

Abstract
Autoimmune diseases affect 10% of the world’s population, and 1 in 200 people worldwide suffer from either multiple sclerosis (MS) or type 1 diabetes (T1D). While the targeted organ systems are different, MS and T1D share similarities in terms of autoreactive immune cells playing a critical role in pathogenesis. Both diseases can be managed only symptomatically without curative remission, and treatment options are limited and non-specific. Most current therapies cause some degree of systemic immune suppression, leaving the patients susceptible to opportunistic infections and other complications. Thus, there is considerable interest in the development of immunotherapies not associated with generalized immune suppression for these diseases. In this review we present current and preclinical strategies for MS and T1D treatment, emphasizing those aimed to modulate the immune response, including the most recent strategies for tolerance induction. A central focus is on the emerging approaches using nano- and microparticle (NMP) platforms, their evolution as immunotherapeutic carriers, including those incorporating specific antigens to induce tolerance and reduce unwanted generalized immune suppression.
Keywords: Multiple sclerosis, experimental autoimmune encephalomyelitis, type 1 diabetes: immunotherapy, drug delivery, immune modulation, immune engineering, microparticles, nanoparticles, antigen specific
Multiple Sclerosis Burden, Pathology and Clinical Approaches
MS is an autoimmune disease that affects the central nervous system (CNS), has onset typically in the early 30s, and a higher prevalence in women than men [1–3]. Approximately 1 in 250 people are diagnosed with MS in the United States alone, and there has been a 5-fold increase in the prevalence of MS since 1976 [4]. According to the National Multiple Sclerosis Society currently nearly one million people are living with MS in the United States and 2.3 million in the world. MS pathology consists of autoimmune-mediated demyelination whereby immune cells destroy the myelin sheath that surrounds axons and kill myelin producing oligodendrocytes [5, 6]. This leads to clinical symptoms including numbness, tingling, fatigue, and eventually paralysis [7]. Relapsing-remitting multiple sclerosis (RRMS), where clinical disease manifests as a series of relapses and remissions with worsening from baseline subsequent to each relapse, is the most common form of MS, which is diagnosed in over 85% of patients. In primary progressive MS (PPMS), symptoms worsen rapidly following disease onset [7]. The pathogenesis of MS has been partially elucidated. Initially, the blood brain barrier becomes permeable, which is a step involved in immune cell infiltration into the CNS as blood brain barrier permeability is associated with increased trans-endothelial migration of activated immune cells [8]. Although the mechanism leading to permeability is not clear, inflammatory cytokines produced by CNS resident cells are associated with this process through the disruption of cell-cell junctions [9]. To this end, the chemokine receptors CCR2, CCR5 and CCR6 have been linked to the migration of immune cells into the CNS [7]. This infiltration of immune cells results in white matter lesions, that expand with each relapse [7]. It is known that CNS destruction is mediated by proinflammatory T cells, macrophages, activated microglia and astrocytes, with B cells playing a role as well [5]. Resident CNS astrocytes and microglia contribute to disease progression by the production of inflammatory cytokines and neurotoxic factors. The phenotype of infiltrating immune cells varies based on how far the disease has progressed, with higher levels of T cells and B cells early in disease and “a smoldering inflammation”, resulting in the development of tertiary lymphoid structures with activated microglia/macrophages in the CNS during chronic stages [7]. In the CNS microglia, recruited macrophages, dendritic cells (DCs) and B cells present autoantigen to T cells [10, 11]. The CD4+ T cells are typically T helper 17 (Th17) and Th1, and react to autoantigens that are part of the CNS such as myelin oligodendrocyte glycoprotein (MOG) and myelin basic protein (MBP). Th17 cells express the transcription factor RORγt and produce the proinflammatory cytokine interleukin-17a (IL-17a), while Th1 cells express Tbet and produce the proinflammatory cytokine IFN-γ. Numerous T helper cells express both RORγt and Tbet and produce IL17 and IFN-γ as well as GM-CSF [12–17]. T helper cell toxicity can be direct, through the release of neurotoxic cytokines or indirect by activation of macrophages [7]. CD8 T cells can secrete inflammatory cytokines such as GMCSF or directly kill oligodendrocytes through a granzyme b mediated mechanism [7]. The Th1 and Th17 response is hypothesized to be the driving force behind the preclinical mouse model of MS, experimental autoimmune encephalomyelitis (EAE), in a manner consistent with human MS [17, 18]. This is corroborated by the fact that defects in Th1 and Th17 cells prevent EAE onset [15, 16].
Therapeutic options for MS are minimal and there is no cure. Corticosteroids, such as methylprednisolone, can provide transient relief of inflammation during relapse. However, broad immunosuppressants are not viable for long-term management due to poor tolerability, ineffective disease control and susceptibility to opportunistic infections [19–22]. Other approaches seek to limit trafficking of immune cells into the CNS, either by decreasing blood brain barrier trafficking or preventing egress from secondary lymphoid organs [20, 23–26]. Treatment with interferon β, which has known immunoregulatory properties, can be given alone or in conjunction with glatiramer acetate, a random mixture of synthetic peptides highly represented in MBP. Interferon-β treatment results in decreased lymphocyte trafficking across the blood brain barrier and glatiramer acetate binds to MHC II, competing with presentation of actual myelin antigens [23, 27, 28]. However, response rate is low, with only a 30–50% reduction in relapse rate [24, 29]. Natalizumab, a monoclonal antibody targeting integrin α4 is another treatment which impedes leukocyte trafficking into the CNS and resulted in one third the number of relapsed compared to placebo control and 20% relapse rate overall [25, 30]. Conversely, Fingolimod (FTY720), a sphingosine-1-phosphate receptor inhibitor, blocks egress from lymph nodes, stopping autoreactive cells from trafficking to the CNS and treatment dropped relapse rate to 15% over a two year study [26, 31]. However, preventing trafficking with Natalizumab or Fingolimod may cause progressive multifocal leukoencephalopathy, a life-threatening opportunistic viral infection of the CNS [21, 32]. Among therapies with anti-CD20 antibodies, which deplete circulating immature and mature B cells, but not plasma cells, Ocrelizumab, a humanized monoclonal antibody, showed success, including in slowing MS disease progression as relapse rate was 46% lower than in IFN-β treatment [33]. However, Ocrelizumab increases the risk of upper respiratory infections 40% vs 33% over IFN- β treatment, oral herpes virus 2.3% vs 0.4% in placebo, and risk of breast cancer was 2.3% compared to 0.8% in placebo group [34]. Additional antibody therapies have shown promise in treatment of MS, including anti-CD52 (B and T cell depletion), and anti-CD25 (targets IL-2 receptor and Treg cells), but all have side effects related to immune suppression, such as infections in the brain [19, 35, 36]. In terms of antigen-specific treatments, a clinical trial was conducted attempting to treat with MBP83–99, however the trial was halted after disease worsened in some patients [37].
Type 1 Diabetes Burden, Pathology and Clinical Approaches
In T1D autoimmunity often manifests in early childhood, with 490,000 children with T1D under the age of 15 worldwide. Less frequently, onset occurs later in adulthood [38, 39]. Disease incidence is increasing, with current estimates suggesting approximately 40,000 new cases annually in the United States alone [39]. T1D is characterized by hyperglycemia due to lack of insulin, which leads to clinical manifestations such as polyuria, polydipsia, mental obtundation, weight loss, nausea, vomiting, abdominal pain, and fatigue. Sequelae of chronic hyperglycemic state include diabetic neuropathy, retinopathy, nephropathy, ulcers, and vasculopathies that can ultimately end with amputation. Injection of exogenous insulin is the primary treatment used to manage T1D [40]. Although insulin supplementation therapy affords moderate disease management, T1D patients experience a number of comorbid complications, which include chronic, potentially life-threatening, kidney disease in 30% of patients, and a 10 times higher risk of developing cardiovascular diseases [41–43].
T1D is classically characterized by CD4+ and CD8+ T cell mediated destruction of insulin-producing β-cells in the pancreas. The innate immune system also plays a role in the pathogenesis, as macrophages and DCs have been observed surrounding the pancreatic islets and present antigen to autoreactive T cells [40]. Stage 1 is associated with the presence of autoreactive cells and β-cell loss, stage 2, with autoreactive cells, β-cell loss and hyperglycemia, and stage 3 with autoreactive cells, β-cell loss, hyperglycemia and clinical symptoms. Stage 1 and 2 can last for years before symptomatic presentation, making it difficult to detect disease before critical β-cell loss from autoimmune attack. Awareness of genetic risk factors and advances in diagnostic procedures have made preventative treatments conceivable before the destruction of a critical mass of β-cells [44]. In particular, children with the HLA-DR4-DQ8 and HLA-DR3-DQ2 haplotypes are more likely to generate autoantibodies for insulin and glutamic acid decarboxylase 65-kilodalton isoform (GAD65) [40, 41].
Therapeutic options.
To date, there is no cure for T1D. However, a myriad of immunological therapies have been explored with limited success. Numerous clinical trials have sought to establish immune tolerance by repeated administration of T1D autoantigen, often employing the primary autoantigen insulin [45–48]. Other treatment options include monoclonal antibodies, which have been used in multiple clinical trials, including anti-CD20 [49] and anti-CD3 (T cell depletion) [50]. In the clinical trial using anti-CD20, patients were diagnosed with T1D if one circulating autoantibody was present and treatment was initiated 90 days following diagnosis with four total treatments over a one year timeframe. One year following anti-CD20 therapy, C peptide level increased to 0.56 pmol/mL compared to 0.47 pmol/mL in placebo control and patients required lower levels of exogenous insulin than placebo group [49]. The study using anti-CD3 also treated newly diabetic patients defined by the presence of autoantibodies and the need for injection of insulin, with onset of treatment began within 12 weeks of diagnosis. Treatment resulted in slower decline in C-peptide level compared to placebo control with with 40% patients who received anti-CD3 having a preservation of baseline C-peptide levels [50]. Other immunomodulatory therapies included an antibody hybrid consisting of a fusion of anti-CTLA4 (coinhibition) to the Fc region [51]. Similar to anti-CD20 and anti-CD3 studies patients were recently diagnosed with T1D via autoantibody levels and were treated at approximately 90 days post diagnosis. Results of this study showed a delayed reduction in C-peptide level with 32% of patients below the threshold of 0.2nmol/L compared to 43% in placebo control [51]. Additionally, a combination of the anti-thymocyte globulin (ATG), cyclophosphamide and granulocyte-colony stimulating factor (G-CSF) with hematopoietic stem cell transplant has shown some success in patients were recently diagnosed with T1D. Diagnosis took place no more than six weeks prior to enrollment based on anti-GAD65 antibodies and C-peptide in the serum. The treatment regimen was as follows: cyclophosphamide injection followed by ATG treatment for five days, stem cell transplant, and G-CSF injection five days following transplant. Patients became insulin independent following treatment, but after an initial rise C-peptide level eventually decreased, but levels remained higher than at diagnosis and some patients eventually progressed to an insulin-dependent state [52]. It is believed that the mechanism of treatment is ablation and reconstitution the immune system thus inhibiting β-cell destruction resulting in the return to normoglycemia [52]. In a similar study, treating patients who were diabetic for over a year, with ATG and G-CSF showed moderate success in a recent clinical trial, through increased C-peptide levels, with levels of 0.28 nmol/l/min higher than control, indicating improved insulin production, however patients remained dependent on exogenous insulin [53]. A preservation of regulatory T (Treg) cells may be associated with greater β-cell function [53]. A continuation of this study has recently revived optimism as a new two year clinical trial with low dose ATG resulted in preservation of insulin production in patients with new onset T1D defined by the presence of at least one T1D autoantibody [54]. C-peptide levels were significantly higher two years post treatment in the ATG treated patients. Other treatments using transferred cells rather than suppressive factors, including Treg cells and tolerogenic DCs, have shown promise in clinical trials [55, 56]. The study utilizing Treg cells treated newly diabetic patients based on autoantibody level and the majority of treated patients were able to maintain elevated C-peptide levels for over two years following the initiation of the study [55]. In contrast, the clinical trial using tolerogenic DCs treated patients with insulin dependence for more than five years and had no detectable C-peptide at the onset of treatment. Following injection of 10 million DCs, C-peptide was detectable for over two years in some patients and treatment was tolerated over that timespan [56].
Commonalities in multiple sclerosis and type 1 diabetes
There are several similarities in disease progression and pathogenesis of MS and T1D. Often the diagnosis does not take place until late, as the clinical manifestations appear at stage three in T1D, and similarly, MS patients are not typically diagnosed until after their first clinical attack [7, 40]. For this reason, there is often lasting damage caused by the immune system before symptomatic presentation and diagnosis, making the burden of the disease more severe. Co-occurrence of these autoimmune diseases is also common. A nation-wide study in Denmark identified that T1D patients are at a three times greater risk for developing MS than healthy individuals [57]. Along this line, both diseases have genetic risk factors associated with the HLA, although the haplotypes vary by disease [41, 58]. There are also non-HLA associated genetic risk factors such as T-cell alleles IL-2 and protein tyrosine phosphatase, non-receptor type 22 (PTPN22) in MS and T1D, respectively. In addition to genetics, environment also plays a role in disease development, with smoking and viral exposure increasing the risk of both MS and T1D [7, 40].
In terms of pathogenesis, in both diseases autoreactive T cells play an important role, targeting autoantigens on β-cells in the pancreas in T1D, and myelin in the CNS in MS [7, 40]. Autoantibody production indicates the contribution of B cells to both diseases. The contribution of innate immune cells is evidenced by the presence of macrophages and DCs surrounding the islets of Langerhans in T1D and by the infiltrating macrophages playing a role in myelin destruction in MS.
Biomaterials and particle-based drug delivery and immune modulation
Biomaterials have been well established as an approach to mediate controlled release of drugs, peptides, and proteins in the pharmaceutical industry due to their advantages over soluble bolus drug administration [59]. The shortcomings of bolus drugs include rapid renal clearance, short half-life, and potentially fatal off-target side effects. Biomaterial systems can overcome these pitfalls by facilitating sustained release, localization of drug cargo to cells, organs, or systems of interest to improve therapeutic response, minimize drug load required, and mitigate adverse systemic reactions.
Building on this work, biomaterial platforms have been recently extended to immunomodulatory approaches [60]. Specifically, nano- and microparticles (NMPs) based therapies have shown promise in recent years in restoring homeostatic immunity. NMPs can encapsulate or have immunomodulatory factors conjugated to their surface and can be delivered in a diverse manner including systemically or locally. Formulation of NMPs and the route of administration influence the effectiveness and the type of immunomodulation [60–62]. Intravenous and intraperitoneal injections provide better trafficking to the liver and spleen [62, 63], while subcutaneous injection may require the addition of recruitment factors, if the NMP system contains particles that are designed to be phagocytized, but are not small enough to drain passively [64]. With proper trafficking, subcutaneous delivery may be clinically favorable because of the ease and safety of administration compared to repeated bolus intravenous delivery. Furthermore, controlled-release biomaterial systems can extend the window of immunomodulation resulting in a reduced dosing frequency, however using biomaterials can decrease the sterility of the injection via in introduction of endotoxin. In addition, encapsulation allows for greater control of release kinetics. Control of release may be useful if it is desired to give a small dose over a long period of time rather than a quick acting dose, although too slow release could decrease efficacy. Additionally, synthetic biomaterials are tailorable and can be coupled with ligands or antibodies for surface receptors to target delivery of immunomodulatory agents to specific cell subsets, as it has been done with α-CD11c, α-DEC205 [65], α-CD40 for DCs [65, 66] and α-CD4 or α-CD8 for T cells [67, 68].
A specific type of treatment that has benefitted from the use of biomaterials is DC therapy. In this line, a NMP approach can alter DC phenotype in vivo, thus bypassing the need for ex vivo manipulation, which has drawbacks, including high cost, poor yield and low levels of regional lymph node homing following re-administration [69–72]. NMPs are also attractive for intracellular delivery to DCs and other antigen-presenting cells (APCs) given that they can be readily phagocytosed if less than ~ 5 μm in diameter [73].
It has been shown that poly(lactic-co-glycolic acid) (PLGA) NMPs themselves can have immunosuppressive effects in certain contexts via two distinct mechanisms. In this work, bone marrow derived DCs treated with empty PLGA MPs had reduced expression of MHC II, CD80 and CD86 due to degradation byproduct, lactic acid [74]. Furthermore, early intravenous treatment with high molecular weight PLGA NPs, without additional factors, resulted in delay in EAE scores with a concomitant shift of neutrophils away from the CNS to the liver (Table), thus altering trafficking rather than the direct suppression shown by Allen et al. [75].
Table.
Nano and microparticle engineering strategies for treatment of autoimmune disease, multiples sclerosis and type 1 diabetes. Relapsing and remitting (RR), Primary progressive (PP), Streptozotocin induced T1D (STZ), and Mimotope (Mim)
| Material | Drug | Antigen | Route | Regimen | Disease | Model | Citation | |
|---|---|---|---|---|---|---|---|---|
| PLGA | 450 nm | N/A | No | i.v. | Prophylactic | EAE | RR EAE | [75] |
| PLGA | 150 nm | PHCCC (Encapsulated) |
No | s.c. | Prophylactic | EAE | PP EAE | [76] |
| PLGA+ Polymethacrylate | 800 nm | IL-10 Plasmid (Encapsulated) |
No | i.m. | Preventative | T1D | STZ | [77] |
| Zirconium Dioxide | 70 nm | Betamethasone phosphate (Encapsulated) |
No | i.p. | Therapeutic | EAE | PP EAE | [78] |
| Zinc+PEG | 2μm | CD40, C80, and CD86 antisense oligonucleotides | N/A | i.p. or s.c. | Therapeutic | T1D | Adoptive Transfer | [79] |
| PLGA | 500 nm | N/A | PLP139–151 (Surface) | i.v. | Prophylactic & Therapeutic | EAE | RR EAE | [62] |
| PLGA + PEMA | 450 nm | N/A | PLP139–151 (Surface) | i.v. | Prophylactic | EAE | RR EAE | [80, 81] |
| PLGA + PEMA | 500 nm | N/A | BDC2.5 (Mim) (Encapsulated) |
i.v. | Preventative | T1D | Adoptive Transfer | [82] |
| PLC | 200–600 nm | N/A | rhMBP (Encapsulated) |
s.c. | Prophylactic | EAE | PP EAE | [83] |
| RGD+ Mannose Modified Chitosan | 320 nm | N/A | Heat shock protein 65–6xP227 (Encapulated |
Oral | Preventative | T1D | NOD | [84] |
| Iron Oxide | 70 nm | N/A | MHC-IGRP13–23 MHC-BDC2.5 (Mim) MHC-MOG35–55 |
i.v. | Preventative Reversal | T1D | NOD PP EAE | [85, 86] |
| PLGA + PEG | 200 nm | Rapamycin (Encapsulated) |
PLP139–151 Encapsualted |
s.c. | Prophylactic or Therapeutic | EAE | RR EAE | [87] |
| PLGA | 500 nm | TGF-β (Surface) |
PLP139–151 (Encapsulated) |
i.v.or s.c. | Prophylactic | EAE | RR EAE | [88] |
| PLGA | 2 μm | Rapamycin (Encapsulated) |
MOG35–55 (Encapsulated) |
Intranodal | Therapeutic | EAE | PP EAE | [89] |
| PLGA | 200 nm | IL-10 (Encapsulated) |
PLP139–151MOG35–55 (Encapsulated) |
s.c. | Prophylactic & Therapeutic | EAE | RR EAE | [90] |
| PLGA | 1, 30 μm | TGF-β, GM-CSF, VD3 (Encapsulated) |
MOG35–55 (Encapsulated) |
s.c. | Prophylactic | EAE | PP EAE | [64] |
| PLGA | 1 or 30 μm | TGF-β, GM-CSF, VD3 (Encapsulated) |
Insulin B9–23 Or Denatured Insulin (Encapsulated) |
s.c | Preventative & Therapeutic | T1D | NOD | [91, 92] |
| PLGA | 1.5 μm | CpG, GM-CSF (In Hydrogel) |
Denatured Insulin (Encapsulated) |
s.c. | Preventative | T1D | NOD | [93] |
| Gold + PEG | 60 nm | ITE (Surface) |
MOG35–55 (Surface) |
i.p. | Prophylactic | EAE | PP EAE | [94] |
| Gold + PEG | 60 nm | ITE (Surface) |
BDC2.5 (Mim) (Surface) |
i.p. | Preventative | T1D | NOD | [95] |
Efforts to avoid off target effects associated with broad immunosuppression have recently emphasized antigen (Ag) specific approaches. The advantages of such strategies include less systemic immune suppression, the ability to modulate specific immune populations and potential for lower dosing amount and frequency [96, 97]. In this approach only the autoreactive immune cells are targeted. In the case of MS and T1D, such treatments aim to induce tolerance or change primarily the phenotype of Ag-specific T cells. To accomplish an Ag-specific response, treatments have focused on APCs, including DCs and macrophages, given the pivotal role of these cells in promoting the immune response of T cells [61, 66, 79, 93, 98–102]. Tolerogenic DCs have been found to favor generation of Treg cells in specific environments, which further suppress autoreactive T cells, ultimately preventing or reversing disease [103]. In addition, APCs can induce anergy by presenting antigen without the proper costimulatory factors or in the context of coinhibitory receptors such as CTLA-4 and PD-1 [104, 105].
NMP Strategies for MS/EAE and T1D treatment using non-antigen-specific immune modulators
There has been significant effort in developing NMP-based strategies that seek to modulate broadly immune cells, either systemically or locally, creating a suppressive phenotype or delivering factors to block Th1 or Th17 responses, but not in an Ag-specific manner (Fig 1a). Several such strategies have been reported successful in ameliorating or inhibiting EAE, by dampening the Th1 and Th17 responses or increasing the number of Treg cells. Some NMP treatments seek to enhance delivery of immunosuppressants that are currently FDA approved such as glucocorticoids, with the goal of lowering the dose needed, which may reduce off target effects and toxicity. Such a strategy delivered the glucocorticoid betamethasone, using an inorganic-organic NP formulation consisting of a complex of zirconium dioxide, flavin mononucleotide and betamethasone phosphate (Table) [78]. In vitro experiments showed uptake by murine bone marrow derived macrophages and consequent downregulation of MHCII, CD86, TNFα and IL-1β [78]. When cultured with human monocytes an anti-inflammatory phenotypes with decreased IL-1β mRNA and increased arginase 1 expression was observed [78]. When injected intraperitoneally in vivo, betamethasone NPs similarly decreased MHCII and CD86 expression and TNFα secretion on macrophages following lipopolysaccharide challenge, and intraperitoneal injection for three consecutive days of EAE mice at a clinical score of two, halted disease progression in a macrophage-dependent manner [78].
Figure. Representative methods of alleviating autoimmunity with NMPs.
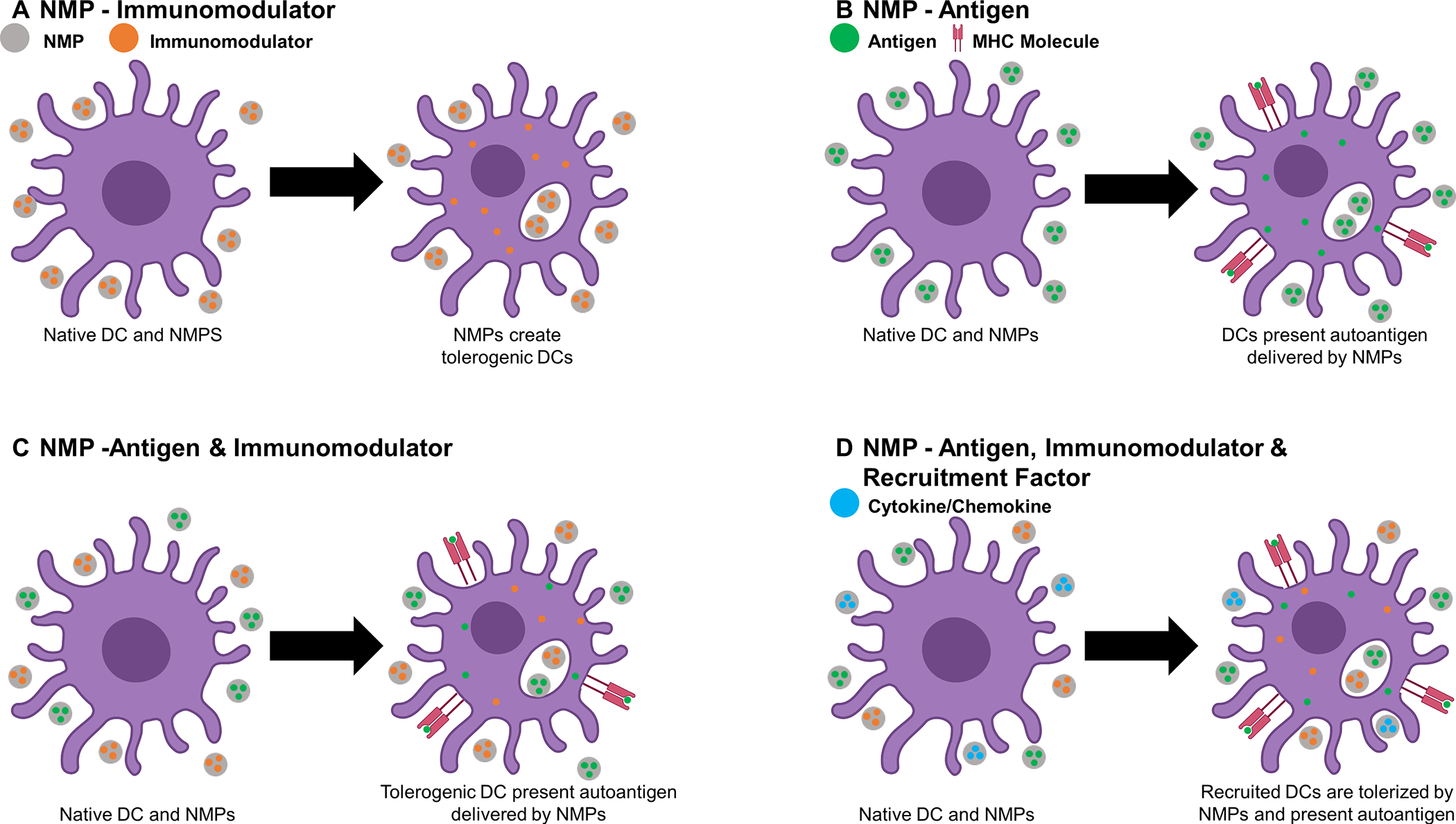
A) NMPs that deliver immunomodulators to DCs in order to create a tolerized phenotype, B) delivery of autoantigen for presentation by DCs, C) delivery of both immunomodulators and antigen, to tolerize DCs which will then present the autoantigen in a tolerogenic context, and D) delivery of recruitment factors to increase DC recruitment to the injection site where autoantigen is taken up and immunomodulators induce a tolerogenic phenotype, after which DCs traffic to draining lymph nodes.
The STING pathway has been also targeted using a polyethylenimine (PEI) NP encapsulating cyclic dinucleotide GMP (c-diGMP) administered i.v. in prophylactic and therapeutic manners in EAE-induced mice, and was found to reduce disease severity [106]. This treatment resulted in production of indoleamine 2, 3 dioxygenase (IDO) in hematopoietic cells and reduced IDO expression in neurons [106].
Another treatment employed the agonist for the metabotropic glutamate receptor-4 (mGluR4) N-phenyl-7-(hydroxyimino)cyclopropaβchromen-1a-carboxamide (PHCCC), encapsulated in PLGA. The treatment slowed EAE development (Table) [76], likely through modulating DC activity, given that mice lacking mGluR4 develop more severe EAE and mGluR4 activation was shown to restrain DC-dependent induction of pathogenic Th17 cells in EAE [107, 108]. High levels of glutamate are seen in EAE and are associated with Th17 response in disease. It is believed that mGluR4 binding limits this response by modulating metabolism, resulting in skewing of T cell phenotype towards Th2 and Treg cells.
Treatments that directly modulate T cell responses have been also explored. An example is the treatment of EAE-induced mice by i.p. administration on the day of induction with hyperforin-conjugated gold NPs. Hyperforin is an herbal compound with anti-inflammatory properties. The treatment resulted in reduced EAE severity with increased Foxp3+ Treg cells and decreased in Th1 and Th17 cells (Table) [109]. A potential drawback is the possibility for liver toxicity following gold NP treatment [110].
A cationic NP using PLGA and a polymethacrylate/dimethylaminoethyl bond has a more neutral zeta potential and improved cellular internalization or endosomal escape [77]. A single intramuscular injection of NPs containing plasmid encoding mouse IL-10, augmented plasma IL-10 levels and reduced IFN-γ levels for up to six weeks in a model of streptozotocin-induced T1D (Table) [77]. In another approach anti-sense oligonucleotides for CD40, CD80, and CD86 were encapsulated in NMPs and blocked the activation of DCs and ameliorated disease in a mouse model of recent-onset T1D (Table) [79]. Draining lymph nodes from the site of injection had increased total numbers of CD25+Foxp3+ Treg cells [79].
NMP strategies to modulate the immune response in a manner independent of the disease-specific autoantigens have shown some promise in preclinical studies both for MS and T1D. Such strategies involved immunomodulators and may cause overall dampening the immune response, but multiple factors dictate whether this is the case. The dose and route of administration can determine if there is immunosuppression as a relatively small dose delivered subcutaneously would not be expected to have systemic effect. The half-life of a therapeutic is also a factor in whether a treatment is systemically immunosuppressive as a treatment cleared rapidly may not have a lasting effect. In addition, delivering NMPs to disease-relevant tissues or lymph nodes can provide a milieu with disease specific antigen to eliminate the need for the delivery of exogenous antigen [79, 111]. However, targeting NMPs to the pancreas or CNS for the localization of treatment represents a substantial challenge. For this reason, the addition of antigen to an immunomodulatory treatment may provide benefit in some therapeutic approaches. Additionally, antigen specificity could potentially reduce the opportunity for systemically dampening the immune response, which is a concern regarding opportunistic infections and off-target effects.
Antigen-Specific NMP strategies for MS/EAE and T1D
While Ag-nonspecific immune modulation shows promise, use of disease-associated antigen may produce generalized immune suppression. The first attempts in this direction were by coupling antigens with NMPs for antigen delivery to extend its circulation time. Further studies employed NMP platforms delivering antigens in combination with immune modulatory factors.
Antigen-Specific NMP strategies for MS/EAE
Initial strategies used NMPs for delivery of antigens alone (Fig 1b), such as proteolipid protein (PLP139–151). PLP139–151 conjugated to PLGA NMP surface and administered i.v. prevented relapses in the relapsing-remitting EAE model in SJL mice, when administered either prophylactically or therapeutically, with the effect being dependent on phagocytosis of peptide-conjugated MPs by MARCO+ marginal zone macrophages in the spleen (Table) [62]. Additionally, increased numbers of Treg cells, and elevated T cell anergy was observed [62]. The same group showed that when PLP139–151 peptide surface conjugated to PLGA/poly(ethylene-alt-maleic anhydride) (PEMA) NPs and administered intravenously, prevented EAE with prophylactic administration and attenuated disease when administered at the peak of EAE (Table) [80]. Although intravenous delivery of this formulation had success in treating relapsing/remitting EAE, subcutaneous administration of the same formulation failed to have the same success, which may be due to differences in NP trafficking [80].
Encapsulation of antigen showed superiority to surface conjugation by avoiding potential for antibody binding to exposed antigen [116]. Furthermore, encapsulation rather than surface conjugation to PLGA/ PEMA NPs was more efficient in treating EAE in prophylactic and therapeutic setups, when administered i.v. [63]. Along this line, encapsulation of recombinant human basic myelin protein (rhMBP) in poly (ε-caprolactone) (PCL) NPs and subcutaneous prophylactic administration resulted in reduction of disease scores of MOG35–55–induced EAE (Table) [83]. The dose of encapsulated peptide can also have an effect, with the high level of encapsulated peptide PLP139–151 having significantly higher efficacy versus low level (Table) [81]. Response to delivered antigen can be a major concern, as there is demonstrated potential to exacerbate disease, as seen in the halted phase II clinical trial treating with MBP83–99 [37].
NMP platforms delivering antigens in combination with immunomodulatory factors have been also explored in EAE (Fig 1c). One example is the surface conjugation of the aryl hydrocarbon receptor (Ahr) ligand 2–1’H-indole-3’-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) to thio-polyethylene glycol (PEG)-coated gold NP, conjugated together with the EAE-specific antigen MOG35–55. The treatment initiated at EAE induction and consisted of weekly intraperitoneal injections effectively suppressed EAE (Table) [94]. It was postulated that the NPs containing myelin antigen and ITE promoted tolerogenic DCs and Treg cell expansion [94], in line with the previous data showing that ITE-Ahr induces tolerogenic DCs and Treg cell differentiation, and inhibits Th1 and Th17 cell function to suppress EAE [112–115].
Other treatments co-encapsulated immunomodulators such as rapamycin with antigen for the treatment of EAE. Rapamycin is known to block the mTOR pathway causing anti-inflammatory effects and blocking B and T cell proliferation [117–121]. Subcutaneous and intravenous injection of PLP139–151 and rapamycin co-encapsulated in PLGA NPs blocked EAE and prevented relapse in an antigen dependent manner in SJL mice (Table) [87]. In another study, a rapamycin-antigen co-encapsulated formulation was explored in MOG35–55/CFA-induced EAE model. Rapamycin and MOG35–55 peptide encapsulated in PLGA MPs and injected intra-nodally into the inguinal lymph nodes prevented and treated EAE in an antigen-specific manner through increased numbers of Foxp3+ Treg cells (Table) [89]. These treatments demonstrate the strong immunomodulatory effect of rapamycin in conjunction with antigen. Another immunomodulator, dexamethasone co-encapsulated with MOG35–55 in acetylated dextran MPs, successfully treated EAE in a therapeutic regimen with decreased splenocyte production of IL-17 [122].
Other immunomodulatory factors such as cytokines have also been explored. EAE was moderately suppressed by subcutaneous administration of PLGA NPs loaded with IL-10 and PLP1139–151, both in prophylactic or therapeutic treatment regimens (Table) [90]. This formulation demonstrates the possibility of suppressing EAE using a subcutaneous delivery of immunomodulatory factors encapsulated in PLGA NPs for controlled release [90]. However, the antigen-specificity of the treatment was not investigated [90]. Another formulation utilized the antigen PLP139–151 conjugated to PLGA NPs, together with cytokine TGF-β to form an antigen coupled PLG-PLP139–151-TGF-β NP system (Table) [88]. In vitro treatment downregulated costimulatory molecules CD80, CD86 and MHCII in vitro, and in vivo administration intravenously or subcutaneously, in prophylactic or therapeutic regimens resulted in reduction of EAE scores [88].
We developed a dual multi-factor MP system consisting of (1) unphagocytosable 30 μm MPs separately loaded with GM-CSF and TGF-β plus (1) phagocytosable 1 μm MPs loaded with Vitamin D3 and MOG35–55 (Fig 1D). This treatment administered subcutaneously blocked EAE progression when administration occurred in a semi-therapeutic fashion post EAE induction (Table) [64]. In the CNS of mice treated with this dual multi-factor MP system there was a reduction in pathogenic T cells expressing both Rorγt and Tbet and lower levels of the cytokines IL-17, IFN-γ and GM-CSF compared to mice treated with unloaded MPs, as well as reduced CD80 on macrophages/microglia [64]. The number and frequency of MHC II+ and CD86+ DCs in the draining lymph nodes were reduced in mice treated with dual multi-factor MP-MOG35–55 versus an irrelevant antigen (OVA323–339) dual multi-factor MP, demonstrating the dependence on antigen. Localized low dose subcutaneous delivery, controlled release of specific factors, and retention at the injection site are all advantages of this dual multi-factor MP system [64], compared to soluble administration. The combination of phagocytosable antigen-MP with the phagocytosable vitamin D3 MP allows intracellular delivery of antigen and vitamin D3 to act on its intracellular receptor and potentially induce tolerization of DCs. Unphagocytosable GM-CSF MP allows the recruitment of DCs and precursors, and TGF-β MP allows tolerization on the recruited DCs, both assuring slow and durable release. In addition, the subcutaneous delivery assures less off-target effects compared to i.v. administration.
NMP formulations that utilize antigen in combination with low doses of tolerogenic factors show promise by avoiding systemic immune suppression. This is likely due to the low doses of immunomodulatory factors and antigen specificity. The ability to avoid rapid release or release at an undesired location is valuable because it reduces the likelihood of off-target effects, which is a concern when administering immunosuppressive factors. However, one potentially major drawback of Ag-specific treatments is difficulty in selecting the most efficacious antigen because not all patients have the same autoreactive response. As it has been shown that some patients have autoantibodies for MBP, while others react to MOG or PLP. To this end, simultaneous NP-mediated delivery of multiple antigens showed higher efficiency in EAE treatment compared to single antigens [123].
Antigen-Specific NMP strategies for T1D treatment
Antigen-Specific NMP platforms have been developed for T1D. Using an artificial antigen presenting approach, one group utilized iron oxide NMPs coated with the islet-specific peptide glucose-6-phosphatase catalytic subunit-related protein (IGRP13–25)-MHCI or II to modulate CD8+ and CD4+ T cells [85, 86]. When 4 week old pre-diabetic, NOD mice were injected intravenously every two weeks for six weeks and then every third week for the remainder of the study, normoglycemia was prolonged (Table) [86]. Additionally, diabetic mice were able to return to normoglycemia following the treatment. Similarly, intravenous injection of surface conjugated IGRP13–25-MHCII NPs in 10-week-old pre-diabetic mice prevented T1D. Mice showed more TR1 cells expressing CD49b, LAG-3, ICOS and TGF-β [85] and the treatment was dependent on the specific antigen and MHCII presentation to maintain efficacy (Table) [85]. The versatility of this NMP platform was highlighted by the multitude of immune-related conditions that can be treated by changing the antigen, including collagen-induced arthritis and EAE, which similarly demonstrated robust mitigation of diseases [85]. In contrast to IGRP13–25, heat shock protein 65–6xP227 loaded in NPs formulated from RGD and mannose modified chitosan. These NPs were able prevent T1D when delivered orally to 4 week old NOD mice and uptake by DCs was seen in the Peyers patches (Table) [84]. In addition, treatment was concomitant with an increase in Treg cells and a decrease in Th1 cells in the pancreatic draining lymph node. This study was notable in the demonstrated efficacy of NMP-based delivery of antigen to the gut [84].
Another T1D associated antigen used in NMPs formulations was BDC2.5 mimotope peptide (p31). Mimotope peptide p31 encapsulated or conjugated to PLGA or PLGA/PEMA NMPs were used to treat NOD.SCID induced with diabetes using adoptive transfer of BDC2.5 and NY8.3 antigen-specific cells. Intravenously delivered NMPs prevented hyperglycemia for up to 50 days post transfer [82]. Intra-islet Foxp3+ Treg cells were identified, which likely tolerized CD4+ and/or CD8+ T cells, through an increase in expression of the coinhibitory molecules PD-1 and CTLA4 [82]. NP-mediated delivery of an insulin hybrid peptide (2.5HIP), consisting of an insulin C-peptide fragment fused to a peptide from chromogranin A (ChgA), known to be recognized by BDC-2.5 T cells, also prevented diabetes in an adoptive transfer model by decreasing IFN-γ producing T cells and increasing FoxP3+ T cells [124].
In addition to administering antigen NMPs alone, combinatorial approaches have been used for T1D [60]. Some approaches have explored antigen delivery in combination with pro-tolerogenic factors to modulate antigen-specific immune tolerance. Co-delivery of the Ahr ligand ITE and proinsulin via PEG-coated gold NMPs showed efficiency in prophylactic treatment of 8-week-old NOD mice (Table) [95]. Prevention overlapped with an increase in Foxp3+ Treg cells in the pancreatic lymph node and diminished expression of Th1 and Th17 gene signatures. DCs showed anti-inflammatory phenotypes with reduced surface expression of CD40, CD80 and MHCII and expression of genes encoding the inflammatory cytokines IL-12 and IL-6 [95].
Other combinatorial treatments were engineered to augment cellular recruitment through incorporation of chemotactic factors. A macroscopic hydrogel loaded with GM-CSF and CpG together with PLGA NMPs encapsulating denatured insulin prevented T1D in 40% of the NOD mice when injected at 8 weeks of age (Table) [93]. The efficacy was dependent on antigen. In a parallel approach, without CpG, an alginate hydrogel containing GM-CSF and PLGA NMPs encapsulating the T1D-specific peptide BDC2.5. show increased Foxp3+ Treg cells in the hydrogel, suggesting that this is a viable approach to augment antigen-specific Treg cell numbers [125]. Using the dual multi-factor MP system consisting of (1) unphagocytosable 30 μm MPs separately loaded with GM-CSF or TGF-β, plus (1) phagocytosable 1 μm MPs loaded with Vitamin D3 or insulin B9–23 (Table) [91], delivered agents intracellularly to the locally recruited DCs. The results demonstrated that two subcutaneous injections of the combinatorial NMP system in 4-week-old NOD mice prevented T1D in 40% of treated NOD mice [91]. A modification of this system increased the amounts of TGF-β and GM-CSF and used denatured insulin as antigen (Table) [92]. Encapsulation, antigen, tolerogenic and recruitment factors were all required, otherwise the treatment was no longer effective. When the new formulation was administered weekly via subcutaneous injection, for three weeks, followed by four monthly booster injections, it was able to delay T1D onset in 8-week-old NOD mice, with 60% of mice remaining diabetes-free [92]. This delay in onset was associated with increased PD-1 on CD4+ and CD8+ T cells as well as an increase in CD11b+ CD11c+ DCs in the draining lymph nodes. Additionally, three subcutaneous administrations during the week of onset, followed by weekly booster injections for three weeks, temporarily reversed T1D in recent onset diabetes, for up to 100 days [92].
Conclusions
The prevalence of the debilitating autoimmune diseases MS and T1D continues to rise and a cure yet to be found. There has been an uptick in exploration of biomaterial strategies for autoimmune disease, with NMP therapies having demonstrated particular promise as vehicles for drug and antigen delivery to specific cells and organs of interest. The studies discussed above suggest that a curative therapy that modulates immunity without the need for systemic immunosuppression is possible, using antigen-specific strategies both in MS and T1D. Clinical trials will be needed to validate the benefit of NMP approaches in treating MS and T1D. Looking forward, coupling immunotherapy with regeneration could reduce the failure rate of treatments. In MS for example, clemastine fumarate, a medication for allergic rhinitis and urticaria, has been shown in a double-blind crossover clinical trial to increase remyelination [126]. Similarly in EAE mice clemastine accelerate remyelination [127–129]. Regeneration may also be necessary in T1D. Along this line, reprograming of α-cells with the transcription factors, pancreas/duodenum homeobox protein 1 (PDX1) and V-maf musculoaponeurotic fibrosarcoma oncogene homolog A (MAFA) to produce insulin is an option that has been explored. When human α-cells are transduced with these transcription factors it enabled these cells to produce insulin [130]. Thus the key to unlocking a durable cure may be through coupling antigen-specific immunotherapies with regeneration.
Acknowledgment:
Support is gratefully acknowledged for R01 AI133623 (BGK and DA), R01 DE027301 (BGK) and T32 DK108736 (JMS). The figure was created in part using biorender.com.
References
- [1].Compston A, Coles A, Multiple sclerosis, Lancet 372(9648) (2008) 1502–17. [DOI] [PubMed] [Google Scholar]
- [2].Kobelt G, Berg J, Atherly D, Hadjimichael O, Costs and quality of life in multiple sclerosis: a cross-sectional study in the United States, Neurology 66(11) (2006) 1696–702. [DOI] [PubMed] [Google Scholar]
- [3].Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, Thompson AJ, Wolinsky JS, Balcer LJ, Banwell B, Barkhof F, Bebo B, Calabresi PA, Clanet M, Comi G, Fox RJ, Freedman MS, Goodman AD, Inglese M, Kappos L, Kieseier BC, Lincoln JA, Lubetzki C, Miller AE, Montalban X, O’Connor PW, Petkau J, Pozzilli C, Rudick RA, Sormani MP, Stüve O, Waubant E, Polman CH, Defining the clinical course of multiple sclerosis: the 2013 revisions, Neurology 83(3) (2014) 278–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wallin MT, Culpepper WJ, Campbell JD, Nelson LM, Langer-Gould A, Marrie RA, Cutter GR, Kaye WE, Wagner L, Tremlett H, Buka SL, Dilokthornsakul P, Topol B, Chen LH, LaRocca NG, The prevalence of MS in the United States: A population-based estimate using health claims data, Neurology 92(10) (2019) e1029–e1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dendrou CA, Fugger L, Friese MA, Immunopathology of multiple sclerosis, Nat Rev Immunol 15(9) (2015) 545–58. [DOI] [PubMed] [Google Scholar]
- [6].Nylander A, Hafler DA, Multiple sclerosis, The Journal of clinical investigation 122(4) (2012) 1180–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Filippi M, Bar-Or A, Piehl F, Preziosa P, Solari A, Vukusic S, Rocca MA, Multiple sclerosis, Nature Reviews Disease Primers 4(1) (2018) 43. [DOI] [PubMed] [Google Scholar]
- [8].Frohman EM, Racke MK, Raine CS, Multiple Sclerosis — The Plaque and Its Pathogenesis, New England Journal of Medicine 354(9) (2006) 942–955. [DOI] [PubMed] [Google Scholar]
- [9].Minagar A, Alexander JS, Blood-brain barrier disruption in multiple sclerosis, Multiple Sclerosis Journal 9(6) (2003) 540–549. [DOI] [PubMed] [Google Scholar]
- [10].Bielekova B, Sung MH, Kadom N, Simon R, McFarland H, Martin R, Expansion and functional relevance of high-avidity myelin-specific CD4+ T cells in multiple sclerosis, J Immunol 172(6) (2004) 3893–904. [DOI] [PubMed] [Google Scholar]
- [11].Hellings N, Barée M, Verhoeven C, D’hooghe MB, Medaer R, Bernard CC, Raus J, Stinissen P, T-cell reactivity to multiple myelin antigens in multiple sclerosis patients and healthy controls, J Neurosci Res 63(3) (2001) 290–302. [DOI] [PubMed] [Google Scholar]
- [12].Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ, IL-23 drives a pathogenic T cell population that induces autoimmune inflammation, J Exp Med 201(2) (2005) 233–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C, A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17, Nat Immunol 6(11) (2005) 1133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR, The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells, Cell 126(6) (2006) 1121–33. [DOI] [PubMed] [Google Scholar]
- [15].Codarri L, Gyülvészi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B, RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation, Nat Immunol 12(6) (2011) 560–7. [DOI] [PubMed] [Google Scholar]
- [16].Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK, Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis, J Exp Med 200(1) (2004) 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Weiner HL, Multiple sclerosis is an inflammatory T-cell-mediated autoimmune disease, Arch Neurol 61(10) (2004) 1613–5. [DOI] [PubMed] [Google Scholar]
- [18].Lavi E, Constantinescu CS, Experimental models of multiple sclerosis, Springer, New York, 2005. [Google Scholar]
- [19].Wingerchuk DM, Carter JL, Multiple sclerosis: current and emerging disease-modifying therapies and treatment strategies, Mayo Clinic proceedings 89(2) (2014) 225–40. [DOI] [PubMed] [Google Scholar]
- [20].Goodin DS, Frohman EM, Garmany GP, Halper J, Likosky WH, Lublin FD, Silberberg DH, Stuart WH, van den Noort S, T.a.T.A.S.o.t.A.A.o.N.a.t.M.C.f.C.P. Guidelines, Disease modifying therapies in multiple sclerosis: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and the MS Council for Clinical Practice Guidelines, Neurology 58(2) (2002) 169–78. [DOI] [PubMed] [Google Scholar]
- [21].Bloomgren G, Richman S, Hotermans C, Subramanyam M, Goelz S, Natarajan A, Lee S, Plavina T, Scanlon JV, Sandrock A, Bozic C, Risk of Natalizumab-Associated Progressive Multifocal Leukoencephalopathy, New England Journal of Medicine 366(20) (2012) 1870–1880. [DOI] [PubMed] [Google Scholar]
- [22].Berger JR, Cree BA, Greenberg B, Hemmer B, Ward BJ, Dong VM, Merschhemke M, Progressive multifocal leukoencephalopathy after fingolimod treatment, Neurology 90(20) (2018) e1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kieseier BC, The Mechanism of Action of Interferon-β in Relapsing Multiple Sclerosis, CNS Drugs 25(6) (2011) 491–502. [DOI] [PubMed] [Google Scholar]
- [24].Goodin DS, Reder AT, Ebers GC, Cutter G, Kremenchutzky M, Oger J, Langdon D, Rametta M, Beckmann K, DeSimone TM, Knappertz V, Survival in MS: a randomized cohort study 21 years after the start of the pivotal IFNβ−1b trial, Neurology 78(17) (2012) 1315–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Goodin DS, Cohen BA, O’Connor P, Kappos L, Stevens JC, T.a.T.A.S.o.t.A.A.o. Neurology, Assessment: the use of natalizumab (Tysabri) for the treatment of multiple sclerosis (an evidence-based review): report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology, Neurology 71(10) (2008) 766–73. [DOI] [PubMed] [Google Scholar]
- [26].Gasperini C, Ruggieri S, Development of oral agent in the treatment of multiple sclerosis: how the first available oral therapy, fingolimod will change therapeutic paradigm approach, Drug Des Devel Ther 6 (2012) 175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ziemssen T, Schrempf W, Glatiramer Acetate: Mechanisms of Action in Multiple Sclerosis, International Review of Neurobiology, Academic Press; 2007, pp. 537–570. [DOI] [PubMed] [Google Scholar]
- [28].Fridkis-Hareli M, Teitelbaum D, Gurevich E, Pecht I, Brautbar C, Kwon OJ, Brenner T, Arnon R, Sela M, Direct binding of myelin basic protein and synthetic copolymer 1 to class II major histocompatibility complex molecules on living antigen-presenting cells--specificity and promiscuity, Proc Natl Acad Sci U S A 91(11) (1994) 4872–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Clerico M, Contessa G, Durelli L, Interferon-β1a for the treatment of multiple sclerosis, Expert Opinion on Biological Therapy 7(4) (2007) 535–542. [DOI] [PubMed] [Google Scholar]
- [30].O’Connor P, Miller D, Riester K, Yang M, Panzara M, Dalton C, Miszkiel K, Khan O, Rice G, Sheremata W, Relapse rates and enhancing lesions in a phase II trial of natalizumab in multiple sclerosis, Multiple Sclerosis Journal 11(5) (2005) 568–572. [DOI] [PubMed] [Google Scholar]
- [31].Kappos L, Radue E-W, O’Connor P, Polman C, Hohlfeld R, Calabresi P, Selmaj K, Agoropoulou C, Leyk M, Zhang-Auberson L, Burtin P, A Placebo-Controlled Trial of Oral Fingolimod in Relapsing Multiple Sclerosis, New England Journal of Medicine 362(5) (2010) 387–401. [DOI] [PubMed] [Google Scholar]
- [32].Berger JR, Cree BA, Greenberg B, Hemmer B, Ward BJ, Dong VM, Merschhemke M, Progressive multifocal leukoencephalopathy after fingolimod treatment, Neurology 90(20) (2018) e1815–e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung H-P, Hemmer B, Lublin F, Montalban X, Rammohan KW, Selmaj K, Traboulsee A, Wolinsky JS, Arnold DL, Klingelschmitt G, Masterman D, Fontoura P, Belachew S, Chin P, Mairon N, Garren H, Kappos L, Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis, New England Journal of Medicine 376(3) (2016) 221–234. [DOI] [PubMed] [Google Scholar]
- [34].Mulero P, Midaglia L, Montalban X, Ocrelizumab: a new milestone in multiple sclerosis therapy, Ther Adv Neurol Disord 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Feinstein A, Freeman J, Lo AC, Treatment of progressive multiple sclerosis: what works, what does not, and what is needed, Lancet Neurol 14(2) (2015) 194–207. [DOI] [PubMed] [Google Scholar]
- [36].Hilas O, Patel PN, Lam S, Disease modifying agents for multiple sclerosis, Open Neurol J 4 (2010) 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, Gran B, Eaton J, Antel J, Frank JA, McFarland HF, Martin R, Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis: Results of a phase II clinical trial with an altered peptide ligand, Nature Medicine 6(10) (2000) 1167. [DOI] [PubMed] [Google Scholar]
- [38].Gale EA, Type 1 diabetes in the young: the harvest of sorrow goes on, Diabetologia 48(8) (2005) 1435–8. [DOI] [PubMed] [Google Scholar]
- [39].Mayer-Davis EJ, Dabelea D, Lawrence JM, Incidence Trends of Type 1 and Type 2 Diabetes among Youths, 2002–2012, N Engl J Med, United States, 2017, p. 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Katsarou A, Gudbjörnsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, Jacobsen LM, Schatz DA, Lernmark Å, Type 1 diabetes mellitus, Nature Reviews Disease Primers 3 (2017) 17016. [DOI] [PubMed] [Google Scholar]
- [41].Bluestone JA, Herold K, Eisenbarth G, Genetics, pathogenesis and clinical interventions in type 1 diabetes, Nature 464(7293) (2010) 1293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Laing SP, Swerdlow AJ, Slater SD, Burden AC, Morris A, Waugh NR, Gatling W, Bingley PJ, Patterson CC, Mortality from heart disease in a cohort of 23,000 patients with insulin-treated diabetes, Diabetologia 46(6) (2003) 760–5. [DOI] [PubMed] [Google Scholar]
- [43].Groop PH, Thomas MC, Moran JL, Waden J, Thorn LM, Makinen VP, Rosengard-Barlund M, Saraheimo M, Hietala K, Heikkila O, Forsblom C, FinnDiane Study G, The presence and severity of chronic kidney disease predicts all-cause mortality in type 1 diabetes, Diabetes 58(7) (2009) 1651–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Watkins RA, Evans-Molina C, Blum JS, DiMeglio LA, Established and emerging biomarkers for the prediction of type 1 diabetes: a systematic review, Translational research : the journal of laboratory and clinical medicine 164(2) (2014) 110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Skyler JS, Krischer JP, Wolfsdorf J, Cowie C, Palmer JP, Greenbaum C, Cuthbertson D, Rafkin-Mervis LE, Chase HP, Leschek E, Effects of oral insulin in relatives of patients with type 1 diabetes: The Diabetes Prevention Trial--Type 1, Diabetes care 28(5) (2005) 1068–76. [DOI] [PubMed] [Google Scholar]
- [46].Nanto-Salonen K, Kupila A, Simell S, Siljander H, Salonsaari T, Hekkala A, Korhonen S, Erkkola R, Sipila JI, Haavisto L, Siltala M, Tuominen J, Hakalax J, Hyoty H, Ilonen J, Veijola R, Simell T, Knip M, Simell O, Nasal insulin to prevent type 1 diabetes in children with HLA genotypes and autoantibodies conferring increased risk of disease: a double-blind, randomised controlled trial, Lancet 372(9651) (2008) 1746–55. [DOI] [PubMed] [Google Scholar]
- [47].Diabetes Prevention Trial--Type G 1 Diabetes Study, Effects of insulin in relatives of patients with type 1 diabetes mellitus, The New England journal of medicine 346(22) (2002) 1685–91. [DOI] [PubMed] [Google Scholar]
- [48].Walter M, Philotheou A, Bonnici F, Ziegler AG, Jimenez R, Group NBIS, No effect of the altered peptide ligand NBI-6024 on beta-cell residual function and insulin needs in new-onset type 1 diabetes, Diabetes care 32(11) (2009) 2036–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, Gottlieb PA, Marks JB, McGee PF, Moran AM, Raskin P, Rodriguez H, Schatz DA, Wherrett D, Wilson DM, Lachin JM, Skyler JS, Type CDSG 1 Diabetes TrialNet Anti, Rituximab, B-lymphocyte depletion, and preservation of beta-cell function, The New England journal of medicine 361(22) (2009) 2143–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sherry N, Hagopian W, Ludvigsson J, Jain SM, Wahlen J, Ferry RJ Jr., Bode B, Aronoff S, Holland C, Carlin D, King KL, Wilder RL, Pillemer S, Bonvini E, Johnson S, Stein KE, Koenig S, Herold KC, Daifotis AG, Protege Trial I, Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial, Lancet 378(9790) (2011) 487–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, Gottlieb PA, Greenbaum CJ, Marks JB, Monzavi R, Moran A, Raskin P, Rodriguez H, Russell WE, Schatz D, Wherrett D, Wilson DM, Krischer JP, Skyler JS, Type G 1 Diabetes TrialNet Abatacept Study, Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial, Lancet 378(9789) (2011) 412–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Snarski E, Milczarczyk A, Torosian T, Paluszewska M, Urbanowska E, Król M, Boguradzki P, Jedynasty K, Franek E, Wiktor-Jedrzejczak W, Independence of exogenous insulin following immunoablation and stem cell reconstitution in newly diagnosed diabetes type I, Bone Marrow Transplantation 46(4) (2011) 562–566. [DOI] [PubMed] [Google Scholar]
- [53].Haller MJ, Gitelman SE, Gottlieb PA, Michels AW, Rosenthal SM, Shuster JJ, Zou B, Brusko TM, Hulme MA, Wasserfall CH, Mathews CE, Atkinson MA, Schatz DA, Anti-thymocyte globulin/G-CSF treatment preserves beta cell function in patients with established type 1 diabetes, J Clin Invest 125(1) (2015) 448–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Haller MJ, Long SA, Blanchfield JL, Schatz DA, Skyler JS, Krischer JP, Bundy BN, Geyer SM, Warnock MV, Miller JL, Atkinson MA, Becker DJ, Baidal DA, DiMeglio LA, Gitelman SE, Goland R, Gottlieb PA, Herold KC, Marks JB, Moran A, Rodriguez H, Russell WE, Wilson DM, Greenbaum CJ, Low-dose Anti-Thymocyte Globulin Preserves C-Peptide and Reduces A1c in New Onset Type 1 Diabetes: Two Year Clinical Trial Data, Diabetes (2019) db190057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, Herold KC, Lares A, Lee MR, Li K, Liu W, Long SA, Masiello LM, Nguyen V, Putnam AL, Rieck M, Sayre PH, Tang Q, Type 1 diabetes immunotherapy using polyclonal regulatory T cells, Sci Transl Med 7(315) (2015) 315ra189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M, Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients, Diabetes care 34(9) (2011) 2026–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Nielsen NM, Westergaard T, Frisch M, Rostgaard K, Wohlfahrt J, Koch-Henriksen N, Melbye M, Hjalgrim H, Type 1 diabetes and multiple sclerosis: A Danish population-based cohort study, Archives of neurology 63(7) (2006) 1001–4. [DOI] [PubMed] [Google Scholar]
- [58].Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, Dilthey A, Su Z, Freeman C, Hunt SE, Edkins S, Gray E, Booth DR, Potter SC, Goris A, Band G, Oturai AB, Strange A, Saarela J, Bellenguez C, Fontaine B, Gillman M, Hemmer B, Gwilliam R, Zipp F, Jayakumar A, Martin R, Leslie S, Hawkins S, Giannoulatou E, D’Alfonso S, Blackburn H, Martinelli Boneschi F, Liddle J, Harbo HF, Perez ML, Spurkland A, Waller MJ, Mycko MP, Ricketts M, Comabella M, Hammond N, Kockum I, McCann OT, Ban M, Whittaker P, Kemppinen A, Weston P, Hawkins C, Widaa S, Zajicek J, Dronov S, Robertson N, Bumpstead SJ, Barcellos LF, Ravindrarajah R, Abraham R, Alfredsson L, Ardlie K, Aubin C, Baker A, Baker K, Baranzini SE, Bergamaschi L, Bergamaschi R, Bernstein A, Berthele A, Boggild M, Bradfield JP, Brassat D, Broadley SA, Buck D, Butzkueven H, Capra R, Carroll WM, Cavalla P, Celius EG, Cepok S, Chiavacci R, Clerget-Darpoux F, Clysters K, Comi G, Cossburn M, Cournu-Rebeix I, Cox MB, Cozen W, Cree BA, Cross AH, Cusi D, Daly MJ, Davis E, de Bakker PI, Debouverie M, D’Hooghe M B, Dixon K, Dobosi R, Dubois B, Ellinghaus D, Elovaara I, Esposito F, Fontenille C, Foote S, Franke A, Galimberti D, Ghezzi A, Glessner J, Gomez R, Gout O, Graham C, Grant SF, Guerini FR, Hakonarson H, Hall P, Hamsten A, Hartung HP, Heard RN, Heath S, Hobart J, Hoshi M, Infante-Duarte C, Ingram G, Ingram W, Islam T, Jagodic M, Kabesch M, Kermode AG, Kilpatrick TJ, Kim C, Klopp N, Koivisto K, Larsson M, Lathrop M, Lechner-Scott JS, Leone MA, Leppa V, Liljedahl U, Bomfim IL, Lincoln RR, Link J, Liu J, Lorentzen AR, Lupoli S, Macciardi F, Mack T, Marriott M, Martinelli V, Mason D, McCauley JL, Mentch F, Mero IL, Mihalova T, Montalban X, Mottershead J, Myhr KM, Naldi P, Ollier W, Page A, Palotie A, Pelletier J, Piccio L, Pickersgill T, Piehl F, Pobywajlo S, Quach HL, Ramsay PP, Reunanen M, Reynolds R, Rioux JD, Rodegher M, Roesner S, Rubio JP, Ruckert IM, Salvetti M, Salvi E, Santaniello A, Schaefer CA, Schreiber S, Schulze C, Scott RJ, Sellebjerg F, Selmaj KW, Sexton D, Shen L, Simms-Acuna B, Skidmore S, Sleiman PM, Smestad C, Sorensen PS, Sondergaard HB, Stankovich J, Strange RC, Sulonen AM, Sundqvist E, Syvanen AC, Taddeo F, Taylor B, Blackwell JM, Tienari P, Bramon E, Tourbah A, Brown MA, Tronczynska E, Casas JP, Tubridy N, Corvin A, Vickery J, Jankowski J, Villoslada P, Markus HS, Wang K, Mathew CG, Wason J, Palmer CN, Wichmann HE, Plomin R, Willoughby E, Rautanen A, Winkelmann J, Wittig M, Trembath RC, Yaouanq J, Viswanathan AC, Zhang H, Wood NW, Zuvich R, Deloukas P, Langford C, Duncanson A, Oksenberg JR, Pericak-Vance MA, Haines JL, Olsson T, Hillert J, Ivinson AJ, De Jager PL, Peltonen L, Stewart GJ, Hafler DA, Hauser SL, McVean G, Donnelly P, Compston A, Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis, Nature 476(7359) (2011) 214–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Langer R, Biomaterials in drug delivery and tissue engineering: one laboratory’s experience, Accounts of chemical research 33(2) (2000) 94–101. [DOI] [PubMed] [Google Scholar]
- [60].Stewart JM, Keselowsky BG, Combinatorial drug delivery approaches for immunomodulation, Adv Drug Deliv Rev 114 (2017) 161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Bracho-Sanchez E, Xia CQ, Clare-Salzler MJ, Keselowsky BG, Micro and Nano Material Carriers for Immunomodulation, Am J Transplant 16(12) (2016) 3362–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZN, Yap WT, Getts MT, Pleiss M, Luo X, King NJ, Shea LD, Miller SD, Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis, Nat Biotechnol 30(12) (2012) 1217–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].McCarthy DP, Yap JW, Harp CT, Song WK, Chen J, Pearson RM, Miller SD, Shea LD, An antigen-encapsulating nanoparticle platform for TH1/17 immune tolerance therapy, Nanomedicine : nanotechnology, biology, and medicine 13(1) (2017) 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Cho JJ, Stewart JM, Drashansky TT, Brusko MA, Zuniga AN, Lorentsen KJ, Keselowsky BG, Avram D, An antigen-specific semi-therapeutic treatment with local delivery of tolerogenic factors through a dual-sized microparticle system blocks experimental autoimmune encephalomyelitis, Biomaterials 143 (2017) 79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Cruz LJ, Rosalia RA, Kleinovink JW, Rueda F, Lowik CW, Ossendorp F, Targeting nanoparticles to CD40, DEC-205 or CD11c molecules on dendritic cells for efficient CD8(+) T cell response: a comparative study, Journal of controlled release : official journal of the Controlled Release Society 192 (2014) 209–18. [DOI] [PubMed] [Google Scholar]
- [66].Lewis JS, Zaveri TD, Crooks CP 2nd, Keselowsky BG, Microparticle surface modifications targeting dendritic cells for non-activating applications, Biomaterials 33(29) (2012) 7221–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].McHugh MD, Park J, Uhrich R, Gao W, Horwitz DA, Fahmy TM, Paracrine co-delivery of TGF-beta and IL-2 using CD4-targeted nanoparticles for induction and maintenance of regulatory T cells, Biomaterials 59 (2015) 172–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Schmid D, Park CG, Hartl CA, Subedi N, Cartwright AN, Puerto RB, Zheng Y, Maiarana J, Freeman GJ, Wucherpfennig KW, Irvine DJ, Goldberg MS, T cell-targeting nanoparticles focus delivery of immunotherapy to improve antitumor immunity, Nature communications 8(1) (2017) 1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Zhang Y, Ma B, Zhou Y, Zhang M, Qiu X, Sui Y, Zhang X, Fan Q, Dendritic cells fused with allogeneic breast cancer cell line induce tumor antigen-specific CTL responses against autologous breast cancer cells, Breast Cancer Res Treat 105(3) (2007) 277–86. [DOI] [PubMed] [Google Scholar]
- [70].Zhou J, Weng D, Zhou F, Pan K, Song H, Wang Q, Wang H, Li Y, Huang L, Zhang H, Huang W, Xia J, Patient-derived renal cell carcinoma cells fused with allogeneic dendritic cells elicit anti-tumor activity: in vitro results and clinical responses, Cancer Immunol Immunother 58(10) (2009) 1587–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kim JH, Lee Y, Bae YS, Kim WS, Kim K, Im HY, Kang WK, Park K, Choi HY, Lee HM, Baek SY, Lee H, Doh H, Kim BM, Kim CY, Jeon C, Jung CW, Phase I/II study of immunotherapy using autologous tumor lysate-pulsed dendritic cells in patients with metastatic renal cell carcinoma, Clin Immunol 125(3) (2007) 257–67. [DOI] [PubMed] [Google Scholar]
- [72].Höltl L, Zelle-Rieser C, Gander H, Papesh C, Ramoner R, Bartsch G, Rogatsch H, Barsoum AL, Coggin JH, Thurnher M, Immunotherapy of metastatic renal cell carcinoma with tumor lysate-pulsed autologous dendritic cells, Clin Cancer Res 8(11) (2002) 3369–76. [PubMed] [Google Scholar]
- [73].Champion JA, Walker A, Mitragotri S, Role of particle size in phagocytosis of polymeric microspheres, Pharmaceutical research 25(8) (2008) 1815–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Allen R, Bolandparvaz A, Ma JA, Manickam VA, Lewis JS, Latent, Immunosuppressive Nature of Poly(lactic-co-glycolic acid) Microparticles, (2018). [DOI] [PMC free article] [PubMed]
- [75].Saito E, Kuo R, Pearson RM, Gohel N, Cheung B, King NJC, Miller SD, Shea LD, Designing drug-free biodegradable nanoparticles to modulate inflammatory monocytes and neutrophils for ameliorating inflammation, Journal of Controlled Release 300 (2019) 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Gammon JM, Tostanoski LH, Adapa AR, Chiu YC, Jewell CM, Controlled delivery of a metabolic modulator promotes regulatory T cells and restrains autoimmunity, J Control Release 210 (2015) 169–78. [DOI] [PubMed] [Google Scholar]
- [77].Basarkar A, Singh J, Poly (lactide-co-glycolide)-polymethacrylate nanoparticles for intramuscular delivery of plasmid encoding interleukin-10 to prevent autoimmune diabetes in mice, Pharmaceutical research 26(1) (2009) 72–81. [DOI] [PubMed] [Google Scholar]
- [78].Montes-Cobos E, Ring S, Fischer HJ, Heck J, Strauss J, Schwaninger M, Reichardt SD, Feldmann C, Luhder F, Reichardt HM, Targeted delivery of glucocorticoids to macrophages in a mouse model of multiple sclerosis using inorganic-organic hybrid nanoparticles, J Control Release 245 (2017) 157–169. [DOI] [PubMed] [Google Scholar]
- [79].Phillips B, Nylander K, Harnaha J, Machen J, Lakomy R, Styche A, Gillis K, Brown L, Lafreniere D, Gallo M, Knox J, Hogeland K, Trucco M, Giannoukakis N, A microsphere-based vaccine prevents and reverses new-onset autoimmune diabetes, Diabetes 57(6) (2008) 1544–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Hunter Z, McCarthy DP, Yap WT, Harp CT, Getts DR, Shea LD, Miller SD, A biodegradable nanoparticle platform for the induction of antigen-specific immune tolerance for treatment of autoimmune disease, ACS Nano 8(3) (2014) 2148–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Kuo R, Saito E, Miller SD, Shea LD, Peptide-Conjugated Nanoparticles Reduce Positive Co-stimulatory Expression and T Cell Activity to Induce Tolerance, Mol Ther 25(7) (2017) 1676–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Prasad S, Neef T, Xu D, Podojil JR, Getts DR, Shea LD, Miller SD, Tolerogenic Ag-PLG nanoparticles induce tregs to suppress activated diabetogenic CD4 and CD8 T cells, J Autoimmun (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Al-Ghobashy MA, ElMeshad AN, Abdelsalam RM, Nooh MM, Al-Shorbagy M, Laible G, Development and Pre-Clinical Evaluation of Recombinant Human Myelin Basic Protein Nano Therapeutic Vaccine in Experimental Autoimmune Encephalomyelitis Mice Animal Model, Scientific reports 7 (2017) 46468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Chen Y, Wu J, Wang J, Zhang W, Xu B, Xu X, Zong L, Targeted delivery of antigen to intestinal dendritic cells induces oral tolerance and prevents autoimmune diabetes in NOD mice, Diabetologia 61(6) (2018) 1384–1396. [DOI] [PubMed] [Google Scholar]
- [85].Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, Tsai S, Wang J, Garabatos N, Izquierdo C, Agrawal S, Keough MB, Yong VW, James E, Moore A, Yang Y, Stratmann T, Serra P, Santamaria P, Expanding antigen-specific regulatory networks to treat autoimmunity, Nature 530(7591) (2016) 434–40. [DOI] [PubMed] [Google Scholar]
- [86].Tsai S, Shameli A, Yamanouchi J, Clemente-Casares X, Wang J, Serra P, Yang Y, Medarova Z, Moore A, Santamaria P, Reversal of autoimmunity by boosting memory-like autoregulatory T cells, Immunity 32(4) (2010) 568–80. [DOI] [PubMed] [Google Scholar]
- [87].Maldonado RA, LaMothe RA, Ferrari JD, Zhang AH, Rossi RJ, Kolte PN, Griset AP, O’Neil C, Altreuter DH, Browning E, Johnston L, Farokhzad OC, Langer R, Scott DW, von Andrian UH, Kishimoto TK, Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance, Proc Natl Acad Sci U S A 112(2) (2015) E156–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Casey LM, Pearson RM, Hughes KR, Liu JMH, Rose JA, North MG, Wang LZ, Lei M, Miller SD, Shea LD, Conjugation of Transforming Growth Factor Beta to Antigen-Loaded Poly(lactide-co-glycolide) Nanoparticles Enhances Efficiency of Antigen-Specific Tolerance, Bioconjug Chem (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Tostanoski LH, Chiu YC, Gammon JM, Simon T, Andorko JI, Bromberg JS, Jewell CM, Reprogramming the Local Lymph Node Microenvironment Promotes Tolerance that Is Systemic and Antigen Specific, Cell Rep 16(11) (2016) 2940–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Cappellano G, Woldetsadik AD, Orilieri E, Shivakumar Y, Rizzi M, Carniato F, Gigliotti CL, Boggio E, Clemente N, Comi C, Dianzani C, Boldorini R, Chiocchetti A, Reno F, Dianzani U, Subcutaneous inverse vaccination with PLGA particles loaded with a MOG peptide and IL-10 decreases the severity of experimental autoimmune encephalomyelitis, Vaccine 32(43) (2014) 5681–9. [DOI] [PubMed] [Google Scholar]
- [91].Lewis JS, Dolgova NV, Zhang Y, Xia CQ, Wasserfall CH, Atkinson MA, Clare-Salzler MJ, Keselowsky BG, A combination dual-sized microparticle system modulates dendritic cells and prevents type 1 diabetes in prediabetic NOD mice, Clin Immunol (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Lewis JS, Stewart JM, Marshall GP, Carstens MR, Zhang Y, Dolgova NV, Xia C, Brusko TM, Wasserfall CH, Clare-Salzler MJ, Atkinson MA, Keselowsky BG, Dual-Sized Microparticle System for Generating Suppressive Dendritic Cells Prevents and Reverses Type 1 Diabetes in the Nonobese Diabetic Mouse Model, ACS Biomaterials Science & Engineering (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Yoon YM, Lewis JS, Carstens MR, Campbell-Thompson M, Wasserfall CH, Atkinson MA, Keselowsky BG, A combination hydrogel microparticle-based vaccine prevents type 1 diabetes in non-obese diabetic mice, Scientific reports 5 (2015) 13155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Yeste A, Nadeau M, Burns EJ, Weiner HL, Quintana FJ, Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis, Proceedings of the National Academy of Sciences of the United States of America 109(28) (2012) 11270–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Yeste A, Takenaka MC, Mascanfroni ID, Nadeau M, Kenison JE, Patel B, Tukpah AM, Babon JA, DeNicola M, Kent SC, Pozo D, Quintana FJ, Tolerogenic nanoparticles inhibit T cell-mediated autoimmunity through SOCS2, Sci Signal 9(433) (2016) ra61. [DOI] [PubMed] [Google Scholar]
- [96].Serra P, Santamaria P, Nanoparticle-based approaches to immune tolerance for the treatment of autoimmune diseases, European Journal of Immunology 48(5) (2018) 751–756. [DOI] [PubMed] [Google Scholar]
- [97].Serda RE, Particle platforms for cancer immunotherapy, International journal of nanomedicine 8 (2013) 1683–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Schlosser E, Mueller M, Fischer S, Basta S, Busch DH, Gander B, Groettrup M, TLR ligands and antigen need to be coencapsulated into the same biodegradable microsphere for the generation of potent cytotoxic T lymphocyte responses, Vaccine 26(13) (2008) 1626–37. [DOI] [PubMed] [Google Scholar]
- [99].Elamanchili P, Lutsiak CM, Hamdy S, Diwan M, Samuel J, “Pathogen-mimicking” nanoparticles for vaccine delivery to dendritic cells, J Immunother 30(4) (2007) 378–95. [DOI] [PubMed] [Google Scholar]
- [100].Singh A, Nie H, Ghosn B, Qin H, Kwak LW, Roy K, Efficient modulation of T-cell response by dual-mode, single-carrier delivery of cytokine-targeted siRNA and DNA vaccine to antigen-presenting cells, Mol Ther 16(12) (2008) 2011–21. [DOI] [PubMed] [Google Scholar]
- [101].Jhunjhunwala S, Raimondi G, Thomson AW, Little SR, Delivery of rapamycin to dendritic cells using degradable microparticles, J Control Release 133(3) (2009) 191–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Reddy ST, Swartz MA, Hubbell JA, Targeting dendritic cells with biomaterials: developing the next generation of vaccines, Trends Immunol 27(12) (2006) 573–9. [DOI] [PubMed] [Google Scholar]
- [103].Morelli AE, Thomson AW, Dendritic cells: regulators of alloimmunity and opportunities for tolerance induction, Immunol Rev 196 (2003) 125–46. [DOI] [PubMed] [Google Scholar]
- [104].Fife BT, Guleria I, Gubbels Bupp M, Eagar TN, Tang Q, Bour-Jordan H, Yagita H, Azuma M, Sayegh MH, Bluestone JA, Insulin-induced remission in new-onset NOD mice is maintained by the PD-1-PD-L1 pathway, J Exp Med 203(12) (2006) 2737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Eagar TN, Karandikar NJ, Bluestone JA, Miller SD, The role of CTLA-4 in induction and maintenance of peripheral T cell tolerance, Eur J Immunol 32(4) (2002) 972–81. [DOI] [PubMed] [Google Scholar]
- [106].Lemos H, Huang L, Chandler PR, Mohamed E, Souza GR, Li L, Pacholczyk G, Barber GN, Hayakawa Y, Munn DH, Mellor AL, Activation of the STING adaptor attenuates experimental autoimmune encephalitis, J Immunol 192(12) (2014) 5571–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Maj M, Bruno V, Dragic Z, Yamamoto R, Battaglia G, Inderbitzin W, Stoehr N, Stein T, Gasparini F, Vranesic I, Kuhn R, Nicoletti F, Flor PJ, (−)-PHCCC, a positive allosteric modulator of mGluR4: characterization, mechanism of action, and neuroprotection, Neuropharmacology 45(7) (2003) 895–906. [DOI] [PubMed] [Google Scholar]
- [108].Fallarino F, Volpi C, Fazio F, Notartomaso S, Vacca C, Busceti C, Bicciato S, Battaglia G, Bruno V, Puccetti P, Fioretti MC, Nicoletti F, Grohmann U, Di Marco R, Metabotropic glutamate receptor-4 modulates adaptive immunity and restrains neuroinflammation, Nat Med 16(8) (2010) 897–902. [DOI] [PubMed] [Google Scholar]
- [109].Nosratabadi R, Rastin M, Sankian M, Haghmorad D, Mahmoudi M, Hyperforin-loaded gold nanoparticle alleviates experimental autoimmune encephalomyelitis by suppressing Th1 and Th17 cells and upregulating regulatory T cells, Nanomedicine : nanotechnology, biology, and medicine 12(7) (2016) 1961–1971. [DOI] [PubMed] [Google Scholar]
- [110].Hwang JH, Kim SJ, Kim Y-H, Noh J-R, Gang G-T, Chung BH, Song NW, Lee C-H, Susceptibility to gold nanoparticle-induced hepatotoxicity is enhanced in a mouse model of nonalcoholic steatohepatitis, Toxicology 294 (2012) 27–35. [DOI] [PubMed] [Google Scholar]
- [111].Balmert SC, Donahue C, Vu JR, Erdos G, Falo LD, Little SR, In vivo induction of regulatory T cells promotes allergen tolerance and suppresses allergic contact dermatitis, Journal of Controlled Release 261 (2017) 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Gandhi R, Kumar D, Burns EJ, Nadeau M, Dake B, Laroni A, Kozoriz D, Weiner HL, Quintana FJ, Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells, Nat Immunol 11(9) (2010) 846–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL, Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor, Nature 453(7191) (2008) 65–71. [DOI] [PubMed] [Google Scholar]
- [114].Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah AM, Burns EJ, Weiner HL, An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis, Proceedings of the National Academy of Sciences of the United States of America 107(48) (2010) 20768–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, Fujii-Kuriyama Y, Kishimoto T, Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism, Proc Natl Acad Sci U S A 107(46) (2010) 19961–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Smarr CB, Yap WT, Neef TP, Pearson RM, Hunter ZN, Ifergan I, Getts DR, Bryce PJ, Shea LD, Miller SD, Biodegradable antigen-associated PLG nanoparticles tolerize Th2-mediated allergic airway inflammation pre- and postsensitization, Proceedings of the National Academy of Sciences of the United States of America 113(18) (2016) 5059–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Dumont FJ, Su QX, Mechanism of action of the immunosuppressant rapamycin, 1995, pp. 373–395. [DOI] [PubMed]
- [118].Aagaard-Tillery KM, Jelinek DF, Inhibition of Human B Lymphocyte Cell Cycle Progression and Differentiation by Rapamycin, Cellular Immunology 156(2) (1994) 493–507. [DOI] [PubMed] [Google Scholar]
- [119].Sehgal SN, Bansbach CC, Rapamycin: In Vitro Profile of a New Immunosuppressive Macrolide, Annals of the New York Academy of Sciences 685(1) (1993) 58–67. [DOI] [PubMed] [Google Scholar]
- [120].Araki K, Ellebedy AH, Ahmed R, TOR in the immune system, Current opinion in cell biology 23(6) (2011) 707–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Powell JD, Pollizzi KN, Heikamp EB, Horton MR, Regulation of immune responses by mTOR, Annual review of immunology 30 (2012) 39–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Peine KJ, Guerau-de-Arellano M, Lee P, Kanthamneni N, Severin M, Probst GD, Peng H, Yang Y, Vangundy Z, Papenfuss TL, Lovett-Racke AE, Bachelder EM, Ainslie KM, Treatment of experimental autoimmune encephalomyelitis by codelivery of disease associated Peptide and dexamethasone in acetalated dextran microparticles, Molecular pharmaceutics 11(3) (2014) 828–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Pearson RM, Casey LM, Hughes KR, Wang LZ, North MG, Getts DR, Miller SD, Shea LD, Controlled Delivery of Single or Multiple Antigens in Tolerogenic Nanoparticles Using Peptide-Polymer Bioconjugates, Molecular therapy : the journal of the American Society of Gene Therapy 25(7) (2017) 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Jamison BL, Neef T, Goodspeed A, Bradley B, Baker RL, Miller SD, Haskins K, Nanoparticles Containing an Insulin-ChgA Hybrid Peptide Protect from Transfer of Autoimmune Diabetes by Shifting the Balance between Effector T Cells and Regulatory T Cells, J Immunol 203(1) (2019) 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Verbeke CS, Gordo S, Schubert DA, Lewin SA, Desai RM, Dobbins J, Wucherpfennig KW, Mooney DJ, Multicomponent Injectable Hydrogels for Antigen-Specific Tolerogenic Immune Modulation, Adv Healthc Mater 6(6) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Green AJ, Gelfand JM, Cree BA, Bevan C, Boscardin WJ, Mei F, Inman J, Arnow S, Devereux M, Abounasr A, Nobuta H, Zhu A, Friessen M, Gerona R, von Budingen HC, Henry RG, Hauser SL, Chan JR, Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): a randomised, controlled, double-blind, crossover trial, Lancet 390(10111) (2017) 2481–2489. [DOI] [PubMed] [Google Scholar]
- [127].Mei F, Lehmann-Horn K, Shen YA, Rankin KA, Stebbins KJ, Lorrain DS, Pekarek K, S AS, Xiao L, Teuscher C, von Budingen HC, Wess J, Lawrence JJ, Green AJ, Fancy SP, Zamvil SS, Chan JR, Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery, Elife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Li Z, He Y, Fan S, Sun B, Clemastine rescues behavioral changes and enhances remyelination in the cuprizone mouse model of demyelination, Neurosci Bull 31(5) (2015) 617–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Liu J, Dupree JL, Gacias M, Frawley R, Sikder T, Naik P, Casaccia P, Clemastine Enhances Myelination in the Prefrontal Cortex and Rescues Behavioral Changes in Socially Isolated Mice, 2016, pp. 957–962. [DOI] [PMC free article] [PubMed]
- [130].Furuyama K, Chera S, van Gurp L, Oropeza D, Ghila L, Damond N, Vethe H, Paulo JA, Joosten AM, Berney T, Bosco D, Dorrell C, Grompe M, Raeder H, Roep BO, Thorel F, Herrera PL, Diabetes relief in mice by glucose-sensing insulin-secreting human alpha-cells, Nature 567(7746) (2019) 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]