

SUMMARY
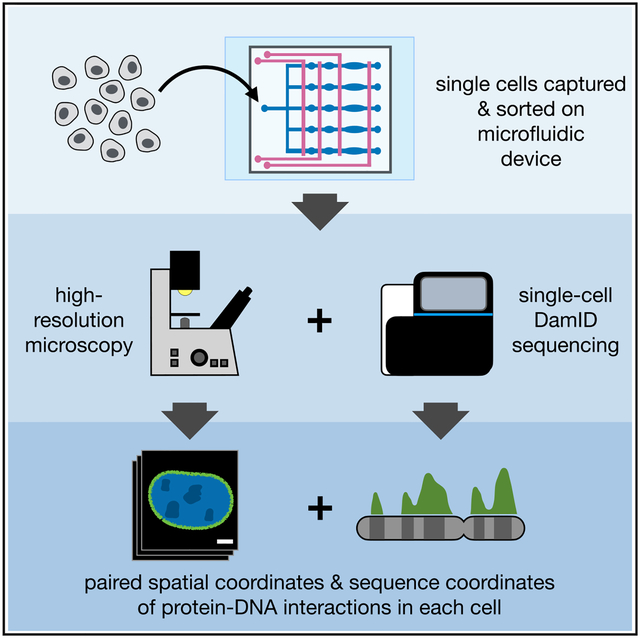
DNA adenine methyltransferase identification (DamID) measures a protein’s DNA-binding history by methylating adenine bases near each protein-DNA interaction site and then selectively amplifying and sequencing these methylated regions. Additionally, these interactions can be visualized using m6A-Tracer, a fluorescent protein that binds to methyladenines. Here, we combine these imaging and sequencing technologies in an integrated microfluidic platform (μDamID) that enables single-cell isolation, imaging, and sorting, followed by DamID. We use μDamID and an improved m6A-Tracer protein to generate paired imaging and sequencing data from individual human cells. We validate interactions between Lamin-B1 protein and lamina-associated domains (LADs), observe variable 3D chromatin organization and broad gene regulation patterns, and jointly measure single-cell heterogeneity in Dam expression and background methylation. μDamID provides the unique ability to compare paired imaging and sequencing data for each cell and between cells, enabling the joint analysis of the nuclear localization, sequence identity, and variability of protein-DNA interactions. A record of this paper’s transparent peer review process is included in the Supplemental Information.
Graphical Abstract
In Brief
Each cell’s behavior depends largely on protein-DNA interactions that regulate gene expression. Here, the authors designed and built a microfluidic device enabling the user to sort and isolate live single cells, image the spatial location of specific protein-DNA interactions at high resolution, and then amplify and sequence DNA from these interaction sites. This provides, for the first time, paired imaging and sequencing data that reveal both the spatial location and sequence identity of protein-DNA interactions across the genome within single cells.
INTRODUCTION
Complex life depends on the protein-DNA interactions that constitute and maintain the epigenome, including interactions with histone proteins, transcription factors, DNA (de)methylases, and chromatin remodeling complexes, among others. These interactions enable the static DNA sequence inside the nucleus to dynamically execute different gene expression programs that shape the cell’s identity and behavior. Methods for measuring protein-DNA interactions have proven indispensable for understanding the epigenome, though, to date, most of this knowledge has been derived from experiments in bulk cell populations. By requiring large numbers of cells, these bulk methods can fail to capture critical epigenomic processes that occur in small numbers of dividing cells, including processes that influence embryo development, developmental diseases, stem cell differentiation, and certain cancers. By averaging together populations of cells, bulk methods also fail to capture important epigenomic dynamics occurring in asynchronous single cells during differentiation or the cell cycle. Because of this, bulk methods can overlook important biological heterogeneity within a tissue. It also remains difficult to pair bulk biochemical data with imaging data, which inherently provide information in single cells, and which can reveal the spatial location of protein-DNA interactions within the nuclei of living cells. These limitations underline the need for high-sensitivity single-cell methods for measuring protein-DNA interactions.
Most approaches for mapping protein-DNA interactions rely on immunoaffinity purification, in which protein-DNA complexes are physically isolated using a high-affinity antibody against the protein, then purified by washing and de-complexed so the interacting DNA can be amplified and measured. The most widely used among these methods is chromatin immunoprecipitation sequencing (ChIP-seq) (Barski et al., 2007; Johnson et al., 2007), which has formed the backbone of several large epigenome mapping projects (Celniker et al., 2009; ENCODE Consortium, 2012; Kundaje et al., 2015). One drawback of ChIP-seq is that protein-DNA complexes, which are often fragile, must survive the shearing or digestion of the surrounding DNA, as well as several intermediate washing and purification steps, in order to be amplified and sequenced. This results in a loss of sensitivity, especially when using a small amount of starting material. More recent immunoaffinity-based methods have reduced the high input requirements of ChIP-seq, but they recover relatively few interactions in small numbers of cells or single cells (Wu et al., 2012; Shen et al., 2015; Jakobsen et al., 2015; Rotem et al., 2015; Zhang et al., 2016; Skene et al., 2018; Harada et al., 2019; Kaya-Okur et al., 2019; Carter et al., 2019; Grosselin et al., 2019).
An alternative method for probing protein-DNA interactions, called DNA adenine methyltransferase identification (DamID), relies not on physical separation of protein-DNA complexes (as in ChIP-seq) but on a sort of “chemical recording” of protein-DNA interactions onto the DNA itself, which can later be selectively amplified (Figure 1A; van Steensel and Henikoff, 2000; Vogel et al., 2007; Aughey and Southall, 2016). This method utilizes a small enzyme from E. coli called DNA adenine methyltransferase (Dam). When genetically fused to the protein of interest, Dam de posits methyl groups near the protein-DNA contacts at the N6 positions of adenine bases (m6A) within GATC sequences (which occur once every 270 bp on average across the human genome). That is, wherever the protein contacts DNA throughout the genome, m6A marks are left at the GATC sites in its trail. These m6A marks are highly stable in eukaryotic cells, which do not tend to methylate (or demethylate) adenines (O’Brown et al., 2019). Dam expression has been shown to have no discernable effect on gene expression in a human cell line, and its m6A marks were shown to be passed to daughter cells, halving in quantity each generation after Dam is inactivated (Park et al., 2019). These properties allow even transient protein-DNA interactions to be recorded as stable, biologically orthogonal chemical signals on the DNA, useful for integrating protein-DNA interactions over time, up to the length of a cell cycle.
Figure 1. μDamID Device Design and Function.
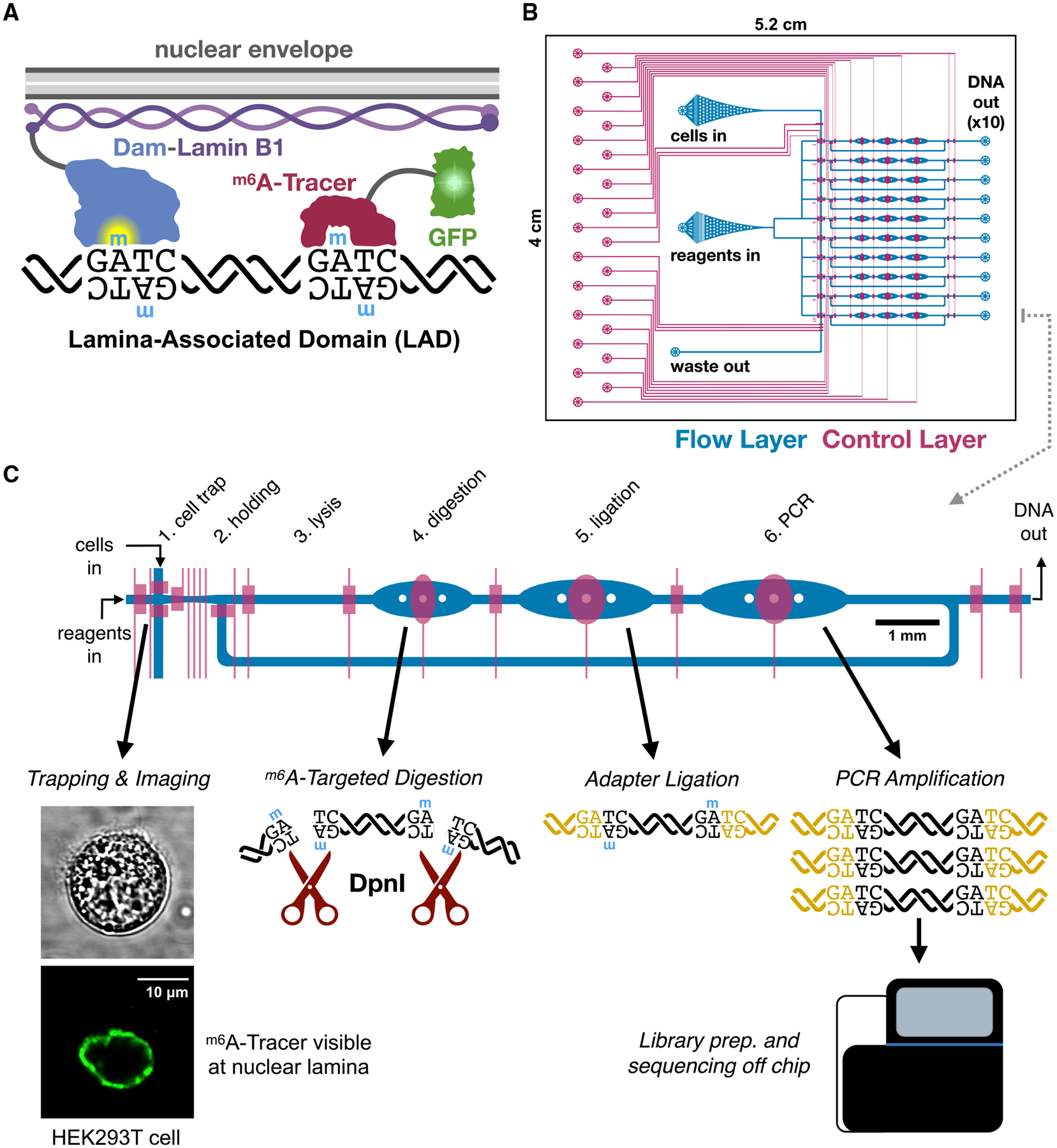
(A) Overview of DamID (van Steensel and Henikoff, 2000) and m6A-Tracer (Kind et al., 2013) technologies applied to study interactions between DNA and nuclear lamina proteins.
(B) The overall design of the 10-cell device, showing the flow layer (blue, where cells and reagents enter channels) and the control layer (red, where elastomeric valves overlap the flow layer to control the flow of liquids).
(C) A closer view of one lane explaining the DamID protocol and the function of each chamber of the device. 10 cells are trapped, imaged, and selected serially, one per lane, then all 10 cells are lysed and processed in parallel.
DamID reads out these chemical recordings of protein-DNA interactions by specifically amplifying and then sequencing fragments of DNA containing the interaction site. First, genomic DNA is purified and digested with DpnI, a restriction enzyme that exclusively cleaves Gm6ATC sites (Figure 1). Then, universal adapters are ligated onto the fragment ends to allow for amplification using universal primers. Only regions with a high density of m6A produce DNA fragments short enough to be amplified by polymerase chain reaction (PCR) and quantified by microarray or high-throughput sequencing (Wu et al., 2016). DamID has been used to explore dynamic regulatory protein-DNA interactions such as transcription factor binding (Orian et al., 2003) and RNA polymerase binding (Southall et al., 2013) as well as protein-DNA interactions that maintain large-scale genome organization. One frequent application of DamID is to study large DNA domains associated with proteins at the nuclear lamina, near the inner membrane of the nuclear envelope (Pickersgill et al., 2006; Guelen et al., 2008, reviewed by van Steensel and Belmont, 2017). Because DamID avoids the limitations of antibody binding, physical separations, or intermediate purification steps, it lends itself to single-cell applications. DamID has been successfully applied to sequence lamina-associated domains (LADs) in single cells in a one-pot reaction, recovering hundreds of thousands of unique fragments per cell (Kind et al., 2015).
While DamID maps the sequence positions of protein-DNA interactions throughout the genome, the spatial location of these interactions in the nucleus can also play an important role in genome regulation (Bickmore and van Steensel, 2013). A method related to DamID can be applied to specifically label and visualize protein-DNA interactions using fluorescence microscopy, revealing their spatial location within the nucleus in live cells (Kind et al., 2013). Visualization requires co-expression of a different fusion protein called m6A-Tracer, which contains green fluorescent protein (GFP) and a domain that binds specifically to methylated GATC sites. This imaging technology has been applied to visualize the dynamics of LADs within single cells (Kind et al., 2013). Both imaging and sequencing protein-DNA interactions can provide useful single-cell epigenomic information, but despite recent advances in single-cell sequencing technologies, it remains fundamentally difficult to track individual cells and pair their sequencing data with other measurements such as imaging. While other DamID studies have performed imaging and sequencing in parallel (Kind et al., 2015; Borsos et al., 2019), they do not provide linked imaging and sequencing data for individual cells. Pairing imaging and sequencing data could be applied to study, for example, how the dynamic remodeling of chromatin proteins across the genome in developing cells relates to the localization of those proteins in the nucleus. Imaging prior to sequencing also allows for the identification and sorting of complex cytological phenotypes in cells, such as the presence of micronuclei and other nuclear abnormalities that would be difficult or impossible using common fluorescence-activated sorting methods.
Here, we aimed to pair DamID with m6A-Tracer imaging to produce coupled imaging and sequencing measurements of protein-DNA interactions in the same single cells. To achieve this, we engineered an integrated microfluidic device that enables single-cell isolation, imaging, selection, and DamID processing, which we call “μDamID,” for microfluidic DamID. We applied our device to image and map nuclear lamina interactions in a transiently transfected human cell line co-expressing m6A-Tracer, and we validated our measurements against bulk DamID data from the same cell line as well as other human cell lines (Lenain et al., 2017; Kind et al., 2015). We discuss the advantages and potential applications of our device as well as future improvements to this system.
RESULTS
Device Design and Operation
We designed and fabricated a polydimethylsiloxane (PDMS) microfluidic device with integrated elastomeric valves to facilitate the various reaction stages of the DamID protocol in a single liquid phase within the same device to avoid sample loss prior to DNA amplification (Figure 1). The device is compatible with high-magnification imaging on inverted microscopes, enabling imaging prior to cell lysis. Each device was designed to process 10 cells in parallel, each in an individual reaction lane fed from a common set of inlets to avoid sample cross-contamination. Valves are controlled by pneumatic actuators operated electronically via a programmable computer interface (White and Streets, 2018).
Device operation was modified from our previous single-cell RNA sequencing platform (Streets et al., 2014). A suspension of single cells is loaded into the cell inlet (Figure 1B), and cells are directed toward a trapping region by a combination of pressure-driven flow and precise peristaltic pumping. Cells enter the device in a wide filter region where dozens can be visualized and screened at once as they move toward a narrow channel leading to the trapping regions. As a cell crosses one of the 10 trapping regions, valves are actuated to immobilize the cell for imaging (Figure S1). The cell is imaged by confocal fluorescence microscopy to visualize the localization of m6A-Tracer, and after image acquisition, the user can choose whether to select the cell for DamID processing, or to reject it and send it out the waste outlet (Figure 1B).
Selected cells are injected from the trapping region into a holding chamber using pressure-driven flow from the reagent inlet (Figures 1C and S1). Once all 10 holding chambers are filled with imaged cells, processing proceeds in parallel for all 10 cells by successively adding the necessary reagents for each step of the single-cell DamID protocol (Kind et al., 2015) and dead-end filling each of the subsequent reaction chambers. Reaction temperatures are controlled by placing the device on a custom-built thermoelectric control unit for dynamic thermal cycling. Enzymes are heat inactivated between each step (Kind et al., 2015), and a low concentration of mild detergent was added to all reactions to prevent enzyme adhesion to PDMS (Streets et al., 2014).
In the first reaction stage, a buffer containing detergent and proteinase pushes the cell into the lysis chamber, where its membranes are lysed and its proteins, including m6A-Tracer, are digested away (Figure 1C). Next, a DpnI reaction mix is added to digest the genomic DNA at Dam-methylated GATC sites in the digestion chamber. Then, a mix of DamID universal adapter oligonucleotides and DNA ligase is added to the ligation chamber. Finally, a PCR mix is added containing primers that anneal to the universal adapters and all valves within the lane are opened, creating a 120-nl cyclic reaction chamber. Contents are thoroughly mixed by peristaltic pumping around the reaction ring, then PCR is carried out on-device by thermocycling. Amplified DNA is collected from each individual lane outlet, and sequencing library preparation is carried out off-device.
Mapping Human Lamina-Associated Domains
We evaluated the performance of this platform by mapping the sequence and spatial location of LADs in a human cell line, allowing us to compare our data with previously published LAD maps from DamID experiments in human cell lines (Kind et al., 2015; Lenain et al., 2017). LADs are large (median 500 kb) and comprise up to 30% of the genome in human cells (Guelen et al., 2008). LADs serve both a structural function, acting as a scaffold that underpins the three-dimensional architecture of the genome in the nucleus, and a regulatory function, as LADs tend to be gene-poor, more heterochromatic, and transcriptionally less active (reviewed by van Steensel and Belmont, 2017 and Buchwalter et al., 2019). m6A-Tracer has previously been applied to visualize LADs, which appear as a characteristic ring around the nuclear periphery in confocal fluorescence microscopy images (Kind et al., 2013; Figure 1C).
We carried out experiments in HEK293T cells for their ease of growth, transfection, suspension, and isolation. To enable rapid expression of Dam and m6A-Tracer transgenes, we transiently transfected cells with DNA plasmids containing genes for a drug-inducible Dam-LMNB1 fusion protein as well as constitutively expressed m6A-Tracer. We then induced Dam-LMNB1 expression, optimizing the expression times to maximize the proportion of cells with fluorescent laminar rings (an example of which is visible in Figure 1C). Because transient transfection yields a heterogeneous population of cells, each with potentially variable copies of the transgenes, it was important for us to be able to take high-resolution confocal images of cells and select only those with visible laminar rings, which were more likely to have the correct expression levels, and which were unlikely to be in the mitosis phase of the cell cycle. This type of complex sorting would not be possible with sorting methods like fluorescence-activated cell sorting (FACS) but is straightforward in our microfluidic platform.
In addition to processing Dam-LMNB1 cells, we transfected cells with the Dam gene alone, not fused to LMNB1, to provide a negative control demonstrating where the unfused Dam enzyme would mark the genome if not tethered to the nuclear lamina (Vogel et al., 2007). This control is useful for estimating the background propensity for each genomic region to be methylated, since Dam preferentially methylates more accessible regions of the genome, including gene-rich regions (Singh and Klar, 1992; Lenain et al., 2017; Aughey et al., 2018). We selected Dam-only cells that had high fluorescence levels across the nucleus and did not appear mitotic. We also performed bulk DamID (Vogel et al., 2007) in populations of transiently transfected HEK293T cells for validation. We used a mutant of Dam (V133A; Elsawy and Chahar, 2014), which is predicted to have weaker methylation activity than the wild-type allele on unmethylated DNA, and we hypothesized that it would reduce background methylation, similar to weakened Dam mutants previously engineered to improve methylation specificity (Park et al., 2019). To test this, we performed bulk DamID experiments comparing the mutant and wild-type alleles and found that the V133A mutant allele provides more than 2-fold greater signal-to-background compared to the wild-type allele (Figure S2). We also performed RNA sequencing in bulk cells that were untreated or transfected with Dam-only, Dam-LMNB1, or m6A-Tracer, and we found only two differentially expressed genes (Figure S2). This corroborates similar published findings by others showing that Dam expression and adenine methylation have little or no effect on gene expression in HEK293T cells (Park et al., 2019).
We first ran three devices containing 25 imaged cells total, with five empty lanes left as negative controls that did not yield sequenceable quantities of DNA. From these 25 cells, we selected a batch of 18 cells for multiplexed sequencing based on imaging quality and DNA yield in order to achieve a desired level of coverage per cell. To increase throughput modestly, we built a second microfluidic control system, enabling us to run two devices in parallel, processing up to 20 cells in one experiment. Using four additional devices, we processed a second batch of 40 Dam-LMNB1 cells, with several experimental changes discussed below. We found that 34 of 38 cells with visible laminar m6A-Tracer “rings” yielded sequenceable DNA quantities (89% yield), and we proceeded to sequence those 34 cells plus two more: one cell with no ring and one cell with a ring but low sequencing yield (D09 and D10, respectively, Figure S3). In total we sequenced 54 cells from both batches. We obtained a mean of 4 million raw read pairs per cell (range 300k–8 M), covering a mean of 140,000 unique DpnI fragments per cell (10k–370k), which falls in the range of previous DamID results from single cells (Kind et al., 2015; Figure S4).
Validating Single-Cell LAD Maps
To assess whether our single-cell μDamID sequencing data provide accurate measurements of LADs, we first compared our single-cell results with those we obtained from bulk DamID in the same cell line. DamID results are reported as a difference or log ratio between the observed coverage from Dam-LMNB1-expressing cells and the expected coverage from background methylation, estimated using coverage from Dam-only-expressing cells (see STAR Methods). This measure is computed within fixed 100-kb bins across the genome, as reported previously (Kind et al., 2015). For each cell, we made binary calls of whether a bin was in contact with the lamina in that cell (STAR Methods), and the broad-scale organization of these single-cell binary LAD maps largely agrees with the bulk data (Figure 2A). By aggregating the raw coverage from our Dam-LMNB1 expressing single cells, we found strong correspondence with the bulk coverage obtained from millions of cells (Figure 2B, r = 0.84).
Figure 2. Validation of μDamID Sequencing Data.
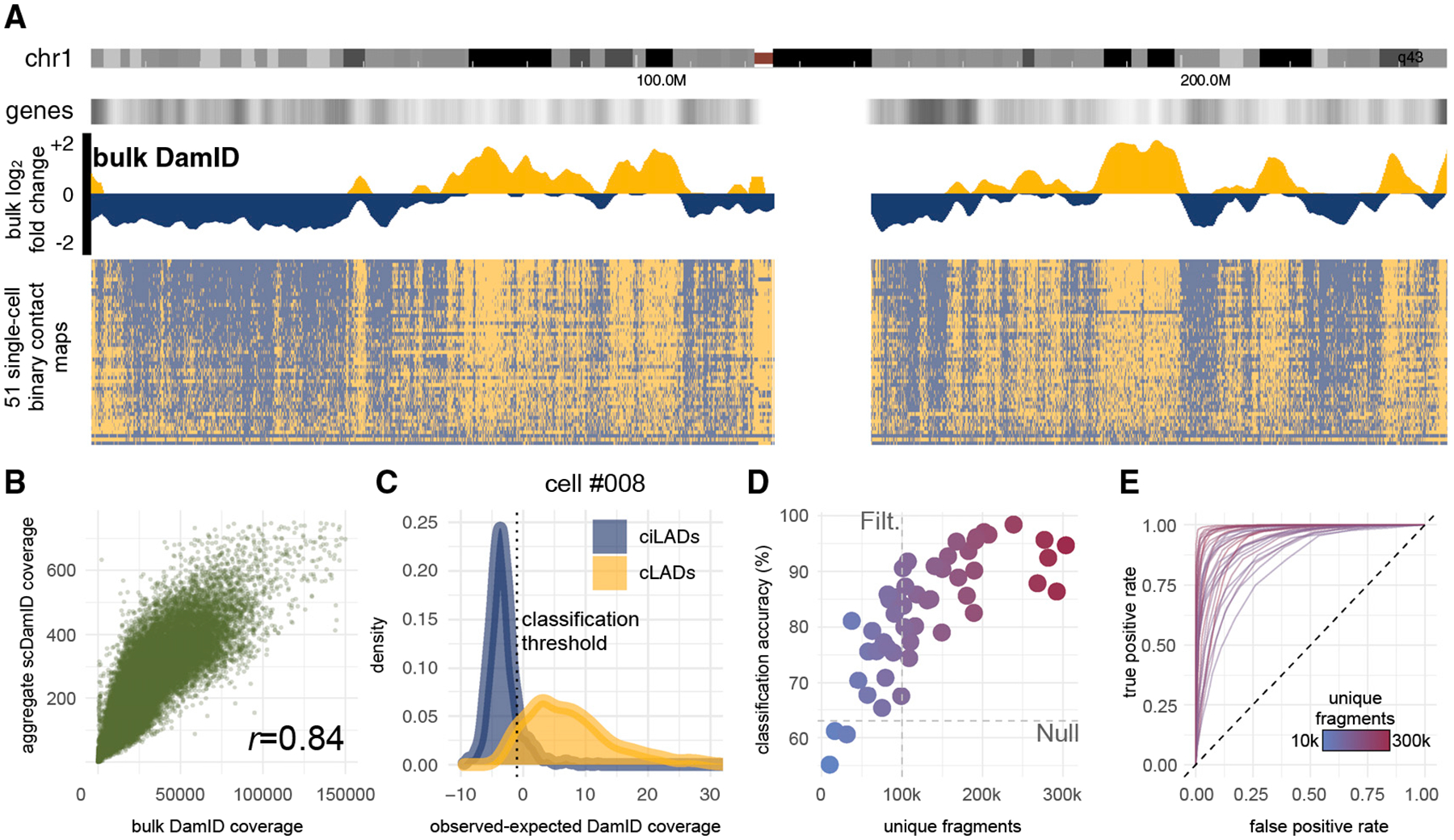
(A) Comparison of bulk and single-cell LMNB1 DamID sequencing data across all of human chromosome 1 (chr1 ideogram in first track from top), normalized to bulk Dam-only data. Positive values (gold) represent regions associated with the nuclear lamina, which tend to have lower relative gene density (shown in second track). The bulk data (third track) are shown as log2-fold-change values between Dam-LMNB1 and Dam-only samples. Each row of the binary contact map (fourth track) represents a single cell, sorted from top to bottom by genome-wide control set classification accuracy.
(B) Scatterplot comparing raw Dam-LMNB1 sequencing coverage in bulk versus aggregated single-cell samples in 100-kb bins.
(C) Normalized coverage distributions within positive (cLADs, gold) and negative (ciLADs, blue) control sets in one cell (#008) expressing Dam-LMNB1. The threshold that distinguishes these sets with maximal accuracy is shown as a vertical dotted line.
(D) The maximum control set classification accuracy for each of 50 Dam-LMNB1 cells versus the number of unique DpnI fragments sequenced for each cell (also indicated by color gradient; outlier cell #007 was excluded). A coverage threshold of 100k fragments used for downstream analyses is indicated, as well as the null accuracy achieved after scrambling values in all bins across the genome (63%).
(E) Receiver-operator characteristic curves for 31 Dam-LMNB1 cells above the 100k coverage threshold.
In order to create the binary contact maps across the genome within single cells, we trained a classifier on a set of stringent positive and negative controls: regions confidently known to be strongly associated with the lamina or strongly unassociated with the lamina based on bulk DamID data from our study and others (Lenain et al., 2017; see STAR Methods). Positive controls were derived from 100-kb bins across the genome that were previously annotated in other human cell lines to be strongly associated with the nuclear lamina (referred to as constitutive LADs or cLADS) and further filtered to have the highest bulk DamID scores in HEK293T cells. These bins are therefore most likely to have high contact frequencies (CFs) in individual cells (Kind et al., 2015; Figure S4). Negative controls were similarly determined using bulk data to be consistently not associated with the nuclear lamina across cell types and in our cells (referred to as constitutive inter-LADs or ciLADS), making them most likely to have low CFs in individual cells (Kind et al., 2015; Figure S4). These stringent control sets constitute roughly 4% of the genome each.
For each single cell expressing Dam-LMNB1, we computed the distribution of its normalized sequencing coverage in bins from the positive (cLAD) and negative (ciLAD) control regions (Figure 2C), with the expectation that cLADs have high coverage, and ciLADs have little or no coverage in each cell. Given these control distributions, we chose a coverage threshold to maximally separate the known cLADs and ciLADs. Across the 51 Dam-LMNB1 cells, we determined thresholds that distinguish the known cLADs and ciLADs with a median accuracy of 85% before any filtering (versus 63% if all bins are scrambled), which correlates positively with the number of unique DpnI fragments sequenced per cell, a measure of library complexity (Figures 2D and S4). Because we used a transient transfection system, expression levels of Dam-LMB1 varied widely from cell to cell, reducing classification accuracy in some cells with high noise levels due to background methylation. We filtered higher-noise cells using a threshold of unique covered fragments, leaving 31 Dam-LMNB1 cells with a median classification accuracy of 90% (range 74%–98%, Figure 2D). Our classification approach enables inference of expected error rates for each bin’s coverage level in each cell, providing a framework for data normalization, interpretation, and further inference. These error rates can be represented with receiver operating characteristic (ROC) curves for each cell, showing the empirical trade-off between false-positive and false-negative classifications at varying normalized coverage thresholds (Figure 2E).
We next computed pairwise correlations between the raw coverage for all single cells with each other, with the bulk data, with aggregated published single-cell DamID data (from Kind et al., 2015), and with the number of annotated genes across 100-kb bins genome wide. After removing low-complexity cells, we performed unsupervised hierarchical clustering on these datasets and produced a heatmap of their pairwise correlations (Figure 3A). We found that the 3 Dam-only single cells cluster with each other, along with the bulk Dam-only data, with the Kind et al., Dam-only data, and with the number of genes, as expected. The Dam-LMNB1 cells cluster separately with each other, with the bulk Dam-LMNB1 data, and with the Kind et al. Dam-LMNB1 data, confirming that these sequencing data are measuring meaningful biological patterns in single cells. These clusters also reflect expected nuclear spatial distributions of methylation reported by m6A-Tracer fluorescence (Figures 3B–3D). Notably, one Dam-LMNB1 cell with unexpectedly high fluorescence signal in the nuclear interior contained a methylation profile that appeared intermediate between the Dam-only and other Dam-LMNB1 cells, perhaps owing to high Dam-LMNB1 expression (Figure 3A). This illustrates how spatial information can be used to validate DamID with joint single-cell imaging and sequencing measurements.
Figure 3. Genome-wide Comparisons of Sequencing Data and Relation to Imaging Data.
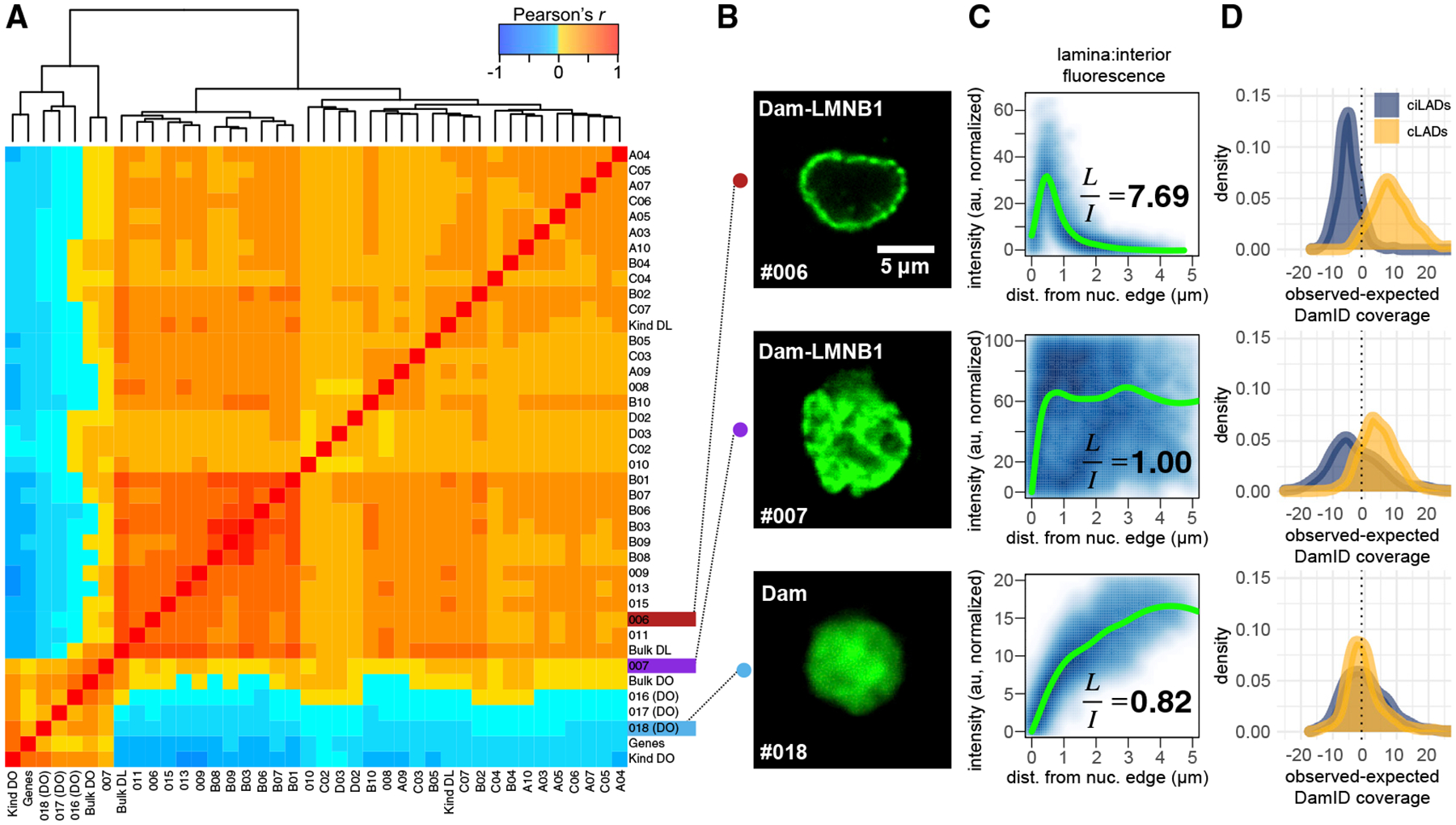
(A) Pairwise cell-cell Pearson correlation heatmap for raw sequencing coverage in 100-kb bins genome-wide, with dendrogram indicating hierarchical clustering results. Cell identifiers label each row (first batch 00**, second batch A–D**). DL, Dam-LMNB1; DO, Dam-only; Genes, number of RefSeq genes in each bin; Kind, aggregated single-cell data from Kind et al. (2015); Bulk, bulk HEK293T DamID data from this study.
(B) Confocal fluorescence microscopy images of m6A-Tracer GFP signal from 3 cells: one expressing Dam-only (#018), one expressing Dam-LMNB1 but showing high interior fluorescence (#007), and one expressing Dam-LMNB1 and showing the expected ring-like fluorescence at the nuclear lamina (#006).
(C) Normalized pixel intensity values plotted as a function of their distance from the nuclear edge (blue), with a fitted loess curve overlaid (green). Ratios of the mean normalized pixel intensities in the lamina (<1 μm from the edge) versus the Interior (>3.5 μm from the edge) are printed on each plot.
(D) DamID sequencing coverage distributions for each of the cLAD or ciLAD control sets (as in Figure 2C).
Identifying Variable LADs
In any given cell, only a subset of potential LADs come into contact with the lamina, and this subset can vary stochastically between cells (Kind et al., 2013). While most LADs at the lamina appear to remain in stable contact with the lamina throughout interphase, some LADs have been shown to move dynamically short distances toward and away from the lamina within the same cell over time (Kind et al., 2013), also potentially contributing to cell-to-cell variability in LADs. Single-cell DamID pro vides a unique opportunity to identify LADs that vary within a population of cells of the same type.
To measure this variability, at each bin in the genome, we counted the number of Dam-LMNB1 cells (out of 31 total cells) in which that bin was classified as having laminar contact to estimate its contact frequency (CF) (Kind et al., 2015), and we developed a method for propagating measurement and sampling uncertainty when inferring the true CF of each bin (STAR Methods; Figures 4A, 4B, and S5). As expected, bins belonging to the cLAD control sets have high CFs and lower gene expression while those in the ciLAD control sets have low CFs and higher gene expression (Figures 4A, 4C, 4D, and 5A). Furthermore, we found that CFs for each bin correlated well overall with published single-cell CFs from a different cell line, KBM7 (r = 0.8, Figure 5A; Kind et al., 2015).
Figure 4. Identification and Characterization of Variable LADs in HEK293T Cells.
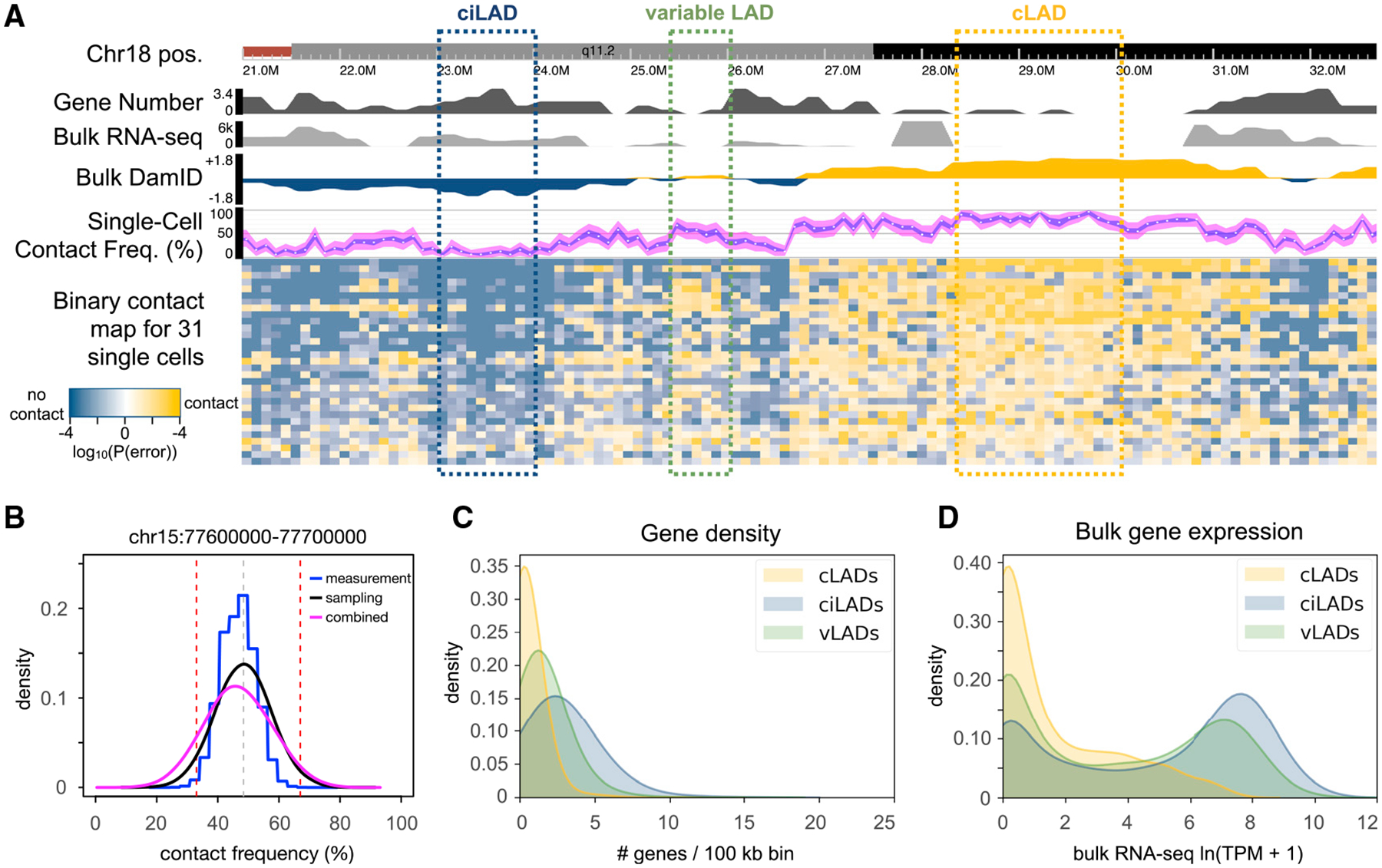
(A) A browser screenshot from chr18:21–33 Mb. The first track shows the chromosome ideogram and coordinates. The second track reports the number of RefSeq genes falling in each 100-kb bin. The third track reports the mean transcripts per million (TPM) value for each gene within each bin from bulk RNA sequencing data from untreated HEK293T cells. The fourth track reports the bulk DamID log2 fold change values as in Figure 2A. The fifth track indicates the CF estimate for each bin (white point), with a blue ribbon indicating the 95% confidence interval for the sample CF (measurement error), and the magenta ribbon indicating the 95% confidence interval for the population CF (measurement + sampling error). The sixth track shows binary contact calls for each bin (columns) in each cell (rows). Shades of gold and blue indicate bins classified as having lamina contact or no lamina contact, respectively, with darker shades indicating higher confidence in the classification (smaller measurement error probability). Annotated cLADs and ciLADs are indicated by gold and blue boxes, respectively, with a variable LAD region (vLAD) in green.
(B) For one bin in a different region, a comparison of measurement (blue) and sampling (black) distributions, along with a combined distribution (magenta) used for CF inference with propagated measurement uncertainty (as shown in A track 5). The gray vertical dotted line is the point estimate for that bin, and red dotted vertical lines are drawn at the vLAD CF thresholds (33% and 66%).
(C and D) Distributions of the number of genes (C) or mean TPM per gene (D) per 100-kb bin for each of the sets of cLADs, ciLADs, or vLADs.
Figure 5. Comparing Single-Cell CFs between Cell Types.
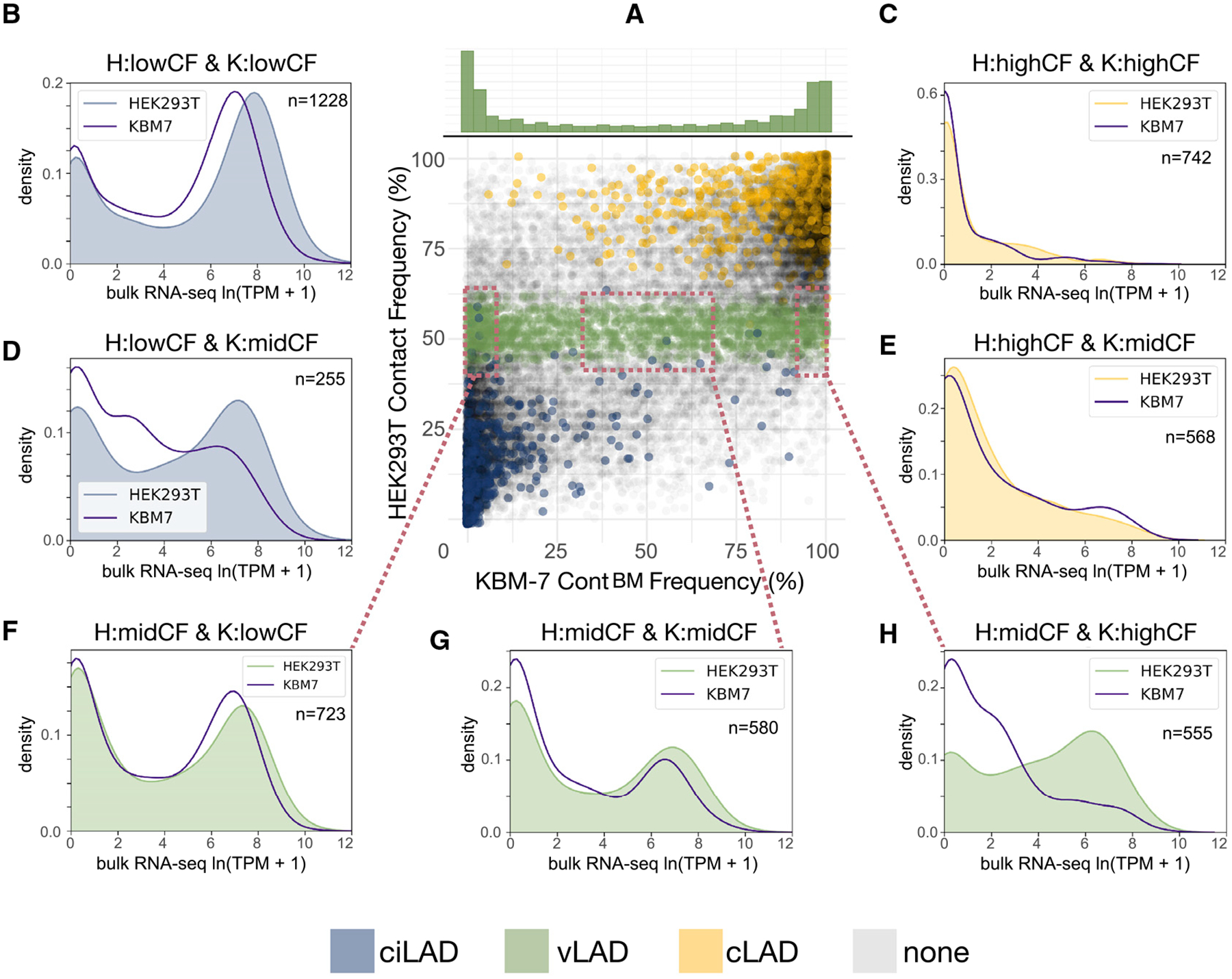
(A) A scatterplot of the CF estimates in HEK293T cells (this study) versus KBM-7 cells (Kind et al., 2015) across all bins in the genome. Each point is colored if the corresponding bin belongs to the cLAD (gold), ciLAD (blue), or vLAD (green) sets defined in HEK293T. Above the scatterplot is a histogram showing the KBM-7 CF distribution for all bins defined as vLADs in HEK293T, illustrating vLAD differences between cell types.
(B–H) Density plots indicating the relative distributions of bulk RNA sequencing coverage (transcripts per million) in each cell type, within bins classified as low CF (<5% CF, with high expression), middle CF (33%–66% CF, with intermediate expression), or high CF (>90% CF, with low expression) in each cell type, allowing for comparison of cell-type specific expression levels in bins that have low CF in both HEK293T cells and KBM7 cells (B), high CF in both HEK293T and KBM7 (C), low CF in HEK293T and middle CF in KBM7 (D), high CF in HEK293T and middle CF in KBM7 (E), middle CF in HEK293T and low CF in KBM7 (F), middle CF in both HEK293T and KBM7 (G), and middle CF in HEK293T and high CF in KBM7 (H).
To identify variable LADs, we defined a conservative set of bins with intermediate CFs between 33% and 66% (STAR Methods; Figure S5). We hypothesized that these stringently defined regions, which comprise 8% of the genome, would be more gene rich and have higher gene expression than cLADs, given their dynamic positioning in cells. Indeed, these variable LADs show intermediate gene density and intermediate bulk gene expression levels compared with the control sets of cLADs and ciLADs (Figures 4C and 4D), consistent with these regions being variably active within different cells.
We then explored whether these variable LADs were conserved in another human cell type. We found that the CFs of bins containing variable LADs identified in HEK293T cells varied widely in KBM7 cells (Figure 5A), suggesting only a small subset of these LADs are variable in both cell types, consistent with prior observations that regions with intermediate CFs are more likely to have different bulk DamID signals across cell types (Kind et al., 2015). Comparison of bulk RNA expression levels in bins that were classified as high, intermediate, or low CF in each cell type corroborated the inverse relationship between single-cell CF and bulk gene expression observed previously (Kind et al., 2015; Figures 5B–5H). For example, as regions shift from intermediate CFs to high CFs in one cell type as compared with the other, we observe a corresponding decrease of geneexpression (Figures 5E and 5H). These observations support the notion that the nuclear lamina serves as a dynamic regulatory element, not only between cell types but within a given cell type (Rooijers et al., 2019).
Imaging LADs in the μDamID Device Using m6A-Tracer-NES
We aimed to use fluorescence microscopy to quantify the spatial distribution of LADs in the μDamID device prior to DamID processing. In the first batch of 18 cells, we imaged m6A-Tracer to identify the localization of lamina-interacting DNA in the nucleus. We selected Dam-LMNB1-expressing cells that had laminar rings consistent with effective LAD methylation, as well as one anomalous Dam-LMNB1 cell with high signal in the nuclear interior (Figure 3C). We also observed fairly uniform fluorescence across the nucleus in cells expressing untethered Dam. These imaging patterns were largely predictive of their respective sequencing coverage distributions (Figure 3D). However, this investigation revealed an important limitation of the m6A-Tracer technology, which is that the m6A-Tracer protein localizes to the nucleus even in cells expressing no Dam (Figures 6A and 6B). One consequence is that cells with Dam and cells without Dam are nearly indistinguishable (Figure 6B), and cells with over-expressed m6A-Tracer show high background fluorescence levels in the nuclear interior even when co-expressing Dam-LMNB1 (Figure 6B). The only way to prevent this background issue is to carefully tune the expression level of m6A-Tracer so that the copy number of m6A-Tracer proteins does not exceed the number of available methylated GATC sites. This tuning would have to occur separately for any new Dam fusion protein. In a heterogeneous expression system like the one used here, since m6A-Tracer and Dam are expressed from separate plasmids, only a small fraction of cells have the correct ratios of expression to produce sharp laminar rings with low background in the nuclear interior (Figure 6B).
Figure 6. Improved Imaging of Protein-DNA Interactions with m6A-Tracer-NES.
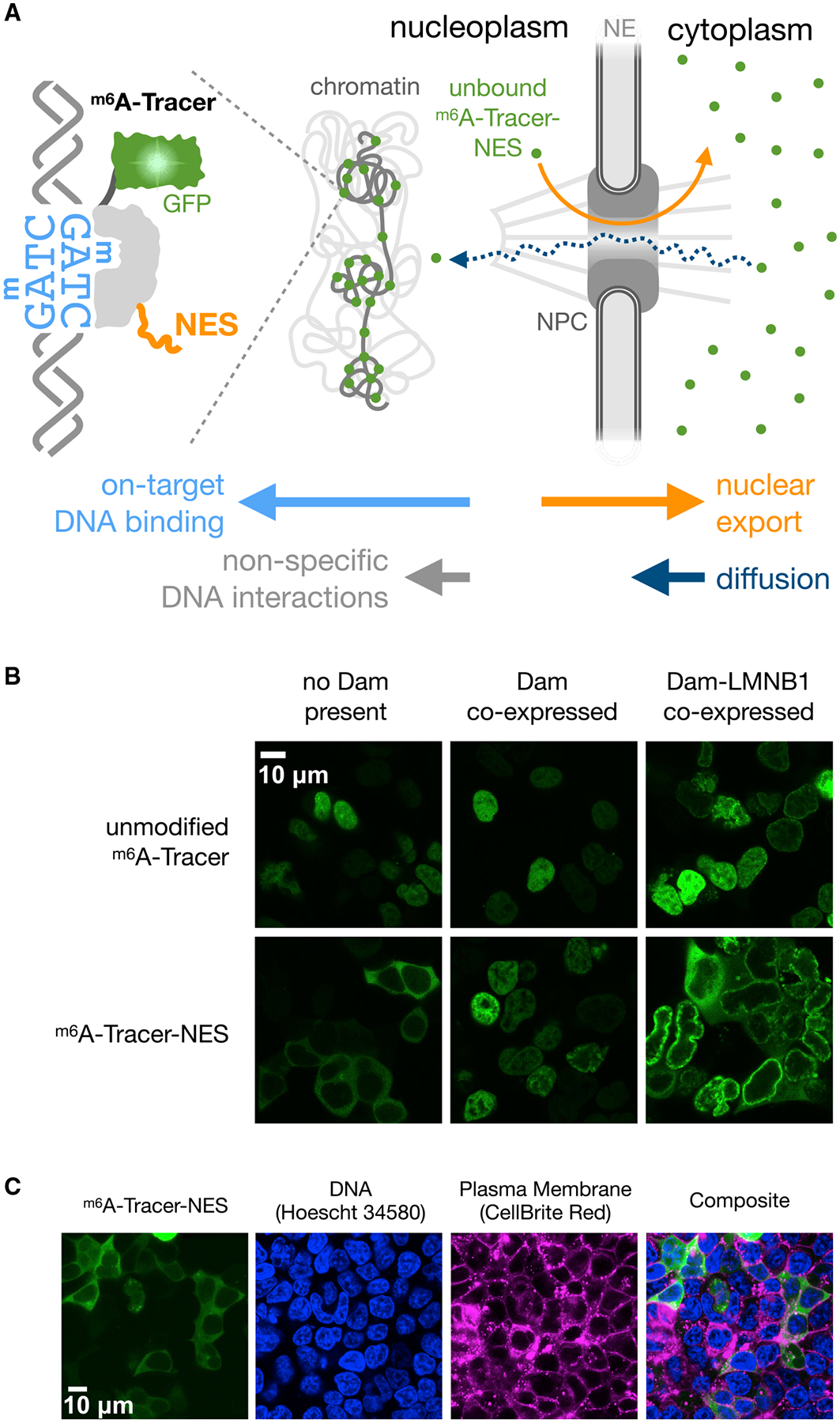
(A) Illustration of potential mechanism by which m6A-Tracer-NES (m6A-Tracer with a C-terminal HIV-1 Rev Nuclear Export Signal) reduces background fluorescence in the nucleus caused by nonspecific DNA interactions, due to the relative rates of export, diffusion, and DNA binding (indicated by horizontal arrows). NPC, nuclear pore complex; NE, nuclear envelope.
(B) Confocal images of m6A-Tracer-NES expressing cells co-stained with Hoescht 34580 to label DNA and CellBrite Red to label plasma membranes, showing cytoplasmic localization when Dam is not co-expressed.
(C) Confocal fluorescent microscope images revealing the different localization patterns of m6A-Tracer (Kind et al., 2013) with or without a NES and with or without Dam or Dam-LMNB1 co-expression.
No cryptic nuclear localization sequences were detected in m6A-Tracer (STAR Methods), nor are human cells likely to contain any significant background levels of m6A without Dam (O’Brown et al., 2019). Instead, its default nuclear localization may arise from a weak interaction between genomic DNA and the DNA-binding domain of m6A-Tracer, combined with the ability of m6A-Tracer to diffuse freely through nuclear pores given its small size (Figure 6A). We hypothesized that adding a nuclear export signal (NES) to m6A-Tracer might overcome its weak affinity for DNA and keep any unbound copies of the protein sequestered in the cytoplasm. We found that the HIV-1 Rev NES sequence fused to either terminus resulted in robust localization of m6A-Tracer to the cytoplasm in cells not expressing Dam (Figures 6C and S6), and for downstream experiments, we proceeded to use the C-terminal fusion, which we call m6A-Tracer-NES.
While the NES appears to prevent nonspecific m6A-Tracer interactions with DNA, it does not overcome on-target binding to Dam-methylated DNA. When Dam was co-expressed, the localization of m6A-Tracer-NES shifted almost entirely from the cytoplasm to the nucleus (Figure 6B). When Dam-LMNB1 was co-expressed, m6A-Tracer-NES shifted to the nuclear lamina, with excess copies remaining in the cytoplasm in a subset of cells with especially high expression (Figures 6B and S6). This shift in localization began within 2–3 h of Dam-LMNB1 induction and produced visible rings in the majority of transfected cells within 5 h (Figure S6). Because m6A-Tracer-NES only binds methylated sites in the nucleus, it solves two major problems: (1) m6A-Tracer fluorescence in the nucleus is no longer ambiguous and can be interpreted as a signal of methylation, and (2) high contrast between the nuclear lamina and the nuclear interior can be achieved for a much wider range of m6A-Tracer expression levels.
μDamID Enables Joint Imaging and Sequencing Analysis
μDamID enables the joint analysis of the nuclear localization and sequence identity of protein-DNA interactions within each cell and between cells. Because the nuclear localization of LADs is well characterized, one could generate and test hypotheses about the sequencing data given the imaging data for each cell in this study. Because the first batch of cells expressed unmodified m6A-Tracer, it is possible that high m6A-Tracer expression could explain why some anomalous Dam-LMNB1 cells have high fluorescence in the nuclear interior (Figure 3B). Furthermore, because the first batch of cells lacked any direct readouts of Dam-LMNB1 expression levels, excessive Dam-LMNB1 expression could explain why some cells have high and unexpected sequencing coverage in ciLADs, leading to lower classification accuracy. To test this, in our second batch of cells we tagged the Dam-LMNB1 fusion protein with the red fluorescing protein tdTomato to enable monitoring of relative expression levels and the precise location of the nuclear lamina, and we used m6A-Tracer-NES to track the physical locations of lamina-associated DNA in the nucleus (Figure 7A).
Figure 7. Joint Image and Sequence Analysis.
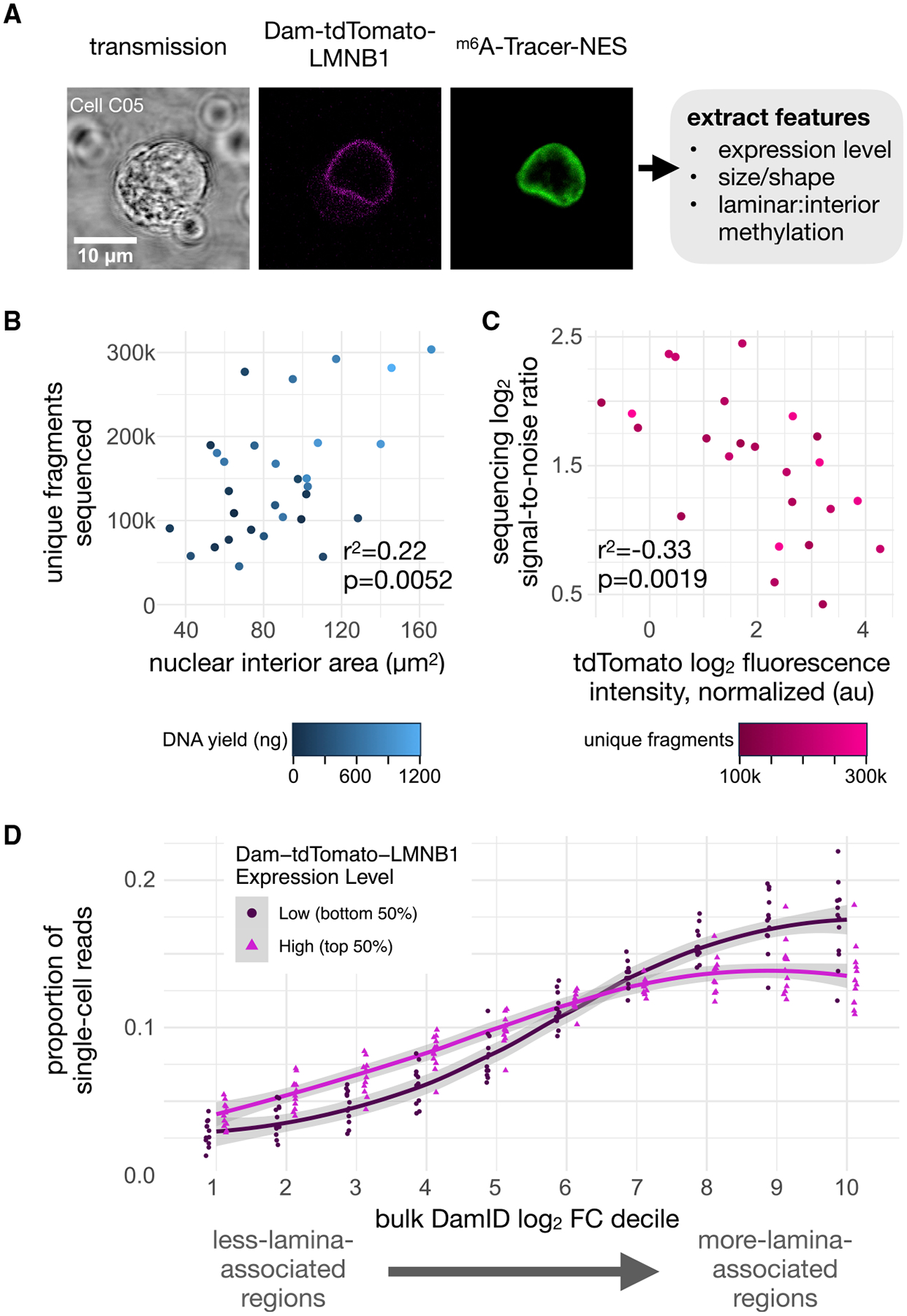
(A) An example of raw imaging data acquired for a single-cell co-expressing m6A-Tracer-NES and Dam-tdTomato-LMNB1 used for imaging feature extraction. Integrated tdTomato intensity in the cell serves as a measure of relative Dam-LMNB1 expression between cells.
(B) Scatterplot of the number of unique DpnI fragments covered by sequencing data for each cell compared with its nuclear area determined from its imaging data, colored by the library DNA yield for that cell (n = 30 batch 2 cells with detectable tdTomato signal). r2 and p (slope > 0) are provided from an ordinary least squares linear model.
(C) Scatterplot of the sequencing signal:noise ratio for each cell (computed using coverage in control regions) compared with its relative Dam-tdTomato-LMNB1 expression (determined by imaging), colored by the number of unique fragments for that cell (n = 24 batch 2 cells with detectable tdTomato signal and >100k unique fragments). r2 and p (slope < 0) are provided as in (B).
(D) A comparison of the distribution of raw single-cell sequencing coverage across deciles of increasing bulk DamID signal in the genome, for the group of 12 cells with the highest (magenta) or lowest (dark red) Dam-tdTomato-LMNB1 expression levels. Values for individual cells are plotted as points in each decile, and loess curves are overlaid with 99% confidence interval ribbons in gray. Higher coverage in the left-hand side of the plot is consistent with greater background methylation.
For each cell, we extracted a rich set of quantitative features from its images, including nuclear lamina size and roundness, cell size and roundness, overall tdTomato intensity, and m6A-Tracer-NES intensity in each compartment. We then compared these imaging features to sequencing features for each cell: library DNA yield, unique fragment number, signal-to-noise ratio, accuracy on control sets, and raw coverage distribution in genomic bins ranked by lamina association from bulk data. Several strong associations stood out from the data (Figure S7). First, we found that cells with larger nuclei tended to yield more DNA in their libraries and resulted in more unique fragments sequenced, indicating greater library complexity (Figure 7B). This matches expectations, given that larger nuclei in this asynchronous, heterogeneous population are likely to have more DNA, either due to ploidy differences or cell-cycle phase.
Second, we found that, among cells with high library complexity (over 100,000 unique fragments), cells with greater expression of Dam-tdTomato-LMNB1 showed diminished sequencing signal-to-noise ratios in cLADs versus ciLADs (Figure 7C) and generally showed higher coverage in less lamina-associated regions of the genome (Figure 7D). This is consistent with our hypothesis, as higher Dam fusion protein expression is expected to produce higher background methylation that is not specific to the protein-DNA interaction of interest. Notably, among Dam-LMNB1-expressing cells we did not find a strong association between m6A-Tracer-NES signal in the nuclear interior and background methylation (Figure S7). This may be because variation in expression of m6A-Tracer obscures biological variation in methylation at the lamina. Imaging data did reveal, however, that two cells without bright laminar rings produced low-complexity sequencing libraries (cells D05 and D09, Figure S3), and these would be difficult to filter out by other sorting approaches and would lead to low-quality outliers in DamID sequencing data. This series of measurements serves as a proof of principle that our device can be used to sort cells based on visual phenotypes that are correlated with sequencing measurements. Here, we use this capability to predict sequencing information content from imaging phenotypes in single cells.
DISCUSSION
We have demonstrated the use of an integrated microfluidic device for single-cell isolation, imaging, and sorting, followed by DamID. This system enables the acquisition of paired imaging and sequencing measurements of protein-DNA interactions within single cells, giving a readout of both the “geography” and identity of these interactions in the nucleus. Specifically, we tested the device by mapping well-characterized interactions between DNA and proteins found at the nuclear lamina, providing a measure of genome regulation and 3D chromatin organization within the cell, and recapitulating similar maps in other cell types. We also improved the method of imaging protein-DNA interactions with m6A-Tracer by attaching a nuclear export signal. This modification greatly reduces background fluorescence due to nonspecific interactions with unmethylated DNA, providing a more universal readout of the m6A methylation status of the nucleus. m6A-Tracer-NES will allow for more sensitive imaging of other classes of protein-DNA interactions in the nucleus, and it could potentially also be utilized in synthetic genetic and epigenetic circuits (Park et al., 2019) to reduce off-target effects, or to serve as a nuclear localization switch.
μDamID is compatible with any imaging modality that can be implemented with a standard inverted microscope, including two-photon fluorescence and other nonlinear optical microscopy techniques, in addition to many common super-resolution microscope configurations that take advantage of photoactivatable fluorescent proteins (reviewed by Huang et al., 2010). However, the thickness of the PDMS device may be incompatible with optics above the sample, such as condensers, that have short working distances. The device can be mounted on a coverslip and therefore can be imaged using high-NA objectives, as done here. In this study, we demonstrate short-term live cell imaging with μDamID; however, imaging modalities that require fixed cells or nuclei are also compatible, although fixation may affect sequencing yields. The flexibility of integrated microfluidic circuits provides compatibility with imaging techniques that require multiple wash steps such as in situ hybridization (reviewed by Rodriguez-Mateos et al., 2020), as well as time-lapse imaging of live cells prior to DamID processing (Ramalingam et al., 2016). However, these implementations would require modifications to the device design.
This technology can also be applied to study many other types of protein-DNA interactions in single cells, and it could be combined with other sequencing and/or imaging modalities to gather even richer information from each cell. For example, the nuclear localization of specific proteins such as heterochromatin-associated proteins or nucleolus-associated proteins can be visualized by fluorescent tagging, then DamID can be used to sequence and identify nearby genomic regions. This device could readily be applied to study chromatin organization in micronuclei and other abnormal nuclei by imaging and selectively sorting these nuclear phenotypes and performing DamID, which would be infeasible by bulk or FACS-based methods. Recent advances allow for simultaneous DamID and transcriptome sequencing in single cells (Rooijers et al., 2019). The integrated valves and modular reaction chambers in the μDamID device could be leveraged to extend this platform to such multi-omic protocols. This would allow for joint analysis of spatial chromatin organization, protein-DNA interactions, and gene expression within single cells. Further improvements to the DamID protocol may also increase its sensitivity and specificity.
Although the sequencing throughput of this particular design is limited to 10 cells per device, many hundreds of cells can be rapidly screened and rejected from the device by monitoring a wide field of cells entering the input filter area. Thus, even relatively rare cell phenotypes can be enriched and sequenced on the device. We note that the rate-limiting step is often high-resolution image acquisition, which can take minutes per cell depending on the imaging method. The throughput of this platform can be increased to hundreds of cells per device by scaling up the design and incorporating features like multiplexed valve control (Kim et al., 2017) and automated image processing and sorting. To scale this technology further, paired imaging and sequencing data could be obtained using spatially or optically registered DNA barcodes (Cole et al., 2017; Nguyen et al., 2017; Yuan et al., 2018; Chen et al., 2020).
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Aaron Streets (astreets@berkeley.edu).
Materials Availability
Plasmids generated in this study have been deposited to Addgene (https://www.addgene.org/browse/article/28211957/).
Data and Code Availability
The sequencing data generated during this study are available at GEO (accession GSE156150). The imaging data generated during this study are available at FigShare: https://doi.org/10.6084/m9.figshare.12798158. Analysis code, control software, device design files, and plasmid sequences are freely available for download on GitHub: https://github.com/altemose/microDamID. Source data for bulk KBM-7 RNA-seq were obtained from SRA (accession SRP044391), and source data for KBM-7 scDamID were obtained from GEO (accession GSE69423).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HEK293T cells (CRL-3216, ATCC, Manassas, VA; validated by microsatellite typing and mycoplasma tested, at passage number <10) were maintained in DMEM (high glucose, with GlutaMAX, with phenol red, without sodium pyruvate; Gibco 10566–016) supplemented with 10% Fetal Bovine Serum (Seradigm Select Grade USDA approved origin; Avantor 89510–186) and 1% Pen Strep (Gibco 15140–122) at 37°C in 5% CO2. This cell line does not contain a Y chromosome.
METHOD DETAILS
Cell Transfection and Harvesting
HEK293T cells were seeded in 24-well plates at 50000 cells per well in 0.5-ml media (see above for culturing and media details). The next day, cells were transfected using FuGene HD transfection reagent according to their standard protocol for HEK293 cells (Promega, Madison, WI). DNA plasmids were cloned in Dam-negative E. coli (New England Biolabs, Ipswitch, MA) to reduce sequencing reads originating from plasmid. Dam-LMNB1 and m6A-Tracer plasmids were obtained from Bas van Steensel (from Kind et al., 2013); Dam-LMNB1 was modified to replace GFP with mCherry and to produce a Dam-only version, as well as to create a Dam-tdTomato-LMNB1 fusion for batch 2 experiments; their sequences are available in the accompanying GitHub repository. 250 ng Dam construct DNA plus 250 ng m6A-Tracer DNA were used per well. As controls to validate transfection, additional wells were left untransfected, transfected with m6A-Tracer only, or transfected with Dam construct only. The following day, successful transfection was validated by widefield fluorescence microscopy, seeing GFP signal in wells containing m6A-Tracer, and mCherry signal in all wells containing Dam construct only. Cells were harvested 72 hours after transfection. 20 hours before harvesting, the media was replaced and 0.5 μl Shield-1 ligand (0.5 mM stock, Takara Bio USA, Inc., Mountain View, CA) was added to each well to stabilize protein expression. Cells transfected with Dam-LMNB1 were inspected by fluorescence microscopy to look for the characteristic signal at the nuclear lamina, indicating proper expression and protein activity. To harvest the cells and prepare them for loading on the device, the cells were washed with PBS, then incubated at room temperature with 1X TrypLE Select (ThermoFisher Scientific, Waltham, MA) for 5 minutes to dissociate them from the plate. Cells were pipetted up and down to break up clumps, then centrifuged at 300xg for 5 minutes, resuspended in PBS, centrifuged again, and resuspended in 500 μl Pick Buffer (50 mM Tris-HCl pH 8.3, 75 mM KCl, 3 mM MgCl2, 137 mM NaCl), achieving a final cell concentration of roughly 500,000 cells per ml. Cells were passed through a 40 μm cell strainer before loading onto the device.
Confocal Imaging
Fluorescence confocal imaging of cells was performed in the trapping region using an inverted scanning confocal microscope with a 488 nm Ar/Kr laser (Leica, Germany) for excitation, with a bandpass filter capturing backscattered light from 500–540 nm at the primary photomultiplier tube (PMT), with the pinhole set to 1 Airy unit, with a transmission PMT capturing widefield unfiltered forward-scattered light, and with a 63X 0.7 NA long-working-distance air objective with a correction collar, zoomed by scanning 4X. For batch 2 imaging, a 63X 1.2 NA water immersion objective was used, with a 6X scanning zoom. The focal plane was positioned in the middle of each nucleus, capturing the largest-circumference cross-section, and final images were averaged over 10 frames to remove noise. For batch 2 cells, 10 confocal z slices were taken for each cell, and the slice with the largest nuclear perimeter was selected for image processing. The 3 cells expressing Dam-only that were sequenced in this study were imaged with a widefield CCD camera. Other Dam-only cells were imaged with confocal microscopy and showed similar relatively homogenous fluorescence throughout the nucleus, and never the distinct ‘ring’ shape found in Dam-LMNB1 expressing cells (Kind et al., 2013; Figure 3B). No image enhancement methods were used prior to quantitative image processing. Images in Figures 1 and 3 have been linearly thresholded to diminish background signal.
Mold Fabrication
Molds for casting each layer were fabricated on silicon wafers by standard photolithography. Photomasks for each layer were designed in AutoCAD and printed at 25400 DPI (CAD/Art Services, Inc., Bandon, Oregon). The mask for the thick layer, in this case the flow layer to make push-up valves, was scaled up in size uniformly by 1.5% to account for thick layer shrinkage. A darkfield mask was used for features made out of negative photoresist: the filters on the flow layer and the entire control layer; a brightfield mask was used for all flow layer channels, which were made out of positive photoresist (mask designs available on GitHub; see Data Availability section below). 10 cm diameter, 500 μm thick test-grade silicon wafers (item #452, University Wafer, Boston, MA) were cleaned by washing with 100% acetone, then 100% isopropanol, then DI water, followed by drying with an air gun, and heating at 200°C for 5 minutes.
To make the control layer mold, SU-8 2025 negative photoresist (MicroChem Corp., Westborough, MA) was spin-coated to achieve 25 μm thickness (7 s at 500 rpm with 100 rpm/s ramp, then 30 s at 3500 rpm with 300 rpm/s ramp). All baking temperatures, baking times, exposure dosages, and development times followed the MicroChem data sheet. All baking steps were performed on pre-heated ceramic hotplates. After soft-baking, the wafer was exposed beneath the darkfield control layer mask using a UV aligner (OAI, San Jose, CA). After post-exposure baking and development, the mold was hard-baked at 150°C for 5 minutes.
To make the flow layer mold, first the filters were patterned with SU-8 2025, which was required to make fine, high-aspect-ratio filter features that would not re-flow at high temperatures. SU-8 2025 was spin-coated to achieve 40 μm thickness (as above but with 2000 rpm final speed) and processed according to the MicroChem datasheet as above, followed by an identical hard-bake step. Next, AZ 40XT-11D positive photoresist (Integrated Micro Materials, Argyle, TX) was spin-coated on top of the SU-8 features to achieve 20 μm thickness across the wafer (as above but with 3000 rpm final speed). All baking temperatures, baking times, exposure dosages, and development times followed the AZ 40XT-11D data sheet. After development, the channels were rounded by reflowing the photoresist, achieved by placing the wafer at 65°C for 1 min, then 95°C for 1 min, then 140°C for 1 min followed by cooling at room temperature. In our experience, reflowing for too long, or attempting to hard-bake the AZ 40XT-11D resulted in undesirable ‘beading’ of the resist, especially at channel junctions. Because it was not hard-baked, no organic solvents were used to clean the resulting mold. Any undeveloped AZ 40XT-11D trapped in the filter regions was carefully removed using 100% acetone applied locally with a cotton swab.
Soft Lithography
Devices were fabricated by multilayer soft lithography (Unger et al., 2000). On-ratio 10:1 base:crosslinker RTV615A PDMS (Momentive Performance Materials, Inc., Waterford, NY) was used for both layers, and layer bonding was performed by partial curing, followed by alignment, then full curing (Lai et al., 2019). To prevent PDMS adhesion to the molds, the molds were silanized by exposure to trichloromethlysilane (Sigma-Aldrich, St. Louis, MO) vapor under vacuum for 20 min. PDMS base and crosslinker were thoroughly mixed by an overhead mixer for 2 minutes, then degassed under vacuum for 90 minutes. Degassed PDMS was spin-coated on the control layer mold (for the ‘thin layer’) to achieve a thickness of 55 μm (7 s at 500 rpm with 100 rpm/s ramp, then 60 s at 2000 rpm with 500 rpm/s ramp), then placed in a covered glass petri dish and baked for 10 minutes at 70°C in a forced-air convection oven (Heratherm OMH60, Thermo Fisher Scientific, Waltham, MA) to achieve partial curing. The flow layer mold (for the ‘thick layer’) was placed in a covered glass petri dish lined with foil, and degassed PDMS was poured onto it to a depth of 5 mm. Any bubbles were removed by air gun or additional degassing under vacuum for 5 minutes, then the thick layer was baked for 19 minutes at 70°C. Holes were punched using a precision punch with a 0.69 mm punch tip (Accu-Punch MP10 with CR0420275N19R1 punch, Syneo, Angleton, TX). The thick layer was peeled off the mold, cut to the edges of the device, and aligned manually under a stereoscope on top of the thin layer, which was still on its mold. The layers were then fully cured and bonded together by baking at 70°C for 120 min. After cooling, the devices were peeled off the mold, and the inlets on the thin layer were punched. The final devices were bonded to 1 mm thick glass slides (first batch) or #1 coverglass (second batch), which were first cleaned by the same method as used for silicon wafers above, using oxygen plasma reactive ion etching (20 W for 23 s at 285 Pa pressure; Plasma Equipment Technical Services, Brentwood, CA), followed by heating at 100°C on a ceramic hot plate for 5 minutes.
Device and Control Hardware Setup
Devices were pneumatically controlled by a solenoid valve manifold (Pneumadyne, Plymouth, MN). Each three-way, normally open solenoid valve switched between a regulated and filtered pressure source inlet at 25 psi (172 kPa) or ambient pressure to close or open microfluidic valves, respectively. Solenoid valves were controlled by the KATARA control board and software (White and Streets, 2018). Most operational steps were carried out on inverted microscopes using 4–10X objectives. For incubation steps, the device was placed on a custom-built liquid-cooled thermoelectric temperature control module (TC-36–25-RS232 PID controller with a 36 V 16 A power source and two serially connected VT-199–1.4–0.8P TE modules and an MP-3022 thermistor; TE technologies, Traverse City, MI) controlled by a new KATARA software module (to be made available on github). A layer of mineral oil was applied between the device and the temperature controller to improve thermal conductivity and uniformity. A stereoscope was used to monitor the device while on the temperature controller.
To set up each new device, each pneumatic valve was connected to one control inlet on the microfluidic device by serially connecting polyurethane tubing (3/32” ID, 5/32” OD; Pneumadyne) to Tygon tubing (0.5 mm ID, 1.5 mm OD) to >4 cm PEEK tubing (0.25 mm ID, 0.8 mm OD; IDEX Corporation, Lake Forest, IL). Solenoid valves were energized to de-pressurize the tubing and the tubing was primed by injecting water using a syringe connected to the end of the PEEK tubing, then the primed PEEK tubing was inserted directly into each punched inlet hole on the device. Solenoid valves were de-energized to pressurize the tubing until all control channels on the device were fully dead-end filled, then each microfluidic valve was tested and inspected by switching on and off its corresponding solenoid valve. All valves were opened and the device was passivated by filling all flow-layer channels with syringe-filtered 0.2% (w/w) Pluronic F-127 solution (P2443; Millipore Sigma, St. Louis, MO) from the reagent inlet and incubating at room temperature for 1 hour. The device was then washed by flowing through 0.5 ml of ultra-filtered water, followed by air to dry it.
Device Operation
Initially, all chamber valves and reagent inlet valves were closed. Gel-loading pipette tips were used to inject reagents and cells into the flow channels. To prepare the device for operation, pick buffer was injected into the reagent inlet and pressurized at 5–10 psi to dead-end fill the reagent inlet channels. Negative controls were generated by injecting pure pick buffer into one of the holding chambers before trapping and sorting cells into the other lanes. 50 μl of cell suspension was then loaded into a gel-loading pipette tip, and injected directly into the cell inlet. A high-precision pressure regulator was used to load the single-cell suspension at 1 psi (7 kPa). Cells were observed in the filter region with brightfield and epifluorescence using a 10X objective to identify candidate cells. These were then tracked through the device until they approached the trapping chamber for an empty lane. To trap a candidate cell, the device’s peristaltic pump was operated at 1 Hz to deliver that cell to the trap region. The trap valves (above and below the trap region; see Figure S1) were closed and the cell was imaged with scanning confocal microscopy as described above. If the cell was rejected after imaging, the trap valves were opened and it was flushed to the waste outlet. Otherwise, the cell was injected into the holding chamber by dead-end filling. This process was repeated to fill each lane with single cells for DamID. To test background DNA levels, we filled several lanes with only cell suspension buffer. Nearly undetectable levels of amplified DNA were recovered from these lanes.
After filling all 10 lanes, the reagent inlet and cell trapping channels were flushed with 0.5 ml of water, which exited through the waste outlet and the cell inlet, to remove any remaining Pick buffer or cell debris, then air dried. The same washing and drying was repeated between each reaction step. To inject reagents for each reaction of the DamID protocol, the trap valves were closed, the reagent channels were dead-end filled with freshly prepared and syringe-filtered reagent, then the reagent inlet valves and the valves for the necessary reaction chambers were opened, and each lane was dead-end filled individually to prevent any possible cross-contamination. Reaction contents are described in below under “Reaction buffers and conditions.”
Reagents were mixed by actuating the chamber valves at 5 Hz for 5 minutes. At the PCR step, rotary mixing was achieved by using the chamber valves to make a peristaltic pump driving fluid around the full reaction ring. For each reaction step, the device was placed on the thermal controller and reactions were with times and temperatures described below. PCR thermocycling conditions are described below, under “PCR thermocycling conditions.” To ensure adequate hydration during PCR, all valves were pressurized. Amplified DNA was flushed out of each lane individually using purified water from the reagent inlet, collected into a gel loading tip placed in the lane outlet to a final volume of 5 ml then transferred to a 0.2 ml PCR strip tube.
| Reaction Buffers and Conditions | ||
|---|---|---|
| Reaction Stage | Buffer | Incubation |
| Trapping & Holding | Pick Buffer: | RT |
| 50 mM Tris-HCl pH 8.3 | ||
| 75 mM KCl, 3 mM MgCl2 | ||
| 137 mM NaCl | ||
| Lysis | 10 mM TRIS acetate pH 7.5 | 42 °C for 4 hours then 80 °C for 10 min |
| 10 mM magnesium acetate | ||
| 50 mM potassium acetate | ||
| 0.67% Tween-20 | ||
| 0.67% Igepal | ||
| 0.67 mg/ml proteinase K | ||
| Digestion | mix 7 μl 10X CutSmart buffer | 37 °C for 4 hours then 80 °C for 20 min |
| 1 μl DpnI (NEB) | ||
| 62 μl H2O | ||
| Ligation | mix 6 μl 10X NEB T4 ligase buffer | 16 °C overnight then 65 °C for 10 min |
| 1 μl DamID adapter stock at 25 μM | ||
| 0.2 μl NEB T4 ligase at 400 U/μl | ||
| 21.8 μl H2O | ||
| 1 μl 2% w/v Tween-20 | ||
| PCR | from Takara Advantage 2 kit: | See “PCR thermocycling conditions,” below |
| mix 5 μl 10X PCR buffer | ||
| 1 μl dNTPs at 10 mM each | ||
| 1 μl polymerase mix | ||
| 0.63 μl DamID primer (AdR_PCR) | ||
| 21.37 μl H2O | ||
| 1 μl 2% Tween-20 | ||
| PCR Thermocycling Conditions | |
|---|---|
| PCR Step | Incubation |
| 1 | 68 °C for 10 min |
| 2 | 94 °C for 1 min |
| 3 | 65 °C for 5 min |
| 4 | 68 °C for 15 min |
| 5 | 94 °C for 1 min |
| 6 | 65 °C for 1 min |
| 7 | 68 °C for 10 min |
| 8 | Go to step 5 (x 3) |
| 9 | 94 °C for 1 min |
| 10 | 65 °C for 1 min |
| 11 | 68 °C for 2 min |
| 12 | Go to step 9 (x 22) |
| 13 | Hold 10 °C |
To anneal DamID adapter (from Vogel et al., 2007): we mixed equal volumes of 50 μM AdRt and 50 μM AdRb oligonucleotides in a microcentrifuge tube in TE plus 50 mM NaCl, then fully submerged it in a beaker of boiling water, then allowed the water to equilibrate to room temperature slowly.
Quality Control, Library Prep, and Sequencing
Samples were diluted to 10 μl total volume and two replicates of qPCR were performed using the DamID PCR primer to estimate DNA quantities relative to the pick-buffer-only negative control (1 μl DNA per replicate in 10 μl reaction volume). We also used 1 μl of sample to measure DNA concentration using a Qubit fluorometer with a High-Sensitivity DNA reagent kit (quantitative range 0.2 ng – 100 ng; ThermoFisher Scientific). Samples with the lowest Ct values and highest Qubit DNA measurements were selected for library preparation and sequencing. Library preparation was carried out using an NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB E7645) with dual-indexed multiplex i5/i7 oligo adapters. Size selection was not performed; PCR was carried out for 9 cycles. Libraries were quantified again by Qubit and size profiled on a TapeStation 4200 with a D5000 HS or D1000 HS kit (Agilent, Santa Clara, CA), then mixed to achieve equimolar amounts of each library. DNA was sequenced on an Illumina MiniSeq with a 150-cycle high output kit, to produce paired 75 bp reads, according to manufacturer instructions (Illumina, San Diego, CA). Roughly 13 million read pairs were obtained for batch 1 cells. For batch 2, we performed library preparation using an NEBnext UltraII FS kit (NEB E7805) and obtained 200 million total read pairs with an Illumina NextSeq 550 High Output run, to guarantee sequencing of nearly the full available library complexity.
Bulk DamID
Genomic DNA was isolated from ~3.7 × 106 transfected HEK293T cells using the DNeasy Blood & Tissue kit (Qiagen) following the protocol for cultured animal cells with the addition of RNase A. The extracted gDNA was then precipitated by adding 2 volumes of 100% ethanol and 0.1 volume of 3 M sodium acetate (pH 5.5) and storing at −20 °C for 30 minutes. Next, centrifugation for 30 minutes at 4 °C, >16,000 x g was performed to spin down the gDNA. The supernatant was removed, and the pellet was washed by adding 1 volume of 70% ethanol. Centrifugation for 5 minutes at 4 °C, >16,000 x g was performed, the supernatant was removed, and the gDNA pellets were air-dried. The gDNA was dissolved in 10 mM Tris-HCl pH 7.5, 0.1 mM EDTA to 1 μg/μl, incubating at 55 °C for 30 minutes to facilitate dissolving. The concentration was measured using Nanodrop.
The following DpnI digestion, adaptor ligation, and DpnII digestion steps were all performed in the same tube (Vogel et al., 2007). Overnight DpnI digestion at 37 °C was performed with 2.5 μg gDNA, 10 U DpnI (NEB), 1X CutSmart (NEB), and water to 10 μl total reaction volume. DpnI was then inactivated at 80 °C for 20 minutes. Adaptors were ligated by combining the 10 μl of DpnI-digested gDNA, 1X ligation buffer (NEB), 2 μM adaptor dsAdR, 5 U T4 ligase (NEB), and water for a total reaction volume of 20 μl. Ligation was performed for 2 hours at 16 °C and then T4 ligase was inactivated for 10 minutes at 65 °C. DpnII digestion was performed by combining the 20 μl of ligated DNA, 10 U DpnII (NEB), 1X DpnII buffer (NEB), and water for a total reaction volume of 50 μl. The DpnII digestion was 1 hour at 37 °C followed by 20 minutes at 65 °C to inactivate DpnII.
Next, 10 μl of the DpnII-digested gDNA was amplified using the Takara Advantage 2 PCR Kit with 1X SA PCR buffer, 1.25 μM Primer Adr-PCR, dNTP mix (0.2 mM each), 1X PCR advantage enzyme mix, and water for a total reaction volume of 50 μl. PCR was performed with an initial extension at 68 °C for 10 minutes; one cycle of 94 °C for 1 minute, 65 °C for 5 minutes, 68 °C for 15 minutes; 4 cycles of 94 °C for 1 minute, 65 °C for 1 minute, 68 °C for 10 minutes; 21 cycles of 94 °C for 1 minute, 65 °C for 1 minute, 68 °C for 2 minutes. Post-amplification DpnII digestion was performed by combining 40 μl of the PCR product with 20 U DpnII, 1X DpnII buffer, and water to a total volume of 100 μl. The DpnII digestion was performed for 2 hours at 37 °C followed by inactivation at 65 °C for 20 minutes. The digested product was purified using QIAquick PCR purification kit. The purified PCR product (1 μg brought up to 50 μl in TE) was sheared to a target size of 200 bp using the Bioruptor Pico with 13 cycles with 30”/30” on/off cycle time. DNA library preparation of the sheared DNA was performed using NEBNext Ultra II DNA Library Prep Kit for Illumina using AMPure XP beads (Beckman Coulter Life Sciences, Indianapolis, IN).
Bulk DamID, Comparing Dam Mutants
Bulk DamID for comparing the wild-type allele and V133A mutant allele was performed as outlined in the Bulk DamID section above with the following modifications. Genomic DNA was extracted from ~ 2.4 × 105 transfected HEK293T cells. A cleanup before methylation-specific amplification was included to remove unligated Dam adapter before PCR. The Monarch PCR & DNA Cleanup Kit with 20 μl DpnII-digested gDNA input and an elution volume of 10 μl was used. Shearing with the Bioruptor Pico was performed for 20 total cycles with 30”/30” on/off cycle time. Paired-end 2 × 75 bp sequencing was performed on an Illumina NextSeq with a mid output kit. Approximately 3.8 million read pairs per sample were obtained.
Bulk RNA-seq
RNA was extracted from ~1.9 × 106 transfected HEK293T cells using the Rneasy Mini Kit from Qiagen with the QIAshredder for homogenization. RNA library preparation was performed using the NEBNext Ultra II RNA Library Prep Kit for Illumina with the NEBNext Poly(A) mRNA Magnetic Isolation Module. Paired-end 2 × 150 bp sequencing for both DamID-seq and RNA-seq libraries was performed on 1 lane of a NovaSeq S4 run. Approximately 252 million read pairs were obtained for each DamID-seq sample, and roughly 64 million read pairs for each RNA sample.
m6A-Tracer-NES
To reduce background fluorescence due to m6A-Tracer, we fused its N or C terminus to one of two different nuclear export signals (NES): HIV-1 Rev (LQLPPLERLTLD) or MAPKK (LQKKLEELEL) (Kakar et al., 2007). We compared the localization of each of the 4 resulting constructs by imaging HEK293T cells transiently transfected with m6A-tracer-NES by itself or with Dam. Negative controls included transfection with unmodified m6A-Tracer only or Dam-only, and no transfection. The MAPKK NES did not appreciably reduce nuclear localization of m6A-tracer-NES in the absence of Dam (Figure S6). However, the HIV-1 Rev NES, in either the N- or C-terminal configuration, showed significant improvement in localizing signal to the cytoplasm in the absence of Dam, while permitting nuclear localization in the presence of Dam (Figures 6B and S6). We proceeded to use the C-terminal HIV-1 Rev m6A-Tracer construct for downstream experiments. Co-transfection with Dam-LMNB1 resulted in a greater proportion of transiently transfected cells having visible laminar rings than with unmodified m6A-Tracer. Timelapse imaging of the same field of Dam-LMNB1 + m6A-Tracer-NES cells over time or different fields at each timepoint (Figure S6) demonstrated that laminar rings become visible within 2–3 hours and reach full intensity around 5 hours after Dam-LMNB1 induction with Shield-1 ligand. To test the possibility that unmodified m6A-Tracer localizes to the nucleus due to a cryptic Nuclear Localization Signal, we searched for NLS motifs using NLSdb (Bernhofer et al., 2018) but found no matches.
QUANTIFICATION AND STATISTICAL ANALYSIS
Bulk RNA-seq
Adapters were trimmed using trimmomatic (v0.39; Bolger et al., 2014; ILLUMINACLIP:adapters-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36, where adapters-PE.fa is:
>PrefixPE/1 TACACTCTTTCCCTACACGACGCTCTTCCGATCT >PrefixPE/2 GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT).
Transcript quantification was performed using Salmon (Patro et al., 2017) with the GRCh38 transcript reference. Differential expression analysis was performed using the voom function in limma (Ritchie et al., 2015). Differential expression was called based on logFC significantly greater than 1 and adjusted p-value < 0.01.
For KBM7 bulk gene expression analysis, publicly available single-end RNA sequencing data (SRA accession SRP044391, Essletzbichler et al., 2014) from two replicates were processed. For adapter trimming, trimmomatic was used in the SE mode with the adapter file ILLUMINACLIP:TruSeq3-SE. All other trimmomatic parameters were the same as were used in the HEK293T RNA-seq data processing, and Salmon was used for transcript quantification in single-end mode.
Bulk and Single-cell DamID
Bulk and single-cell DamID reads were demultiplexed using Illumina’s BaseSpace platform to obtain fastq files for each sample. DamID and Illumina adapter sequences were trimmed off using trimmomatic (v0.39; Bolger et al. (2014); ILLUMINACLIP:adapters-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:20, where adapters-PE.fa is:
>PrefixPE/1 TACACTCTTTCCCTACACGACGCTCTTCCGATCT >PrefixPE/2 GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT >Dam GGTCGCGGCCGAGGA >Dam_rc TCCTCGGCCGCGACC
Trimmed reads were aligned to a custom reference (hg38 reference sequence plus the Dam-LMNB1 and m6A-Tracer plasmid sequences) using BWA-MEM (v0.7.15-r1140, Li, 2013). Alignments with mapping quality 0 were discarded using samtools (v1.9, Li et al., 2009). The hg38 reference sequence was split into simulated DpnI digestion fragments by reporting all intervals between GATC sites (excluding the GATC sites themselves), yielding 7180359 possible DpnI fragments across the 24 chromosome assemblies. The number of reads overlapping each fragment was counted using bedtools (v2.28; Quinlan and Hall, 2010). For single-cell data, the number of DpnI fragments with non-zero coverage was reported within each non-overlapping bin in the genome (28163 total 100 kb bins, after excluding unmappable regions with zero coverage in any cell). For bulk data, the number of read pairs overlapping each 100 kb bin was reported. The same exact pipeline was applied to the raw reads from Kind et al. (2015) (GEO accession GSE69423). RefSeq gene positions were downloaded from the UCSC Genome Browser and counted in each bin. For bulk data, Dam-LMNB1 vs DamOnly enrichment was computed using Deseq2 in each 100 kb bin (Love et al., 2014). For single-cell data, the expected background coverage in each bin was computed as n(m/t), where n is the number of unique fragments sequenced from that cell, m is the number of bulk Dam-only read pairs mapping to that bin, and t is the total number of mapped bulk Dam-only read pairs. Single-cell normalization was computed either as a ratio of observed to expected coverage (for browser visualization and comparison to bulk data), or as their difference (for classification and coverage distribution plotting). Positive and negative control sets of cLAD and ciLAD bins were defined under the assumption that genomic regions that have high bulk DamID signal and that are lamina associated across many cell types are likely to be in contact with the lamina in the vast majority of single cells, which is supported by previous scDamID data (Kind et al., 2015; Figure S4). Specifically, we defined them as bins with a bulk Dam-LMNB1:Dam-only DESeq2 p-value smaller than 0.05/28760, that intersected published cLADs and ciLADs in other cell lines (Lenain et al., 2017), and that were among the top 1200 most differentially enriched bins in either direction (positive or negative log fold change for cLADs and ciLADs, respectively). Normalized coverage thresholds for LAD/iLAD (i.e. contact vs. no contact) classification were computed for each cell to maximize accuracy on the cLAD and ciLAD control sets. To examine whether using the full control sets to set thresholds and define classification error was resulting in substantial overfitting, we split the control sets into training and test sets for threshold setting and accuracy determination, respectively, and only observed a 0.7% mean drop in accuracy relative to using the full sets. Signal-to-noise ratios were computed for each cell using the normalized coverage distributions in the cLAD and ciLAD control sets as (μcLAD – μciLAD)/σciLAD. For most downstream analyses, we chose to exclude 20 cells with fewer than 100,000 unique covered fragments, which includes cells with poor laminar rings and lower DNA yields (Figures 2D and S4). For any given application of μDamID, this threshold will depend on the level of noise due to background methylation in the biological system being used, which is expected to depend in part on the expression level of the Dam fusion protein. In a transiently transfected cell population, this expression level is expected to vary widely, which motivated the use of data to explore this as a cause of variable classification accuracy between cells. The remaining 31 Dam-LMNB1 cells had a median classification accuracy of 90% (range 74%−98%).
Calling vLADs
Variable LADs were defined as bins called as LADs in 33–66% of cells and conservatively filtered to remove regions resulting from sampling error. This was done by computing, for each bin and for each cell, the probability that the true sample contact frequency lies outside the interval (33%, 66%). We estimated this probability using a Poisson-binomial distribution, a generalization of the binomial distribution allowing individual samples to have varying success probabilities. Specifically, each bin in the genome has k cells called as LADs and n-k cells called as iLADs, with n=31 in this study. For the k LADs we generated a vector of k false-positive probabilities, with each probability estimated as the fraction of negative-control ciLADs with coverage greater than the observed coverage in that bin. We used this probability vector to parameterize a Poisson-binomial distribution with k draws, providing the distribution of false-positive calls in the bin. We repeated this for the n-k iLAD bins, with each false-negative probability estimated as the fraction of positive-control cLADs with coverage lower than the observed coverage in that bin. These two distributions were combined into a single density by reflecting the false-positive distribution about the y axis, scaling each one according to its mean, and adding k to produce the plots in Figures S5A–S5C). Only regions with p<10−3 for both tails were called as variable LADs. We then generated 10,000 samples of the sample contact frequency, c, from this distribution and used each one to generate a single binomial (n=31, p=c/31) sample, generating a combined measurement and sampling distribution with greater variance than either alone (Figure 4B), from which we generated 95% confidence intervals for the population contact frequency in each bin (Figure 4A). Statistical analyses and plots were made in R (v4.0.0) using the ggplot2 (v3.3.0), gplots (v3.0.3), colorRamps (v2.3), reshape2 (v1.4.4), ggextra (v0.9) and poisbinom (v1.0.1) packages. Browser figures were generated using the WashU Epigenome Browser (Li et al., 2019).
Image Processing
Images were processed in R (v4.0.0) and plots were produced using the reshape2 (v1.4.3), SDMTools (v1.1–221.1), spatstat (v1.59–0), magick (v2.0), and ggplot2 (v3.3.0) packages. Grayscale images were converted to numeric matrices and edge detection was performed using Canny edge detection using the image_canny function in magick, varying the geometry parameters manually for each cell. The center of mass of all edge points was obtained, and all edge points were plotted in Cartesian coordinated with this center of mass as the origin. Noise was removed by removing points with a nearest neighbor more than 2 microns away. Edge point coordinates were converted to polar coordinates, and the farthest points from the origin in each 10 degree arc were reported. Within each 10 degree arc, all pixel intensities from the original image within the edges of the nucleus were reported as a function of their distance from the farthest edge point in that arc to make Figure 3C. For each cell a loess curve (span 0.3) was fitted to the data after subtracting the minimum intensity value within 3.5 microns of the edge. The Lamina:Interior ratio was computed as the ratio of mean intensity of pixels within 1 micron of the edge to the mean intensity of pixels more than 3.5 microns from the edge, after subtracting the minimum value of the loess curve for that cell. To provide an additional metric, we computed the distance from the laminar edge where the fluorescence intensity decays to 10% of the peak laminar intensity. A nuclear mask was created by drawing a polygon with vertices as the farthest point in each arc from the center of mass, and a similar cell mask polygon was generated using the transmission image—these masks enabled the computation of cell and nuclear area and perimeter as well as the mean fluorescence intensity in different compartments of the cell. For batch 2 cells, we used the tdTomato image for each cell to infer the laminar boundary by thresholding the images iteratively and performing morphological dilations until a closed loop formed, providing a new nuclear mask. As a measure of nuclear roundness, we computed the inverse isoperimetric quotient for each cell: the ratio of the area of a circle with the equivalent perimeter to the observed area of the nucleus, computed as P2/(4πA) (domain 1 to infinity). All image processing steps are deterministic and reproducible, with all R code and necessary metadata files published in our github repository.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Dam−/dcm− Competent E. Coli | New England Biolabs | Cat#C2925I |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM with GlutaMAX | Gibco | Cat#10566–016 |
| Pen Strep | Gibco | Cat#15140–122 |
| FBS (Seradigm Select Grade) | Avantor | Cat#89510–186 |
| FuGene HD | Promega | Cat#E2311 |
| Shield-1 Ligand | Takara Bio USA, Inc. | Cat#632189 |
| TrypLE Select | Gibco | Cat#12563–011 |
| SU-8 2025 negative photoresist | Microchem | SU-8 2025 |
| AZ 40XT-11D positive photoresist | Integrated Micro Materials | AZ40XT-11D |
| PDMS | Momentive Performance Materials | RTV615A |
| Trichloromethylsilane | Millipore Sigma | Cat#M85301–5G |
| Pluronic F-127 | Millipore Sigma | Cat#P2443 |
| Proteinase K | New England Biolabs | Cat#P8107S |
| common salts & detergents: KCl, MgCl2, NaCl, TRIS-HCl, TRIS acetate, magnesium acetate, potassium acetate, sodium acetate, EDTA, Tween-20, IGEPAL | Various | Various |
| T4 DNA ligase and T4 ligase buffer | New England Biolabs | Cat#M0202S |
| DpnI and CutSmart buffer | New England Biolabs | Cat#R0176S |
| Takara Advantage 2 PCR Kit | Takara Bio USA, Inc. | Cat#639207 |
| DpnII and DpnII Buffer | New England Biolabs | Cat#R0543S |
| DNeasy Blood & Tissue Kit | Qiagen | Cat#69504 |
| QIAquick PCR Purification Kit | Qiagen | Cat#28104 |
| RNeasy Mini Kit with QIAshredder | Qiagen | Cat#74104 |
| AMPure XP magnetic SPRI beads | Beckman Coulter | Cat#A63880 |
| Monarch PCR & DNA Cleanup Kit | New England Biolabs | Cat#T1030S |
| solvents: SU-8 developer, AZ 300MIF developer, molecular biology grade 100% ethanol, IPA, acetone | Various | Various |
| RNase A | Qiagen | Cat#19101 |
| Critical Commercial Assays | ||
| Qubit dsDNA HS Assay Kit | ThermoFisher Scientific | Cat#Q32851 |
| TapeStation D5000 HS Ladder, Reagents, and ScreenTape | Agilent | Cat#5067–5594, Cat#5067–5593, Cat#5067–5592 |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | New England Biolabs | Cat#E7645 |
| NEBnext Ultra II FS DNA Library Prep Kit for Illumina | New England Biolabs | Cat#E7805 |
| NEBNext Ultra II RNA Library Prep Kit for Illumina | New England Biolabs | Cat#E7770S |
| NEBNext Poly(A) mRNA Magnetic Isolation Module | New England Biolabs | Cat#E7490S |
| Deposited Data | ||
| KBM-7 bulk RNA-seq | Essletzbichler et al., 2015 | SRA: SRP044391 |
| KBM-7 single-cell DamID | Kind et al., 2015 | GEO: GSE69423 |
| HEK293T cell bulk & single-cell DamID sequencing reads and bulk RNA-seq reads | this study | GEO: GSE156150 |
| HEK293T cell raw images | this study | available on FigShare: https://doi.org/10.6084/m9.figshare.12798158 |
| Experimental Models: Cell Lines | ||
| HEK293T cells | ATCC | Cat#CRL-3216; RRID:CVCL_0063 |
| Oligonucleotides | ||
| AdRt: CTAATACGACTCACTATAGGGCAGCGTGGTCGCGGCCGAGGA | Integrated DNA Technologies | CustomOrder |
| AdRb: TCCTCGGCCG | Integrated DNA Technologies | CustomOrder |
| AdR_PCR: NNNNGTGGTCGCGGCCGAGGATC | Integrated DNA Technologies | CustomOrder |
| NEBNext Multiplex Oligos for Illumina (96 Unique Dual Index Primer Pairs) | New England Biolabs | Cat#E6440S |
| Recombinant DNA | ||
| plasmid: DD-DamV133A-LMNB1-IRES2-mCherry | this study (modified from a gift from Bas van Steensel) | Deposited to Addgene (#159599), sequence also on GitHub https://github.com/altemose/microDamID |
| plasmid: DD-DamV133A-IRES2-mCherry | this study (modified from a gift from Bas van Steensel) | Deposited to Addgene (#159600), sequence also on GitHub https://github.com/altemose/microDamID |
| plasmid: DD-DamWT-LMNB1-IRES2-mCherry | this study (modified from a gift from Bas van Steensel) | Deposited to Addgene (#159601), sequence also on GitHub https://github.com/altemose/microDamID |
| plasmid: DD-DamWT-IRES2-mCherry | this study (modified from a gift from Bas van Steensel) | Deposited to Addgene (#159602), sequence also on GitHub https://github.com/altemose/microDamID |
| plasmid: DD-DamV133A-tdTomato-LMNB1 | this study (modified from a gift from Bas van Steensel) | Deposited to Addgene (#159604), sequence also on GitHub https://github.com/altemose/microDamID |
| plasmid: m6A-Tracer | gift from Bas van Steensel | Kind et al., 2013 |
| plasmid: m6A-Tracer-NES | this study | Deposited to Addgene (#159607), sequence also on GitHub https://github.com/altemose/microDamID |
| Software and Algorithms | ||
| trimmomatic v0.39 | Bolger et al., 2014 | |
| Salmon | Patro et al., 2017 | |
| limma (R package) | Ritchie et al., 2015 | |
| BWA-MEM v0.7.15-r1140 | Li 2013 | |
| samtools v1.9 | Li et al., 2009 | |
| bedtools v2.28 | Quinlan et al., 2010 | |
| Deseq2 | Love et al., 2014 | |
| R (v4.0.0) | The R Project for Statistical Computing | |
| Additional R packages: ggplot2 (v3.3.0), gplots (v3.0.3), colorRamps (v2.3), reshape2 (v1.4.4), ggextra (v0.9), poisbinom (v1.0.1), SDMTools (v1.1–221.1), spatstat (v1.59–0), magick (v2.0) | CRAN & Bioconductor repositories | |
| Wash U Epigenome Browser | Li et al., 2019 | |
| In-house code for file parsing (bash, perl, & python) and data analysis (python & R) | this study | All code is available on this study’s GitHub repository: https://github.com/altemose/microDamID |
| Other | ||
| 10 cm diameter, 500 μm thick test-grade silicon wafers | University Wafer | Cat#452 |
| Photomasks (25400 DPI) | CAD/Art Services | Design files are available on this study’s GitHub repository: https://github.com/altemose/microDamID |
| Gold SEAL 48×65 mm No.1 coverglass | ThermoFisher Scientific | Cat#3335 |
| PEEK tubing (0.25 mm ID, 0.8 mm OD) | IDEX Corporation | Cat#1581 |
| Gel loading tips | Cole-Parmer | Cat#UX-25713–12 |
Highlights.
New microfluidic device images and sorts single cells then performs single-cell DamID
Improved m6A-Tracer method images protein-DNA interactions with less background
μDamID identifies LADs that vary between single cells and across cell types
Imaging can be used to select cells with greater DamID sequencing signal to noise
ACKNOWLEDGMENTS
We would like to thank Anushka Gupta, Gabriel Dorlhiac, Zoë Steier, Adam Gayoso, Tyler Chen, Xinyi Zhang, and Sophie Dumont for their helpful feedback on this work. We are grateful to Carolyn de Graaf for providing us with LAD coordinates, to Bo Huang and Bianxiao Cui for providing guidance and materials, and to Bas van Steensel for providing us with plasmids. N.A. is supported by a Howard Hughes Medical Institute Gilliam Fellowship for Advanced Study. This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health (grant number R35GM124916). A.S. is a Chan Zuckerberg Biohub Investigator.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cels.2020.08.015.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aughey GN, Estacio Gomez AE, Thomson J, Yin H, and Southall TD (2018). CATaDa reveals global remodelling of chromatin accessibility during stem cell differentiation in vivo. eLife 7, e32341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aughey GN, and Southall TD (2016). Dam it’s good! DamID profiling of protein-DNA interactions. Wiley Interdiscip. Rev Dev. Biol 5, 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, Wei G, Chepelev I, and Zhao K (2007). High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837. [DOI] [PubMed] [Google Scholar]
- Bernhofer M, Goldberg T, Wolf S, Ahmed M, Zaugg J, Boden M, and Rost B (2018). NLSdb-major update for database of nuclear localization signals and nuclear export signals. Nucleic Acids Res 46, D503–D508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickmore WA, and van Steensel B (2013). Genome architecture: domain organization of interphase chromosomes. Cell 152, 1270–1284. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsos M, Perricone SM, Schauer T, Pontabry J, de Luca KL, de Vries SS, Ruiz-Morales ER, Torres-Padilla ME, and Kind J (2019). Genome–lamina interactions are established de novo in the early mouse embryo. Nature 569, 729–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchwalter A, Kaneshiro JM, and Hetzer MW (2019). Coaching from the sidelines: the nuclear periphery in genome regulation. Nat. Rev. Genet 20, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter B, Ku WL, Kang JY, Hu G, Perrie J, Tang Q, and Zhao K (2019). Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq). Nat. Commun 10, 3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celniker SE, Dillon LAL, Gerstein MB, Gunsalus KC, Henikoff S, Karpen GH, Kellis M, Lai EC, Lieb JD, MacAlpine DM, et al. ; modENCODE Consortium (2009). Unlocking the secrets of the genome. Nature 459, 927–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TN, Gupta A, Zalavadia M, and Streets AM (2020). mCB-seq: microfluidic cell bar coding and sequencing for high-resolution imaging and sequencing of single cells. bioRxiv 10.1101/2020.02.18.954974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole RH, Tang SY, Siltanen CA, Shahi P, Zhang JQ, Poust S, Gartner ZJ, and Abate AR (2017). Printed droplet microfluidics for on demand dispensing of picoliter droplets and cells. Proc. Natl. Acad. Sci. USA 114, 8728–8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsawy H, and Chahar S (2014). Increasing DNA substrate specificity of the EcoDam DNA-(adenine N(6))-methyltransferase by site-directed mutagenesis. Biochemistry Mosc 79, 1262–1266. [DOI] [PubMed] [Google Scholar]
- ENCODE Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essletzbichler P, Konopka T, Santoro F, Chen D, Gapp BV, Kralovics R, Brummelkamp TR, Nijman SMB, and Bürckstümmer T (2014). Megabase-scale deletion using CRISPR/Cas9 to generate a fully haploid human cell line. Genome Res 24, 2059–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosselin K, Durand A, Marsolier J, Poitou A, Marangoni E, Nemati F, Dahmani A, Lameiras S, Reyal F, Frenoy O, et al. (2019). High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat. Genet 51, 1060–1066. [DOI] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, and Steensel BV (2008). Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453, 948–951. [DOI] [PubMed] [Google Scholar]
- Harada A, Maehara K, Handa T, Arimura Y, Nogami J, Hayashi-Takanaka Y, Shirahige K, Kurumizaka H, Kimura H, and Ohkawa Y (2019). A chromatin integration labelling method enables epigenomic profiling with lower input. Nat. Cell Biol 21, 287–296. [DOI] [PubMed] [Google Scholar]
- Huang B, Babcock H, and Zhuang X (2010). Breaking the diffraction barrier: super-resolution imaging of cells. Cell 143, 1047–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsen JS, Bagger FO, Hasemann MS, Schuster MB, Frank AK, Waage J, Vitting-Seerup K, and Porse BT (2015). Amplification of pico-scale DNA mediated by bacterial carrier DNA for small-cell-number transcription factor ChIP-seq. BMC Genomics 16, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, and Wold B (2007). Genome-wide mapping of in vivo protein-DNA interactions. Science 316, 1497–1502. [DOI] [PubMed] [Google Scholar]
- Kakar M, Davis JR, Kern SE, and Lim CS (2007). Optimizing the protein switch: altering nuclear import and export signals, and ligand binding domain. J. Control. Release 120, 220–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya-Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, Henikoff JG, Ahmad K, and Henikoff S (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun 10, 1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, De Jonghe J, Kulesa AB, Feldman D, Vatanen T, Bhattacharyya RP, Berdy B, Gomez J, Nolan J, Epstein S, and Blainey PC (2017). High-throughput automated microfluidic sample preparation for accurate microbial genomics. Nat. Commun 8, 13919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind J, Pagie L, de Vries SS, Nahidiazar L, Dey SS, Bienko M, Zhan Y, Lajoie B, de Graaf CA, Amendola M, et al. (2015). Genome-wide maps of nuclear lamina interactions in single human cells. Cell 163, 134–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind J, Pagie L, Ortabozkoyun H, Boyle S, de Vries SS, Janssen H, Amendola M, Nolen LD, Bickmore WA, and van Steensel B (2013). Single-cell dynamics of genome-nuclear lamina interactions. Cell 153, 178–192. [DOI] [PubMed] [Google Scholar]
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al. ; Roadmap Epigenomics Consortium (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A, Altemose N, White JA, and Streets AM (2019). On-ratio PDMS bonding for multilayer microfluidic device fabrication. J. Micromech. Microeng 29, 107001. [Google Scholar]
- Lenain C, de Graaf CA, Pagie L, Visser NL, de Haas M, de Vries SS, Peric-Hupkes D, van Steensel B, and Peeper DS (2017). Massive reshaping of genome–nuclear lamina interactions during oncogene-induced senescence. Genome Res 27, 1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Hsu S, Purushotham D, Sears RL, and Wang T (2019). WashU epigenome Browser update 2019. Nucleic Acids Res 47, W158–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv https://arxiv.org/abs/1303.3997v2. [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HQ, Baxter BC, Brower K, Diaz-Botia CA, DeRisi JL, Fordyce PM, and Thorn KS (2017). Programmable microfluidic synthesis of over one thousand uniquely identifiable spectral codes. Adv. Opt. Mater 5, 1600548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brown ZK, Boulias K, Wang J, Wang SY, O’Brown NM, Hao Z, Shibuya H, Fady PE, Shi Y, He C, et al. (2019). Sources of artifact in measurements of 6mA and 4mC abundance in eukaryotic genomic DNA. BMC Genomics 20, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orian A, van Steensel B, Delrow J, Bussemaker HJ, Li L, Sawado T, Williams E, Loo LW, Cowley SM, Yost C, et al. (2003). Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev 17, 1101–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Patel N, Keung AJ, and Khalil AS (2019). Engineering epigenetic regulation using synthetic read-write modules. Cell 176, 227–238.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickersgill H, Kalverda B, de Wit E, Talhout W, Fornerod M, and van Steensel B (2006). Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat. Genet 38, 1005–1014. [DOI] [PubMed] [Google Scholar]
- Quinlan ARA, and Hall IMI (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalingam N, Fowler B, Szpankowski L, Leyrat AA, Hukari K, Maung MT, Yorza W, Norris M, Cesar C, Shuga J, et al. (2016). Fluidic logic used in a systems approach to enable integrated single-cell functional analysis. Front. Bioeng. Biotechnol 4, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Mateos P, Azevedo NF, Almeida C, and Pamme N (2020). FISH and chips: a review of microfluidic platforms for FISH analysis. Med. Microbiol. Immunol 209, 373–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooijers K, Markodimitraki CM, Rang FJ, de Vries SS, Chialastri A, de Luca KL, Mooijman D, Dey SS, and Kind J (2019). Simultaneous quantification of protein–DNA contacts and transcriptomes in single cells. Nat. Biotechnol 37, 766–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotem A, Ram O, Shoresh N, Sperling RA, Goren A, Weitz DA, and Bernstein BE (2015). Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol 33, 1165–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Jiang D, Fu Y, Wu X, Guo H, Feng B, Pang Y, Streets AM, Tang F, and Huang Y (2015). H3K4me3 epigenomic landscape derived from ChIP-Seq of 1,000 mouse early embryonic cells. Cell Res 25, 143–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, and Klar AJ (1992). Active genes in budding yeast display enhanced in vivo accessibility to foreign DNA methylases: a novel in vivo probe for chromatin structure of yeast. Genes Dev 6, 186–196. [DOI] [PubMed] [Google Scholar]
- Skene PJ, Henikoff JG, and Henikoff S (2018). Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc 13, 1006–1019. [DOI] [PubMed] [Google Scholar]
- Southall TD, Gold KS, Egger B, Davidson CM, Caygill EE, Marshall OJ, and Brand AH (2013). Cell-type-specific profiling of gene expression and chromatin binding without cell isolation: assaying RNA Pol II occupancy in neural stem cells. Dev. Cell 26, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streets AM, Zhang X, Cao C, Pang Y, Wu X, Xiong L, Yang L, Fu Y, Zhao L, Tang F, et al. (2014). Microfluidic single-cell whole-transcriptome sequencing. Proc Natl Acad Sci U S A 111, 7048–7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger MA, Chou HP, Thorsen T, Scherer A, and Quake SR (2000). Monolithic microfabricated valves and pumps by multilayer soft lithography. Science 288, 113–116. [DOI] [PubMed] [Google Scholar]
- van Steensel B, and Belmont AS (2017). Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, and Henikoff S (2000). Identification of in vivo DNA targets of chromatin proteins using tethered dam methyltransferase. Nat. Biotechnol 18, 424–428. [DOI] [PubMed] [Google Scholar]
- Vogel MJ, Peric-Hupkes D, and van Steensel B (2007). Detection of in vivo protein–DNA interactions using DamID in mammalian cells. Nat. Protoc 2, 1467–1478. [DOI] [PubMed] [Google Scholar]
- White JA, and Streets AM (2018). Controller for microfluidic large-scale integration. HardwareX 3, 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu AR, Kawahara TLA, Rapicavoli NA, van Riggelen J, Shroff EH, Xu L, Felsher DW, Chang HY, and Quake SR (2012). High throughput automated chromatin immunoprecipitation as a platform for drug screening and antibody validation. Lab Chip 12, 2190–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Olson BG, and Yao J (2016). DamID-seq: genome-wide mapping of protein-DNA interactions by high throughput sequencing of adenine-methylated DNA fragments. J. Vis. Exp 107, e53620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Sheng J, and Sims PA (2018). SCOPE-seq: a scalable technology for linking live cell imaging and single-cell RNA sequencing. Genome Biol 19, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Zheng H, Huang B, Li W, Xiang Y, Peng X, Ming J, Wu X, Zhang Y, Xu Q, et al. (2016). Allelic reprogramming of the histone modification H3K4me3 in early mammalian development. Nature 537, 553–557. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data generated during this study are available at GEO (accession GSE156150). The imaging data generated during this study are available at FigShare: https://doi.org/10.6084/m9.figshare.12798158. Analysis code, control software, device design files, and plasmid sequences are freely available for download on GitHub: https://github.com/altemose/microDamID. Source data for bulk KBM-7 RNA-seq were obtained from SRA (accession SRP044391), and source data for KBM-7 scDamID were obtained from GEO (accession GSE69423).