

ABSTRACT
Decades of studies have established that nuclear lamin polymers form the nuclear lamina, a protein meshwork that supports the nuclear envelope structure and tethers heterochromatin to the nuclear periphery. Much less is known about unpolymerized nuclear lamins in the nuclear interior, some of which are now known to undergo specific phosphorylation. A recent finding that phosphorylated lamins bind gene enhancer regions offers a new hypothesis that lamin phosphorylation may influence transcriptional regulation in the nuclear interior. In this review, we discuss the regulation, localization, and functions of phosphorylated lamins. We summarize kinases that phosphorylate lamins in a variety of biological contexts. Our discussion extends to laminopathies, a spectrum of degenerative disorders caused by lamin gene mutations, such as cardiomyopathies and progeria. We compare the prevailing hypothesis for laminopathy pathogenesis based on lamins’ function at the nuclear lamina with an emerging hypothesis based on phosphorylated lamins’ function in the nuclear interior.
KEYWORDS: Nuclear lamin, lamin A/C, phosphorylation, nuclear interior, laminopathies, enhancer, chromatin, chromosome, lamina-associated domain, mitosis, c-Jun, farnesylation, interphase, cdk1, progeria, cardiomyopathies, lmna, muscular dystrophy
Introduction
The nuclear lamina is a protein meshwork that covers the nuclear side of the inner nuclear membrane in animal cells. Nuclear lamins are a class of intermediate filament proteins and constitute the nuclear lamina by polymerizing and assembling into filaments. The nuclear lamina provides structural integrity to the nucleus and serves as a scaffold for interphase chromosomes by tethering heterochromatin domains to the nuclear periphery [1–3]. In addition, nuclear lamins are thought to participate in various cellular processes [4] including transcriptional regulation [5,6], chromosome organization [3], DNA damage response [7], cell signaling [8,9], cell cycle regulation [10], and mechanotransduction [11,12]. Mutations in genes encoding nuclear lamins cause a spectrum of human disorders collectively called laminopathies, including cardiomyopathies, muscular dystrophies, and the premature aging disorder Hutchinson-Gilford progeria [13]. The molecular mechanisms by which nuclear lamins participate in various biological processes remain elusive, as do the pathogenic mechanisms underlying laminopathies.
There are two nuclear lamin types, A-type and B-type [14]. A-type lamins include Lamin A and Lamin C (Lamin A/C), two splice isoforms encoded by LMNA in humans. B-type lamins include Lamin B1 encoded by LMNB1 and Lamin B2 encoded by LMNB2 in humans. The A-type lamin gene arose in vertebrate evolution from the ancestral B-type lamin genes, which are conserved across metazoans [15]. Each lamin subtype forms separate lamin polymers and filaments in the nuclear lamina [6,16]. A-type lamins are expressed robustly in differentiated cells but nearly undetectable in pluripotent stem cells and during early embryogenesis [17–20]. In contrast, B-type lamins are thought to be expressed in every cell [21,22]. The biological significance of the cell-type specificity and species specificity of different lamin subtypes is poorly understood.
Nuclear lamins have also been observed in the interior of the nucleus of interphase cells [6,23–25]. Nuclear-interior lamins were originally thought to constitute a macromolecular ‘nuclear matrix’, a hypothetical chromatin scaffold in the nuclear interior [24,26]. However, recent studies have found that at least some fraction of nuclear-interior lamins are soluble, mobile, and unpolymerized [6,27–30]. Thus, nuclear-interior lamins exhibit molecular features starkly different from those of polymer lamins at the nuclear lamina.
Phosphorylation of nuclear-peripheral lamins provides the mechanistic basis for nuclear lamina disassembly during the mitosis phase of the cell cycle. Nuclear lamin phosphorylation causes lamin depolymerization at the onset of mitosis for nuclear envelope breakdown [31–33]. At the end of mitosis, nuclear lamins are dephosphorylated and reassembled into polymers in the nuclear lamina. Lamin phosphorylation has also been observed in interphase [34,35], but the molecular details of interphase-phosphorylated lamins had been obscure until recently. Recent studies found that interphase phosphorylation marks a fraction of nuclear lamins in the nuclear interior [27–29]. Furthermore, some phosphorylated lamins in the nuclear interior bind to genomic regions characteristic of gene enhancers in the human genome [28]. Thus, a focus on phosphorylation of nuclear lamins has opened a new avenue for investigating nuclear lamin functions in the cell.
In this review, we summarize the current understanding of molecular features, localization, regulation, and functions of phosphorylated nuclear lamins. We distinguish the various cellular pathways through which lamins are phosphorylated. We discuss our recent observation suggesting that phosphorylated lamins act as transcriptional activators at enhancers in the nuclear interior. Finally, we extend our discussion to the ways in which laminopathy-causing mutations might influence lamin phosphorylation and the functions of phosphorylated lamins, offering new hypotheses for the pathogenesis of laminopathies.
Lamin phosphorylation and nuclear lamina disassembly during mitosis
Nuclear lamins are composed of three structural domains: the short N-terminal head domain (aa1-33 in human Lamin A/C; amino acid position in UniProtKB P02545), the central rod domain (aa34-383 in Lamin A/C), and the C-terminal tail domain (aa384-646 in Lamin A) [36–40] Figure 1a, B. The tail domain includes an immunoglobulin (Ig) fold domain (aa436-544) that harbors various protein and DNA interacting sites [41]. The central rod domains of two lamin molecules interact in parallel to form dimers [38]. Lamin dimers then interact in a head-to-tail fashion to form polymers, with the tail domain being protruded out of the polymer axis [38,42,43]. Lamin polymers further interact in an antiparallel fashion to form tetrameric filaments [44].
Figure 1.
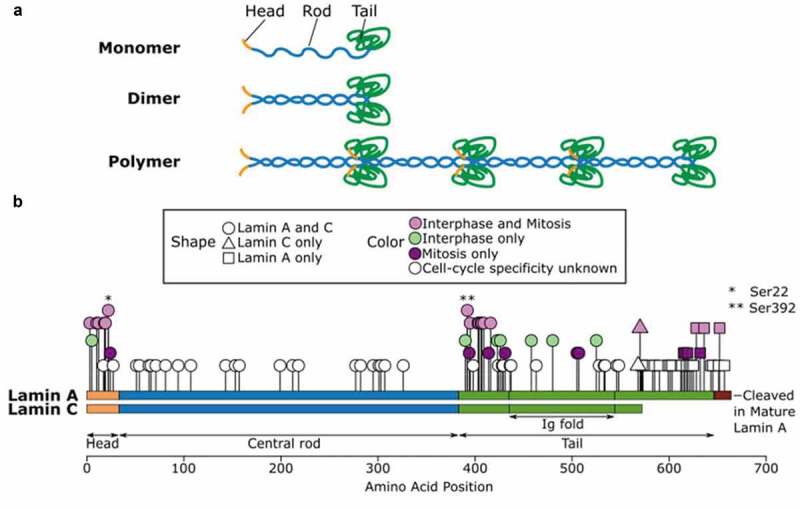
Lamin polymerization and phosphorylation. (a) Schematic representation of lamin polymerization. Lamins form dimers through rod domain interactions. Lamin dimers associate longitudinally into polar head-to-tail polymers. (b) Distribution of phosphorylation sites in Lamin A/C. Phosphorylation sites are stratified by the cell-cycle phase in which the residue is reported to be phosphorylated
Phosphorylation of nuclear lamins reaches the highest level at the onset of the mitosis phase of the cell cycle to disassemble the lamin polymers [42,43,45]. Mitotic lamin phosphorylation predominantly occurs at two residues flanking either side of the central rod domain, often called ‘mitotic sites’, which are Ser22 and Ser392 in Lamin A/C Figure 1b, Ser23 and Ser393 in Lamin B1 (amino acid position in UniProtKB P20700), and Thr34, Ser37, and/or Ser405 in Lamin B2 (amino acid position in UniProtKB Q03252) [31,32]. Consistent with the presence of these pairs of mitotic sites, Lamin A, Lamin B, Lamin C have approximately 2 moles of associated phosphate per mole of lamin during mitosis [46]. Evidence suggests that every Lamin A/C molecule is phosphorylated at Ser22 during mitosis [28]. Phosphorylation at the two mitotic sites induces lamin depolymerization in vitro [33,47] and is required for nuclear lamina disassembly in vivo [48]. Evidence also suggests that depolymerized lamins are dimers during mitosis [16]. Conversely, dephosphorylation of the mitotic sites is required for nuclear lamin polymerization in vitro [47] and nuclear lamina assembly in vivo [49]. In addition to the two canonical mitotic sites, 28 other serine and threonine residues in Lamin A/C have been reported to exhibit increased phosphorylation during mitosis [27,35,50] (Supplementary Table 1). Many of these residues also flank the rod domain Figure 1b, although the contribution of these additional phosphorylations to lamin depolymerization during mitosis is not well understood.
There are several interesting differences between A-type and B-type lamins in their localization during mitosis. Lamin A/C are dissociated from the nuclear membrane and dispersed throughout the mitotic cytoplasm upon phosphorylation and depolymerization. In contrast, B-type lamins remain associated with the remnants of the nuclear membrane [46,51]. The association of B-type lamins with the remnants of the nuclear membrane is thought to be mediated by their C-terminal farnesylation, which is absent in Lamin A/C. Lack of farnesylation in Lamin A is due to the protease-mediated cleavage of the C-terminus during Lamin A maturation, and this cleavage site is encoded in an exon acquired during LMNA gene evolution in vertebrates [15]. Lamin C lacks the farnesylation site altogether. Toward the end of mitosis, both A-type and B-type lamins accumulate on the surface of condensed telophase chromatin, but in different ways [52,53]. Lamin A/C accumulation starts at the central region of telophase chromatin (called the ‘core’ region) and this process depends on the prior localization of Lamin A/C-interacting protein BAF (Barrier-to-Autointegration Factor) at the core region [52,53]. In contrast, Lamin B1 accumulation does not begin at the core and the process is independent of BAF [52,53]. Lamin A/C remain phosphorylated when localized to telophase chromatin [54], and evidence suggests that Lamin A/C are dephosphorylated on the telophase chromatin surface for repolymerization [55]. Whether B-type lamins are also dephosphorylated on the telophase chromatin surface has not been explored. These differences of mitotic localization between A-type and B-type lamins might be related to the observation that B-type lamins promote assembly of the mitotic spindles during mitosis, while A-type lamins appear to lack this function [56]. Whether A-type lamins have specific functions during mitosis is not known.
Nuclear lamin phosphorylation in interphase
The first report that nuclear lamins are phosphorylated in interphase dates to 1980 [34], although the biological significance of interphase phosphorylation had long been obscure until recently. One study estimated that the level of interphase lamin phosphorylation is 4–7 times lower than their mitotic phosphorylation level (therefore 0.3–0.5 moles of phosphates per mole of lamin) [46], suggesting that only a subset of lamins undergo phosphorylation during interphase. Compared to Lamin A/C, interphase phosphorylation of B-type lamins has been much less investigated [57,58]. Reviewing the literature, we identified 92 total phosphorylation sites reported for Lamin A and/or Lamin C in any cell cycle stage [27,35,50,59–64] (Figure 1b; Supplementary Table 1). Of the 92 phosphorylation sites in Lamin A/C, 25 are known to be phosphorylated during interphase in human HeLa or murine A9 cell lines [27,35]. Eighteen of the 25 interphase phosphorylation sites in Lamin A/C are also reported to be phosphorylated during mitosis, including Ser22 and Ser392, the canonical mitotic sites. In fibroblasts, Ser22-phosphorylated Lamin A/C is observed in G1, S, and G2 phases of interphase, with some variability in the Ser22 phosphorylation level between interphase cells [28]. Consistent with the notion that Ser22 phosphorylation drives lamin depolymerization, Ser22-phosphorylated Lamin A/C in interphase are localized in the nuclear interior, not at the nuclear periphery [27,28]. Lamin A with phospho-mimetic Ser22Asp or phospho-mimetic Ser392Asp substitutions are highly mobile in interphase nuclei [27], suggesting that Ser22 and Ser392-phosphorylated Lamin A/C in interphase represent unpolymerized Lamin A/C. Unlike during mitosis, however, the nuclear lamina appears intact in interphase cells with Ser22-phosphorylated Lamin A/C present in the nuclear interior [28]. Furthermore, the Ser22-phosphorylated population appears to represent a small fraction of all Lamin A/C molecules in the interphase nucleus [28]. Thus, interphase Ser22-phosphorylation occurs in a small fraction of Lamin A/C and does not induce depolymerization of the entire nuclear lamina in normal cells. Phospho-mimetic substitution of Lamin A/C at Ser390, Ser404, or Ser407 also promotes relocalization of Lamin A/C to the nuclear interior, similarly to Ser392 and Ser22 phosphorylation [27]. Evidence suggests that Ser403 phosphorylation promotes nuclear import of Lamin A/C, while Ser628 phosphorylation restricts nuclear import [27,65]. Given the large overlap between interphase and mitotic phosphorylation sites, some phosphorylated nuclear lamins in interphase might be carryovers from lamin phosphorylation in mitosis. Alternatively, nuclear lamins may be phosphorylated de novo in interphase by kinases active in interphase.
We recently observed that Lamin C is more strongly phosphorylated at Ser22 than Lamin A in interphase fibroblasts. This high degree of Lamin C phosphorylation may be related to the previous observation that Lamin C is more soluble than Lamin A in interphase cells [16]. What makes Lamin C more susceptible to phosphorylation than Lamin A? Unlike Lamin A and B-type lamins, Lamin C does not undergo farnesylation, and consequently, newly synthesized Lamin C is thought to populate the nucleoplasm first before becoming incorporated into the lamina. On the other hand, newly synthesized precursor Lamin A and B-type lamins are directly incorporated into the nuclear lamina through the contiguous ER membrane/nuclear membrane structure to which they are tethered by farnesylation [66]. A prediction based on this difference in the lamina incorporation pathways is that Lamin A and B-type Lamins build the foundation of the nuclear lamina meshwork, on which Lamin C meshwork assembles. While exact localization of Lamin C within the nuclear lamina has not yet been defined, a recent study using super-resolution microscopy reported that localization of Lamin A/C (detected by an antibody recognizing both Lamin A and C) is closer to the nuclear interior than Lamin B1 within the nuclear lamina meshwork [67]. Thus, one possibility is that Lamin C is most accessible by kinases of all lamin subtypes within the nuclear lamina by virtue of their differential localization within the nuclear lamina.
In summary, the nuclear-interior localization of phosphorylated forms of lamins presents the exciting possibility that phosphorylated lamins may have unexplored functions distinct from nuclear-peripheral polymerized lamins. In the next section, we discuss regulators of lamin phosphorylation.
Kinases and phosphatases for nuclear lamins
Numerous kinases and phosphatases for nuclear lamins have been identified in a variety of biological contexts (Figure 2; Supplementary Table 1). Cyclin-Dependent Kinase 1 (CDK1) and Protein Kinase C (PKC) phosphorylate nuclear lamins at the onset of mitosis. CDK1 becomes active specifically at the onset of mitosis after forming a complex with Cyclin B. The CDK1-Cyclin B complex phosphorylates Thr19, Ser22, and Ser392 of Lamin A/C, Ser23, and Ser393 of Lamin B1, and Thr34, Ser37, and Ser405 of Lamin B2 for lamin depolymerization in mitosis [31,57,68,69]. PKC phosphorylates Ser395 and Ser405 of Lamin B1 during mitosis [70], and evidence suggests that PKC also phosphorylates Ser5, Ser395, Thr416, and Ser572 of Lamin A/C [35]. While neither the CDK1-Cyclin B1 complex nor PKC is known to accumulate specifically at the nuclear envelope at the onset of mitosis [71–73], there is little doubt that lamin phosphorylation predominantly takes place at the nuclear periphery because phosphorylation is required for depolymerization of nuclear-peripheral lamins [31–33]. The current model suggests that phosphorylation of nuclear lamins as well as other nuclear envelope proteins by CDK1, PKC, and other kinases culminates in nuclear envelope breakdown in mitosis [66]. Unlike CDK1 [74], PKC activity itself is not restricted to mitosis [75]. Nuclear lamins are dephosphorylated at the end of mitosis (telophase) for nuclear lamina reformation, and this process is mediated by phosphatases. Phosphatases PP1 and PP2A dephosphorylate Lamin A/C at Thr19 and Ser22 [76] and Lamin B2 [77] and are required for nuclear envelope reassembly upon mitotic exit [78,79]. PP1 is known to accumulate on the surface of telophase chromatin, as are lamins [55,79]. Therefore, lamins are likely dephosphorylated on the chromatin surface for repolymerization. The activity of PP1/PP2A and CDK1 is mutually antagonistic [80], providing the mechanistic basis for the phosphorylation–dephosphorylation cycle of nuclear lamins during mitosis. Beyond the mitotic exit, PP1 and PP2A become active in various other biological contexts such as glycogen [81] and sphingolipid metabolism [82], as well as the DNA damage response [83], potentially contributing to regulation of lamin phosphorylation in non-mitotic contexts.
Figure 2.
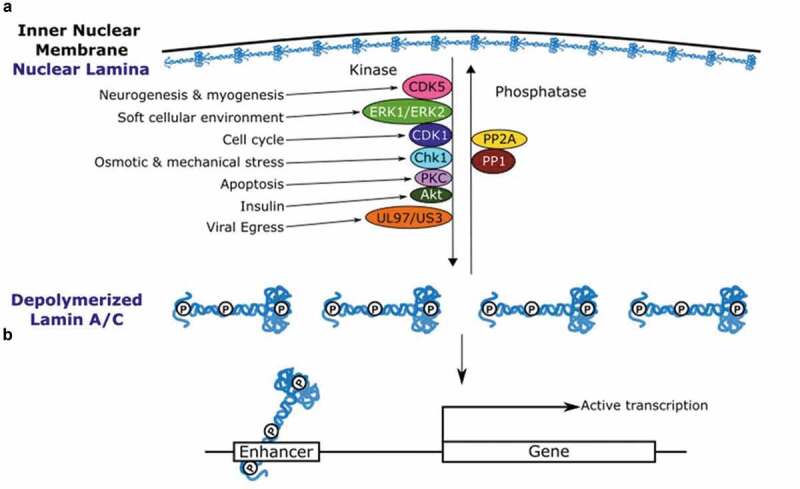
Regulators and functions of lamin phosphorylation. (a) Kinases and phosphatases known to regulate lamin phosphorylation are shown. Phosphorylation of lamins could cause depolymerization and localization to the nuclear interior. (b) Phosphorylated Lamin A/C bind to enhancers of active genes in the nuclear interior
Among lamin kinases active in interphase are ERK1 and ERK2 (also known as MAP kinases), two closely related kinases active under various extracellular stimuli. ERK1 and ERK2 phosphorylate Lamin A/C at Ser22 [84]. Ser22 of Lamin A/C is within both the CDK and ERK recognition sequence motifs. ERK1 and ERK2 can directly interact with Lamin A and Lamin C in vitro and in vivo [85]. An interesting possibility is that ERK1 and ERK2 phosphorylate Lamin A/C for nuclear-interior localization in interphase in response to various conditions triggering cellular stress. One such condition may be the type of soft extracellular environment in which Ser22 and Ser392 phosphorylation is known to be promoted [29,86]. In this context, nuclear-interior localization of phosphorylated Lamin A/C is thought to facilitate rounding of the nucleus in response to rounding of the cell under soft microenvironments [29,86]. Another lamin kinase active in interphase is Akt (also known as Protein Kinase B), active in many cellular processes including glucose metabolism [87]. Akt phosphorylates Lamin A/C at Ser404 in the mouse myoblast C2C12 cell line, and this phosphorylation is promoted by insulin [88,89]. Akt also phosphorylates Ser458 of Lamin A/C in muscle tissues isolated from LMNA-related myopathy patients [90]. In addition, a proteome study identified Thr10, Ser406, Ser407, Thr409, Ser414, Thr416, and Thr548 of Lamin A/C as substrates of Akt and ribosomal S6 kinases in cancer cell lines [62].
Several lamin kinases are known to promote extensive modulation of the nuclear lamina in interphase cells. PKC-δ phosphorylates B-type lamins for nuclear lamina disassembly during apoptosis [91,92]. During sea urchin fertilization, PKC in oocytes phosphorylates Lamin B1 of the sperm nuclei, leading to dissolution of the sperm nuclear lamina required for male pronucleus formation [93]. CDK5, a member of the cyclin-dependent kinase family that does not require cyclins for activation [94], phosphorylates Ser22 and Ser392 of Lamin A/C and Ser23 and Ser393 of Lamin B1 in the mouse neuronal HT22 cell line [95]. Evidence suggests that an aberrant increase of CDK5 activity in neuronal cells causes nuclear dispersion through lamin disassembly, resulting in neuronal death in Alzheimer’s disease [95]. CDK5 activity has also been reported in non-neuronal cells, such as in muscle, in which CDK5 promotes differentiation of myoblasts [94], although the relationship between lamin phosphorylation and myogenesis is unclear. Chk1, a kinase that coordinates cell-cycle arrest with DNA damage response, phosphorylates Ser307 of Lamin A/C [96]. Chk1 is known to localize at the nuclear periphery upon mechanical and osmotic stress and is thought to contribute to structural alteration of the nuclear envelope [97]. Therefore, Ser307 phosphorylation of Lamin A/C may contribute to the structural change of the nuclear lamina upon mechanical or osmotic stress.
Finally, nuclear lamin phosphorylation can be catalyzed by viral kinases for virus egress. UL97, a CDK1-like kinase of human cytomegalovirus (HCMV), phosphorylates Lamin A/C at Ser22 [98]. Consistently, HCMV infection dramatically increases Ser22-phosphorylated Lamin A/C levels in the nuclear interior [99]. UL97-mediated Ser22 phosphorylation is recognized by prolyl isomerase PIN1 to promote Lamin A/C depolymerization [99]. The US3 kinase of herpes simplex virus type 1 (HSV-1) also phosphorylates Lamin A/C [100], and a study suggests the target residues are Ser22 and Ser392 [99].
In summary, a number of kinases and phosphatases operate on nuclear lamins within diverse biological contexts, sometimes at identical residues. It is plausible that these kinases and phosphatases regulate the equilibrium between polymer lamins and unpolymerized lamins in the interphase nuclei (Figure 2a). What remain to be defined are the subcellular locations at which lamin phosphorylation and dephosphorylation take place during interphase. Furthermore, the functional significance of lamin phosphorylation is poorly understood apart from structural modulation of the nuclear lamina. In the next section, we discuss a new direction of research suggesting that phosphorylated lamins have specific functions in gene regulation in the nuclear interior.
Phosphorylated lamin c at enhancers
Nuclear lamins exhibit a high affinity to DNA and chromatin, with a nano-molar range dissociation constant (KD) for interactions between the C-terminal domain of Lamin A/C and DNA or nucleosomes [101]. Polymer nuclear lamins at the nuclear periphery interact with large heterochromatin domains called lamina-associated domains (LADs), which contain mostly transcriptionally inactive genes [102–106]. By tethering LADs to the nuclear periphery, nuclear lamins influence the spatial organization of chromosomal regions [1]. Evidence suggests that nuclear lamins also promote transcriptional repression of some of the genes embedded in LADs [107–109]. Given the chromatin binding property of nuclear lamins, one hypothesis had been that phosphorylated lamins in the nuclear interior might also bind chromatin, but at different locations than LADs.
We recently investigated the genomic distribution of Ser22-phosphorylated Lamin A/C in interphase human fibroblast cells [28]. Using an antibody specific to Ser22 phosphorylation of Lamin A/C in chromatin immunoprecipitation coupled with high-throughput sequencing (ChIP-seq), we observed that Ser22-phosphorylated Lamin A/C interacts with numerous genomic sites with features of active enhancers, near genes undergoing active transcription. The enhancer-like features of Ser22-phosphorylated Lamin A/C-binding sites are in stark contrast to transcriptionally-inactive, megabase-wide heterochromatin features of LADs [1,102–106]. As described earlier, Ser22-phosphorylated Lamin C is more abundant than Ser22-phosphorylated Lamin A in interphase fibroblasts. Consistent with this observation, Lamin C with phospho-mimetic Ser22Asp and Ser392Asp substitutions binds more strongly to putative enhancers than Lamin A with the same phospho-mimetic substitutions [28]. These observations suggested that Lamin C is the primary form binding to putative enhancers upon phosphorylation. Thus, phosphorylated Lamin C may act as a transcriptional activator directly regulating transcription at enhancers in the nuclear interior Figure 2b.
In what biological context might the enhancer binding of Ser22-phosphorylated Lamin C be promoted? Our study revealed that Ser22-phosphorylated Lamin C-bound sites overlap almost exclusively with the genomic sites occupied by the AP-1 transcription factor c-Jun [28]. c-Jun is activated by multiple kinases including JNKs (Jun-N-terminal kinases) and ERK upon various extracellular stimuli [110]. While direct interaction between Lamin A/C and c-Jun has not been reported, Lamin A/C is known to interact with c-Fos, the binding partner of c-Jun in the AP-1 transcription factor complex, at the nuclear periphery [85,111]. Evidence suggests that c-Fos is unphosphorylated and inactive when interacting with Lamin A/C at the nuclear periphery, and ERK2-dependent phosphorylation of c-Fos at the nuclear lamina relocalizes c-Fos to the nuclear interior for DNA binding [85]. One hypothesis based on these observations is that Lamin C and c-Jun/c-Fos might be phosphorylated together at the nuclear periphery by ERK2 and directed to AP1-target enhancers. Overall, an attractive model is that various cellular conditions that promote lamin phosphorylation result in a transcriptional response directly mediated by phosphorylated Lamin C binding to gene enhancers Figure 2.
The lamin A/C-LAP2α complex
Several studies have utilized chromatin fractionation to probe chromatin regions associated with nuclear-interior lamins [112–114]. Lund et al. performed ChIP-seq using an antibody that detects total Lamin A/C but in a soluble fraction obtained by micrococcal nuclease digestion, a mild lysis condition that enriches unpolymerized lamins [112]. This procedure identified low-level continuous enrichment of Lamin A/C across large regions outside of LADs [112]. These Lamin A/C-associated regions were over-represented for histone modifications associated with transcriptional repression [112]. A similar observation was made via total Lamin A/C ChIP-seq in a soluble fraction obtained by mild mechanical DNA shearing [113]. Lamin A/C-associated regions found via this procedure overlapped genomic regions bound by LAP2α, a Lamin A/C-binding protein exclusively localized in the nuclear interior [113,115,116]. Therefore, LAP2α-associated nuclear-interior Lamin A/C likely bind to transcriptionally inactive regions outside of LADs. The difference in genomic localization profiles between LAP2α-associated Lamin A/C and Ser22-phosphorylated Lamin C (which binds to putative active enhancers) suggests that LAP2α-associated Lamin A/C and Ser22-phosphorylated Lamin C represent distinct pools of nuclear-interior Lamin A/C. The interaction between Lamin A/C and LAP2α is mediated by the C-terminal region of Lamin A/C (amino acids 319–572), which harbors numerous interphase phosphorylation sites [116]. One possibility is that phosphorylation and dephosphorylation within the C-terminal region affects the Lamin A/C–LAP2α interaction, thereby regulating an exchange between LAP2α-associated and non-associated Lamin A/C in the nuclear interior.
The function of the LAP2α has been investigated extensively [117,118]. LAP2α binds Lamin A/C and is thought to retain Lamin A/C in the nuclear interior during the G1 cell-cycle phase [119,120]. Lamin A/C and LAP2α interact with Retinoblastoma Protein (RB) [121,122], a repressor of the E2F-mediated G1-to-S cell cycle transition [123]. Lamin A/C deletion results in reduction of RB abundance, presumably due to an increased susceptibility of RB to proteasome-mediated degradation [124,125], and promotes cell-cycle progression into the S-phase [124]. Similarly, LAP2α-knockout cells are defective in cell cycle arrest [119]. These studies suggest that LAP2α-associated nuclear-interior Lamin A/C protects RB from degradation, thereby negatively regulating cell proliferation. Whether Ser22-phosphorylated Lamin A/C participates in regulation of RB has not been explored.
Laminopathies
There are over 200 known autosomal-dominant point mutations in LMNA that cause human disease. These diseases, collectively called laminopathies, include cardiomyopathies, muscular dystrophies, lipodystrophies, peripheral neuropathies, and Hutchinson-Gilford progeria [126]. Pathogenic mutations in LMNB1 and LMNB2 are much less frequent [127], possibly due to the perinatal requirement of LMNB1 and LMNB2 as opposed to postnatal requirement of LMNA [128–130]. The laminopathy mutations in LMNA cause nonsynonymous substitutions in the vast majority of cases. There is no apparent relationship between the amino acid locations of the mutations and disease phenotypes except in a few cases [14]. There is a phenotypic overlap among certain laminopathies such as Emery-Dreifuss muscular dystrophy type 2 (EDMD2) and dilated cardiomyopathy type 1A (CMD1A), both of which affect the cardiac muscle [131]. Although the mechanisms by which LMNA mutations cause laminopathies remain unknown, a number of molecular changes have been reported in cells or tissues derived from laminopathy patients or in animal or cellular models of laminopathies. These molecular changes include abnormal gene expression [132], abnormal cell signaling [133], increased DNA damage [134], abnormal localization of telomeres [135], altered nuclear shape [136], and altered response to mechanical stress [137]. A challenge in finding treatment for laminopathies has been to identify the molecular changes that trigger their pathogenesis and distinguish these upstream molecular changes from downstream molecular changes.
The prevailing hypotheses for laminopathies
There have been two prevailing, non-mutually exclusive hypotheses for the pathogenic mechanism underlying laminopathies, both based on lamins’ functions at the nuclear periphery [13]. The gene expression hypothesis states that laminopathy mutations disrupt interactions between the nuclear lamina and LADs, leading to abnormal gene expression. The structural hypothesis states that laminopathy mutations render the nuclear envelope structurally defective, causing dysregulation of intranuclear processes.
The gene expression hypothesis has been studied extensively in the context of Hutchinson-Gilford progeria syndrome (HGPS), a premature aging syndrome caused by heterozygous LMNA mutations [138]. HGPS is caused predominantly by a mutation within the Lamin A-specific region of the LMNA gene that activates cryptic splicing and results in a mutant Lamin A protein called ‘progerin’ [138]. Progerin lacks the C-terminal cleavage site that is used to detach the farnesylated C-terminal end in normal Lamin A processing, thus being permanently farnesylated [138]. Progerin accumulates at the nuclear periphery due to this permanent farnesylation, and progerin accumulation has been hypothesized to cause disorganization of heterochromatin at lamina-associated domains (LADs) [139]. Supporting this hypothesis, in progeria-patient fibroblasts, some LADs lose interactions with nuclear-peripheral Lamin A, and the losses of LADs coincide with losses of histone H3 trimethylation at lysine 9 and lysine 27, two modifications that mark heterochromatin [28,140]. Similar losses of heterochromatin have been observed by immunofluorescence and electron microscopy in progeria-patient cells [140,141].
Recently, several groups, including ours, have conducted detailed analyses to define whether losses of heterochromatin are responsible for dysregulated gene expression in laminopathies. Our parallel analysis of gene expression, lamina–chromatin interaction, and histone modifications revealed that only a very small number of dysregulated genes are located within lost LADs in progeria-patient fibroblasts, although LAD losses do accompany losses of heterochromatin-associated histone modifications [28]. Similar observations have been reported for other laminopathies. Lee et al. found increased chromatin accessibility within LADs, indicative of losses of heterochromatin, in the cardiomyocytes differentiated from the induced pluripotent stem cells (iPSC) derived from LMNA-related dilated cardiomyopathy patients [142]. However, increased chromatin accessibility within LADs was not directly responsible for abnormal activation of platelet-derived growth factor (PDGF) signaling that caused arrhythmic phenotypes of the mutant cardiomyocytes [142]. Bertero et al. performed chromatin conformation analysis in cardiomyocytes differentiated from other LMNA-related cardiomyopathy-patient iPSCs and identified genomic regions that lose heterochromatin [143]. Again, the heterochromatin change did not explain most gene expression alterations in the mutant cardiomyocytes [143]. Together, these independent studies suggest that the alteration of LADs is unlikely to be a major contributor to abnormal gene expression changes in laminopathies.
Phosphorylated lamin C–enhancer binding is altered in progeria
An emerging new hypothesis, drawing upon evidence provided by recent reports on phosphorylated lamin activity, is that impaired functions of phosphorylated Lamin A/C in the nuclear interior underlie the pathogenesis of laminopathies. We therefore recently examined whether interactions between Ser22-phosphorylated Lamin C and enhancers are altered in the fibroblasts derived from progeria patients. This investigation led us to observe that a specific subset of enhancer-like elements either gain or lose interactions with Ser22-phosphorylated Lamin C in progeria [28]. Consistent with the hypothesis that Ser22-phosphorylated Lamin C acts as a transcriptional activator, gains of Ser22-phosphorylated Lamin C binding were correlated with acquisition of histone acetylation and c-Jun binding at the binding sites, and losses with reduction of histone acetylation and c-Jun binding in progeria-patient cells [28]. Furthermore, gains and losses of Ser22-phosphorylated Lamin C binding were accompanied by increased and decreased expression of nearby genes in progeria-patient cells, respectively. In particular, we found that abnormally activated genes with nearby gains of Ser22-phosphorylated Lamin C are important in the pathophysiology of progeria [28]. In these progeria-patient cells, progerin itself was not phosphorylated at Ser22, and the phosphorylation level of Lamin C and subnuclear localization of Ser22-phosphorylated Lamin C did not appear to change. Thus, it remains unclear how Ser22-phosphorylated Lamin C is misdirected in progeria. Given the observation that progerin directly interacts with Lamin C [144], one possibility is that the progerin–Lamin C interaction alters the binding specificity of Lamin C. Several chemical compounds known to inhibit the interaction between Lamin A/C and progerin [144] may be useful for examining this possibility.
Laminopathy mutations that affect lamin phosphorylation
There are multiple ways in which laminopathy mutations could affect nuclear lamin phosphorylation. First, laminopathy mutations could lead to nonsynonymous substitutions of the residues subject to phosphorylation. Such pathogenic substitutions in Lamin A/C include Thr10Ile associated with lipodystrophy [145], Ser22Leu associated with dilated cardiomyopathy [146], Ser27Ile associated with limb-girdle muscular dystrophy with dilated cardiomyopathy [147], Ser143Phe associated with congenital muscular dystrophy [148], and Thr528Lys associated with Emery-Dreifuss muscular dystrophy [59,149] (Supplementary Table 1). Whether these mutations affect assembly or localization of Lamin A/C has not to date been characterized. Pathogenic missense substitutions in Lamin A/C are apparently not over-represented among phosphorylation sites, potentially due to the critical role of Lamin A/C phosphorylation during mitosis (pathogenic substitutions overlap 22% of phosphorylation sites vs. 42% of non-phosphorylation sites. Pathogenic site data from http://www.umd.be/LMNA/). Second, pathogenic mutations may alter kinase-recognition motifs surrounding phosphorylation sites. While this scenario has not been explored in detail, Lin et al. predicted that pathogenic LMNA mutations decrease Lamin A/C phosphorylation overall in an in silico analysis [150]. Third, pathogenic mutations may alter the accessibility of kinases or phosphatases to target residues through protein conformation changes. Mitsuhashi et al. reported that Ser458, a site within the Ig fold domain, is phosphorylated in the muscle tissues of LMNA-related muscular-dystrophy patients only when mutations are located within the Ig fold [90]. In contrast, Ser458 was not phosphorylated in neuromuscular disorders unrelated to LMNA mutations or cells expressing mutant Lamin A that causes non-myopathic laminopathies [90]. The authors found that the kinase Akt1 phosphorylates Ser458 and speculated that myopathy-causing LMNA mutations render Ser458 accessible to Akt1 through a conformational change of the Ig fold domain [90]. However, Ser458 phosphorylation has later been reported in LMNA-wild-type HeLa cells [27]. Finally, there are reports that laminopathy mutations are associated with changes in Lamin A/C phosphorylation at undefined residues. Cenni et al. reported strong reduction of overall Lamin A/C phosphorylation in myoblasts and myotubes derived from various muscular dystrophy patients with LMNA gene mutations [89]. Lin et al. reported that R60G substitution, which causes dilated cardiomyopathy, renders this mutant protein more resistant to phosphorylation by p38 MAPK at undefined sites [150]. These studies highlight multiple ways in which pathogenic LMNA mutations could affect Lamin A/C phosphorylation. Overall, however, only a small number of studies have investigated the impact of the laminopathy mutations on lamin phosphorylation. Therefore, an unbiased survey of the phosphorylation state of Lamin A/C in tissues affected in laminopathies is warranted.
Conclusion & outlook
Our review catalogs an extensive repertoire of phosphorylation sites in nuclear lamins as well as kinases and phosphatases that regulate lamin phosphorylation (Supplementary Table 1). We recognize a wide variety of biological contexts that promote lamin phosphorylation during interphase Figure 2. Instead of phosphorylated lamins solely existing as byproducts of lamin disassembly during mitosis, an emerging hypothesis posits that phosphorylated lamins have specific functions in the nuclear interior in interphase cells. One example of such a function is the binding of Ser22-phosphorylated lamin C to the genomic regions characteristic of active enhancers near transcriptionally active genes [28]. Recent studies highlight multiple ways in which laminopathy mutations are predicted to affect lamin phosphorylation and functions of phosphorylated lamins. Thus, lamin phosphorylation presents a new avenue to investigate lamin functions in the cell and the molecular basis for the pathogenesis of laminopathies.
Our review also clarifies a number of important questions that have been left unanswered. These questions concern the biological contexts in which nuclear lamin phosphorylation is regulated by specific kinases and phosphatases; the subcellular locations at which these kinases and phosphatases operate on lamins; the functions of phosphorylated nuclear lamins at chromatin and in other cellular processes; and the mechanisms by which laminopathy mutations affect lamin phosphorylation and the functions of phosphorylated lamins. Addressing these questions will require the development of new tools and techniques, such as phosphorylation-specific antibodies, manipulation of phosphorylation states in vivo, and a proteome-wide survey of lamin phosphorylation in normal and disease tissues. Finally, it should be noted that other post-translational modifications, such as sumoylation, acetylation, and ADP-ribosylation, have been reported for lamins but not studied as extensively as phosphorylation [41,151]. We anticipate a new endeavor to characterize functions and regulatory mechanisms of various post-translational modifications of nuclear lamins in the coming years.
Supplementary Material
Acknowledgments
We thank Megan Rowton, Sebastian Pott, and Omar Almakki for critical reading of this paper. S.Y.L. and K.I. are supported by NIH R21/R33 AG054770.
Funding Statement
This work was supported by the National Institute on Aging [AG054770].
Author contributions
K.I. and S.Y.L. wrote the manuscript.
Disclosure statement
The authors declare no competing interests.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].van Steensel B, Belmont AS.. Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell. 2017;169(5):780–791. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Dechat T, Adam SA, Taimen P, et al. Nuclear lamins. Cold Spring Harb Perspect Biol. 2010;2(11):a000547. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dechat T, Pfleghaar K, Sengupta K, et al. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008;22(7):832–853. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].de Leeuw R, Gruenbaum Y, Medalia O. Nuclear lamins: thin filaments with major functions. Trends Cell Biol. 2018;28(1):34–45. [Internet]. [DOI] [PubMed] [Google Scholar]
- [5].Paddy MR, Belmont AS, Saumweber H, et al. Interphase nuclear envelope lamins form a discontinuous network that interacts with only a fraction of the chromatin in the nuclear periphery. Cell. 1990;62(1):89–106. [Internet]. [DOI] [PubMed] [Google Scholar]
- [6].Shimi T, Pfleghaar K, Kojima S-I, et al. The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes Dev. 2008;22(24):3409–3421. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Graziano S, Kreienkamp R, Coll-Bonfill N, et al. Causes and consequences of genomic instability in laminopathies: replication stress and interferon response. Nucleus. 2018;9(1):258–275. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Muchir A, Pavlidis P, Decostre V, et al. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in emery-dreifuss muscular dystrophy. J Clin Invest. 2007;117(5):1282–1293. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Muchir A, Shan J, Bonne G, et al. Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum Mol Genet. 2009;18:241–247. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kennedy BK, Pennypacker JK. RB and lamins in cell cycle regulation and aging. Adv Exp Med Biol. 2014;773:127–142. [Internet]. [DOI] [PubMed] [Google Scholar]
- [11].Lammerding J, Schulze PC, Takahashi T, et al. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest. 2004;113(3):370–378. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fedorchak GR, Kaminski A, Lammerding J. Cellular mechanosensing: getting to the nucleus of it all. Prog Biophys Mol Biol. 2014;115(2–3):76–92. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Worman HJ, Fong LG, Muchir A, et al., Laminopathies and the long strange trip from basic cell biology to therapy. J Clin Invest. 2009;119(7):1825–1836. [Internet]. Available from. : http://www.jci.org/articles/view/37679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dittmer TA, Misteli T. The lamin protein family. Genome Biol. 2011;12(5):222. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Peter A, Stick R. Evolution of the lamin protein family: what introns can tell. Nucleus. 2012;3(1):44–59. [Internet]. [DOI] [PubMed] [Google Scholar]
- [16].Kolb T, Maass K, Hergt M, et al. Lamin A and lamin C form homodimers and coexist in higher complex forms both in the nucleoplasmic fraction and in the lamina of cultured human cells. Nucleus. 2011;2(5):425–433. [Internet]. [DOI] [PubMed] [Google Scholar]
- [17].Röber RA, Weber K, Osborn M. Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study. Development. 1989;105:365–378. [Internet]. Available from. : https://www.ncbi.nlm.nih.gov/pubmed/2680424 [DOI] [PubMed] [Google Scholar]
- [18].Stewart C, Burke B. Teratocarcinoma stem cells and early mouse embryos contain only a single major lamin polypeptide closely resembling lamin B. Cell. 1987;51(3):383–392. [Internet]. [DOI] [PubMed] [Google Scholar]
- [19].Eckersley-Maslin MA, Bergmann JH, Lazar Z, et al. Lamin A/C is expressed in pluripotent mouse embryonic stem cells. Nucleus. 2013;4(1):53–60. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Constantinescu D, Gray HL, Sammak PJ, et al. Lamin A/C expression is a marker of mouse and human embryonic stem cell differentiation. Stem Cells. 2006;24:177–185. [Internet]. [DOI] [PubMed] [Google Scholar]
- [21].Schatten G, Maul GG, Schatten H, et al. Nuclear lamins and peripheral nuclear antigens during fertilization and embryogenesis in mice and sea urchins. Proc Natl Acad Sci U S A. 1985;82(14):4727–4731. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lehner CF, Stick R, Eppenberger HM, et al. Differential expression of nuclear lamin proteins during chicken development. J Cell Biol. 1987;105(1):577–587. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Naetar N, Ferraioli S, Foisner R. Lamins in the nuclear interior - life outside the lamina. J Cell Sci. 2017;130(13):2087–2096. Internet. [DOI] [PubMed] [Google Scholar]
- [24].Hozák P, Sasseville AM, Raymond Y, et al. Lamin proteins form an internal nucleoskeleton as well as a peripheral lamina in human cells J Cell Sci. 1995;108(Pt 2):635–644. [Internet]. Available from. : https://www.ncbi.nlm.nih.gov/pubmed/7769007. [DOI] [PubMed] [Google Scholar]
- [25].Neri LM, Raymond Y, Giordano A, et al. Lamin A is part of the internal nucleoskeleton of human erythroleukemia cells. J Cell Physiol. 1999;178(3):284–295. [Internet]. [DOI] [PubMed] [Google Scholar]
- [26].Barboro P, D’Arrigo C, Diaspro A, et al., Unraveling the organization of the internal nuclear matrix: RNA-dependent anchoring of NuMA to a lamin scaffold. Exp Cell Res. 2002;279(2):202–218. [Internet]. Available from. : https://www.ncbi.nlm.nih.gov/pubmed/12243746 [DOI] [PubMed] [Google Scholar]
- [27].Kochin V, Shimi T, Torvaldson E, et al. Interphase phosphorylation of lamin A. J Cell Sci. 2014;127(12):2683–2696. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ikegami K, Secchia S, Almakki O, et al. Phosphorylated Lamin A/C in the Nuclear Interior Binds Active Enhancers Associated with Abnormal Transcription in Progeria. Dev Cell. 2020;52(6):699–713.e11. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Swift J, Ivanovska IL, Buxboim A, et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341(6149):1240104. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Broers JL, Machiels BM, van Eys GJ, et al. Dynamics of the nuclear lamina as monitored by GFP-tagged A-type lamins J Cell Sci. 1999;112(Pt 20):3463–3475. [Internet]. Available from. : http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=10504295&retmode=ref&cmd=prlinks. [DOI] [PubMed] [Google Scholar]
- [31].Peter M, Nakagawa J, Dorée M, et al., In vitro disassembly of the nuclear lamina and M phase-specific phosphorylation of lamins by cdc2 kinase. Cell. 1990;61(4):591–602. [Internet]. Available from. : https://www.ncbi.nlm.nih.gov/pubmed/2188731 [DOI] [PubMed] [Google Scholar]
- [32].Heald R, McKeon F, Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell. 1990;61(4):579–589. [Internet]. Available from. : https://www.ncbi.nlm.nih.gov/pubmed/2344612 [DOI] [PubMed] [Google Scholar]
- [33].Ward GE, Kirschner MW, Identification of cell cycle-regulated phosphorylation sites on nuclear lamin C. Cell. 1990;61(4):561–577. [Internet]. Available from. : https://www.ncbi.nlm.nih.gov/pubmed/2188730 [DOI] [PubMed] [Google Scholar]
- [34].Gerace L, Blobel G, The nuclear envelope lamina is reversibly depolymerized during mitosis. Cell. 1980;19(1):277–287. [Internet]. Available from. : https://www.ncbi.nlm.nih.gov/pubmed/7357605 [DOI] [PubMed] [Google Scholar]
- [35].Eggert M, Radomski N, Linder D, et al. Identification of novel phosphorylation sites in murine A-type lamins. Eur J Biochem. 1993;213(2):659–671. [Internet]. [DOI] [PubMed] [Google Scholar]
- [36].Parry DA, Conway JF, Steinert PM. Structural studies on lamin. Similarities and differences between lamin and intermediate-filament proteins. Biochem J. 1986;238(1):305–308. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fisher DZ, Chaudhary N, Blobel G. cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc Natl Acad Sci U S A. 1986;83(17):6450–6454. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Stuurman N, Heins S, Aebi U. Nuclear lamins: their structure, assembly, and interactions. J Struct Biol. 1998;122(1–2):42–66. [Internet]. [DOI] [PubMed] [Google Scholar]
- [39].Dhe-Paganon S, Werner ED, Chi Y-I, et al. Structure of the globular tail of nuclear lamin. J Biol Chem. 2002;277(20):17381–17384. [Internet]. [DOI] [PubMed] [Google Scholar]
- [40].Krimm I, Ostlund C, Gilquin B, et al. The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure. 2002;10(6):811–823. [Internet]. [DOI] [PubMed] [Google Scholar]
- [41].Simon DN, Wilson KL. Partners and post-translational modifications of nuclear lamins. Chromosoma. 2013;122:13–31. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Heitlinger E, Peter M, Lustig A, et al. The role of the head and tail domain in lamin structure and assembly: analysis of bacterially expressed chicken lamin A and truncated B2 lamins. J Struct Biol. 1992;108(1):74–89. [Internet]. [DOI] [PubMed] [Google Scholar]
- [43].Heitlinger E, Peter M, Häner M, et al. Expression of chicken lamin B2 in Escherichia coli: characterization of its structure, assembly, and molecular interactions. J Cell Biol. 1991;113(3):485–495. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Turgay Y, Eibauer M, Goldman AE, et al. The molecular architecture of lamins in somatic cells. Nature. 2017;543(7644):261–264. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ottaviano Y, Gerace L. Phosphorylation of the nuclear lamins during interphase and mitosis. J Biol Chem. 1985;260:624–632. [Internet]. Available from. : https://www.ncbi.nlm.nih.gov/pubmed/3965465 [PubMed] [Google Scholar]
- [46].Gerace L, Comeau C, Benson M. Organization and modulation of nuclear lamina structure. J Cell Sci Suppl. 1984;1(Supplement 1):137–160. [Internet]. [DOI] [PubMed] [Google Scholar]
- [47].Peter M, Heitlinger E, Häner M, et al., Disassembly of in vitro formed lamin head-to-tail polymers by CDC2 kinase. Embo J. 1991;10(6):1535–1544. [Internet]. Available from. : https://www.ncbi.nlm.nih.gov/pubmed/1851086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Heald R, McKeon F. Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell. 1990;61(4):579–589. [Internet]. [DOI] [PubMed] [Google Scholar]
- [49].Nigg EA. Targets of cyclin-dependent protein kinases. Curr Opin Cell Biol. 1993;5(2):187–193. [Internet]. [DOI] [PubMed] [Google Scholar]
- [50].Olsen JV, Vermeulen M, Santamaria A, et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 2010;3(104):ra3. [Internet]. [DOI] [PubMed] [Google Scholar]
- [51].Georgatos SD, Pyrpasopoulou A, Theodoropoulos PA. Nuclear envelope breakdown in mammalian cells involves stepwise lamina disassembly and microtubule-drive deformation of the nuclear membrane J Cell Sci. 1997;110(Pt 17):2129–2140. [Internet]. Available from: https://www.ncbi.nlm.nih.gov/pubmed/9378763. [DOI] [PubMed] [Google Scholar]
- [52].Lee KK, Haraguchi T, Lee RS, et al. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J Cell Sci. 2001;114:4567–4573. [Internet]. Available from: https://www.ncbi.nlm.nih.gov/pubmed/11792821 [DOI] [PubMed] [Google Scholar]
- [53].Haraguchi T, Kojidani T, Koujin T, et al. Live cell imaging and electron microscopy reveal dynamic processes of BAF-directed nuclear envelope assembly. J Cell Sci. 2008;121(15):2540–2554. [Internet]. [DOI] [PubMed] [Google Scholar]
- [54].Moriuchi T, Kuroda M, Kusumoto F, et al. Lamin a reassembly at the end of mitosis is regulated by its SUMO-interacting motif. Exp Cell Res. 2016;342(1):83–94. [Internet]. [DOI] [PubMed] [Google Scholar]
- [55].Trinkle-Mulcahy L, Andrews PD, Wickramasinghe S, et al. Time-lapse imaging reveals dynamic relocalization of PP1gamma throughout the mammalian cell cycle. Mol Biol Cell. 2003;14(1):107–117. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tsai M-Y, Wang S, Heidinger JM, et al. A mitotic lamin B matrix induced by RanGTP required for spindle assembly. Science. 2006;311(5769):1887–1893. [Internet]. [DOI] [PubMed] [Google Scholar]
- [57].Kuga T, Nozaki N, Matsushita K, et al. Phosphorylation statuses at different residues of lamin B2, B1, and A/C dynamically and independently change throughout the cell cycle. Exp Cell Res. 2010;316(14):2301–2312. [Internet]. [DOI] [PubMed] [Google Scholar]
- [58].Boubriak II, Malhas AN, Drozdz MM, et al. Stress-induced release of Oct- 1from the nuclear envelope is mediated by JNK phosphorylation of lamin B1. PLoS One. 2017;12(5):e0177990. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ochoa D, Jarnuczak AF, Viéitez C, et al. The functional landscape of the human phosphoproteome. Nat Biotechnol. 2020;38(3):365–373. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Olsen JV, Blagoev B, Gnad F, et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127(3):635–648. [Internet]. [DOI] [PubMed] [Google Scholar]
- [61].Rigbolt KTG, Prokhorova TA, Akimov V, et al. System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci Signal. 2011;4(164):rs3. [Internet]. [DOI] [PubMed] [Google Scholar]
- [62].Moritz A, Li Y, Guo A, et al. Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci Signal. 2010;3(136):ra64. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ren L, Li C, Shao W, et al. TiO2 with tandem fractionation (TAFT): an approach for rapid, deep, reproducible, and high-throughput phosphoproteome analysis. J Proteome Res. 2018;17(1):710–721. [Internet]. [DOI] [PubMed] [Google Scholar]
- [64].Lee C-P, Huang Y-H, Lin S-F, et al. Epstein-Barr virus BGLF4 kinase induces disassembly of the nuclear lamina to facilitate virion production. J Virol. 2008;82(23):11913–11926. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Haas M, Jost E. Functional analysis of phosphorylation sites in human lamin A controlling lamin disassembly, nuclear transport and assembly. Eur J Cell Biol. 1993;62:237–247. [Internet]. Available from: https://www.ncbi.nlm.nih.gov/pubmed/7925482 [PubMed] [Google Scholar]
- [66].Güttinger S, Laurell E, Kutay U. Orchestrating nuclear envelope disassembly and reassembly during mitosis. Nat Rev Mol Cell Biol. 2009;10:178–191. [Internet]. [DOI] [PubMed] [Google Scholar]
- [67].Nmezi B, Xu J, Fu R, et al. Concentric organization of A- and B-type lamins predicts their distinct roles in the spatial organization and stability of the nuclear lamina. Proc Natl Acad Sci U S A. 2019;116(10):4307–4315. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Georgatos SD, Pyrpasopoulou A, Theodoropoulos PA. Nuclear envelope breakdown in mammalian cells involves stepwise lamina disassembly and microtubule-drive deformation of the nuclear membrane J Cell Sci. 1997;110(Pt 17):2129–2140. [Internet]. Available from. : http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=9378763&retmode=ref&cmd=prlinks. [DOI] [PubMed] [Google Scholar]
- [69].Chen J-T, Ho C-W, Chi L-M, et al. Identification of the lamin A/C phosphoepitope recognized by the antibody P-STM in mitotic HeLa S3 cells. BMC Biochem. 2013;14(1):18. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Goss VL, Hocevar BA, Thompson LJ, et al. Identification of nuclear beta II protein kinase C as a mitotic lamin kinase. J Biol Chem. 1994;269:19074–19080. [Internet]. Available from: https://www.ncbi.nlm.nih.gov/pubmed/8034666 [PubMed] [Google Scholar]
- [71].Jackman M, Firth M, Pines J, Human cyclins B1 and B2 are localized to strikingly different structures: B1 to microtubules, B2 primarily to the Golgi apparatus. Embo J. 1995;14(8):1646–1654. [Internet]. Available from: https://www.ncbi.nlm.nih.gov/pubmed/7737117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Jackman M, Lindon C, Nigg EA, et al. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat Cell Biol. 2003;5(2):143–148. [Internet]. [DOI] [PubMed] [Google Scholar]
- [73].Mall M, Walter T, Gorjánácz M, et al. Mitotic lamin disassembly is triggered by lipid-mediated signaling. J Cell Biol. 2012;198(6):981–990. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Lohka MJ, Hayes MK, Maller JL. Purification of maturation-promoting factor, an intracellular regulator of early mitotic events. Proc Natl Acad Sci U S A. 1988;85(9):3009–3013. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Black AR, Black JD. Protein kinase C signaling and cell cycle regulation. Front Immunol. 2012;3:423. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kauko O, Imanishi SY, Kulesskiy E, et al. Phosphoproteome and drug-response effects mediated by the three protein phosphatase 2A inhibitor proteins CIP2A, SET, and PME-1. J Biol Chem. 2020;295(13):4194–4211. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Thompson LJ, Bollen M, Fields AP. Identification of protein phosphatase 1 as a mitotic lamin phosphatase. J Biol Chem. 1997;272(47):29693–29697. [Internet]. [DOI] [PubMed] [Google Scholar]
- [78].Schmitz MHA, Held M, Janssens V, et al. Live-cell imaging RNAi screen identifies PP2A-B55alpha and importin-beta1 as key mitotic exit regulators in human cells. Nat Cell Biol. 2010;12(9):886–893. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Steen RL, Martins SB, Taskén K, et al. Recruitment of protein phosphatase 1 to the nuclear envelope by A-kinase anchoring protein AKAP149 is a prerequisite for nuclear lamina assembly. J Cell Biol. 2000;150:1251–1262. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Nilsson J. Protein phosphatases in the regulation of mitosis. J Cell Biol. 2019;218(2):395–409. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Printen JA, Brady MJ, Saltiel AR. PTG, a protein phosphatase 1-binding protein with a role in glycogen metabolism. Science. 1997;275(5305):1475–1478. [Internet]. [DOI] [PubMed] [Google Scholar]
- [82].Oaks J, Ogretmen B. Regulation of PP2A by sphingolipid metabolism and signaling. Front Oncol. 2014;4:388. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Freeman AK, Monteiro AN. Phosphatases in the cellular response to DNA damage. Cell Commun Signal. 2010;8(1):27. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Carlson SM, Chouinard CR, Labadorf A, et al. Large-scale discovery of ERK2 substrates identifies ERK-mediated transcriptional regulation by ETV3. Sci Signal. 2011;4(196):rs11. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].González JM, Navarro-Puche A, Casar B, et al. Fast regulation of AP-1 activity through interaction of lamin A/C, ERK1/2, and c-Fos at the nuclear envelope. J Cell Biol. 2008;183(4):653–666. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Buxboim A, Swift J, Irianto J, et al. Matrix elasticity regulates lamin-A,C phosphorylation and turnover with feedback to actomyosin. Curr Biol. 2014;24(16):1909–1917. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–1274. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Cenni V, Bertacchini J, Beretti F, et al. Lamin A Ser404 is a nuclear target of Akt phosphorylation in C2C12 cells. J Proteome Res. 2008;7(11):4727–4735. [Internet]. [DOI] [PubMed] [Google Scholar]
- [89].Cenni V, Sabatelli P, Mattioli E, et al. Lamin A N-terminal phosphorylation is associated with myoblast activation: impairment in Emery-Dreifuss muscular dystrophy. J Med Genet. 2005;42(3):214–220. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Mitsuhashi H, Hayashi YK, Matsuda C, et al. Specific phosphorylation of Ser458 of A-type lamins in LMNA-associated myopathy patients. J Cell Sci. 2010;123(22):3893–3900. [Internet]. [DOI] [PubMed] [Google Scholar]
- [91].Cross T, Griffiths G, Deacon E, et al. PKC-delta is an apoptotic lamin kinase. Oncogene. 2000;19(19):2331–2337. [Internet]. [DOI] [PubMed] [Google Scholar]
- [92].Shimizu T, Cao CX, Shao RG, et al. Lamin B phosphorylation by protein kinase calpha and proteolysis during apoptosis in human leukemia HL60 cells. J Biol Chem. 1998;273(15):8669–8674. [Internet]. [DOI] [PubMed] [Google Scholar]
- [93].Collas P, Thompson L, Fields AP, et al. Protein kinase C-mediated interphase lamin B phosphorylation and solubilization. J Biol Chem. 1997;272(34):21274–21280. [Internet]. [DOI] [PubMed] [Google Scholar]
- [94].Sharma S, Sicinski P. A kinase of many talents: non-neuronal functions of CDK5 in development and disease. Open Biol. 2020;10(1):190287. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Chang K-H, Multani PS, Sun K-H, et al. Nuclear envelope dispersion triggered by deregulated Cdk5 precedes neuronal death. Mol Biol Cell. 2011;22(9):1452–1462. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Blasius M, Forment JV, Thakkar N, et al. A phospho-proteomic screen identifies substrates of the checkpoint kinase Chk1. Genome Biol. 2011;12(8):R78. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Kumar A, Mazzanti M, Mistrik M, et al. ATR mediates a checkpoint at the nuclear envelope in response to mechanical stress. Cell. 2014;158(3):633–646. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Hamirally S, Kamil JP, Ndassa-Colday YM, et al. Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog. 2009;5(1):e1000275. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Milbradt J, Hutterer C, Bahsi H, et al. The prolyl isomerase pin1 promotes the herpesvirus-induced phosphorylation-dependent disassembly of the nuclear lamina required for nucleocytoplasmic egress. PLoS Pathog. 2016;12(8):e1005825. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Mou F, Forest T, Baines JD. US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J Virol. 2007;81(12):6459–6470. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Bruston F, Delbarre E, Ostlund C, et al. Loss of a DNA binding site within the tail of prelamin A contributes to altered heterochromatin anchorage by progerin. FEBS Lett. 2010;584(14):2999–3004. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Guelen L, Pagie L, Brasset E, et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453(7197):948–951. [Internet]. [DOI] [PubMed] [Google Scholar]
- [103].Meuleman W, Peric-Hupkes D, Kind J, et al. Constitutive nuclear lamina-genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res. 2012;23(2):270–280. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Ikegami K, Egelhofer TA, Strome S, et al. Caenorhabditis elegans chromosome arms are anchored to the nuclear membrane via discontinuous association with LEM-2. Genome Biol. 2010;11(12):R120. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Lund E, Oldenburg AR, Collas P. Enriched domain detector: a program for detection of wide genomic enrichment domains robust against local variations. Nucleic Acids Res. 2014;42(11):e92. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Pickersgill H, Kalverda B, de Wit E, et al. Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat Genet. 2006;38(9):1005–1014. [Internet]. [DOI] [PubMed] [Google Scholar]
- [107].Leemans C, van der Zwalm MCH, Brueckner L, et al. Promoter-intrinsic and local chromatin features determine gene repression in LADs. Cell. 2019;177(4):852–64.e14. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Reddy KL, Zullo JM, Bertolino E, et al. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature. 2008;452(7184):243–247. [Internet]. [DOI] [PubMed] [Google Scholar]
- [109].Finlan LE, Sproul D, Thomson I, et al. Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet. 2008;4(3):e1000039. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Johnson GL, Nakamura K. The c-jun kinase/stress-activated pathway: regulation, function and role in human disease. Biochim Biophys Acta. 2007;1773(8):1341–1348. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Ivorra C, Kubicek M, González JM, et al. A mechanism of AP-1 suppression through interaction of c-Fos with lamin A/C. Genes Dev. 2006;20(3):307–320. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Lund EG, Duband-Goulet I, Oldenburg A, et al. Distinct features of lamin A-interacting chromatin domains mapped by ChIP-sequencing from sonicated or micrococcal nuclease-digested chromatin. Nucleus. 2015;6(1):30–39. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Gesson K, Rescheneder P, Skoruppa MP, et al. A-type lamins bind both hetero- and euchromatin, the latter being regulated by lamina-associated polypeptide 2 alpha. Genome Res. 2016;26(4):462–473. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Pascual-Reguant L, Blanco E, Galan S, et al. Lamin B1 mapping reveals the existence of dynamic and functional euchromatin lamin B1 domains. Nat Commun. 2018;9(1):3420. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Dechat T, Gotzmann J, Stockinger A, et al. Detergent–salt resistance of LAP2α in interphase nuclei and phosphorylation-dependent association with chromosomes early in nuclear assembly implies functions in nuclear structure dynamics. Embo J. 1998;17(16):4887–4902. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Dechat T, Korbei B, Vaughan OA, et al. Lamina-associated polypeptide 2alpha binds intranuclear A-type lamins. J Cell Sci. 2000;113 Pt 19:3473–3484. [Internet]. Available from: https://www.ncbi.nlm.nih.gov/pubmed/10984438 [DOI] [PubMed] [Google Scholar]
- [117].Gesson K, Vidak S, Foisner R. Lamina-associated polypeptide (LAP)2α and nucleoplasmic lamins in adult stem cell regulation and disease. Semin Cell Dev Biol. 2014;29:116–124. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Naetar N, Ferraioli S, Foisner R, Lamins in the nuclear interior − life outside the lamina. J Cell Sci. 2017;130(13):2087–2096. [Internet]. Available from: https://jcs.biologists.org/content/130/13/2087.short [DOI] [PubMed] [Google Scholar]
- [119].Naetar N, Korbei B, Kozlov S, et al. Loss of nucleoplasmic LAP2α–lamin A complexes causes erythroid and epidermal progenitor hyperproliferation. Nat Cell Biol. 2008;10(11):1341–1348. [Internet]. [DOI] [PubMed] [Google Scholar]
- [120].Naetar N, Foisner R. Lamin complexes in the nuclear interior control progenitor cell proliferation and tissue homeostasis. Cell Cycle. 2009;8(10):1488–1493. [Internet]. [DOI] [PubMed] [Google Scholar]
- [121].Markiewicz E, Dechat T, Foisner R, et al. Lamin A/C binding protein LAP2α Is required for nuclear anchorage of retinoblastoma protein. MBoC. 2002;13(12):4401–4413. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Dorner D, Vlcek S, Foeger N, et al. Lamina-associated polypeptide 2alpha regulates cell cycle progression and differentiation via the retinoblastoma-E2F pathway. J Cell Biol. 2006;173(1):83–93. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25(38):5220–5227. [Internet]. [DOI] [PubMed] [Google Scholar]
- [124].Johnson BR, Nitta RT, Frock RL, et al. A-type lamins regulate retinoblastoma protein function by promoting subnuclear localization and preventing proteasomal degradation. Proc Natl Acad Sci U S A. 2004;101(26):9677–9682. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Frock RL, Kudlow BA, Evans AM, et al. Lamin A/C and emerin are critical for skeletal muscle satellite cell differentiation. Genes Dev. 2006;20(4):486–500. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Kang S-M, Yoon M-H, Park B-J. Laminopathies; Mutations on single gene and various human genetic diseases. BMB Rep. 2018;51(7):327–337. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Worman HJ, Bonne G, “Laminopathies”: a wide spectrum of human diseases. Exp Cell Res. 2007;313(10):2121–2133. [Internet]. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0014482707001279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Kim Y, Zheng Y, Generation and characterization of a conditional deletion allele for Lmna in mice. Biochem Biophys Res Commun. 2013;440(1):8–13. [Internet]. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0006291X13014290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Kubben N, Voncken JW, Konings G, et al. Post-natal myogenic and adipogenic developmental: defects and metabolic impairment upon loss of A-type lamins. Nucleus. 2011;2(3):195–207. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Lee JM, Jung H-J, Fong LG, et al. Do lamin B1 and lamin B2 have redundant functions? Nucleus. 2014;5(4):287–292. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Lu JT, Muchir A, Nagy PL, et al. LMNA cardiomyopathy: cell biology and genetics meet clinical medicine. Dis Model Mech. 2011;4(5):562–568. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Osmanagic-Myers S, Foisner R, Kozminski KG. The structural and gene expression hypotheses in laminopathic diseases-not so different after all. Mol Biol Cell. 2019;30(15):1786–1790. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Maraldi NM, Capanni C, Cenni V, et al. Laminopathies and lamin-associated signaling pathways. J Cell Biochem. 2011;112(4):979–992. [Internet]. [DOI] [PubMed] [Google Scholar]
- [134].Gonzalo S. DNA damage and lamins. Adv Exp Med Biol. 2014;773:377–399. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Burla R, La Torre M, Saggio I. Mammalian telomeres and their partnership with lamins. Nucleus. 2016;7(2):187–202. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Vigouroux C, Bonne G. Laminopathies: one gene, two proteins, five diseases. In: Madame Curie Bioscience Database. [Internet]. cited 2020 May19. Austin (TX): Landes Bioscience; 2000–2013. Available from: https://www.ncbi.nlm.nih.gov/books/NBK6151/ [Google Scholar]
- [137].Osmanagic-Myers S, Dechat T, Foisner R. Lamins at the crossroads of mechanosignaling. Genes Dev. 2015;29(3):225–237. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Eriksson M, Brown WT, Gordon LB, et al. Recurrent de novo point mutations in lamin A cause hutchinson-gilford progeria syndrome. Nature. 2003;423(6937):293–298. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Gordon LB, Rothman FG, López-Otín C, et al. Progeria: a paradigm for translational medicine. Cell. 2014;156(3):400–407. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].McCord RP, Nazario-Toole A, Zhang H, et al. Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res. 2012;23(2):260–269. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Shumaker DK, Dechat T, Kohlmaier A, et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A. 2006;103(23):8703–8708. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Lee J, Termglinchan V, Diecke S, et al. Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature. 2019;572(7769):335–340. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Bertero A, Fields PA, Smith AST, et al. Chromatin compartment dynamics in a haploinsufficient model of cardiac laminopathy. J Cell Biol. 2019;218(9):2919–2944. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Lee S-J, Jung Y-S, Yoon M-H, et al. Interruption of progerin-lamin A/C binding ameliorates Hutchinson-Gilford progeria syndrome phenotype. J Clin Invest. 2016;126(10):3879–3893. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Hussain I, Patni N, Ueda M, et al. A novel generalized lipodystrophy-associated progeroid syndrome due to recurrent heterozygous LMNA p.T10I mutation. J Clin Endocrinol Metab. 2018;103:1005–1014. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Pethig K, Genschel J, Peters T, et al. LMNA mutations in cardiac transplant recipients. Cardiology. 2005;103(2):57–62. [Internet]. [DOI] [PubMed] [Google Scholar]
- [147].Ruggiero L, Fiorillo C, Tessa A, et al. Muscle fiber type disproportion (FTD) in a family with mutations in the LMNA gene. Muscle Nerve. 2015;51(4):604–608. [Internet]. [DOI] [PubMed] [Google Scholar]
- [148].Kirschner J, Brune T, Wehnert M, et al. p.S143F mutation in lamin A/C: a new phenotype combining myopathy and progeria. Ann Neurol. 2005;57(1):148–151. [Internet]. [DOI] [PubMed] [Google Scholar]
- [149].Bonne G, Mercuri E, Muchir A, et al. Clinical and molecular genetic spectrum of autosomal dominant emery-dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann Neurol. 2000;48(2):170–180. [Internet]. Available from: https://www.ncbi.nlm.nih.gov/pubmed/10939567 [PubMed] [Google Scholar]
- [150].Lin EW, Brady GF, Kwan R, et al. Genotype-phenotype analysis of LMNA-related diseases predicts phenotype-selective alterations in lamin phosphorylation. Faseb J. 2020;347:9051–9073. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Snider NT, Omary MB. Post-translational modifications of intermediate filament proteins: mechanisms and functions. Nat Rev Mol Cell Biol. 2014;15(3):163–177. [Internet]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.