

Abstract
The National Institute on Aging in conjunction with the Alzheimer's Association (NIA‐AA) recently proposed a biological framework for defining the Alzheimer's disease (AD) continuum. This new framework is based upon the key AD biomarkers (amyloid, tau, neurodegeneration, AT[N]) instead of clinical symptoms and represents the latest understanding that the pathological processes underlying AD begin decades before the manifestation of symptoms. By using these same biomarkers, individuals with Down syndrome (DS), who are genetically predisposed to developing AD, can also be placed more precisely along the AD continuum. The A/T(N) framework is therefore thought to provide an objective manner by which to select and enrich samples for clinical trials. This new framework is highly flexible and allows the addition of newly confirmed AD biomarkers into the existing AT(N) groups. As biomarkers for other pathological processes are validated, they can also be added to the AT(N) classification scheme, which will allow for better characterization and staging of AD in DS. These biological classifications can then be merged with clinical staging for an examination of factors that impact the biological and clinical progression of the disease. Here, we leverage previously published guidelines for the AT(N) framework to generate such a plan for AD among adults with DS.
Keywords: Alzheimer's disease, biomarkers, Down syndrome
1. INTRODUCTION
A core purpose for the generation of the amyloid, tau, neurodegeneration (AT[N]) framework was to “enable a more precise approach to interventional trials where specific pathways can be targeted in the disease progress and in the appropriate people.” 1 Recently, the National Institute on Aging and Alzheimer's Association (NIA‐AA) proposed a “research framework” based on the AT(N) model 2 for observational and interventional research on Alzheimer's disease (AD). 3 Differently from the prior NIA‐AA diagnostic criteria, 4 this framework defines AD as a biological rather than a clinical construct, characterized by extracellular deposits of amyloid‐beta peptide (Aβ; “A”), intraneuronal aggregates of hyperphosphorylated tau (“T”) and neurodegeneration (“[N]”). The “N” is placed in parentheses to emphasize that the biomarkers in the (N) group are fundamentally different from “A” and “T” biomarkers because they are: (1) not specific for neurodegeneration due to AD, (2) may be attributed to other possible comorbid conditions, and (3) do not map onto neuropathologic findings used to diagnose AD. The AT(N) model considers A, T, and (N) status relatively independent from one another with a known sequential order. However, the model then combines the clinically defined diagnostic classifications with AT(N) biomarker status for consistent terminology for research use.
Briefly, negative amyloid and tau along with the absence of neurodegeneration (A−T−[N]–) defines the normal biomarker profile, and amyloid negativity with either positivity for tau or presence of neurodegeneration corresponds to suspected non‐Alzheimer's pathology. β‐Amyloidosis (A+) is sufficient to identify the Alzheimer's continuum. Within this continuum, A+T−(N)– denotes Alzheimer's pathologic change (preclinical AD), while A+T+ (with or without [N]+) establishes definite AD. The AT(N) framework has the potential to enrich clinical trials with individuals who show objective evidence that they are on the AD continuum while also permitting the staging of individual patients and providing prognosis as well as stratification for precision‐based clinical trials. The AT(N) classification system has been studied by multiple groups and has demonstrated utility in classifying individuals with late‐onset sporadic AD on the basis of biomarkers. 5 , 6 , 7
In 2013, the Food and Drug Administration (FDA) released draft guidance on drug development for AD. The guidance built on the understanding that AD is a progressive disease with clinical symptoms of dementia appearing decades after the AD pathophysiological process has begun and proposed a disease classification that acknowledged three stages of AD: the preclinical, prodromal, and dementia stages. 8 In 2018, the FDA revised the draft guidance and expanded the taxonomy of AD by recognizing four stages. 9 These include: Stage 1: “Preclinical AD”; Stage 2: “Preclinical/ Prodromal AD”; Stage 3: “Prodromal AD”; and Stage 4: “AD dementia.” We are now poised to study these stages of AD in Down syndrome (DSAD) using the most advanced AD biomarkers available to refine the AT(N) classification for use in this population.
2. APPLICATION OF AT(N) TO THE DS POPULATION
To date, the AT(N) framework has been applied in limited ways to other populations that are at risk for AD as a method to expand this model and to enrich clinical trials for AD. 10 Given the unique features described below of AD among adults with DS, the framework may have utility for rapid advancement of precision medicine approaches to novel clinical trials in this population. DS is, by definition, a genetically determined form of AD as recognized in the International Work Group on Criteria for AD (IWG‐2). 11 In DS, one of the most common forms of intellectual disability, the underlying genetic link between trisomy 21 and AD has been convincingly established. 12 , 13 , 14 , 15 By age 40 years, all adults with DS exhibit some degree of elevated brain amyloid. 16 , 17 , 18 The leading explanation for this link is tied to the triplication of chromosome 21 (trisomy 21) and the resulting overexpression of the amyloid precursor protein (APP) gene coded on this chromosome. 19 The excessive production of Aβ as a result is key to the pathogenesis of AD in adults with DS. 20 Although other genes coded on chromosome 21 may contribute to the early emergence of dementia and the phenomenon of accelerated aging seen in adults with DS, 21 forms of partial trisomy 21 which do not result in triplication of APP (ie, the APP‐containing portion of chromosome 21 is not present in the third copy) are not associated with clinical and pathological signs of AD. 14 , 15
Despite these consistent AD neuropathologic changes, the timing of the development of dementia as part of AD in DS is quite variable, 22 suggesting the presence of other genetic and environmental risk and protective factors. Individuals with DS have a lifetime risk for dementia in excess of 90%, and DS is now acknowledged to be a genetic form of AD similar to the much less common autosomal‐dominant causes of AD. 23 , 24 Although the development of dementia is not inevitable in all adults with DS, the risk increases incrementally with age. 25 Furthermore, as in the late‐onset form in the general population, the AT(N) classification of adults with DS will be strongly influenced by the age of the individual.
Identifying cognitive impairment at an early stage of the AD continuum has become an increasingly important goal in AD research, as it is widely believed that the greatest chance for therapeutic success will be obtained by intervening early in the disease, before widespread and irreversible neurodegeneration has occurred. 26 As a result, the AT(N) framework describes AD across its full spectrum (ie, preclinical to dementia) in terms of biomarker positivity/negativity and is agnostic with respect to clinical symptoms. As more longitudinal data are collected in DS, correlations between the distinct AT(N) classifications with clinical and cognitive status will be possible as well as a richer understanding of the rates of change in each biomarker category: amyloid, tau, and neurodegeneration across the AD conitnuum in DS. This more precise assessment will facilitate primary, secondary, and tertiary prevention trials for ADin individuals with DS. 27 , 28 , 29
In the general population 30 , 31 , 32 as well as in DS 33 , 34 , 35 , 36 the construct of mild cognitive impairment (ie, prodromal AD) as well as the identification of disease in the preclinical stage (eg, accumulation of amyloid in a cognitively stable individual) is central to the clinical diagnostic formulation of AD. Mild cognitive impairment (MCI) in the general population, as well as in DS, is generally regarded as the borderland between the cognitive changes of aging and early dementia where there is measurable decline in memory as well as some decline on instrumental activities of daily living (iADLs) but preservation of basic activities of daily functioning. 22 , 37 , 38 , 39 , 40 The characterization of preclinical and prodromal AD is now possible with the advancement of state‐of‐the‐art biomarker modalities such as amyloid and tau assessment using positron emission tomography (PET) imaging and cerebrospinal fluid (CSF) measures as well as emerging plasma biomarkers such as mass spectral Aβ assays 41 , 42 in the absence of or minimal cognitive decline.
RESEARCH IN CONTEXT
Systematic review: Alzheimer's disease (AD) is highly prevalent in Down syndrome (DS). The National Institute on Aging in conjunction with the Alzheimer's Association (NIA‐AA) recently proposed a biological framework for defining the AD continuum. This new framework is based on key AD biomarkers (amyloid, tau, neurodegeneration, AT[N]) instead of clinical symptoms and represents the latest understanding that the pathological processes underlying AD begin decades before the manifestation of symptoms.
Interpretation: These biological classifications can then be merged with clinical staging for an examination of factors that impact the biological and clinical progression of the disease. We leverage previously published guidelines for the AT(N) framework to generate such a plan for AD among adults with DS.
Future directions: Further work on longitudinal AD biomarkers in DS should help clarify whether the AT(N) classification system can be applied to individuals with DS both for clinical trial stratification as well as for use as a potential staging and prognostic tool in the clinic, representing a fundamental tool for precision medicine.
Here we propose an application of the AT(N) framework for the full characterization of the AD continuum in DS using both state‐of‐the‐art biomarkers and clinical assessments. Given the variability in cognitive assessments, the inclusion of biomarkers may facilitate the evaluation of potential efficacy of therapy in this population. Forthcoming data from the Alzheimer's Biomarker Consortium–Down Syndrome (ABC‐DS) 43 and the European Horizon21 consortium 44 will inform the diagnostic accuracy and prognostic potential of AT(N) in DS.
3. CLINICAL ASSESSMENT OF COGNITIVE STATUS
The defining feature of all causes of dementia is a decline from the baseline level of function and performance of daily skills. Although this may be straightforward to establish in the general population, it can be a much more complicated task in adults with DS because of lifelong intellectual impairment and significant variance in baseline cognitive functioning. 45 , 46 This is especially true for older adults with DS due to various factors impacting living arrangements in which there may be poor record keeping since childhood, lack of continuity in staff members supervising adults with DS over time, and a large number of physicians/health‐care providers throughout his or her life span. In the absence of a personal historian who can accurately and comprehensively attest to an individual's baseline level of functioning, the assessment of a reported cognitive and behavioral change may be exponentially more difficult. 47 , 48 The early signs of dementia in adults with DS can be subtle and often require an astute observer to identify these changes. Often, individuals with DS are served by numerous caregivers throughout their lifetime, and often newly involved caregivers will presume that the current level of observed ability represents the individual's baseline level of functioning and, thus, miss signs of early decline that has already occurred.
In the clinical setting, accurate assessment of cognition and function depend upon a comprehensive history, which for individuals with DS must be done in the context of knowledge and quantification of their historic level of intellectual disability. 49 It is important that a thorough history be obtained to compile evidence consistent with an emerging cognitive impairment while probing for potential factors contributing to decline. Pertinent historical information is useful from personal accounts of caregivers and family members who have known the individual for an extended length of time. 50 In addition, other sources of information, such as previous neuropsychological testing or school Individual Education Plan information, can greatly assist in accurately characterizing an individual's baseline level of functioning. In addition, medical history, medications, family history, social history, review of systems, laboratory evaluations, and brain imaging will be essential to rule out comorbidities that can masquerade as AD‐related cognitive impairment. Objective evidence of memory decline will be essential for the diagnosis of MCI‐DS and dementia. A number of cognitive assessment instruments are currently being evaluated in natural history studies of AD in DS, including the ABC–DS. 43 At this time, there is no single cognitive instrument that has been longitudinally validated in the context of AD biomarkers in DS but many are being presently intensely researched. 51 , 52 , 53 , 54 , 55
Once arriving at the suspected clinical diagnosis of MCI or dementia, the AT(N) framework can be used to stage an individual with DS along the AD continuum with respect to extent of underlying biomarker changes (Table 1). This staging can be used to provide expected clinical prognosis, including an estimated duration of independent functioning, time to dementia, and to also enrich for more homogenous samples in clinical trials. The proposed clinical staging of the cognitive continuum was adapted from previously published guidelines for preclinical AD, 56 MCI, 57 and AD dementia. 58
TABLE 1.
Clinical staging of cognitive continuum—Diagnostic recommendations
|
Abbreviations: AD, Alzheimer's disease; ADL, activities of daily living; DS, Down syndrome; iADL, instrumental activities of daily living.
The difficulties with MCI diagnoses in the general population are well established. MCI in adults with DS (MCI‐DS) is an even more challenging diagnosis and cross‐sectional assessments can be unreliable. Therefore, longitudinal assessments are optimal and required. Additional work is needed to determine optimal psychometric assessment instruments, cutoff scores, and/or combinations of instruments in this population for refinement of the MCI designation. Specifically, the following points will need to be considered as the concept of MCI‐DS evolves and will be informed by forthcoming data from ABC–DS: (1) Identification of the most informative cognitive assessment instruments for MCI varying based on severity of ID. (2) Quantification of decline needed to represent a clinically meaningful change. (3) Relationship between cognitive assessments and rates of change in various AD biomarkers. In order to confirm that MCI‐DS is in fact prodromal AD, the use of biomarkers to confirm AD as the underlying etiology will be required.
4. BIOMARKER ASSESSMENTS OF AMYLOID, TAU, AND NEURODEGENERATION IN DS
Over the past few years, substantial progress has been made in elucidating the natural history of AD in people with DS using the latest biomarkers including amyloid and tau PET imaging, volumetric brain MRI, as well as biofluid markers in CSF and plasma. 59 There exist remarkable similarities between AD biomarkers in DS and other populations with AD. 60 , 61 , 62 , 63 , 64 Greater hippocampal atrophy is associated with a greater amyloid load. 61 Cognitive and functional measures do not correlate as strongly with amyloid deposition as they do with abnormalities on 18F‐fluourodeoxyglucose (FDG) and tau PET. 61 , 65 , 66
4.1. Amyloid (A)
Amyloid PET positivity as observed using PET imaging in DS seems to resemble autosomal dominant AD more closely than sporadic AD. Specifically, Pittsburgh compound B (PIB) demonstrates an early and predominant basal ganglia signal 60 , 62 , 67 , 68
although other tracers (eg, florbetabir) have shown a pattern more similar to sporadic AD. 61 The similarity of AD in DS and ADAD is thought to result from overproduction of Aβ. APP overproduction in DS leads to baseline plasma levels of Aβ40 and Aβ42 and Aβ42/Aβ40 ratios which are higher than those in non‐DS individuals. 64 , 69 , 70 A positive correlation of tau and a negative correlation of CSF Aβ1‐42 have been reported with age 71 and several studies have documented correlations of the changes in amyloid in DS with AD. 72 , 73 , 74 , 75 Higher levels of Aβ42 or the Aβ42/Aβ40 ratio appear to be associated with the onset of AD in DS, 76 , 77 although this is not entirely consistent in the literature. 78 CSF Aβ42 levels are first increased in early life and then become lower with age, representing deposition of Aβ into plaques. 64 , 79 , 80 Most studies seem to suggest that as with sporadic and autosomal‐dominant AD, pathophysiological changes associated with AD in DS occur approximately two decades before the onset of symptoms of dementia.
Blood‐based biomarkers have clear advantages as biomarkers as they are easily accessible. Individuals with DS have higher baseline plasma Aβ1‐42 and Aβ1‐40 concentrations compared to individuals without DS 81 due to the extra copy of the APP gene and the resulting overproduction of APP and Aβ. There have been a limited number of CSF studies in individuals with DS which show elevated levels of Aβ42 early in life, but with age, CSF Aβ42 levels decline (as expected with their deposition into plaques) while CSF tau levels progressively increase. 64 , 81
4.2. Tau (T)
Neurofibrillary tangles (NFTs)which are comprised of abnormal tau, are a key pathological hallmark of AD and correlate with the emergence of clinical symptoms more closely than amyloid plaques. This relationship has also been demonstrated in post mortem pathology of DS brains, where NFTs correlate with cognitive decline. 82 Tau PET signal in the DS brain appears to be similar to sporadic AD and can be assessed using standard Braak staging. 66 Specifically, tau deposition in adults with DS has been studied using the PET tracer (18F) AV‐1451. 66 Abnormal tau distribution (in the form of NFTs) first involves the medial temporal cortices and then spreads posteriorly, 66 similar in manner to that observed in sporadic AD. More recently, plasma and CSF tau have been studied in individuals with DS, with their levels correlating with AD dementia in DS. 64 , 78 , 83 , 84 , 85
4.3. Neurodegeneration (N)
Markers of AD‐specific neurodegeneration include regional hypometabolism on FDG PET9 86 , 87 , 88 or hippocampal atrophy 89 , 90 , 91 have been studied in DS and parallel findings from the sporadic and autosomal dominant forms of AD. More recently, plasma neurofilament light chain (NfL) levels (also a marker of neurodegeneration) have been shown to correlate with clinical status of AD in DS 92 as well as standard AD biomarkers such as amyloid PET and tau PET. 93 Specifically, plasma NfL levels appear to increase with age in but can still distinguish between normal aging and AD 81 , 92 . Plasma NfL levels have also been shown to correlate with other markers of neurodegeneration such as hypometabolism on FDG PET and hippocampal atrophy, as well as cognitive and functional decline. 93
Thus, various biomarker modalities (eg, imaging, biofluids) can be used to characterize individuals with DS as exhibiting amyloid, tau, or neurodegeneration “positivity” in the AT(N) classification scheme. The biomarkers currently being used to characterize individuals with DS are listed in Table 2.
TABLE 2.
Biomarker classification AT(N) pathology among adults with DS
|
Abbreviations: Aβ, amyloid beta; AT(N), amyloid, tau, neurodegeneration; CSF, cerebrospinal fluid; FDG, 18F‐fluorodeoxyglucose; MRI, magnetic resonance imaging; PET, positron emission tomography
The AT(N) classification scheme can then be applied to such individuals as depicted in Table 3. Briefly, individuals with DS with stable cognition who have no elevations in brain amyloid or tau and no evidence of neurodegeneration would be classified as A−/T−/N−. Therefore, they would not be on the AD continuum. However, an individual with stable cognition who has elevated brain amyloid but no evidence of elevated tau or any neurodegeneration (A+/T−/N−) would be categorized as preclinical AD. An individual who has symptoms consistent with MCI‐DS who has elevated brain amyloid but no elevated tau or evidence of neurodegeneration (A+/T+/N) would be classified as Prodromal AD. Finally, an individual with MCI‐DS but who is A−/T+/N− would be considered as having a neurodegenerative disease other than AD (non‐AD) as the basis for their symptoms. Therefore, by use of the A/T(N) framework, it is anticipated that we will be able to conduct clinical trials in a more finely characterized participant sample.
TABLE 3.
AT(N) Framework for adults with Down syndrome
| Combined syndromal cognitive and biomarker categorization | |||||
|---|---|---|---|---|---|
| AT(N) profiles | Biomarker category | Part of Alzheimer's continuum (Y/N) | Stable cognition | MCI | Dementia |
| A‐/T−/(N)− | Normal biomarkers | No | Stable cognition + normal AD biomarkers | MCI‐DS + normal AD biomarkers | Dementia + normal AD biomarkers |
| A+/T−/(N)− | AD pathological change | Yes | Preclinical AD pathological change | MCI‐DS + AD pathological change | Dementia + AD pathological change |
| A+/T+/(N)− | AD | Yes | Preclinical AD | Prodromal AD | AD + dementia |
| A+/T+/(N)+ | AD | Yes | Preclinical AD | Prodromal AD | AD + dementia |
| A+/T−/(N)+ | AD and concomitant suspected non‐ AD pathological change | Yes | Preclinical AD* | Prodromal AD* | AD* + dementia |
| A−/T+/(N)− | Non‐AD pathological Change | No | Preclinical non‐AD | MCI not due to AD | Non‐AD dementia |
| A−/T−/(N)+ | Non‐AD pathological Change | No | Preclinical non‐AD | MCI not due to AD | Non‐AD dementia |
| A−/T+/(N)+ | Non‐AD pathological Change | No | Preclinical non‐AD | MCI not due to AD | Non‐AD dementia |
NOTE: AD* = AD and concomitant suspected non‐AD pathological changes
Abbreviations: AT(N), amyloid, tau, neurodegeneration; AD, Alzheimer's disease; MCI, mild cognitive impairment.
5. ESTIMATING A+/T+(N)+ PREVALENCE IN DS
We intend to look at AT(N) classification across the different clinical diagnostic categories, that is, Cognitively Stable, Mild Cognitive Impairment, and Dementia in the ABC‐DS Study to calculate A+/T+/(N)+ prevalence and to correlate the various classifications with clinical and cognitive status. Based on a review of the literature, 20 , 36 , 41 , 42 , 59 , 60 , 61 , 62 , 63 , 64 , 65 , 66 , 67 , 68 , 69 , 70 , 71 , 72 , 73 , 74 , 75 , 76 , 77 , 78 , 79 , 80 , 81 , 82 , 83 , 84 , 85 , 86 , 87 , 88 , 89 , 90 , 91 , 92 , 93 we estimate that, between ages 35 to 55 years, there will be 80% A+, 40% T+, and 10% (N)+ for cognitively stable adults with DS; 80% A+, 60% T+, and 20% (N)+ for MCI‐DS; and 80% A+, 80% T+, and 60% (N)+ for dementia in the DS group (Figure 1).
FIGURE 1.
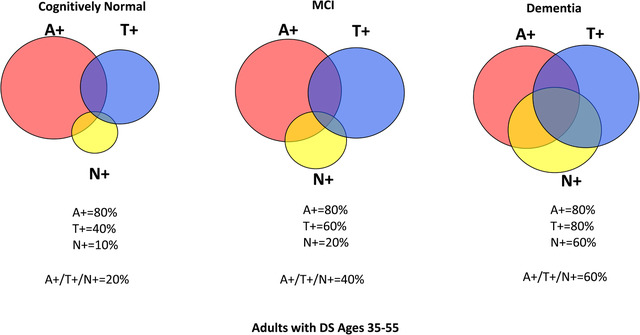
Estimated prevalence of A/T(N) positivity across clinical diagnoses. We estimate that between ages 35 to 55 years, there will be 80% A+, 40% T+, and 10% (N)+ for cognitively stable adults with Down syndrome (DS); 80% A+, 60% T+, and 20% (N)+ for mild cognitive impairment (MCI)‐DS; and 80% A+, 80% T+, and 60% (N)+ for the dementia in DS group. A+ = elevated brain amyloid, T+ = tau pathology present, (N) = neurodegeneration present
There may be limitations specific to the A/T(N) classification system. For example, amyloid imaging may underestimate true amyloid positivity. In addition, some biomarkers indicating tau pathology may become positive at different stages of the disease (ie, CSF becoming abnormal before PET imaging). And, there appears to be a potential discrepancy between timing of positive MRI indicators of atrophy (and hence neurodegeneration) versus increased levels of plasma NfL. Finally, these differences indicate that dichotomization may potentially decrease sensitivity to changes in cognition.
As longitudinal data become available, the utility of the AT(N) classification scheme will be compared with each individual's clinical status over time. We will test if there are differences between the biochemical and neuroimaging measures of AT(N) . We will confirm prevalence of AD biomarker positivity across different ages and clinical diagnoses. Additionally, we will assess how best we can operationalize the biomarker binarization to ensure the external validity of the results. Finally, we will evaluate the impact of cerebrovascular disease, including microhemorrhages related to cerebral amyloid angiopathy; neuroinflammation; and, as post mortem data accrue, other pathologies such as TDP‐43 and Lewy bodies on A/T(N)'s accuracy in staging disease and predicting clinical status. A similar longitudinal AD biomarker study (Horizon21) is ongoing in Europe with plans to harmonize some elements with ABC‐DS going forward. 44
6. CONCLUSIONS
Recent work on the AT(N) frameworkin the general population suggests that individuals exhibiting abnormalities on all three biomarkers are at the greatest risk of developing AD dementia. The AT(N) model considers A, T, and (N) status relatively independent from one another with a known sequential order. The current framework is proposed as a starting point for use of A/T(N) classificationin the DS population. It is understood that this is not a final model and as new data emerge, the framework will be revised and updated accordingly in order to parallel the state of current knowledge. As with the original AT(N) Framework, this staging system is intended to aid in the refinement of clinical trials and to facilitate a better understanding of the biology ofAD in adults with DS. This research framework is not intended for clinical use at this time.
Further work on longitudinal AD biomarkers in DS should help clarify whether the AT(N) classification system can be applied to individuals with DS both for clinical trial stratification as well as for use as a potential staging and prognostic tool in the clinic, representing a fundamental tool for precision medicine.
FUNDING INFORMATION
Michael S. Rafii is supported by 1R61AG066543.
CONFLICTS OF INTEREST
The authors report no conflicts of interest that are relevant for this manuscript.
Rafii MS, Ances BM, Schupf N, et al. The AT(N) framework for Alzheimer's disease in adults with Down syndrome. Alzheimer's Dement. 2020;12:e12062 10.1002/dad2.12062
REFERENCES
- 1. Knopman DS, Haeberlein SB, Carrillo MC, et al. The National Institute on Aging and the Alzheimer's Association Research Framework for Alzheimer's disease: perspectives from the research roundtable. Alzheimers Dement. 2018;14(4):563‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jack CR, Jr , Bennett DA, Blennow K, et al. AT(N): an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology. 2016;87(5):539‐547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jack CR, Jr , Bennett DA, Blennow K, et al. NIA‐AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14(4):535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jack CR, Jr , Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):257‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ekman U, Ferreira D, Westman E. The AT(N) biomarker scheme and patterns of brain atrophy assessed in mild cognitive impairment. Scientific Reports. 2018;8(1):8431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kern S, Zetterberg H, Kern J, et al. Prevalence of preclinical Alzheimer disease: comparison of current classification systems. Neurology. 2018;90(19):e1682‐e1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Roe CM, Babulal GM, Stout SH, et al. Using the AT(N) framework to examine driving in preclinical AD. Geriatrics (Basel). 2018;3(2). pii: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Food and Drug Administration . Draft Guidance for Industry. Alzheimer's disease: developing drugs for the treatment of early stage disease. 2013. http://www.fda.gov.libproxy2.usc.edu/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM338287.pdf
- 9. Food and Drug Administration . Early Alzheimer's disease: developing drugs for treatment guidance for industry. 2018. https://www-fda-gov.libproxy2.usc.edu/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM596728.pdf
- 10. Wolz R, Schwarz AJ, Gray KR, Yu P, Hill DL. Alzheimer's Disease Neuroimaging Initiative . Enrichment of clinical trials in MCI due to AD using markers of amyloid and neurodegeneration. Neurology. 2016;87(12):1235‐1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dubois B, Feldman HH, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG‐2 criteria [published correction appears in Lancet Neurol. 2014 Aug;13(8):757]. Lancet Neurol. 2014;13(6):614‐629. [DOI] [PubMed] [Google Scholar]
- 12. Wisniewski KE, Wisniewski HM, Wen GY. Occurrence of neuropathological changes and dementia of Alzheimer's disease in Down's syndrome. Ann Neural. 1985;17:278‐282. [DOI] [PubMed] [Google Scholar]
- 13. Rumble B, Retallack R, Hilbich C, et al. Amyloid A4 and its precursor in Down's syndrome and Alzheimer's disease. New Engl J Med. 1989;320:1446‐1462. [DOI] [PubMed] [Google Scholar]
- 14. Prasher VP, Farrer MJ, Kessling AM, Fisher EM, West RJ, Barber PC. Molecular mapping of Alzheimer‐type dementia in Down's syndrome. Ann Neurol. 1998;43:380‐383.9506555 [Google Scholar]
- 15. Doran E, Keator D, Head E, et al. Down syndrome, partial trisomy 21, and absence of Alzheimer's disease: the role of APP. J Alzheimers Dis. 2017;56(2):459‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mann DM, Alzheimer's disease and Down's syndrome. Histopathology. 1988;13(2):125‐137. [DOI] [PubMed] [Google Scholar]
- 17. Mann DM, Brown A, Prinja D, et al. An analysis of the morphology of senile plaques in Down's syndrome patients of different ages using immunocytochemical and lectin histochemical techniques. Neuropathol Appl Neurobiol. 1989;15(4):317‐329. [DOI] [PubMed] [Google Scholar]
- 18. Antonarakis SE, Skotko BG, Rafii MS, et al. Down syndrome. Nat Rev Dis Primers. 2020;6(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wiseman FK, Al‐Janabi T, Hardy J, et al. A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat Rev Neurosci. 2015;16(9):564‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Teller JK, Russo C, DeBusk LM, et al. Presence of soluble amyloid beta‐peptide precedes amyloid plaque formation in Down's syndrome. Nat Med. 1996;2(1):93‐95. [DOI] [PubMed] [Google Scholar]
- 21. Park J, Oh Y, Chung KC. Two key genes closely implicated with the neuropathological characteristics in Down syndrome: dYRK1A and RCAN1. BMB Rep. 2009;42(1):6‐15. [DOI] [PubMed] [Google Scholar]
- 22. Lautarescu BA, Holland AJ, Zaman SH. The early presentation of dementia in people with down syndrome: a systematic review of longitudinal studies. Neuropsychol Rev. 2017;27(1):31‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McCarron M, McCallion P, Reilly E, Mulryan N. A prospective 14‐year longitudinal follow‐up of dementia in persons with Down syndrome. J Intellect Disabil Res. 2014;58:61‐70. [DOI] [PubMed] [Google Scholar]
- 24. McCarron M, McCallion P, Reilly E, Dunne P, Carroll R, Mulryan N. A prospective 20‐year longitudinal follow‐up of dementia in persons with Down syndrome. J Intellect Disabil Res. 2017;61:843‐852. [DOI] [PubMed] [Google Scholar]
- 25. Sinai A, Mokrysz C, Bernal J, Bohnen I, Bonell S, Courtenay K. Predictors of age of diagnosis and survival of Alzheimer's disease in Down syndrome. J Alzheimers Dis. 2018;61:717‐728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rafii MS, Aisen PS. Alzheimer's disease clinical trials: moving toward successful prevention. CNS Drugs. 2019;33(2):99‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ness S, Rafii M, Aisen P, Krams M, Silverman W, Manji H. Down's syndrome and Alzheimer's disease: towards secondary prevention. Nat Rev Drug Discov. 2012;11(9):655‐656. [DOI] [PubMed] [Google Scholar]
- 28. Rafii MS. Pro: are we ready to translate Alzheimer's disease modifying therapies to people with Down syndrome? Alzheimers Res Ther. 2014;6(5‐8):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rafii MS. Improving memory and cognition in individuals with Down syndrome. CNS Drugs. 2016;30(7):567‐573. [DOI] [PubMed] [Google Scholar]
- 30. Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome (published correction appears in Arch Neurol 1999 Jun;56(6):760). Arch Neurol. 1999;56(3):303‐308. [DOI] [PubMed] [Google Scholar]
- 31. Ward A, Tardiff S, Dye C, Arrighi HM. Rate of conversion from prodromal Alzheimer's disease to Alzheimer's dementia: a systematic review of the literature. Dement Geriatr Cogn Dis Extra. 2013;3(1):320‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sperling R, Mormino E, Johnson K. The evolution of preclinical Alzheimer's disease: implications for prevention trials. Neuron. 2014;84(3):608‐622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Krinsky‐McHale SJ, Devenny DA, Kittler P, Silverman W. Selective attention deficits associated with mild cognitive impairment and early stage Alzheimer's disease in adults with Down syndrome. Am J Ment Retard. 2008;113(5):369‐386. [DOI] [PubMed] [Google Scholar]
- 34. García‐Alba J, Ramírez‐Toraño F, Esteba‐Castillo S, et al. Neuropsychological and neurophysiological characterization of mild cognitive impairment and Alzheimer's disease in Down syndrome. Neurobiol Aging. 2019;84:70‐79. [DOI] [PubMed] [Google Scholar]
- 35. Jenkins EC, Ye L, Velinov M, et al. Mild cognitive impairment identified in older individuals with Down syndrome by reduced telomere signal numbers and shorter telomeres measured in microns. Am J Med Genet B Neuropsychiatr Genet. 2012;159B(5):598‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mapstone M, Gross TJ, Macciardi F, et al. Metabolic correlates of prevalent mild cognitive impairment and Alzheimer's disease in adults with Down syndrome. Alzheimers Dement (Amst). 2020;12(1):e12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lawton MP, Brody EM. Assessment of older people: self‐maintaining and instrumental activities of daily living. Gerontologist. 1969;9(3 Part 1):179‐186. [PubMed] [Google Scholar]
- 38. Schoufour JD, Mitnitski A, Rockwood K, Hilgenkamp TI, Evenhuis HM, Echteld MA. Predicting disabilities in daily functioning in older people with intellectual disabilities using a frailty index. Res Dev Disabil. 2014;35(10):2267‐2277. [DOI] [PubMed] [Google Scholar]
- 39. Ballard C, Mobley W, Hardy J, Williams G, Corbett A. Dementia in Down's syndrome. Lancet Neurol. 2016;15(6):622‐636. [DOI] [PubMed] [Google Scholar]
- 40. Hithersay R, Hamburg S, Knight B, Strydom A. Cognitive decline and dementia in Down syndrome. Curr Opin Psychiatry. 2017;30(2):102‐107. [DOI] [PubMed] [Google Scholar]
- 41. Nakamura A, Kaneko N, Villemagne VL, et al. High performance plasma amyloid‐β biomarkers for Alzheimer's disease. Nature. 2018;554(7691):249‐254. [DOI] [PubMed] [Google Scholar]
- 42. Schindler SE, Bollinger JG, Ovod V, et al. High‐precision plasma β‐amyloid 42/40 predicts current and future brain amyloidosis. Neurology. 2019;93(17):e1647‐e1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Handen BL. The search for biomarkers of Alzheimer's disease in down syndrome. Am J Intellect Dev Disabil. 2020;125(2):97‐99. [DOI] [PubMed] [Google Scholar]
- 44. Strydom A, Coppus A, Blesa R, et al. Alzheimer's disease in Down syndrome: an overlooked population for prevention trials. Alzheimers Dement (N Y). 2018;4:703‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sheehan R, Sinai A, Bass N, et al. Dementia diagnostic criteria in Down syndrome. Int J Geriatr Psychiatry. 2015;30(8):857‐863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smith D, Chicoine B. Difficulties of diagnosing and managing dementia in people with Down syndrome. Br J Psychiatry. 2018;213(5):668‐669. [DOI] [PubMed] [Google Scholar]
- 47. Prasher VP, Sachdeva N, Tarrant N. Diagnosing dementia in adults with Down's syndrome. Neurodegener Dis Manag. 2015;5(3):249‐256. [DOI] [PubMed] [Google Scholar]
- 48. Dekker AD, Strydom A, Coppus AM, et al. Behavioural and psychological symptoms of dementia in Down syndrome: early indicators of clinical Alzheimer's disease? Cortex. 2015;73:36‐61. [DOI] [PubMed] [Google Scholar]
- 49. Moran JA, Rafii MS, Keller SM, et al. The National Task Group on intellectual disabilities and dementia practices consensus recommendations for the evaluation and management of dementia in adults with intellectual disabilities. Mayo Clin Proc. 2013;88(8):831‐840. [DOI] [PubMed] [Google Scholar]
- 50. Holst G, Johansson M, Ahlström G. Signs in people with intellectual disabilities: interviews with managers and staff on the identification process of dementia. Healthcare (Basel). 2018;6(3):103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Firth NC, Startin CM, Hithersay R, et al. Aging related cognitive changes associated with Alzheimer's disease in Down syndrome. Ann Clin Transl Neurol. 2018;5(6):741‐751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Startin CM, Hamburg S, Hithersay R, et al. Cognitive markers of preclinical and prodromal Alzheimer's disease in Down syndrome. Alzheimers Dement. 2019;15(2):245‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sabbagh M, Edgin J. Clinical assessment of cognitive decline in adults with down syndrome. Curr Alzheimer Res. 2016;13(1):30‐34. [DOI] [PubMed] [Google Scholar]
- 54. Hartley SL, Handen BL, Devenny DA, et al. Cognitive functioning in relation to brain amyloid‐β in healthy adults with Down syndrome. Brain. 2014;137(Pt 9):2556‐2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lessov‐Schlaggar CN, Del Rosario OL, Morris JC, Ances BM, Schlaggar BL, Constantino JN. Adaptation of the clinical dementia rating scale for adults with Down syndrome. J Neurodev Disord. 2019;11(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):280‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):270‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee NC, Chien YH, Hwu WL. A review of biomarkers for Alzheimer's disease in Down syndrome. Neurol Ther. 2017;6(Suppl 1):69‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Handen BL, Cohen AD, Channamalappa U, et al. Imaging brain amyloid in nondemented young adults with Down syndrome using Pittsburgh compound B. Alzheimers Dement. 2012;8(6):496‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rafii MS, Wishnek H, Brewer JB, et al. The down syndrome biomarker initiative (DSBI) pilot: proof of concept for deep phenotyping of Alzheimer's disease biomarkers in Down syndrome. Front Behav Neurosci. 2015;9:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lao PJ, Handen BL, Betthauser TJ, et al. Longitudinal changes in amyloid positron emission tomography and volumetric magnetic resonance imaging in the nondemented Down syndrome population. Alzheimers Dement (Amst). 2017;9:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Abrahamson EE, Head E, Lott IT, et al. Neuropathological correlates of amyloid PET imaging in Down syndrome. Dev Neurobiol. 2019;79(7):750‐766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fortea J, Vilaplana E, Carmona‐Iragui M, etal. Clinical and biomarker changes of Alzheimer's disease in adults with Down syndrome: a cross‐sectional study. Lancet (in Press). 2020;395 10242: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lao PJ, Handen BL, Betthauser TJ, et al. Alzheimer‐like pattern of hypometabolism emerges with elevated amyloid‐β burden in Down syndrome. J Alzheimers Dis. 2018;61(2):631‐644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rafii MS, Lukic AS, Andrews RD, et al, Down Syndrome Biomarker Initiative and the Alzheimer's Disease Neuroimaging Initiative . PET imaging of tau pathology and relationship to amyloid, longitudinal MRI, and cognitive change in down syndrome: results from the down syndrome biomarker initiative (DSBI). J Alzheimers Dis. 2017;60(2):439‐450. [DOI] [PubMed] [Google Scholar]
- 67. Klunk WE, Price JC, Mathis CA, et al. Amyloid deposition begins in the striatum of presenilin‐1 mutation carriers from two unrelated pedigrees. J Neurosci. 2007;27(23):6174‐6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hartley SL, Handen BL, Devenny D, et al. Cognitive decline and brain amyloid‐β accumulation across 3 years in adults with Down syndrome. Neurobiol Aging. 2017;58:68‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Schupf N, Patel B, Silverman W, et al. Elevated plasma amyloid beta‐peptide 1‐42 and onset of dementia in adults with Down syndrome. Neurosci Lett. 2001;301(3):199‐203. [DOI] [PubMed] [Google Scholar]
- 70. Mehta PD, Mehta SP, Fedor B, Patrick BA, Emmerling M, Dalton AJ. Plasma amyloid beta protein 1‐42 levels are increased in old Down syndrome but not in young Down syndrome. Neurosci Lett. 2003;342(3):155‐158. [DOI] [PubMed] [Google Scholar]
- 71. Tapiola T, Soininen H, Pirttila T. CSF tau and Abeta42 levels in patients with Down's syndrome. Neurology. 2001;56(7):979‐980. [DOI] [PubMed] [Google Scholar]
- 72. Coppus AM, Schuur M, Vergeer J, et al. Plasma beta amyloid and the risk of Alzheimer's disease in Down syndrome. Neurobiol Aging. 2012;33(9):1988‐1994. [DOI] [PubMed] [Google Scholar]
- 73. Schupf N, Zigman WB, Tang MX, et al. Change in plasma Abeta peptides and onset of dementia in adults with Down syndrome. Neurology. 2010;75(18):1639‐1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jones EL, Hanney M, Francis PT, Ballard CG. Amyloid beta concentrations in older people with Down syndrome and dementia. Neurosci Lett. 2009;451(2):162‐164. [DOI] [PubMed] [Google Scholar]
- 75. Head E, Doran E, Nistor M, et al. Plasma amyloid‐beta as a function of age, level of intellectual disability, and presence of dementia in Down syndrome. J Alzheimer's Dis. 2011;23(3):399‐409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Matsuoka Y, Andrews HF, Becker AG, et al. The relationship of plasma Abeta levels to dementia in aging individuals with Down syndrome. Alzheimer Dis Assoc Disord. 2009;23(4):315‐318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lee NC, Yang SY, Chieh JJ, et al. Blood beta‐amyloid and tau in Down syndrome: a comparison with Alzheimer's disease. Front Aging Neurosci. 2016;8:316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Alhajraf F, Ness D, Hye A, Strydom A. Plasma amyloid and tau as dementia biomarkers in Down syndrome: systematic review and meta‐analyses. Dev Neurobiol. 2019;79(7):684‐698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tamaoka A, Sekijima Y, Matsuno S, Tokuda T, Shoji S, Ikeda SI. Amyloid beta protein species in cerebrospinal fluid and in brain from patients with Down's syndrome. Ann Neurol. 1999;46(6):933. [DOI] [PubMed] [Google Scholar]
- 80. Portelius E, Holtta M, Soininen H, et al. Altered cerebrospinal fluid levels of amyloid beta and amyloid precursor‐like protein 1 peptides in Down's syndrome. Neuromol Med. 2014;16(2):510‐516. [DOI] [PubMed] [Google Scholar]
- 81. Fortea J, Carmona‐Iragui M, Benejam B, et al. Plasma and CSF biomarkers for the diagnosis of Alzheimer's disease in adults with Down syndrome: a cross‐sectional study. Lancet Neurol. 2018;17(10):860‐869. [DOI] [PubMed] [Google Scholar]
- 82. Margallo‐Lana ML, Moore PB, Kay DW, et al. Fifteen‐year follow‐up of 92 hospitalized adults with Down's syndrome: incidence of cognitive decline, its relationship to age and neuropathology. J Intellect Disabil Res. 2007;51(Pt. 6):463‐477. [DOI] [PubMed] [Google Scholar]
- 83. Kasai T, Tatebe H, Kondo M, et al. Increased levels of plasma total tau in adult Down syndrome. PLoS One. 2017;12(11):e0188802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Startin CM, Ashton NJ, Hamburg S, et al. Plasma biomarkers for amyloid, tau, and cytokines in Down syndrome and sporadic Alzheimer's disease. Alzheimers Res Ther. 2019;11(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Fang WQ, Hwu WL, Chien YH, et al. Composite scores of plasma tau and β‐amyloids correlate with dementia in Down syndrome. ACS Chem Neurosci. 2020;11(2):191‐196. [DOI] [PubMed] [Google Scholar]
- 86. Head E, Powell DK, Schmitt FA. Metabolic and vascular imaging biomarkers in Down syndrome provide unique insights into brain aging and Alzheimer disease pathogenesis. Front Aging Neurosci. 2018;10:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Matthews DC, Lukic AS, Andrews RD, et al. Dissociation of Down syndrome and Alzheimer's disease effects with imaging. Alzheimers Dement (N Y). 2016;2(2):69‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schapiro MB, Ball MJ, Grady CL, Haxby JV, Kaye JA, Rapoport SI. Dementia in Down's syndrome: cerebral glucose utilization, neuropsychological assessment, and neuropathology. Neurology. 1988;38(6):938‐942. [DOI] [PubMed] [Google Scholar]
- 89. Teipel SJ, Schapiro MB, Alexander GE, et al. Relation of corpus callosum and hippocampal size to age in nondemented adults with Down's syndrome. Am J Psychiatry. 2003;160(10):1870‐1878. [DOI] [PubMed] [Google Scholar]
- 90. Aylward EH, Li Q, Honeycutt NA, et al. MRI volumes of the hippocampus and amygdala in adults with Down's syndrome with and without dementia. Am J Psychiatry. 1999;156(4):564‐568. [DOI] [PubMed] [Google Scholar]
- 91. Krasuski JS, Alexander GE, Horwitz B, Rapoport SI, Schapiro MB. Relation of medial temporal lobe volumes to age and memory function in nondemented adults with Down's syndrome: implications for the prodromal phase of Alzheimer's disease. Am J Psychiatry. 2002;159(1):74‐81. [DOI] [PubMed] [Google Scholar]
- 92. Strydom A, Heslegrave A, Startin CM, et al. Neurofilament light as a blood biomarker for neurodegeneration in Down syndrome. Alzheimers Res Ther. 2018;10(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Rafii MS, Donohue MC, Matthews DC, et al. Plasma neurofilament light and Alzheimer's disease biomarkers in down syndrome: results from the Down syndrome biomarker initiative (DSBI). J Alzheimers Dis. 2019;70(1):131‐138. [DOI] [PubMed] [Google Scholar]