molecule (FGF21xC-PEG27-(tvAY)3) exhibited the lowest Tden value, equal to 50.5 °C.
Table 1. Biophysical Properties of FGF2 and Conjugates.
| preparation | MW [kDa] | HMWa [kDa] | Tden [°C] |
|---|---|---|---|
| FGF2 | 17.2 | 17.4 | 54.5 |
| FGF23xC-(vcMMAE)3 | 21.8 | 16.6 | |
| FGF23xC-(tvAY)3 | 21.5 | 26.5 | 50.9 |
| FGF23xC-(PEG4-tvAY)3 | 23.1 | 30.6 | 57.5 |
| FGF23xC-(PEG27-tvAY)3 | 26.2 | 58.6 | 54.1 |
| FGF21xC-PEG27-(tvAY)3 | 23.8 | 33.3 | 50.5 |
Equivalent protein molecular weights determined by SEC.
To verify how cytotoxic payloads affect FGF2 aggregation, we analyzed the change of light back-reflection during the thermal unfolding of conjugates. None of the tested samples aggregated under the applied experimental conditions (Figure 3C).
Since it is well-known that PEGylation increases the hydrodynamic radius of a protein in a nonlinear manner,41 we performed a size-exclusion chromatography (SEC) analysis to determine the size of FGF2 conjugates (Figure 3D) and to estimate their equivalent molecular weights (Table 1). In contrast to the hydrophobic auristatin derivative (MMAE),20 triple substitutions of FGF2 with hydrophilic tvAY increased the hydrodynamic size of FGF2 protein (Figure 3D, Table 1). The addition of PEG molecules further enlarged the hydrodynamic radius of conjugates up to the equivalent protein molecular weight of 58.6 kDa, in the case of FGF23xC-(PEG27-tvAY)3. Additionally, we analyzed the stability of FGF2 and FGF2 conjugates over time in human serum in the absence of heparin (Figure 2E). We observed that all conjugates were intact after 120-h incubation in human serum.
Affinity of FGF2 Conjugates to FGFR1
PEGylation is a useful method to reduce nonspecific interactions of biomolecules.41−43 However, in some cases, PEGylated proteins exhibit lower affinity to their molecular targets.
To examine the impact of introduced payloads on the interaction of FGF2-based conjugates with the extracellular domain of FGFR1, we performed biolayer interferometry (BLI) measurements.
The coupling of three tvAY molecules to FGF23xC (FGF23xC-(tvAY)3) slightly decreased the affinity to FGFR1, as compared to FGF2 (Figure 4). However, PEGylated conjugates (FGF23xC-(PEG4-tvAY)3, FGF21xC-PEG27-(tvAY)3, and FGF23xC-(PEG27-tvAY)3) showed a very similar FGFR1 binding profile to FGF2 (Figure 4), indicating that PEGylated tvAY does not affect the interaction of FGF2 with FGFR1. Estimated dissociation constants are in the nanomolar range. We are aware that, due to complex interaction (i.e., poorly defined oligomeric state of FGFR1), the data should be interpreted only qualitatively.
Figure 4.

Binding of FGF2 and FGF2 conjugates to a recombinant extracellular fragment of FGFR1-α-IIIc fused to Fc determined by biolayer interferometry. The biotinylated FGFR1c was immobilized on high-precision streptavidin biosensors (SAX), and then association and dissociation of FGF2 or FGF2 conjugates were monitored for 400 s. The red lines represent fitting curves according to a 1:1 model.
Biological Activities of Proteins and Conjugates
Selective Internalization of Conjugates
To be able to kill the cancer cells, cytotoxic conjugates should be effectively internalized from the cell surface.44 Therefore, we checked whether FGF2 conjugates can be taken up by the FGFR1-α-IIIc-expressing cells. DyLight488-labeled FGF2 or conjugates (green) were incubated with coculture of U2OS cells (FGFR1-negative) stained with CellTracker RED CMTPX (red) and nonstained U2OS cells stably transfected with FGFR1-α-IIIc (U2OS-R1)38 (FGFR1-positive). After 35 min at 37 °C, the cells were fixed and analyzed with confocal microscopy. As shown in Figure 5A, all conjugates were internalized only by U2OS-R1 cells, indicating that conjugates’ endocytosis occurs via an FGFR1-dependent pathway. Additionally, we performed a quantified analysis of the uptake of FGF2 and conjugates by U2OS-R1 and U2OS cells using flow cytometry (Figure 5B,C). The internalization of all tested samples by U2OS-R1 cells occurred at a very high level, in contrast to U2OS cells, where the uptake was at least 10-fold lower. Interestingly, in U2OS-R1 cells, we observed a slightly higher uptake of FGF23xC-(PEG27-tvAY)3 as compared to FGF23xC-(PEG4-tvAY)3. Differences between other preparations were statistically insignificant (Figure 5C).
Figure 5.

Internalization of fluorescence-labeled FGF2 and FGF2 conjugates into U2OS cells expressing FGFR1. (A) Fluorescence microscopy analysis of the uptake of labeled FGF2 and FGF2 conjugates. Equal numbers of U2OS cells prestained with CellTracker Red CMTPX (red) and U2OS-R1 (nonstained) were seeded and then incubated with 500 ng/mL of FGF2 or the conjugates labeled with DyLight488 (green) for 40 min on ice, and then moved to 37 °C for 35 min to allow for internalization. Next, the cells were fixed, stained with DAPI (blue), and analyzed by confocal microscopy. The scale bars correspond to 10 μm. (B) Flow cytometry analysis of internalization efficiency into U2OS-R1 (FGFR1-positive) and U2OS (FGFR1-negative) cell lines. The cells were incubated on ice with 500 ng/mL of FGF2 and FGF2 conjugates labeled with DyLight488 for 40 min. Then cells were moved to 37 °C for 35 min and subsequently analyzed by flow cytometry. (C) Quantitative analysis of internalization. The data shown are mean fluorescence intensities (MFI) from three independent experiments ± SD. Statistical significance: * p < 0.05, ** p < 0.01, *** p < 0.001.
Intracellular Trafficking of FGF2 and Conjugates
After binding of FGF2 to FGFR1, dimerized receptors together with the ligands undergo internalization from the cell membrane, and then the complexes are sorted into endosomes and directed to lysosomes.45 Endosomal and lysosomal proteases hydrolyze peptide bonds of internalized cargo, facilitating, in the case of protein–drug conjugates, the release of the active form of the cytotoxic agent.46,47
To verify whether FGF2 conjugates reach the acidic compartment within the cell, we studied their intracellular localization in U2OS-R1 cells using confocal microscopy. We labeled early endosomes using a green fluorescent protein (GFP) fused to Rab5 (Rab5-GFP) and lysosomes with GFP fusions of lysosomal membrane-associated protein 1 (LAMP1-GFP). Next, the cells were incubated at 37 °C for 35 or 50 min with FGF2 or conjugates labeled with DyLight550. In the case of FGF2 wild type as well as FGF2 conjugates, the red fluorescent signal colocalized with green fluorescence of Rab5-GFP or LAMP1-GFP after 35 or 50 min of incubation, respectively (Figure 6). These experiments show that FGF2 and its conjugates are efficiently internalized into U2OS-R1 cells and traffic via endosomes to lysosomes.
Figure 6.

Confocal microscopy imaging of endocytosed FGF2 and FGF2 conjugates. U2OS-R1 cells were transfected with Rab5-GFP (early endosome marker) or LAMP1-GFP (lysosome marker). Subsequently, the cells were incubated with 500 ng/mL of DyLight550-labeled FGF2 or conjugates on ice for 40 min and then shifted to 37 °C for 35 min in the case of Rab5-GFP-transfected cells or 50 min for LAMP1-GFP-transfected cells. Scale bars correspond to 10 μm. Insets: magnified views of boxed regions in the main images.
Cytotoxic Effect of the Conjugates
To evaluate in vitro selectivity and cytotoxicity of the conjugates, we used a panel of five human cancer cell lines differing in expression level8,48 and splicing isoform of FGFR1. We used two FGFR1-negative cell lines (U2OS and HCC15) and three FGFR1-positive cell lines (U2OS-R1 expressing FGFR1-α-IIIc, DMS114 expressing FGFR1-β-IIIc, and NCI-H520 expressing FGFR1-α-IIIb).49 The cells were treated with FGF2, the conjugates and free AY in the concentration range from 0.003 nM to 273 nM for 96 h. Then, cell viability was assessed with the alamarBlue assay. The EC50 values were calculated from the Hill equation.
All of the FGF2 conjugates showed a strong cytotoxic effect against FGFR1-positive cell lines (U2OS-R1, DMS114, NCI-H520) and were nontoxic to control cell lines (U2OS, HCC15), exhibiting an almost nondetectable level of FGFR18,48 (Figure 7, Table 2). In the case of the U2OS-R1 cell line, the conjugates exhibited EC50 in the subnanomolar range. FGF21xC-PEG27-(tvAY)3 demonstrated an 8.6 and 5.3 times higher EC50 value, as compared with FGF23xC-(tvAY)3 and FGF23xC-(PEG4-tvAY)3, respectively. FGF2 conjugates showed the largest cytotoxic potency in DMS114 cells, presenting the highest natural level of FGFR1 among tested cell lines, with EC50 in the low subnanomolar range. The NCI-H520 cell line, expressing relatively low amounts of FGFR1, was less sensitive to conjugates with EC50 values more than 100 times higher than that observed in U2OS-R1 cells. Notably, in this case, the FGF21xC-PEG27-(tvAY)3 conjugate exhibited 3.1 and 5.7 times higher EC50 values than FGF23xC-(PEG4-tvAY)3 and FGF23xC-(PEG27-tvAY)3 conjugates, respectively.
Figure 7.

Viability of cells treated with the FGF2, FGF2 conjugates, and free AY. FGFR1-positive (U2OS-R1, DMS114, NCI-H520) and FGFR1-negative (U2OS and HCC15) cell lines were treated with indicated agents at six concentrations for 96 h. Then the viability of the cells was assessed using the alamarBlue reagent. Data shown are mean values from three experiments ± SD. The solid lines represent the Hill equation fits.
Table 2. Cytotoxicity of FGF2 Conjugates and Free AY in Different Cell Lines.
| cell
lines |
|||||
|---|---|---|---|---|---|
| U2OS-R1 | DMS114 | NCI-H520 | U2OS | HCC15 | |
| Preparation | EC50 [nM] | ||||
| FGF23xC-(tvAY)3 | 0.11b | 0.035 | 60.1 | nta | nt |
| FGF23xC-(PEG4-tvAY)3 | 0.18b | 0.033 | 50.5b | nt | nt |
| FGF23xC-(PEG27-tvAY)3 | 0.23 | 0.039 | 27.7b | nt | nt |
| FGF21xC-PEG27-(tvAY)3 | 0.95 | 0.019 | 158.0 | nt | nt |
| AY | nt | nt | nt | nt | nt |
nt, nontoxic in the studied concentration range. For statistical analysis between FGF21xC-PEG27-(tvAY)3 and other conjugates, a t test was applied using SigmaPlot software.
p < 0.05 was considered statistically significant; n = 3.
Interestingly, free auristatin Y (AY) was nontoxic to all tested cell lines. We found that the cytotoxicity of FGF2 conjugates is determined by the FGFR1 expression level on the cell surface,8,48 and confirmed that auristatin Y needs to be specifically delivered into the cell interior in order to evoke its toxic effect.
Discussion
Despite continuous progress in the field of targeted cancer therapy, only five ADC-based drugs have been approved by the FDA.21,50 Immunogenicity of mAb, drug resistance (especially P-glycoprotein-mediated efflux of hydrophobic drugs), nonoptimal bioavailability, and off-target toxicity are among major limitations of ADCs.51−57 Thus, the development of novel targeted approaches suitable for cancer treatment remains a vital need.
There are numerous protein scaffolds that could potentially replace monoclonal antibodies as the targeting molecules, including antibody fragments,6,7 DARPins,10,11 knottins,12,13 and affibodies.14,15 Previously, we demonstrated that affibody,58,59 diaffibody,60 scFv-Fc,8,9 FGF1,40,61,62 and FGF236,48,63,64 are effective proteins in terms of specific delivery of the cytotoxic drug vcMMAE to tumor cells.
One of the problems in applying vcMMAE, especially in conjugates with high drug loading, is its hydrophobicity, a factor greatly influencing tissue penetration and specificity of cellular internalization.65−68 We previously found that an FGF2 conjugate containing three vcMMAE molecules (FGF23xC-(vcMMAE)3) at a concentration higher than 125 nM exhibits nonspecific cytotoxicity toward the FGFR1-negative U2OS cell line.36 However, there are numerous chemical derivatives of auristatin, i.e., MMAE, MMAF, MMAU, and AY, which differ in hydrophobicity, charge, and cytotoxicity.20,22,39,69 Taking into account the plan to load FGF2 with three auristatin molecules, we decided to use more hydrophilic AY. Unlike MMAE,39 AY did not show toxicity as a free drug (Figure 7, Table 2). In general, increased hydrophilicity of cytotoxic payloads affects their efflux from the cell interior, which reduces multiple drug resistance (MDR).70 Moreover, the cytotoxic agents coupled with polar amino acids (such as tvAY, Figure 1A) cannot be a substrate of the ABCB1 transporter, which further overcomes MDR.71−74 Concomitantly, cytotoxic payloads with lower hydrophobicity exhibit lower bystander killing,75,76 which can be considered as a possible drawback of AY manifesting in reduced potency against FGFR-negative cells in the tumor environment.
Replacement of hydrophobic vcMMAE with hydrophilic vtAY increased polarity of FGF2 conjugates, which manifested in shorter retention time observed in SEC experiments (Figure 3D, Table 1. Importantly, the FGF23xC-(tvAY)3 conjugate was nontoxic to FGFR1-negative cell lines (Figure 7, Table 2). Finally, in agreement with ADC data,20,22,65 the therapeutic index was increased due to the hydrophilic nature of the (tvAY)3 payload.
To further increase the hydrophilicity of cytotoxic cargo and to increase the hydrodynamic radius of conjugates, we attached PEG linkers in a series of FGF2 conjugates. Both properties play a key role in pharmacokinetics and in vivo potency of conjugates.20,25,65,77,78 The short PEG4 linker introduced between FGF2 and vtAY (FGF23xC-(PEG4-tvAY)3) increased hydrodynamic molecular weight, in agreement with the PEG-hydration model.41 Further, a large increase in the hydrodynamic radius corresponding to a globular protein of 58 kDa was observed when PEG27 was applied (FGF23xC-(PEG27-tvAY)3). Using PEG4 and PEG27 Burke et al. and Simmons et al. observed comparable hydrodynamic radius of classical ADCs;33,79 thus, it is likely that the size of FGF23xC-(PEG27-tvAY)3 exceeds the limit of renal filtration.80,81 However, a further pharmacokinetic study should be performed to confirm this.
FGF23xC-(PEG27-tvAY)3 showed cytotoxic effects comparable to non-PEGylated FGF23xC-(tvAY)3 or a conjugate containing a short PEG4 linker (FGF23xC-(PEG4-tvAY)3). We decided to study whether conjugation via a single cysteine present in the KCK extension at the N-terminus with tvAY3 attached to the PEG27-SRCRCRC scaffold peptide would show comparable cytotoxicity as observed in FGF23xC-(PEG27-tvAY)3. This might be beneficial for loading proteins that contain a single exposed cysteine in their sequence with three (or even more) payloads.
The biophysical properties [FL spectra, denaturation temperature, tendency to the aggregation of FGF21xC-(PEG27-tvAY3) and FGF23xC-(PEG27-tvAY)3] were comparable. Nevertheless, the cytotoxicity of (FGF23xC-(PEG27-tvAY)3) toward U2OS-R1 and NCI-H520 cell lines (Table 2) was about four and six times higher, respectively.
All studied FGF2-based conjugates showed several desirable characteristics, such as homogeneity and defined DPR (Figure 2A), stability in human serum (Figure 3E), resistance to aggregation (Figure 3C), selective affinity to FGFR1-positive cells (Figure 5), specific internalization via FGFR-mediated endocytosis, and trafficking from endosomes to lysosomes (Figure 6). All conjugates were highly efficient in killing FGFR-expressing cancer cells and nontoxic for FGFR-negative cell lines (Figure 7, Table 2). Since free AY cannot easily diffuse across cell membranes, it has to be actively delivered into the cell (Figure 7, Table 2).
So far, the FDA has approved 15 PEGylated protein drugs. Most of them contain heterogenic PEG moieties bigger than 5 kDa, and only four are generated by site-specific PEGylation. The latter show a significant advantage since they are easier to purify and are homogenic.42,82,83 In the ADC field, PEG molecules are used mainly for increasing the hydrophilicity of the payload and the bioavailability of the conjugate. Unfortunately, PEG molecules bring several disadvantages, including the possible loss of biological activity and the decrease in affinity to the molecular target.41,82
FGFRs are abundant in normal adult tissues (e.g., skin, cornea, lung, heart, placenta, kidney, ureter, and retina).84,85 Nonetheless, the level of FGFRs in many cancer cells is significantly elevated.86−88 Therefore, the development of drugs targeting FGFRs is a well-justified anticancer strategy. However, so far, only one ADC targeting the FGF receptor (Aprutumab Ixadotin, BAY 1187982) has been evaluated in a phase 1 clinical study.89 BAY 1187982, targeted at FGFR2-IIIb and FGFR2-IIIc, contained a derivative of auristatin W as a cytotoxic drug, not specifically conjugated to lysine residues via a noncleavable linker.90 The trial was terminated because BAY 1187982 was poorly tolerated. The study suggests that observed side effects were associated with the toxicity of the payload.89 Thus, novel approaches to modify the cytotoxic drugs in therapeutic conjugates are of high importance.
Another important issue is the systemic elimination half-life of conjugates. A significant impact on prolongation of ADCs’ half-life is exerted by the Fc region that binds to the neonatal Fc receptor91 expressed in various tissues, including the endothelial cells, interstitial macrophages, Kupffer cells, alveolar macrophages, enterocytes, and choroid plexus epithelium.92 FGF2-conjugates lack the Fc fragment, but FGF2 protein has a high affinity to heparans occurring on the surface of many types of cells and in body fluids. Interaction with sugar moiety highly stabilizes FGF2 and protects the FGF2-conjugates from degradation. We have shown that FGF2-tvAY conjugates are stable in human plasma for at least 5 days (Figure 3E); thus, coadministration of heparin along with FGF2 conjugates should result in an increased half-life in comparison to other targeting scaffolds that do not have systemic stabilizing/recyclizing partners. Nevertheless, this issue needs to be clarified in a further detailed pharmacokinetic study.
Here, we used small-molecular-weight, homogeneous PEG molecules with a defined chain length to couple auristatin Y to the FGF2 molecule in a site-specific manner. The analysis of the tertiary structure of FGF2 conjugates (Figure 3A), the receptor affinity measurements (Figure 4), cell internalization experiments (Figure 5) and the analysis of the intracellular trafficking (Figure 6) clearly showed that payloads introduced via PEG4 or PEG27 linkers do not alter the biophysical and biological properties of FGF2.
All conjugates were characterized by the defined drug-to-protein ratio (DAR = 3), due to a site-specific attachment of the defined PEGylated derivative of vtAY. They exhibited a highly toxic effect toward FGFR1-positive cell lines, especially those expressing isoform IIIc of FGFR1 specific for FGF2.93,94 The highest cytotoxicity was observed in the case of DMS114 cells, which expressed isoform β of FGFR1, which is correlated with reduced survival of a patient.93,95 The most potent conjugate, FGF23xC-(PEG27-tvAY)3, with highly desirable hydrodynamic properties, possesses very high potential for a further in vivo study in order to validate its feasibility for anticancer targeted therapy.
Acknowledgments
This work was supported by the statutory budget of the Department of Protein Engineering, Faculty of Biotechnology, University of Wroclaw, and by the grant Sonata Bis 2015/18/E/NZ3/00501 from National Science Centre. We would like to acknowledge Marta Minkiewicz and Jakub Szymczyk for excellent assistance in cell culture maintenance. We thank NanoTemper Technologies for providing access to the Prometheus NT.48 device and application specialist Dr. Jakub Nowak and Pawel Kania (NanoTemper Technologies) for their support during protein stability experiments. We thank Dr. Ellen M. Haugsten (from the Institute for Cancer Research) for providing the stably transfected U2OS-R1 cell line.
Glossary
Abbreviations
- AY
auristatin Y
- tvAY
l-Thr-d-Val-AY
- ADC
antibody–drug conjugate
- BLI
biolayer interferometry
- DPR
drug-to-protein ratio
- FGF
fibroblast growth factor
- FGFR
fibroblast growth factor receptor
- MMAE
monomethyl auristatin E
- nanoDSF
nanodifferential scanning fluorimetry
The authors declare no competing financial interest.
References
- van der Veldt A. A. M.; Lubberink M.; Mathijssen R. H. J.; Loos W. J.; Herder G. J. M.; Greuter H. N.; Comans E. F. I.; Rutten H. B.; Eriksson J.; Windhorst A. D.; et al. Toward Prediction of Efficacy of Chemotherapy: A Proof of Concept Study in Lung Cancer Patients Using [11C]Docetaxel and Positron Emission Tomography. Clin. Cancer Res. 2013, 19 (15), 4163–4173. 10.1158/1078-0432.CCR-12-3779. [DOI] [PubMed] [Google Scholar]
- Smith D. A.; Beaumont K.; Maurer T. S.; Di L. Clearance in Drug Design. J. Med. Chem. 2019, 62 (5), 2245–2255. 10.1021/acs.jmedchem.8b01263. [DOI] [PubMed] [Google Scholar]
- Dennis M. S.; Jin H.; Dugger D.; Yang R.; McFarland L.; Ogasawara A.; Williams S.; Cole M. J.; Ross S.; Schwall R. Imaging Tumors with an Albumin-Binding Fab, a Novel Tumor-Targeting Agent. Cancer Res. 2007, 67 (1), 254–261. 10.1158/0008-5472.CAN-06-2531. [DOI] [PubMed] [Google Scholar]
- Deonarain M.; Yahioglu G.; Stamati I.; Pomowski A.; Clarke J.; Edwards B.; Diez-Posada S.; Stewart A. Small-Format Drug Conjugates: A Viable Alternative to ADCs for Solid Tumours?. Antibodies 2018, 7 (2), 16. 10.3390/antib7020016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazzamalli S.; Dal Corso A.; Widmayer F.; Neri D. Chemically Defined Antibody- and Small Molecule-Drug Conjugates for in Vivo Tumor Targeting Applications: A Comparative Analysis. J. Am. Chem. Soc. 2018, 140 (5), 1617–1621. 10.1021/jacs.7b13361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu C.; Terraube V.; Tam A. S. P.; Stochaj W.; Fennell B. J.; Lin L.; Stahl M.; LaVallie E. R.; Somers W.; Finlay W. J. J.; et al. A Combination of Structural and Empirical Analyses Delineates the Key Contacts Mediating Stability and Affinity Increases in an Optimized Biotherapeutic Single-Chain Fv (ScFv). J. Biol. Chem. 2016, 291 (3), 1267–1276. 10.1074/jbc.M115.688010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervé-Aubert K.; Allard-Vannier E.; Joubert N.; Lakhrif Z.; Alric C.; Martin C.; Viaud-Massuard M.-C.; Dimier-Poisson I.; Aubrey N.; Chourpa I. Impact of Site-Specific Conjugation of ScFv to Multifunctional Nanomedicines Using Second Generation Maleimide. Bioconjugate Chem. 2018, 29 (5), 1553–1559. 10.1021/acs.bioconjchem.8b00091. [DOI] [PubMed] [Google Scholar]
- Sokolowska-Wedzina A.; Chodaczek G.; Chudzian J.; Borek A.; Zakrzewska M.; Otlewski J. High-Affinity Internalizing Human ScFv-Fc Antibody for Targeting FGFR1-Overexpressing Lung Cancer. Mol. Cancer Res. 2017, 15 (8), 1040–1050. 10.1158/1541-7786.MCR-16-0136. [DOI] [PubMed] [Google Scholar]
- Borek A.; Sokolowska-Wedzina A.; Chodaczek G.; Otlewski J. Generation of High-Affinity, Internalizing Anti-FGFR2 Single-Chain Variable Antibody Fragment Fused with Fc for Targeting Gastrointestinal Cancers. PLoS One 2018, 13 (2), e0192194 10.1371/journal.pone.0192194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger M. A.; Zbinden R.; Flütsch A.; Gutte P. G. M.; Engeler S.; Roschitzki-Voser H.; Grütter M. G. Design, Construction, and Characterization of a Second-Generation DARPin Library with Reduced Hydrophobicity. Protein Sci. 2013, 22 (9), 1239–1257. 10.1002/pro.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyev S.; Proshkina G.; Ryabova A.; Tavanti F.; Menziani M. C.; Eidelshtein G.; Avishai G.; Kotlyar A. Synthesis, Characterization, and Selective Delivery of DARPin-Gold Nanoparticle Conjugates to Cancer Cells. Bioconjugate Chem. 2017, 28 (10), 2569–2574. 10.1021/acs.bioconjchem.7b00410. [DOI] [PubMed] [Google Scholar]
- Currier N. V.; Ackerman S. E.; Kintzing J. R.; Chen R.; Filsinger Interrante M.; Steiner A.; Sato A. K.; Cochran J. R. Targeted Drug Delivery with an Integrin-Binding Knottin-Fc-MMAF Conjugate Produced by Cell-Free Protein Synthesis. Mol. Cancer Ther. 2016, 15 (6), 1291–1300. 10.1158/1535-7163.MCT-15-0881. [DOI] [PubMed] [Google Scholar]
- Cox N.; Kintzing J. R.; Smith M.; Grant G. A.; Cochran J. R. Integrin-Targeting Knottin Peptide-Drug Conjugates Are Potent Inhibitors of Tumor Cell Proliferation. Angew. Chem., Int. Ed. 2016, 55 (34), 9894–9897. 10.1002/anie.201603488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlova A.; Jonsson A.; Rosik D.; Lundqvist H.; Lindborg M.; Abrahmsen L.; Ekblad C.; Frejd F. Y.; Tolmachev V. Site-Specific Radiometal Labeling and Improved Biodistribution Using ABY-027, A Novel HER2-Targeting Affibody Molecule-Albumin-Binding Domain Fusion Protein. J. Nucl. Med. 2013, 54 (6), 961–968. 10.2967/jnumed.112.110700. [DOI] [PubMed] [Google Scholar]
- Altai M.; Liu H.; Ding H.; Mitran B.; Edqvist P.-H.; Tolmachev V.; Orlova A.; Gräslund T. Affibody-Derived Drug Conjugates: Potent Cytotoxic Molecules for Treatment of HER2 over-Expressing Tumors. J. Controlled Release 2018, 288, 84–95. 10.1016/j.jconrel.2018.08.040. [DOI] [PubMed] [Google Scholar]
- Di Bonaventura I.; Jin X.; Visini R.; Probst D.; Javor S.; Gan B.-H.; Michaud G.; Natalello A.; Doglia S. M.; Köhler T.; et al. Chemical Space Guided Discovery of Antimicrobial Bridged Bicyclic Peptides against Pseudomonas Aeruginosa and Its Biofilms. Chem. Sci. 2017, 8 (10), 6784–6798. 10.1039/C7SC01314K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes C. A.; Pei D. Bicyclic Peptides as Next-Generation Therapeutics. Chem. - Eur. J. 2017, 23 (52), 12690–12703. 10.1002/chem.201702117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feni L.; Parente S.; Robert C.; Gazzola S.; Arosio D.; Piarulli U.; Neundorf I. Kiss and Run: Promoting Effective and Targeted Cellular Uptake of a Drug Delivery Vehicle Composed of an Integrin-Targeting Diketopiperazine Peptidomimetic and a Cell-Penetrating Peptide. Bioconjugate Chem. 2019, 30 (7), 2011–2022. 10.1021/acs.bioconjchem.9b00292. [DOI] [PubMed] [Google Scholar]
- FDA . Clinical Trials. https://clinicaltrials.gov/.
- Lyon R. P.; Bovee T. D.; Doronina S. O.; Burke P. J.; Hunter J. H.; Neff-Laford H. D.; Jonas M.; Anderson M. E.; Setter J. R.; Senter P. D. Reducing Hydrophobicity of Homogeneous Antibody-Drug Conjugates Improves Pharmacokinetics and Therapeutic Index. Nat. Biotechnol. 2015, 33 (7), 733–735. 10.1038/nbt.3212. [DOI] [PubMed] [Google Scholar]
- Hoffmann R. M.; Coumbe B. G. T.; Josephs D. H.; Mele S.; Ilieva K. M.; Cheung A.; Tutt A. N.; Spicer J. F.; Thurston D. E.; Crescioli S.; et al. Antibody Structure and Engineering Considerations for the Design and Function of Antibody Drug Conjugates (ADCs). Oncoimmunology 2018, 7 (3), e1395127 10.1080/2162402X.2017.1395127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekholm F.; Ruokonen S.-K.; Redón M.; Pitkänen V.; Vilkman A.; Saarinen J.; Helin J.; Satomaa T.; Wiedmer S. Hydrophilic Monomethyl Auristatin E Derivatives as Novel Candidates for the Design of Antibody-Drug Conjugates. Separations 2019, 6 (1), 1. 10.3390/separations6010001. [DOI] [Google Scholar]
- Strop P.; Liu S. H.; Dorywalska M.; Delaria K.; Dushin R. G.; Tran T. T.; Ho W. H.; Farias S.; Casas M. G.; Abdiche Y.; et al. Location Matters: Site of Conjugation Modulates Stability and Pharmacokinetics of Antibody Drug Conjugates. Chem. Biol. 2013, 20 (2), 161–167. 10.1016/j.chembiol.2013.01.010. [DOI] [PubMed] [Google Scholar]
- Hamblett K. J.; Senter P. D.; Chace D. F.; Sun M. M. C.; Lenox J.; Cerveny C. G.; Kissler K. M.; Bernhardt S. X.; Kopcha A. K.; Zabinski R. F.; et al. Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody Drug Conjugate. Clin. Cancer Res. 2004, 10 (20), 7063–7070. 10.1158/1078-0432.CCR-04-0789. [DOI] [PubMed] [Google Scholar]
- Strop P.; Delaria K.; Foletti D.; Witt J. M.; Hasa-Moreno A.; Poulsen K.; Casas M. G.; Dorywalska M.; Farias S.; Pios A.; et al. Site-Specific Conjugation Improves Therapeutic Index of Antibody Drug Conjugates with High Drug Loading. Nat. Biotechnol. 2015, 33 (7), 694–696. 10.1038/nbt.3274. [DOI] [PubMed] [Google Scholar]
- Masters J. C.; Nickens D. J.; Xuan D.; Shazer R. L.; Amantea M. Clinical Toxicity of Antibody Drug Conjugates: A Meta-Analysis of Payloads. Invest. New Drugs 2018, 36 (1), 121–135. 10.1007/s10637-017-0520-6. [DOI] [PubMed] [Google Scholar]
- Tsuchikama K.; An Z. Antibody-Drug Conjugates: Recent Advances in Conjugation and Linker Chemistries. Protein Cell 2018, 9 (1), 33–46. 10.1007/s13238-016-0323-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P.; Nayak B.; Dey R. K. PEGylation in Anti-Cancer Therapy: An Overview. Asian J. Pharm. Sci. 2016, 11 (3), 337–348. 10.1016/j.ajps.2015.08.011. [DOI] [Google Scholar]
- Welch R. P.; Lee H.; Luzuriaga M. A.; Brohlin O. R.; Gassensmith J. J. Protein-Polymer Delivery: Chemistry from the Cold Chain to the Clinic. Bioconjugate Chem. 2018, 29 (9), 2867–2883. 10.1021/acs.bioconjchem.8b00483. [DOI] [PubMed] [Google Scholar]
- Liu X.; Sun J.; Gao W. Site-Selective Protein Modification with Polymers for Advanced Biomedical Applications. Biomaterials 2018, 178, 413–434. 10.1016/j.biomaterials.2018.04.050. [DOI] [PubMed] [Google Scholar]
- Russell A. J.; Baker S. L.; Colina C. M.; Figg C. A.; Kaar J. L.; Matyjaszewski K.; Simakova A.; Sumerlin B. S. Next Generation Protein-Polymer Conjugates. AIChE J. 2018, 64 (9), 3230–3245. 10.1002/aic.16338. [DOI] [Google Scholar]
- Ekladious I.; Colson Y. L.; Grinstaff M. W. Polymer-Drug Conjugate Therapeutics: Advances, Insights and Prospects. Nat. Rev. Drug Discovery 2019, 18 (4), 273–294. 10.1038/s41573-018-0005-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke P. J.; Hamilton J. Z.; Jeffrey S. C.; Hunter J. H.; Doronina S. O.; Okeley N. M.; Miyamoto J. B.; Anderson M. E.; Stone I. J.; Ulrich M. L.; et al. Optimization of a PEGylated Glucuronide-Monomethylauristatin E Linker for Antibody-Drug Conjugates. Mol. Cancer Ther. 2017, 16 (1), 116–123. 10.1158/1535-7163.MCT-16-0343. [DOI] [PubMed] [Google Scholar]
- Zhao R. Y.; Wilhelm S. D.; Audette C.; Jones G.; Leece B. A.; Lazar A. C.; Goldmacher V. S.; Singh R.; Kovtun Y.; Widdison W. C.; et al. Synthesis and Evaluation of Hydrophilic Linkers for Antibody-Maytansinoid Conjugates. J. Med. Chem. 2011, 54 (10), 3606–3623. 10.1021/jm2002958. [DOI] [PubMed] [Google Scholar]
- Leal A. D.; Krishnamurthy A.; Head L.; Messersmith W. A. Antibody Drug Conjugates under Investigation in Phase I and Phase II Clinical Trials for Gastrointestinal Cancer. Expert Opin. Invest. Drugs 2018, 27 (11), 901–916. 10.1080/13543784.2018.1541085. [DOI] [PubMed] [Google Scholar]
- Krzyscik M. A.; Zakrzewska M.; Sørensen V.; Sokolowska-Wedzina A.; Lobocki M.; Swiderska K. W.; Krowarsch D.; Wiedlocha A.; Otlewski J. Cytotoxic Conjugates of Fibroblast Growth Factor 2 (FGF2) with Monomethyl Auristatin e for Effective Killing of Cells Expressing FGF Receptors. ACS Omega 2017, 2 (7), 3792–3805. 10.1021/acsomega.7b00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiero A.; Villa C. H.; Bander E.; Rey D. A.; Bergkvist M.; Batt C. A.; Manova-Todorova K.; Deen W. M.; Scheinberg D. A.; McDevitt M. R. Paradoxical Glomerular Filtration of Carbon Nanotubes. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (27), 12369–12374. 10.1073/pnas.0913667107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugsten E. M.; Malecki J.; Bjorklund S. M. S.; Olsnes S.; Wesche J. Ubiquitination of Fibroblast Growth Factor Receptor 1 Is Required for Its Intracellular Sorting but Not for Its Endocytosis. Mol. Biol. Cell 2008, 19 (8), 3390–3403. 10.1091/mbc.e07-12-1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doronina S. O.; Toki B. E.; Torgov M. Y.; Mendelsohn B. A.; Cerveny C. G.; Chace D. F.; DeBlanc R. L.; Gearing R. P.; Bovee T. D.; Siegall C. B.; et al. Development of Potent Monoclonal Antibody Auristatin Conjugates for Cancer Therapy. Nat. Biotechnol. 2003, 21 (7), 778–784. 10.1038/nbt832. [DOI] [PubMed] [Google Scholar]
- Lobocki M.; Zakrzewska M.; Szlachcic A.; Krzyscik M. A.; Sokolowska-Wedzina A.; Otlewski J. High-Yield Site-Specific Conjugation of Fibroblast Growth Factor 1 with Monomethylauristatin e via Cysteine Flanked by Basic Residues. Bioconjugate Chem. 2017, 28 (7), 1850–1858. 10.1021/acs.bioconjchem.7b00158. [DOI] [PubMed] [Google Scholar]
- Veronese F. M.; Mero A.; Pasut G.. Protein PEGylation, Basic Science and Biological Applications. In PEGylated Protein Drugs: Basic Science and Clinical Applications; Birkhäuser Basel: Basel, 2009; pp 11–31. [Google Scholar]
- Ginn C.; Khalili H.; Lever R.; Brocchini S. PEGylation and Its Impact on the Design of New Protein-Based Medicines. Future Med. Chem. 2014, 6 (16), 1829–1846. 10.4155/fmc.14.125. [DOI] [PubMed] [Google Scholar]
- Veronese F. M.; Mero A. The Impact of PEGylation on Biological Therapies. BioDrugs 2008, 22 (5), 315–329. 10.2165/00063030-200822050-00004. [DOI] [PubMed] [Google Scholar]
- Staudacher A. H.; Brown M. P. Antibody Drug Conjugates and Bystander Killing: Is Antigen-Dependent Internalisation Required?. Br. J. Cancer 2017, 117 (12), 1736–1742. 10.1038/bjc.2017.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porebska N.; Latko M.; Kucińska M.; Zakrzewska M.; Otlewski J.; Opaliński Ł. Targeting Cellular Trafficking of Fibroblast Growth Factor Receptors as a Strategy for Selective Cancer Treatment. J. Clin. Med. 2019, 8 (1), 7. 10.3390/jcm8010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brix K.Lysosomal Proteases. In Lysosomes; Springer US: Boston, MA, 2005; pp 50–59. [Google Scholar]
- Müller S.; Dennemärker J.; Reinheckel T. Specific Functions of Lysosomal Proteases in Endocytic and Autophagic Pathways. Biochim. Biophys. Acta, Proteins Proteomics 2012, 1824 (1), 34–43. 10.1016/j.bbapap.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzyscik M. A.; Opaliński Ł.; Otlewski J. Novel Method for Preparation of Site-Specific, Stoichiometric-Controlled Dual Warhead Conjugate of FGF2 via Dimerization Employing Sortase A-Mediated Ligation. Mol. Pharmaceutics 2019, 16 (8), 3588–3599. 10.1021/acs.molpharmaceut.9b00434. [DOI] [PubMed] [Google Scholar]
- Malchers F.; Dietlein F.; Schottle J.; Lu X.; Nogova L.; Albus K.; Fernandez-Cuesta L.; Heuckmann J. M.; Gautschi O.; Diebold J.; et al. Cell-Autonomous and Non-Cell-Autonomous Mechanisms of Transformation by Amplified FGFR1 in Lung Cancer. Cancer Discovery 2014, 4 (2), 246–257. 10.1158/2159-8290.CD-13-0323. [DOI] [PubMed] [Google Scholar]
- FDA . FDA approves first chemoimmunotherapy regimen for patients with relapsed or refractory diffuse large B-cell lymphoma https://www.fda.gov/news-events/press-announcements/fda-approves-first-chemoimmunotherapy-regimen-patients-relapsed-or-refractory-diffuse-large-b-cell?utm_campaign=061019_PR_FDA.
- Perez H. L.; Cardarelli P. M.; Deshpande S.; Gangwar S.; Schroeder G. M.; Vite G. D.; Borzilleri R. M. Antibody-Drug Conjugates: Current Status and Future Directions. Drug Discovery Today 2014, 19 (7), 869–881. 10.1016/j.drudis.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Carrasco-Triguero M. Insights on the Immunogenicity of Antibody-Drug Conjugates. Bioanalysis 2015, 7 (13), 1565–1568. 10.4155/bio.15.86. [DOI] [PubMed] [Google Scholar]
- Loganzo F.; Sung M.; Gerber H.-P. Mechanisms of Resistance to Antibody-Drug Conjugates. Mol. Cancer Ther. 2016, 15 (12), 2825–2834. 10.1158/1535-7163.MCT-16-0408. [DOI] [PubMed] [Google Scholar]
- García-Alonso S.; Ocaña A.; Pandiella A. Resistance to Antibody-Drug Conjugates. Cancer Res. 2018, 78 (9), 2159–2165. 10.1158/0008-5472.CAN-17-3671. [DOI] [PubMed] [Google Scholar]
- Moreno-Gamez S.; Hill A. L.; Rosenbloom D. I. S.; Petrov D. A.; Nowak M. A.; Pennings P. S. Imperfect Drug Penetration Leads to Spatial Monotherapy and Rapid Evolution of Multidrug Resistance. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (22), E2874–E2883. 10.1073/pnas.1424184112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham J.; Salama N. N.; Azab A. K. The Role of P-Glycoprotein in Drug Resistance in Multiple Myeloma. Leuk. Lymphoma 2015, 56 (1), 26–33. 10.3109/10428194.2014.907890. [DOI] [PubMed] [Google Scholar]
- Mahalingaiah P. K.; Ciurlionis R.; Durbin K. R.; Yeager R. L.; Philip B. K.; Bawa B.; Mantena S. R.; Enright B. P.; Liguori M. J.; Van Vleet T. R. Potential Mechanisms of Target-Independent Uptake and Toxicity of Antibody-Drug Conjugates. Pharmacol. Ther. 2019, 200, 110–125. 10.1016/j.pharmthera.2019.04.008. [DOI] [PubMed] [Google Scholar]
- Sochaj-Gregorczyk A. M.; Serwotka-Suszczak A. M.; Otlewski J. A Novel Affibody-Auristatin E Conjugate With a Potent and Selective Activity Against HER2+ Cell Lines. J. Immunother. 2016, 39 (6), 223–232. 10.1097/CJI.0000000000000125. [DOI] [PubMed] [Google Scholar]
- Sochaj-Gregorczyk A.; Ludzia P.; Kozdrowska E.; Jakimowicz P.; Sokolowska-Wedzina A.; Otlewski J. Design and In Vitro Evaluation of a Cytotoxic Conjugate Based on the Anti-HER2 Affibody Fused to the Fc Fragment of IgG1. Int. J. Mol. Sci. 2017, 18 (8), 1688. 10.3390/ijms18081688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serwotka-Suszczak A.; Sochaj-Gregorczyk A.; Pieczykolan J.; Krowarsch D.; Jelen F.; Otlewski J. A Conjugate Based on Anti-HER2 Diaffibody and Auristatin E Targets HER2-Positive Cancer Cells. Int. J. Mol. Sci. 2017, 18 (2), 401. 10.3390/ijms18020401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szlachcic A.; Pala K.; Zakrzewska M.; Jakimowicz P.; Wiedlocha A.; Otlewski J. FGF1-Gold Nanoparticle Conjugates Targeting FGFR Efficiently Decrease Cell Viability upon NIR Irradiation. Int. J. Nanomed. 2012, 7, 5915–5927. 10.2147/IJN.S36575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szlachcic A.; Zakrzewska M.; Lobocki M.; Jakimowicz P.; Otlewski J. Design and Characteristics of Cytotoxic Fibroblast Growth Factor 1 Conjugate for Fibroblast Growth Factor Receptor-Targeted Cancer Therapy. Drug Des., Dev. Ther. 2016, 10, 2547–2560. 10.2147/DDDT.S105896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Świderska K. W.; Szlachcic A.; Czyrek A.; Zakrzewska M.; Otlewski J. Site-Specific Conjugation of Fibroblast Growth Factor 2 (FGF2) Based on Incorporation of Alkyne-Reactive Unnatural Amino Acid. Bioorg. Med. Chem. 2017, 25 (14), 3685–3693. 10.1016/j.bmc.2017.05.003. [DOI] [PubMed] [Google Scholar]
- Świderska K.; Szlachcic A.; Opaliński Ł.; Zakrzewska M.; Otlewski J. FGF2 Dual Warhead Conjugate with Monomethyl Auristatin E and α-Amanitin Displays a Cytotoxic Effect towards Cancer Cells Overproducing FGF Receptor 1. Int. J. Mol. Sci. 2018, 19 (7), 2098. 10.3390/ijms19072098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satomaa T.; Pynnönen H.; Vilkman A.; Kotiranta T.; Pitkänen V.; Heiskanen A.; Herpers B.; Price L.; Helin J.; Saarinen J. Hydrophilic Auristatin Glycoside Payload Enables Improved Antibody-Drug Conjugate Efficacy and Biocompatibility. Antibodies 2018, 7 (2), 15. 10.3390/antib7020015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain N.; Smith S. W.; Ghone S.; Tomczuk B. Current ADC Linker Chemistry. Pharm. Res. 2015, 32 (11), 3526–3540. 10.1007/s11095-015-1657-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushwah V.; Katiyar S. S.; Agrawal A. K.; Saraf I.; Singh I. P.; Lamprou D. A.; Gupta R. C.; Jain S. Implication of Linker Length on Cell Cytotoxicity, Pharmacokinetic and Toxicity Profile of Gemcitabine-Docetaxel Combinatorial Dual Drug Conjugate. Int. J. Pharm. 2018, 548 (1), 357–374. 10.1016/j.ijpharm.2018.07.016. [DOI] [PubMed] [Google Scholar]
- Ritchie M.; Tchistiakova L.; Scott N. Implications of Receptor-Mediated Endocytosis and Intracellular Trafficking Dynamics in the Development of Antibody Drug Conjugates. MAbs 2013, 5 (1), 13–21. 10.4161/mabs.22854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson M. P.; Maaheimo H.; Ekholm F. S. New Insight on the Structural Features of the Cytotoxic Auristatins MMAE and MMAF Revealed by Combined NMR Spectroscopy and Quantum Chemical Modelling. Sci. Rep. 2017, 7 (1), 15920. 10.1038/s41598-017-15674-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H.; Liu Y.; Yu Z.; Sun M.; Lin L.; Liu W.; Han Q.; Wei M.; Jin Y. The Analysis of Key Factors Related to ADCs Structural Design. Front. Pharmacol. 2019, 10, 373. 10.3389/fphar.2019.00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shefet-Carasso L.; Benhar I. Antibody-Targeted Drugs and Drug Resistance—Challenges and Solutions. Drug Resist. Updates 2015, 18, 36–46. 10.1016/j.drup.2014.11.001. [DOI] [PubMed] [Google Scholar]
- Beck A.; Goetsch L.; Dumontet C.; Corvaïa N. Strategies and Challenges for the next Generation of Antibody-Drug Conjugates. Nat. Rev. Drug Discovery 2017, 16 (5), 315–337. 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- Nasiri H.; Valedkarimi Z.; Aghebati-Maleki L.; Majidi J. Antibody-Drug Conjugates: Promising and Efficient Tools for Targeted Cancer Therapy. J. Cell. Physiol. 2018, 233 (9), 6441–6457. 10.1002/jcp.26435. [DOI] [PubMed] [Google Scholar]
- Kovtun Y. V.; Audette C. A.; Mayo M. F.; Jones G. E.; Doherty H.; Maloney E. K.; Erickson H. K.; Sun X.; Wilhelm S.; Ab O.; et al. Antibody-Maytansinoid Conjugates Designed to Bypass Multidrug Resistance. Cancer Res. 2010, 70 (6), 2528–2537. 10.1158/0008-5472.CAN-09-3546. [DOI] [PubMed] [Google Scholar]
- Kovtun Y. V.; Audette C. A.; Ye Y.; Xie H.; Ruberti M. F.; Phinney S. J.; Leece B. A.; Chittenden T.; Blättler W. A.; Goldmacher V. S. Antibody-Drug Conjugates Designed to Eradicate Tumors with Homogeneous and Heterogeneous Expression of the Target Antigen. Cancer Res. 2006, 66 (6), 3214–3221. 10.1158/0008-5472.CAN-05-3973. [DOI] [PubMed] [Google Scholar]
- Okeley N. M.; Miyamoto J. B.; Zhang X.; Sanderson R. J.; Benjamin D. R.; Sievers E. L.; Senter P. D.; Alley S. C. Intracellular Activation of SGN-35, a Potent Anti-CD30 Antibody-Drug Conjugate. Clin. Cancer Res. 2010, 16 (3), 888–897. 10.1158/1078-0432.CCR-09-2069. [DOI] [PubMed] [Google Scholar]
- Pabst M.; McDowell W.; Manin A.; Kyle A.; Camper N.; De Juan E.; Parekh V.; Rudge F.; Makwana H.; Kantner T.; et al. Modulation of Drug-Linker Design to Enhance in Vivo Potency of Homogeneous Antibody-Drug Conjugates. J. Controlled Release 2017, 253, 160–164. 10.1016/j.jconrel.2017.02.027. [DOI] [PubMed] [Google Scholar]
- Viricel W.; Fournet G.; Beaumel S.; Perrial E.; Papot S.; Dumontet C.; Joseph B. Monodisperse Polysarcosine-Based Highly-Loaded Antibody-Drug Conjugates. Chem. Sci. 2019, 10 (14), 4048–4053. 10.1039/C9SC00285E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons J. K.; Burke P. J.; Cochran J. H.; Pittman P. G.; Lyon R. P. Reducing the Antigen-Independent Toxicity of Antibody-Drug Conjugates by Minimizing Their Non-Specific Clearance through PEGylation. Toxicol. Appl. Pharmacol. 2020, 392, 114932. 10.1016/j.taap.2020.114932. [DOI] [PubMed] [Google Scholar]
- Ruggiero A.; Villa C. H.; Bander E.; Rey D. A.; Bergkvist M.; Batt C. A.; Manova-Todorova K.; Deen W. M.; Scheinberg D. A.; McDevitt M. R. Paradoxical Glomerular Filtration of Carbon Nanotubes. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (27), 12369–12374. 10.1073/pnas.0913667107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox M. E.; Szoka F. C.; Fréchet J. M. J. Soluble Polymer Carriers for the Treatment of Cancer: The Importance of Molecular Architecture. Acc. Chem. Res. 2009, 42 (8), 1141–1151. 10.1021/ar900035f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y.; Lu H. Protein PEPylation: A New Paradigm of Protein-Polymer Conjugation. Bioconjugate Chem. 2019, 30 (6), 1604–1616. 10.1021/acs.bioconjchem.9b00236. [DOI] [PubMed] [Google Scholar]
- Hensler K.PEGylated Protein Drugs: Basic Science and Clinical Applications; Veronese F. M., Ed.; Birkhäuser Basel: Basel, 2009. [Google Scholar]
- Hughes S. E. Differential Expression of the Fibroblast Growth Factor Receptor (FGFR) Multigene Family in Normal Human Adult Tissues. J. Histochem. Cytochem. 1997, 45 (7), 1005–1019. 10.1177/002215549704500710. [DOI] [PubMed] [Google Scholar]
- Cornish E. E.; Natoli R. C.; Hendrickson A.; Provis J. M. Differential Distribution of Fibroblast Growth Factor Receptors (FGFRs) on Foveal Cones: FGFR-4 Is an Early Marker of Cone Photoreceptors. Mol. Vis. 2004, 10, 1–14. [PubMed] [Google Scholar]
- Haugsten E. M.; Wiedlocha A.; Olsnes S.; Wesche J. Roles of Fibroblast Growth Factor Receptors in Carcinogenesis. Mol. Cancer Res. 2010, 8 (11), 1439–1452. 10.1158/1541-7786.MCR-10-0168. [DOI] [PubMed] [Google Scholar]
- Wesche J.; Haglund K.; Haugsten E. M. Fibroblast Growth Factors and Their Receptors in Cancer. Biochem. J. 2011, 437 (2), 199–213. 10.1042/BJ20101603. [DOI] [PubMed] [Google Scholar]
- Weinstein J. N; Collisson E. A; Mills G. B; Shaw K. R M.; Ozenberger B. A; Ellrott K.; Shmulevich I.; Sander C.; Stuart J. M; et al. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45 (10), 1113–1120. 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.-B.; Meric-Bernstam F.; Kalyan A.; Babich A.; Liu R.; Tanigawa T.; Sommer A.; Osada M.; Reetz F.; Laurent D.; et al. First-in-Human Phase I Study of Aprutumab Ixadotin, a Fibroblast Growth Factor Receptor 2 Antibody-Drug Conjugate (BAY 1187982) in Patients with Advanced Cancer. Target. Oncol. 2019, 14 (5), 591–601. 10.1007/s11523-019-00670-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer A.; Kopitz C.; Schatz C. A.; Nising C. F.; Mahlert C.; Lerchen H.-G.; Stelte-Ludwig B.; Hammer S.; Greven S.; Schuhmacher J.; et al. Preclinical Efficacy of the Auristatin-Based Antibody-Drug Conjugate BAY 1187982 for the Treatment of FGFR2-Positive Solid Tumors. Cancer Res. 2016, 76 (21), 6331–6339. 10.1158/0008-5472.CAN-16-0180. [DOI] [PubMed] [Google Scholar]
- Mackness B. C.; Jaworski J. A.; Boudanova E.; Park A.; Valente D.; Mauriac C.; Pasquier O.; Schmidt T.; Kabiri M.; Kandira A.; et al. Antibody Fc Engineering for Enhanced Neonatal Fc Receptor Binding and Prolonged Circulation Half-Life. MAbs 2019, 11 (7), 1276–1288. 10.1080/19420862.2019.1633883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latvala S.; Jacobsen B.; Otteneder M. B.; Herrmann A.; Kronenberg S. Distribution of FcRn Across Species and Tissues. J. Histochem. Cytochem. 2017, 65 (6), 321–333. 10.1369/0022155417705095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt M. K.; Taylor M. A.; Schiemann B. J.; Sossey-Alaoui K.; Schiemann W. P. Fibroblast Growth Factor Receptor Splice Variants Are Stable Markers of Oncogenic Transforming Growth Factor B1 Signaling in Metastatic Breast Cancers. Breast Cancer Res. 2014, 16 (2), 3381. 10.1186/bcr3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner S.; Duan D. S.; de Vries C.; Peters K. G.; Johnson D. E.; Williams L. T. Differential Splicing in the Extracellular Region of Fibroblast Growth Factor Receptor 1 Generates Receptor Variants with Different Ligand-Binding Specificities. Mol. Cell. Biol. 1992, 12 (1), 82–88. 10.1128/MCB.12.1.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luqmani Y. A.; Mortimer C.; Yiangou C.; Johnston C. L.; Bansal G. S.; Sinnett D.; Law M.; Charles Coombes R. Expression of 2 Variant Forms of Fibroblast Growth Factor Receptor 1 in Human Breast. Int. J. Cancer 1995, 64 (4), 274–279. 10.1002/ijc.2910640411. [DOI] [PubMed] [Google Scholar]