

Abstract
Background and Purpose
Arctigenin, a major bioactive component of Fructus arctii, has been reported to have antidepressant‐like effects. However, the mechanisms underlying these effects are still unclear. Neuroinflammation can be caused by excessive production of proinflammatory cytokines in microglia via high‐mobility group box 1 (HMGB1)/TLR4/NF‐κB and TNF‐α/TNFR1/NF‐κB signalling pathways, leading to depression. In this study, we have investigated the antidepressant mechanism of arctigenin by conducting in vitro and in vivo studies.
Experimental Approach
The effects of chronic unpredictable mild stress (CUMS) on wild‐type (WT) and TLR4−/− mice were examined. Antidepressant‐like effects of arctigenin were tested using the CUMS‐induced model of depression in WT mice. The effects of arctigenin were assessed on the HMGB1/TLR4/NF‐κB and TNF‐α/TNFR1/NF‐κB signalling pathways in the prefrontal cortex (PFC) of mouse brain and HMGB1‐ or TNF‐α‐stimulated primary cultured microglia. The interaction between HMGB1 and TLR4 or TNF‐α and TNFR1 with or without arctigenin was examined by localized surface plasmon resonance (LSPR) and co‐immunoprecipitation assays.
Key Results
The immobility times in the tail suspension test (TST) and forced swimming test (FST) were reduced in TLR4−/− mice, compared with WT mice. Arctigenin exhibited antidepressant‐like effects. Arctigenin also inhibited microglia activation and inflammatory responses in the PFC of mouse brain. Arctigenin inhibited HMGB1 and TLR4 or TNF‐α and TNFR1 interactions, and suppressed both HMGB1/TLR4/NF‐κB and TNF‐α/TNFR1/NF‐κB signalling pathways.
Conclusions and Implications
Arctigenin has antidepressant‐like effects by attenuating excessive microglial activation and neuroinflammation through the HMGB1/TLR4/NF‐κB and TNF‐α/TNFR1/NF‐κB signalling pathways. This suggests that arctigenin has potential as a new drug candidate suitable for clinical trials to treat depression.
Keywords: antidepressant, arctigenin, HMGB1, microglia, neuroinflammation, TNF‐α
Abbreviations
- CMC‐Na
carboxymethyl cellulose sodium
- Co‐IP
co‐immunoprecipitation
- CUMS
chronic unpredictable mild stress
- FST
forced swimming test
- HMGB1
high‐mobility group box 1
- Iba‐1
ionized calcium binding adaptor molecule 1
- IDO
indoleamine 2,3‐dioxygenase
- LSPR
localized surface plasmon resonance
- MDD
major depressive disorder
- MTS
3‐(4, 5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium
- MyD88
myeloid differentiation primary response gene 88
- OFT
open field test
- PFC
prefrontal cortex
- RAGE
receptors advanced glycation end products
- RIP
receptor interacting protein
- SPF
specific pathogen free
- SPT
sucrose preference test
- SSRIs
selective serotonin reuptake inhibitors
- TLR
Toll‐like receptor
- TNFR
TNF receptor
- TRAF2
TNFR‐associated factor 2
- TST
tail suspension test
What is already known
Inflammation is potentially important in the pathophysiology of depression.
Arctigenin has anti‐inflammatory, neuroprotective, antidepressant‐ and anxiolytic‐like effects.
What this study adds
Arctigenin exerts antidepressant‐like effects by attenuating microglial activation and neuroinflammation.
These effects of arctigenin result from inhibiting the HMGB1/TLR4/NF‐κB and TNF‐α/TNFR1/NF‐κB signalling pathways.
What is the clinical significance
Arctigenin may be considered as a promising new drug to treat depression.
1. INTRODUCTION
Depression is a complex disease characterized by a feeling of sadness, anhedonia, avolition, worthlessness and hopelessness (Ribeiro, Ribeiro, & von Doellinger, 2018). In the United States, the lifetime prevalence of major depressive disorder (MDD) was 16.2% and for 12 months, was 6.6% (Kessler et al., 2003) and was 3.3% and 2.3%, respectively, in China (Gu et al., 2013). Depression is of huge social and economic importance worldwide and many researchers are trying to elucidate the mechanisms for treating MDD.
Microglia are dispersed throughout the CNS and play a major role in the immune defence system of the CNS (Habib & Beyer, 2015). However, chronic unpredictable mild stress (CUMS) induces microglia activation (Duan et al., 2020) and promotes the production of inducible NOS (iNOS) and proinflammatory cytokines (TNF‐α IL‐1β and IL‐6) in the hippocampal tissues of mice (Lou, Wang, & Wang, 2019). Proinflammatory cytokines also increase the activity of the enzyme indoleamine 2,3‐dioxygenase (IDO) resulting in depletion of 5‐HT levels (Grohmann, Fallarino, & Puccetti, 2003) that are associated with depressed mood, anhedonia, sleep disturbance, anorexia, decreased libido and fatigue (Leonard, 2014).
As a damage‐associated molecular pattern, the protein known as extracellular high‐mobility group box 1 (HMGB1) binds to a variety of cell surface receptors, including Toll‐like receptor 4 (TLR4) TLR2 and the receptor for advanced glycation end products (RAGE) (Kang et al., 2014). TLR4, a pattern recognition receptor, is expressed in a range of cells, such as microglia, astrocytes and neurons, and plays an important role in regulating the inflammatory response to stress (Guo et al., 2019). HMGB1 and TLR4 form a ligand–receptor pair that is involved in diseases characterized by cell death and injury (Guo et al., 2019), including depression (Guo et al., 2019; Xu et al., 2020) and liver injury (Lu et al., 2020). In addition, TNF‐α, as a key inflammatory factor, can disrupt the integrity of the blood–brain barrier (Cheng et al., 2018) and induce depression‐like behaviour in mice (Qian, Wang, Tao, Long, & Wang, 2017). Meanwhile, as the main receptor of TNF‐α, TNF receptor 1 (TNFR1) is also involved in the pathophysiology of depression (Dellarole et al., 2014). Su et al. (2017) reported that the NF‐κB was activated in the hippocampi of wild‐type (WT) mice after CUMS exposure by regulating the expression of cytokines. Depression‐like behaviours caused by stress are known to be dependent on HMGB1/TLR4/NF‐κB (Liu et al., 2019) and TNF‐α/TNFR1/NF‐κB signalling pathways in CUMS‐exposed mice (Lu et al., 2019). Therefore, the inhibition of the inflammatory reactions activated by the HMGB1/TLR4/NF‐κB and TNF‐α/TNFR1/NF‐κB signalling pathways might provide effective targets for pharmacological intervention treating depression‐like behaviours.
Several antidepressant drugs are already available to treat patients with depression, including selective serotonin reuptake inhibitors (SSRIs), monoamine reuptake inhibitors, and MAO inhibitors (Chandrasekhar, Ramya, Navya, Phani Kumar, & Anilakumar, 2017). However, the current antidepressants have many side effects and adverse drug reactions (Givens, 2016).
Arctiin and arctigenin are the major bioactive components of Fructus arctii, the dry ripe fruit of Compositae Arctium lappa L. Arctiin can be metabolized to arctigenin by intestinal microflora after oral administration (Wang et al., 2013). We recently found that arctiin has antidepressive effects by inhibiting the activation of NF‐κB mediated by the HMGB1/TLR4‐ and TNF‐α/TNFR1‐pathways, which consequently attenuates microglial activation and neuroinflammation (Xu et al., 2020). Du et al. (2019) reported that arctigenin has antidepressant‐ and anxiolytic‐like effects. However, the precise mechanisms of arctigenin antidepressant action are still unclear. Therefore, in this study, we examined inflammatory pathways to evaluate possible mechanisms by which arctigenin induces the antidepressant effects in rodent models of depression. The mechanisms underlying the actions of arctigenin were further studied in vitro through the examination of primary microglia responses to HMGB1 or TNF‐α stimulation and the effects on the HMGB1/TLR4/NF‐κB and TNF‐α/TNFR1/NF‐κB signalling pathways in mouse brain.
2. METHODS
2.1. Animals
All animal care and experimental procedures were carried out in accordance with the guidelines of the Animal Care Committee of Yanbian University (resolution number 201501022) and were approved by the Animal Research Committee of Yanbian University, China. The animals were treated humanely, and all efforts were made to minimize the animals' suffering and the animal numbers. Animal studies are reported in compliance with the ARRIVE guidelines (Percie du Sert et al., 2020) and with the recommendations made by the British Journal of Pharmacology (Lilley et al., 2020).
Adult male wild‐type C57BL/6 (WT B6) mice (8–10 weeks old, 18–22 g, RRID:MGI:5656552) were provided by the Liaoning Changsheng Biotechnology Co. Ltd. (specific pathogen free [SPF], SCXK [LIAO] Liaoning, China, 2015‐001) and Japan SLC (SPF, Hamamatsu, Japan). TLR4‐deficient (TLR4−/−) male mice with C57BL/6 background (SPF, RRID:MGI:3588776) were generously provided by Dr Shizuo Akira (Graduate School of Medicine, Osaka University) (Aosai et al., 2002) which were used at 8 to 10 weeks of age. The mice were housed under standard conditions (12/12‐h light–dark cycle, temperature 25°C, 55% humidity and under SPF conditions). The mice were housed four or five per cage in plastic cages with soft bedding and supplied with standard food and water ad libitum. Behavioural evaluation sessions took place during the light phase (9:00 a.m. to 5:00 p.m.) in a quiet room maintained at 25°C. A total of 118 WT B6 mice and 8 TLR4−/− mice were used in these experiments.
2.2. CUMS procedure
The CUMS procedure was adopted with slight modifications (Lu et al., 2019; Willner, Towell, Sampson, Sophokleous, & Muscat, 1987). During this experiment, the mice in the normal group were left undisturbed in the home cages in a separate room with exception of general handing (e.g., regular cage cleaning) that was matched by that for the CUMS groups. Mice were subjected once a day for 6 weeks to one of the following stressors: food or water deprivation for 24 h, cage tilting (45°) for 24 h, damp bedding (200‐ml water in 100‐g sawdust bedding) for 24 h, tail clamp (1 cm from the beginning of the tail) for 1 min, swimming in cold water (4°C) for 5 min, and light/dark perversion for 24 h. To prevent habituation and provide an unpredictable feature to the stressors, all the stressors were randomly scheduled over a week and repeated throughout the 6‐week experiment.
2.3. Experimental designs
After 7 days of environmental adaptation, 50 mice were randomly divided into five groups (n = 10): a control group, not subjected to any treatment; three groups treated with arctigenin (25, 50, or 100 mg·kg−1 body weight) and a positive control group treated with sertraline (10 mg·kg−1 body weight), as a clinically used SSRI antidepressant drug. Sertraline or arctigenin was orally administered once daily, for consecutive 2 weeks for the treatment groups. The control group received the same volume of 0.5% (w/v) carboxymethyl cellulose sodium (CMC‐Na). The behavioural evaluation in the tail suspension test (TST), forced swimming test (FST), and open field test (OFT) was performed on days 12 to 14. After behavioural evaluation, mice were anaesthetized with isoflurane and killed by rapid cervical dislocation. The time‐course of this protocol is shown in Figure 1b.
FIGURE 1.
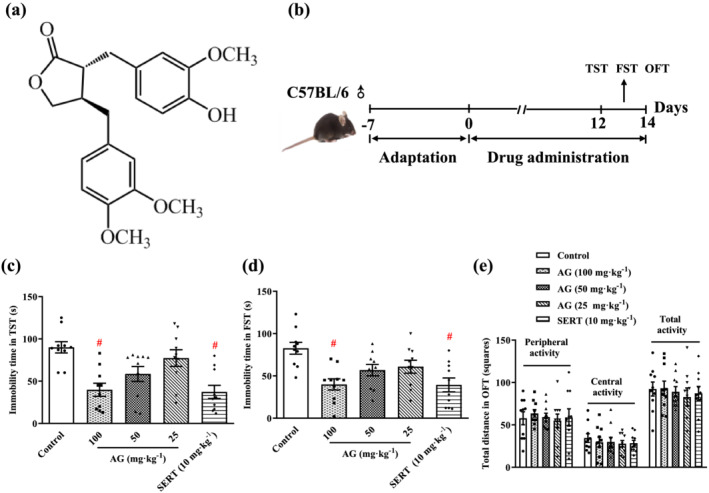
Arctigenin produces antidepressant‐like effects in the TST and FST. (a) The chemical structure of arctigenin. (b) Schematic timeline of experimental procedures. arctigenin (AG; 25, 50, or 100 mg·kg−1) or sertraline (SERT; 10 mg·kg−1) decreased the immobility time in the TST (c) and FST (d) in a dose‐dependent manner. (e) Arctigenin (25, 50, or 100 mg·kg−1) or sertraline (10 mg·kg−1) had no effect on the spontaneous locomotor activity in the OFT. The data are expressed as means ± SEM (n = 10). # P < 0.05, significantly different from control group; one‐way ANOVA followed by post hoc Tukey's test
After 7 days of adaptation to the environment, 60 mice were randomly divided into six groups (n = 10), including control (not subjected to CUMS or treatment) group, vehicle (CUMS plus vehicle) group, CUMS plus arctigenin (25, 50, or 100 mg·kg−1 body weight) groups, and CUMS plus sertraline (10 mg·kg−1 body weight) group. Drug administration and CUMS exposure were started at the initiation of the study which lasted for 6 weeks. Mice in the control and vehicle groups received the same volume of 0.5% (w/v) CMC‐Na. The behavioural evaluation in the sucrose preference test (SPT), TST, and FST was performed on days 35 to 41. On day 42, 1 h after the last drug administration, five mice from each group were anaesthetized with isoflurane, blood was collected by direct cardiac puncture, and serum were separated for elisa. Mice were then killed by rapid cervical dislocation, and the brain was removed followed by separation of prefrontal cortex (PFC). Five PFC tissues were used for western blot assay, antibody array and elisa, while the other five PFC tissues were used for immunohistochemistry or Nissl staining (Figure 2a).
FIGURE 2.
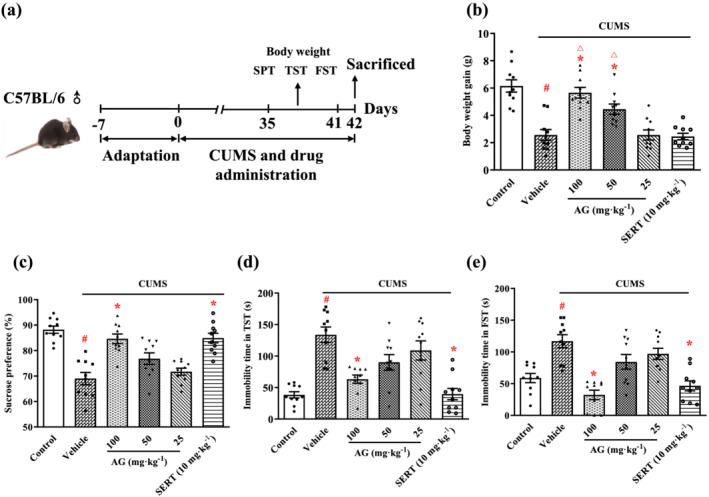
Arctigenin reverses depression‐like behaviours in the CUMS‐exposed mice. (a) Schematic timeline of the experimental procedures. (b) Antidepressant‐like effects of arctigenin (AG; 25, 50, or 100 mg·kg−1) or sertraline (SERT; 10 mg·kg−1) on body weight gain of CUMS‐exposed mice. (c) Antidepressant‐like effects of arctigenin (25, 50, or 100 mg·kg−1) or sertraline (10 mg·kg−1) on the SPT of CUMS‐exposed mice. Antidepressant‐like effects of arctigenin (25, 50, or 100 mg·kg−1) or sertraline (10 mg·kg−1) on the TST (d) and FST (e) of CUMS‐exposed mice. The data are expressed as means ± SEM (n = 10). # P < 0.05, significantly different from control group; *P < 0.05, significantly different from vehicle group; △ P < 0.05, significantly different from sertraline group; one‐way ANOVA followed by post hoc Tukey's test
2.4. Tail suspension test
The TST was performed according to the previously reported methods (Jiang et al., 2012). In brief, mice were suspended 50 cm above the floor by adhesive tape placed approximately 1 cm from the tip of the tail. All animals were suspended for 6 min: the first 2 min was an adaption phase, and the duration of immobility was recorded during the latter 4 min. Mice were considered immobile only when they hung passively and were completely motionless. Any mouse that did climb were removed from the experimental analysis. The observers were unaware of the treatment of the mice.
2.5. Forced swimming test
The FST was performed according to the previously reported methods (Jiang et al., 2017). In brief, each mouse was placed into a transparent cylinder (45 cm in height, 20 cm in diameter) filled with 15‐cm‐high water (25°C). The water was replaced after each trial. All animals were forced to swim for 6 min: the first 2 min was an adaption phase, and the duration of immobility was recorded during the later 4 min. The immobility time was measured when the mice are floating in the water without struggling and only making movements necessary to keep their heads above the water. The observers were unaware of the treatment of the mice.
2.6. Open field test
Locomotor activity was studied using OFT, and the open field apparatus was similar to those described previously (Jiang et al., 2017; Jiang et al., 2012). The mice were placed individually in the dark in an open field apparatus (100 × 100 × 40 cm) with the floor divided into 25 (5 × 5) squares. The apparatus was illuminated with a red bulb (50 W) on the ceiling. The squares each mouse crossed were counted over a 5‐min period under dim light conditions. The open field apparatus was thoroughly cleaned after each trial. The observers were unaware of the treatment of the mice.
2.7. Sucrose preference test
The SPT was performed according to the previously reported method (Gao et al., 2019). In brief, the mice in each group were given the choice to drink from two bottles in individual cages, one with 1% sucrose solution and the other with water. All of the mice were acclimatized to the two‐bottle choice condition for 2 days. To prevent potential location preference of drinking, the position of the two bottles was changed every 6 h. The mice were deprived of food and water for 24 h prior to the test. On each test, bottles were pre‐weighed and the position of the bottles was interchanged. One hour later, the amount of sucrose solution or water consumed was determined by weighing the bottles again. Sucrose preference (%) = consumption × 100/(sucrose consumption + water consumption). The observers were unaware of the treatment of the mice.
2.8. Immunohistochemical staining and Nissl staining
The mice were deeply anaesthetized with pentobarbital sodium and perfused transcardially with 4% paraformaldehyde in 0.01‐M PBS 1 h after the stressor and drug exposure. The brain was removed and fixed for 24 h and then dehydrated with 30% sucrose solution. After that, coronal PFC sections (8 μm) were cut with a freezing microtome (CM1900; Leica Microsystems, Wetzlar, Germany) and collected serially.
For immunohistochemical staining, the selected PFC sections were fixed in methanol 20 min, after washing in PBS, blocked with 0.01‐M PBS containing 5% BSA, 0.3% Triton X‐100 at room temperature for 1 h, and then followed by incubation with primary antibody specific for rabbit anti‐Annexin V (1:200, Abcam, Cat# ab14196, RRID:AB_300979), mouse anti‐NeuN (1:400, Abcam, Cat# ab104224, RRID:AB_10711040), goat anti‐ionized calcium binding adaptor molecule 1 (Iba‐1) (1:200, Abcam, Cat# ab48004, RRID:AB_870576), rabbit anti‐HMGB1 (1:100, Abcam, Cat# ab18256, RRID:AB_444360), rabbit anti‐TLR4 (1:200, Abcam, Cat# ab13556, RRID:AB_300457), and rabbit anti‐phospho‐NF‐κB p65 (p‐NF‐κB p65) (1:200, Abcam, Cat# ab106129, RRID:AB_10902018) at 4°C overnight. After washing in 0.01‐M PBS, the sections were subsequently incubated for 1 h at room temperature with Alexa Fluor® 488 goat anti‐mouse IgG (H + L) (1:500, Abcam, Cat# ab150113) to localize NeuN, Alexa Fluor® 488 donkey anti‐goat IgG (H + L) (1:500, Abcam, Cat# ab150129, RRID:AB_2576208) to localize Iba‐1, and Alexa Fluor® 594 donkey anti‐rabbit IgG (H + L) (1:10, Thermo Fisher Scientific, Cat# R37119, RRID:AB_2556547) to localize TLR4 (p‐NF‐κB p65, HMGB1, Annexin V). Finally, the sections were counterstained with DAPI and were analysed with Olympus IX83 microscope (Olympus, Tokyo, Japan). The number of Annexin V‐positive neurons was counted using ImageJ software (V1.8.0, NIH, Bethesda, MD, USA, RRID:SCR_003070). The Immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology (Alexander et al., 2018).
For Nissl staining, the selected PFC sections were stained with Nissl staining solution at 50°C and then dehydrated through a graded alcohol series (70%, 80%, 90%, and 100%). Then the sections were washed with xylene and covered with a coverslip. Images were examined with an Olympus IX83 microscope (Olympus). The number of Nissl‐stained neurons was counted using ImageJ software (V1.8.0, NIH, RRID:SCR_003070).
2.9. Antibody array
Quantibody® Mouse Interleukin Array 1 was performed according to the manufacturer's protocol (RayBiotech, Cat# QAM‐INT‐1‐4, RRID:AB_1228369) on the PFC tissue lysate from the individual animals.
2.10. ELISA
The concentrations of IL‐1β, TNF‐α, 5‐HT and dopamine in the serum, 5‐HT, dopamine, and IDO in the PFC of the mice, and the production of HMGB1 in the culture supernatants of primary cultured microglia were determined using elisa Kits (MLBIO, Shanghai, China), according to the manufacturer's instructions.
2.11. NO assay
The nitrite accumulation in serum was assessed by the Griess reaction. Briefly, an aliquot of the serum (50 μl) was mixed with 150 μl of Griess reagent (1% sulfanilamide in water and 0.1% N‐1‐naphthylethylenediamine dihydrochloride in 5% phosphoric acid) and incubated at room temperature for 20 min. The absorbance at 540 nm was measured by Multiskan GO (Thermo Scientific, Vantaa, Finland).
2.12. Primary microglia cultures
To explore the precise mechanism of the effects shown in the in vivo experiment and to simulate the natural environment of depression in the brain, we used primary cultures of microglia from the PFC of mice for the in vitro experiments. Quantitative analysis of double labelling with rabbit anti‐Iba‐1 (1:100, Abcam, Cat# ab178847, RRID:AB_2832244) and DAPI nuclear dye showed that the purity of primary cultured microglia was ≥98% (Figure 7a). Primary microglia were prepared as described previously (Dou et al., 2018). Briefly, 1‐day old C57BL/6 mouse pups of both sexes were rapidly decapitated, minimizing suffering, discomfort, or stress. After removing the meninges, the PFC was collected into HBSS in sterile containers. After the meninges were totally removed from cerebral cortex, tissues were chopped and transferred into 0.125% trypsin–EDTA for 15 min at 37°C; then trypsinization was stopped with 10% FBS. After mechanical triturating and further centrifugation, the cells were resuspended in the DMEM/F12 with 10% FBS and plated at a final density of 3.5 × 106 cells per flask in 75‐cm2 flasks containing 100 μg·ml−1 of poly‐l‐lysine solution. After 14 days, the mixed glial cultures were shaken on an orbital shaker (250 rpm for 2 h at 37°C) to dislodge microglia, and cells were cultured in fresh DMEM/F12 containing 10% FBS and 1% penicillin–streptomycin and were maintained in a 5% CO2 incubator under a humidified atmosphere of 95%.
FIGURE 7.
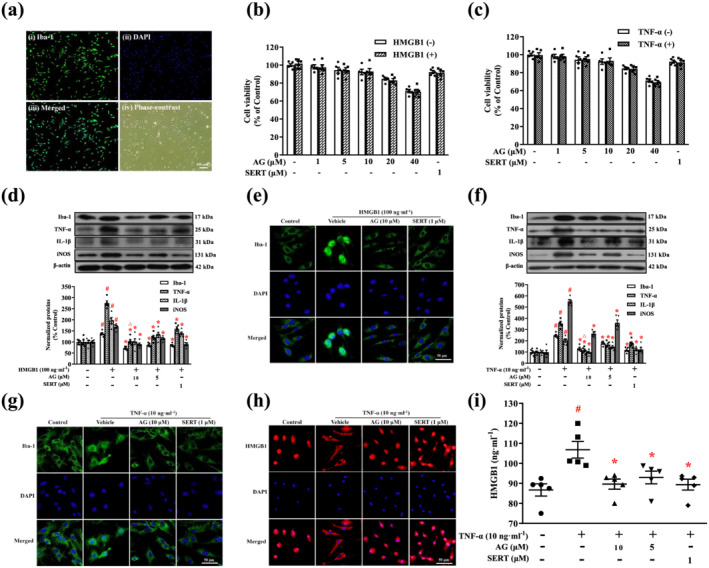
Arctigenin inhibited HMGB1‐ or TNF‐α‐stimulated microglial activation and production of inflammatory mediators in microglia. (a) Representative images showing microglial purity. Microglia were fixed and immunostained for Iba‐1 (i). Microglial nuclei were counterstained with DAPI (ii). Merged image of Iba‐1 and DAPI (iii). Phase‐contrast image of microglia (iv). (b, c) Microglia were treated with the indicated concentrations of arctigenin (AG; 0, 1, 5, 10, 20, or 40 μM) or sertraline (SERT; 1 μM) for 12 h prior to treatment with or without HMGB1 (100 ng·ml−1) or TNF‐α (10 ng·ml−1) for 24 h. Cell viability was assessed by MTS assay, and the results are expressed as the ratio, expressed as a percentage, of surviving cells to control cells. (d, f) Representative western blot of Iba‐1, TNF‐α, IL‐1β, and iNOS expression in HMGB1‐ or TNF‐α‐stimulated microglia. The protein levels were normalized to the level of β‐actin. (e, g) Representative microscopic images showed Iba‐1 (green) and DAPI (blue) in HMGB1‐ or TNF‐α‐stimulated microglia. (h) Representative microscopic images showed HMGB1 (red) and DAPI (blue) in TNF‐α‐stimulated microglia. (i) HMGB1 production by TNF‐α‐stimulated microglia was measured by elisa. The data are expressed as means ± SEM (n = 5). # P < 0.05, significantly different from control group; *P < 0.05, significantly different from vehicle group; △ P < 0.05, significantly different from sertraline group; one‐way ANOVA followed by post hoc Tukey's test
2.13. Cell viability assay
Cell viability was analysed by the 3‐(4, 5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium (MTS) reduction assay. Briefly, primary microglia were seeded in 96‐well plates (7.5 × 104 cells per well) for 24 h. On the next day, the cells were treated with different concentration (0–40 μM) of arctigenin for 12 h prior to incubation with HMGB1 (100 ng·ml−1) or TNF‐α (10 ng·ml−1) for 24 h at 37°C. Finally, 10‐μl MTS solution (CellTiter 96® AQueous One Solution Reagent, Promega, Madison, WI, USA) was added into each well, and the cells were cultured for an additional 3 h. All incubations were performed in a humidified incubator at 37°C, 5% CO2. Absorbance was measured at 490 nm using a Multiskan GO (Thermo Electron Corp., Marietta, OH, USA).
2.14. Immunofluorescence staining
Primary microglia were cultured in 24‐well plates containing 100 μg·ml−1 of poly‐l‐lysine solution and pretreated with arctigenin (10 μM) or sertraline (1 μM) for 12 h and then exposed to HMGB1 (100 ng·ml−1) or TNF‐α (10 ng·ml−1) for 30 min or 24 h. Cells were fixed with ice‐cold methanol for 20 min after removing the culture media and permeabilized with 0.2% Triton X‐100 for 15 min followed by blocking with 5% BSA for 30 min at room temperature. Cells were incubated with primary antibody specific for rabbit anti‐NF‐κB p65 (1:400, Cell Signaling Technology, Cat# 8242, RRID:AB_10859369), rabbit anti‐Iba‐1 (1:100, Abcam, Cat# ab178847, RRID:AB_2832244), rabbit anti‐HMGB1 (1:400, Abcam, Cat# ab18256, RRID:AB_444360), and rabbit anti‐TLR4 (1:200, Abcam, Cat# ab13556, RRID:AB_300457) primary antibody in PBS containing 1% BSA overnight at 4°C and then Alexa Fluor® 488 donkey anti‐rabbit IgG (1:100, Thermo Fisher Scientific, Cat# A‐21206, RRID:AB_2535792) to localize Iba‐1 or NF‐κB p65 and Alexa Fluor® 594 donkey anti‐rabbit IgG (H + L) (1:10, Thermo Fisher Scientific, Cat# R37119, RRID:AB_2556547) to localize HMGB1 or TLR4 for 30 min at room temperature. Finally, the sections were counterstained with DAPI and were analysed with Olympus IX83 microscope (Olympus).
2.15. Molecular docking
Molecular docking study was performed to investigate the binding mode between the arctigenin or sertraline and the human TLR4 or TNFR1 using AutoDock Vina 1.1.2 (AutoDock, RRID:SCR_012746) (Trott & Olson, 2010). The three‐dimensional (3D) structure of the human TLR4 (PDB ID: 3FXI) and human TNFR1 (PDB ID: 1TNR) was downloaded from Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB, RRID:SCR_012820) (http://www.rcsb.org/pdb/). The 3D structure of arctigenin was drawn by ChemBioDraw Ultra 14.0 and ChemBio3D Ultra 14.0 software. The AutoDockTools 1.5.6 package (Morris et al., 2009; Sanner, 1999) was employed to generate the docking input files. The search grid of the TLR4 was identified as center_x: 21.383, center_y: −14.226, and center_z: 16.565 with dimensions size_x: 15, size_y: 15, and size_z: 15. The search grid of the TNFR1 was identified as center_x: 32.653, center_y: 15.745, and center_z: 42.933 with dimensions size_x: 15, size_y: 18, and size_z: 15. The value of exhaustiveness was set to 20. For Vina docking, the default parameters were used if it was not mentioned. The best‐scoring pose as judged by the Vina docking score was chosen and visually analysed using PyMoL 1.7.6 software (PyMOL, RRID:SCR_000305) (http://www.pymol.org/).
2.16. Localized surface plasmon resonance assay
The protein‐small molecule and protein–protein interactions were measured using an OpenSPR localized surface plasmon resonance (LSPR) biosensor. All the steps were performed according to previously described protocol (Lu et al., 2019; Song et al., 2019). Briefly, the protein TLR4 or TNFR1 was covalently immobilized on COOH‐sensor chips (Nicoya, Canada) by the EDC/NHS chemistry. Then the protein (HMGB1 [200 μl; 0–20 nM] and TNF‐α [200 μl; 0–40 nM]) or small molecule (arctigenin, 200 μl; 0–500 μM or sertraline, 200 μl; 0–10 μM) was continuously diluted into several different concentrations using the running buffer and injected into the chip from low to high concentrations. Meanwhile, BSA was used as a negative control. In each cycle, the binding time was 240 s, the disassociation time was 180 s, and the flow rate was 20 μl·min−1. After detection, 0.25% SDS was added to dissociate the peptides from target protein. Finally, the kinetic parameters of the binding reactions were calculated and analysed by Trace Drawer software (Ridgeview Instruments AB, Sweden). TLR4 or TNFR1 was dissolved in PBS, and the concentration was diluted to 500 μg·ml−1. HMGB1 or TNF‐α was dissolved in HBS‐EP buffer (10‐mM HEPES, 150‐mM NaCl, 3‐mM EDTA, 0.005% Tween‐20), and the initial concentration was diluted to 10 μM. arctigenin and sertraline were dissolved in DMSO, and the initial concentrations were diluted to 25 and 10 mM, respectively. This fit yielded the association rate (K a), the dissociation rate (K d), and the equilibrium dissociation constant (also known as the affinity constant) (assuming the relationship K D = K d/K a).
2.17. Western blotting
Primary microglia were pretreated with arctigenin (5 or 10 μM) or sertraline (1 μM) for 12 h and then exposed to HMGB1 (100 ng·ml−1) or TNF‐α (10 ng·ml−1) for 30 min or 24 h and were collected. The total proteins were extracted from PFC (20 mg) of mice or pellets of primary microglia using RIPA containing 1% protease inhibitor (Solarbio, Beijing, China). Protein concentration was determined with the colourimetric BCA assay (BestBio, Shanghai, China). Equal quantities of protein (20 μg) were separated using SDS‐PAGE and transferred onto PVDF membranes (Merck Millipore Ltd., Tullagreen, Carrigtwohill, County Cork, Ireland). The membrane was blocked with 5% (w/v) skimmed dry milk constituted in 1× PBS Tween‐20 for 1 h at room temperature and then followed by incubation with primary antibody specific for rabbit anti‐TLR4 (1:1,000, Cell Signaling Technology, Cat# 14358, RRID:AB_27984601), rabbit anti‐MyD88 (1:1,000, Cell Signaling Technology, Cat# 4283, RRID:AB_10547882), rabbit anti‐TNFR1 (1:1,000, Cell Signaling Technology, Cat# 13377, RRID:AB_2798194), rabbit anti‐NF‐κB p65 (1:1,000, Cell Signaling Technology, Cat# 8242, RRID:AB_10859369), rabbit anti‐TNF‐α (1:1,000, Cell Signaling Technology, Cat# 3707, RRID:AB_2240625), rabbit anti‐β‐actin (1:2,000, Cell Signaling Technology, Cat# 4967, RRID:AB_330288), and rabbit anti‐α‐tubulin (1:1,000, Cell Signaling Technology, Cat# 2125, RRID:AB_2619646), rabbit anti‐p‐NF‐κB p65 (1:1,000, Cell Signaling Technology, Cat# 3033, RRID:AB_331284), rabbit anti‐Iba‐1 (1:1,000, Abcam, Cat# ab178847, RRID:AB_2832244), rabbit anti‐HMGB1 (1:1,000, Abcam, Cat# ab18256, RRID:AB_444360), rabbit anti‐IκB‐α (1:1,000, Abcam, Cat# ab49978, RRID:AB_881506), rabbit anti‐p‐IκB‐α (1:1,000, Abcam, Cat# ab133462, RRID:AB_2801653), rabbit anti‐IL‐1β (1:1,000, Abcam, Cat# ab150777, RRID:AB_2861416), and rabbit anti‐iNOS (1:1,000, Abcam, Cat# ab178945, RRID:AB_2861417), rabbit anti‐TNFR‐associated factor 2 (TRAF2) (1:1,000, Santa Cruz Biotechnology, Cat# sc‐877, RRID:AB_632534), and rabbit anti‐receptor interacting protein (RIP) (1:500, Santa Cruz Biotechnology, Cat# sc‐7881, RRID:AB_2178118) at 4°C overnight. After the membranes were washed with 1× PBS containing 0.05% Tween‐20, they were incubated with goat anti‐rabbit HRP‐conjugated secondary antibody purchased from Proteintech Group (1:2,000, Cat# SA00001‐2, RRID:AB_2722564, Chicago, IL, USA) at room temperature for 1 h. After repeating the washing step, protein bands were visualized using the ECL western blotting analysis kit (Millipore Corporation, Billerica, MA, USA). The density of the immunoreactive bands was analysed using ImageJ software (V1.8.0, NIH, RRID:SCR_003070).
2.18. Co‐immunoprecipitation assay
Primary microglia were pretreated with arctigenin (5 or 10 μM) or sertraline (1 μM) for 12 h and then exposed to HMGB1 (100 ng·ml−1) or TNF‐α (10 ng·ml−1) for 24 h; then cells were collected and lysed in lysis buffer (R0030, Solarbio) for 30 min at 4°C. After centrifugation at 14,800 rpm for 30 min at 4°C, the whole‐cell lysates (400 μg) were incubated with 1 μg of the appropriate normal mouse IgG (Santa Cruz Biotechnology, Cat# sc‐2025, RRID:AB_737182) and 20 μl of protein agarose A/G beads (Santa Cruz Biotechnology, Cat# sc‐2003, RRID:AB_10201400) at 4°C for 30 min to reduce non‐specific combination, followed by transient centrifugation, supernatant was collected and incubated with 2 μg of mouse anti‐TLR4 (Santa Cruz Biotechnology, Cat# sc‐293072, RRID:AB_10611320) or mouse anti‐TNFR1 (Santa Cruz Biotechnology, Cat# sc‐8436, RRID:AB_628377) antibody 2 h at 4°C on a rotating plate, and then 20 μl of protein agarose A/G beads (Santa Cruz Biotechnology, Cat# sc‐2003, RRID:AB_10201400) was added to each sample. Following an additional 12 h of incubation at 4°C, the precipitates were washed three times with lysis buffer and subjected to western blot analyses.
2.19. Luciferase reporter assay
Luciferase reporter assay was performed as previously described (Li et al., 2019; Xing et al., 2018). Primary microglia were cultured in 6‐cm dishes and then transfected with the vectors for NF‐κB luciferase plasmid using FuGENE® HD (E2311, Promega). Following 24‐h transfection, cells were seeded in 96‐well plates, the cells treated with arctigenin (5 or 10 μM) and sertraline (1 μM) for 12 h, and then exposed to HMGB1 (100 ng·ml−1) or TNF‐α (10 ng·ml−1) for 12 h. The luciferase assays were conducted with the Luciferase Assay System (Promega).
2.20. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). All the studies were designed to generate groups of equal sample size, using randomization and blinded analysis. Five animals were used for biochemical analysis, five animals were used for immunohistochemical assays, and 10 animals were used for behavioural experiments in each group. The sample size, the number of independent values, was determined by our earlier experiments (Xu et al., 2020). Therefore, the group size was at least n = 5 to allow the statistical analysis. However, the antibody arrays were not analysed statistically, as the group sizes were smaller (n = 3). The declared group size was the number of independent values. For western blotting, MTS assay, immunohistochemical staining, Nissl staining, and luciferase reporter assay, all values were normalized to the control group and the Y axis of the control group value was set to 100%. This normalization process was used to minimize the background variations derived from different experimental settings. All analyses were performed using GraphPad Prism 8 software (GraphPad Software, San Diego, CA, USA, RRID:SCR_002798), and data are presented as mean ± SEM. The D'Agostino and Pearson omnibus normality test was carried out to assess the normality of data in the animal behavioural tests, and the Shapiro–Wilk normality test was conducted on normalized data from the biochemical tests. The significance of differences was analysed by one‐way ANOVA followed by post hoc Tukey's multiple comparison test (more than two groups) or a Student's unpaired t test (two groups), and post hoc Tukey's multiple comparison test was run only when F was significant. Differences among groups were considered significant at values of P < 0.05. All the outliers in our study were included in the data analysis and presentation.
2.21. Materials
Arctigenin (purity ≥ 98%; MW: 372.41) was obtained from Shanghai Yuanye Bio‐Technology Co., Ltd (Shanghai, China). Sertraline (HY‐B0176A) was obtained from MedChemExpress (MCE, NJ, USA). CMC‐Na was obtained from Shanghai Sangon Biotech Co., Ltd (Shanghai, China). Nissl staining solution (1% cresyl violet) was obtained from Xi'an Hat Biotechnology Co., Ltd (Xi'an, China). HBSS and FBS were from Gibico (Grand Island, NY, USA). DMEM/F‐12 was from Biological Industries (Kibbutz Beit Haemek, Israel). DMSO was purchased from Sigma‐Aldrich (St. Louis, MO, USA). Poly‐l‐lysine solution was purchased from BBI Life Sciences Co., Ltd (Shanghai, China). Recombinant mouse HMGB1 (764006) was obtained from Biolegend Inc. (San Diego, CA, USA). Recombinant human HMGB1 (10326‐H08H), recombinant human TLR4 (10146‐H08B), and recombinant mouse TNFR1 (50496‐M02H) were obtained from Sino Biological (Beijing, China). Recombinant mouse TNF‐α (315‐01A‐20) was obtained from PeproTech Inc. (Rocky Hill, NJ, USA). The above four proteins were used in LSPR assay.
2.22. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in the IUPHAR/BPS Guide to PHARMACOLOGY (http://www.guidetopharmacology.org) and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3. RESULTS
3.1. Antidepressant‐like effects of arctigenin in the TST and FST behaviour tests
For a preliminary evaluation of the antidepressant activity of arctigenin, the effects on behaviour were tested in all the mice, without exposure to stress. After 2 weeks of oral administration of arctigenin, the immobility times of TST and FST were dose‐dependently reduced and the difference became significant at 100 mg·kg−1 arctigenin, comparing with untreated control mice. Sertraline (10 mg·kg−1) significantly shortened the immobility time of TST and FST (Figure 1c,d). Arctigenin or sertraline made no significant difference to the spontaneous peripheral, central and total locomotion activities of OFT (Figure 1e). This suggests that the effects of arctigenin on TST and FST are not related to locomotor hyperactivity. These results indicate that arctigenin has a potential antidepressant effect, which is consistent with the previous report (Du et al., 2019).
3.2. Effects of arctigenin on depression‐like behaviours in CUMS‐exposed mice
As shown in Figure 2b, arctigenin administration resulted in dose‐dependent weight gains in the CUMS mice and at 50 or 100 mg·kg−1, the CUMS mice exhibited the weight gain of control mice. It was interesting that sertraline (10 mg·kg−1) had no effect on body weight gain in CUMS mice. Figure 2c shows that the consumption of sucrose was significantly reduced in the CUMS‐exposed mice compared with the control mice. As expected, treatment with arctigenin (100 mg·kg−1) and sertraline (10 mg·kg−1) restored the preference for sucrose.
The depressive behaviours of mice were measured by recording the immobility time in the TST and FST (Figure 2d,e). The duration of immobility in the TST and FST was clearly prolonged in the CUMS‐exposed mice, compared with the control mice. In contrast, arctigenin dose‐dependently reversed the immobility duration and, at 100 mg·kg−1, arctigenin markedly shortened the immobility time of TST and FST in the CUMS‐exposed mice. The highest dose of arctigenin (100 mg·kg−1) had the optimal antidepressant‐like effect in the CUMS‐model mice. Sertraline (10 mg·kg−1) also significantly suppressed the CUMS‐induced increased immobility time in the TST and FST. These doses of arctigenin and sertraline were therefore selected to use in the subsequent experiments.
3.3. Effect of arctigenin on the activation of microglia and inflammatory mediators in the PFC and serum of CUMS‐exposed mice
The expression of Iba‐1 (a microglia‐specific marker) and HMGB1 in the CUMS‐exposed mice were greatly enhanced compared with the control mice. This enhanced expression was significantly lower after treatment with arctigenin or sertraline (Figure 3a). Double immunofluorescent staining specific for Iba‐1 and HMGB1 was assessed in the PFC of CUMS‐exposed mouse brain. As shown in Figure 3b, stress induced, a greater expression of HMGB1 in the microglia of CUMS‐exposed vehicle group than in the control group, and arctigenin or sertraline reversed these changes. Thus, the effect of arctigenin or sertraline on CUMS‐induced excessive microglia activation and high expression of HMGB1 was confirmed.
FIGURE 3.
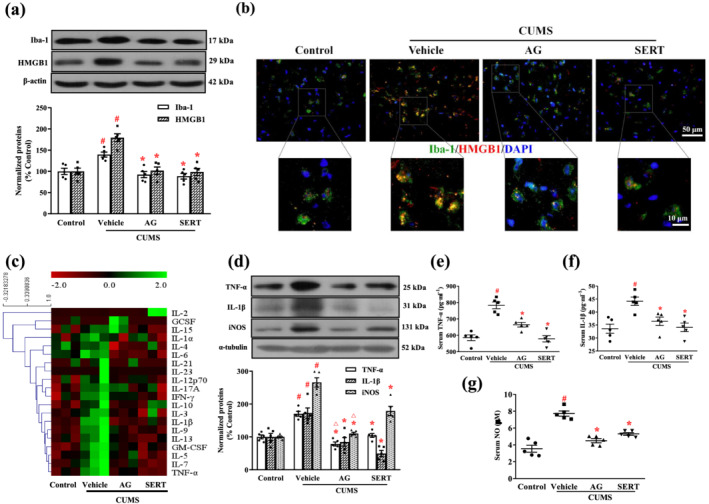
Arctigenin suppressed CUMS‐induced microglial activation and production of inflammatory mediators in the PFC or serum of mice. Effects of arctigenin (AG; 100 mg·kg−1) were compared with those of sertraline (SERT; 10 mg·kg−1) given for 6 weeks. (a) Representative western blotting results for Iba‐1 and HMGB1 in the PFC. Protein levels were normalized to the level of β‐actin (n = 5). (b) Representative microscopic images showed double immunohistochemical staining for Iba‐1 (green) and HMGB1 (red) in the PFC. The smaller areas in the square were shown below at higher magnification (n = 5). (c) Heat map of differentially expressed cytokines in the PFC of mice (n = 3). The colour intensity is proportional to the relative expression level (red: underexpressed; green: overexpressed). (d) Representative western blot results. Protein levels were normalized to the level of α‐tubulin (n = 5). (e) The levels of TNF‐α in the serum of mice (n = 5). (f) The levels of IL‐1β in the serum of mice (n = 5). (g) The levels of NO in the serum of mice (n = 5). The data are expressed as means ± SEM. # P < 0.05, significantly different from control group; *P < 0.05, significantly different from vehicle group; △ P < 0.05, significantly different from sertraline group; one‐way ANOVA followed by post hoc Tukey's test
The effects of arctigenin on neuroinflammatory reaction was first measured by analysing the cytokine expression in the PFC of mice, using an antibody array. As shown in Figure 3c, the levels of TNF‐α, GM‐CSF IL‐1β, IL‐5 IL‐6, IL‐7 IL‐9 IL‐13 and IFN‐γ were increased in the CUMS‐exposed vehicle group, compared with the control group. The CUMS‐induced increases in the levels of TNF‐α, GM‐CSF, IL‐1β, IL‐6, IL‐7, and IL‐9 were inhibited by arctigenin treatment. The CUMS‐induced increases in TNF‐α, GM‐CSF, IL‐1β, IL‐5, IL‐6, IL‐7, IL‐9, IL‐13, and IFN‐γ were also diminished by treatment with sertraline. We further examined the expression of inflammatory mediators in the PFC using western blots (Figure 3d). The expression of TNF‐α, IL‐1β, and iNOS in the CUMS‐exposed vehicle group were significantly elevated compared with the control group, and these elevated levels were significantly reduced after treatment with arctigenin or sertraline. Interestingly, arctigenin treatment was more effective than sertraline in lowering the expression of TNF‐α and iNOS. We also examined the levels of inflammatory mediators in the serum of mice by elisa (Figure 3e–g). The production of TNF‐α, IL‐1β, and NO in CUMS‐exposed mice was much greater than that of unstressed control mice. This elevation was significantly inhibited by treatment with arctigenin or sertraline.
3.4. Effect of arctigenin on neuronal damage and Annexin V expression in the PFC of CUMS‐exposed mice
In the following experiments, Nissl staining was used to observe the effect of arctigenin on the morphological changes in neurons in the PFC of CUMS‐exposed mice. Round cell bodies, visible nucleus and abundant Nissl bodies were observed in the neurons of PFC from the control group. However, in the CUMS‐exposed vehicle group, there was a loss of neurons and, in those remaining, there was Nissl body disintegration. The number of Nissl‐positive cells was significantly lower in the vehicle group than in the control group. However, the severity of neuronal damage and the decrease of Nissl‐positive cell numbers induced by CUMS were markedly ameliorated by arctigenin or sertraline (Figure 4a,b). Double immunohistochemical staining of PFC samples showed that the proportion of Annexin V‐positive neurons significantly increased in the CUMS‐exposed vehicle group, compared with the control group, whereas the CUMS‐induced abundant neuron apoptosis was apparently rescued by the administration of arctigenin or sertraline (Figure 4c,d).
FIGURE 4.
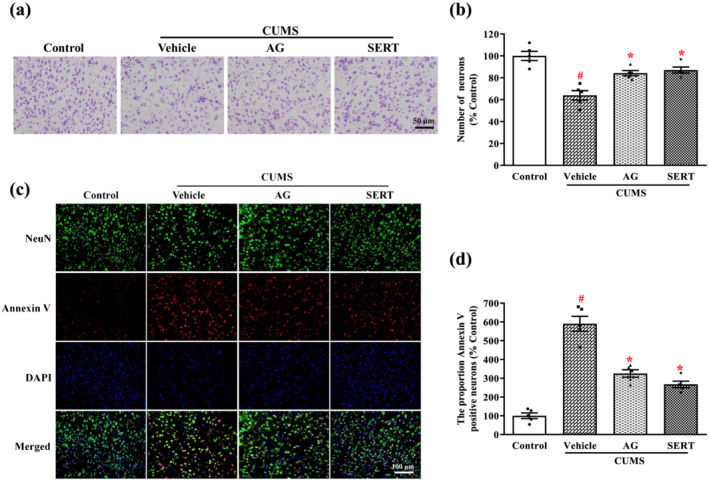
Arctigenin suppressed CUMS‐induced neuron damage in the PFC of mice. Arctigenin (AG;100 mg·kg−1) or sertraline (SERT; 10 mg·kg−1) was administered once daily for 6 weeks. (a) Nissl staining was performed on sections of the PFC. Representative photomicrographs of cresyl violet‐stained sections of the PFC were shown. (b) Density analysis revealed that the CUMS‐induced decrease in neurons in the PFC was significantly inhibited by arctigenin or sertraline. Data shown are means ± SEM (n = 5). (c) Representative microscopic images showed double immunohistochemical staining for NeuN (green) and Annexin V (red) in the PFC. (d) Density analysis revealed that the CUMS‐induced increase in the rate of neuronal apoptosis in the PFC was significantly inhibited by arctigenin or sertraline. The data are expressed as means ± SEM (n = 5). # P < 0.05, significantly different from control group; *P < 0.05, significantly different from vehicle group; one‐way ANOVA followed by post hoc Tukey's test
3.5. Effect of arctigenin on IDO, 5‐HT, and dopamine in the PFC or serum of CUMS‐exposed mice
CUMS exposure resulted in an increased level of IDO in the PFC of the vehicle group, and this increased IDO level was decreased after treatment with arctigenin OR sertraline (Figure 5a). However, the levels of 5‐HT and dopamine in the PFC or serum of CUMS‐exposed mice were significantly lower, compared with unstressed control mice. The lower serum levels of 5‐HT and dopamine in the PFC or serum of CUMS mice were reversed by treatment with arctigenin or sertraline (Figure 5b–e).
FIGURE 5.
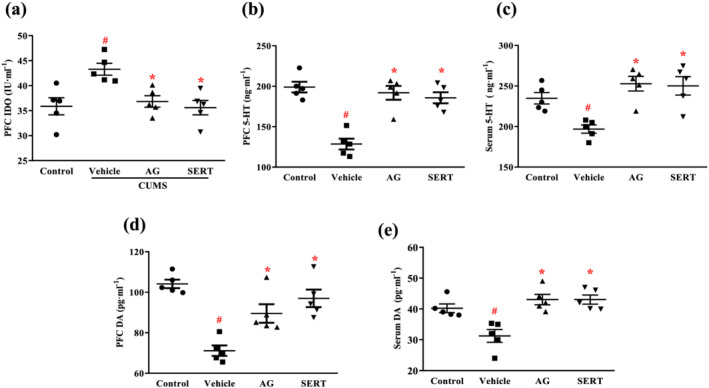
Arctigenin reversed CUMS‐induced IDO increase and monoamine decrease in the PFC or serum of mice. Arctigenin (AG;100 mg·kg−1) or sertraline (SERT; 10 mg·kg−1) was administered once daily for 6 weeks. (a) The levels of IDO in the PFC of mice. The levels of 5‐HT in the PFC (b) and serum (c) of mice. The levels of dopamine (DA) in the PFC (d) and serum (e) of mice. The data are expressed as means ± SEM (n = 5). # P < 0.05, significantly different from control group; *P < 0.05, significantly different from vehicle group; one‐way ANOVA followed by post hoc Tukey's test
3.6. Effect of arctigenin on HMGB1/TLR4/NF‐κB and TNF‐α/TNFR1/NF‐κB signalling pathways in the PFC of CUMS‐exposed mice
To examine the participation of TLR4 in the CUMS‐induced depression‐like behaviours, we next examined the effects of CUMS on TLR4−/− mice (Figure 6a). Immobility times in the TST and FST were reduced in TLR4−/− mice, compared with WT mice, suggesting that TLR4 may play a key role in the development of stress‐induced depression‐like behaviour.
FIGURE 6.
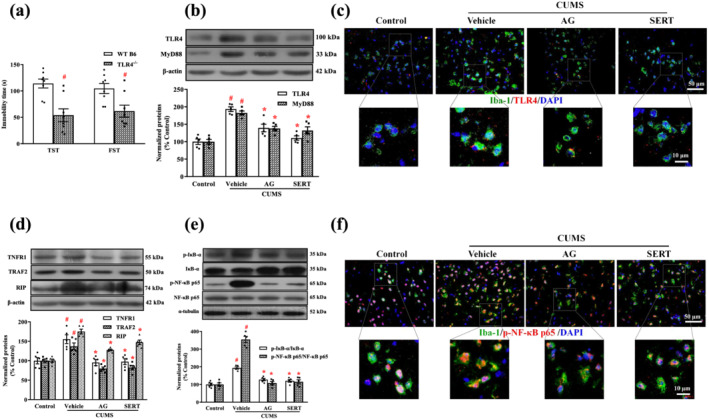
Arctigenin inhibited CUMS‐induced HMGB1/TLR4/NF‐κB or TNF‐α/TNFR1/NF‐κB signalling pathway in the PFC of mice. Arctigenin (AG;100 mg·kg−1) or sertraline (SERT; 10 mg·kg−1) was administered once daily for 6 weeks. (a) WT B6 and TLR4−/− mice were exposed to CUMS for 6 weeks, and despair behaviours in CUMS‐induced WT B6 and TLR4−/− mice were assessed with the TST and FST. The data are expressed as means ± SEM (n = 8). # P < 0.05, significantly different from WT B6 mice; Student's unpaired t test. (b) The expression of TLR4 and MyD88 was analysed by western blot. The protein levels were normalized to the level of β‐actin (n = 5). (c) Representative microscopic images showed double immunohistochemical staining for Iba‐1 (green) and TLR4 (red) in the PFC. The smaller areas in the square were shown below at higher magnification (n = 5). (d) The expression of TNFR1, TRAF2, and RIP was analysed by western blot. The protein levels were normalized to the level of β‐actin. (e) The total p‐IκB‐α and p‐NF‐κB p65 protein levels in the PFC of mice were analysed by western blot. Each immunoreactive band of the phosphorylated protein was normalized against unphosphorylated protein (n = 5). (f) Representative microscopic images showed double immunohistochemical staining for Iba‐1 (green) and p‐NF‐κB p65 (red) in the PFC. The smaller areas in the square were shown below at higher magnification (n = 5). The data are expressed as means ± SEM. # P < 0.05, significantly different from control group; *P < 0.05, significantly different from vehicle group; one‐way ANOVA followed by post hoc Tukey's test
Next, we tested the effects of arctigenin on the HMGB1/TLR4/NF‐κB signalling pathway by analysing its components using western blot and immunohistochemistry. Western blot analyses indicated that TLR4 and MyD88 expression in the PFC of CUMS‐exposed mice increased significantly more than that of unstressed control mice. Treatment with arctigenin or sertraline significantly reversed the CUMS‐induced alterations in the expression of TLR4 and MyD88, compared with the vehicle group (Figure 6b). In the corresponding double immunohistochemical staining, TLR4 molecules were highly expressed in microglia of the PFC in CUMS‐exposed mice, whereas arctigenin or sertraline inhibited their up‐regulated expression (Figure 6c).
To further investigate whether arctigenin affects the CUMS‐induced TNF‐α/TNFR1/NF‐κB signalling pathway, we examined TNFR1 and its mediated protein expression by western blot analysis (Figure 6d). In comparison with the control group, CUMS markedly increased the expression of TNFR1, TRAF2, and RIP. However, arctigenin or sertraline treatment significantly decreased their expression.
Next, we examined IκB‐α and NF‐κB p65 phosphorylation in the PFC by western blotting (Figure 6e). The expressions of p‐IκB‐α and p‐NF‐κB p65 in the CUMS‐exposed mice were significantly higher than in unstressed control mice. In contrast, treatment with either arctigenin or sertraline significantly decreased the phosphorylation of IκB‐α and NF‐κB p65.
We used double immunohistochemical staining to assess the p‐NF‐κB p65 expression in microglia of the PFC. Figure 6f shows that the expression of p‐NF‐κB p65 in microglia of the PFC from CUMS‐exposed mice was enhanced in contrast to the unstressed control group, whereas treatment with arctigenin or sertraline inhibited the expression of p‐NF‐κB p65 in microglia of the PFC, compared with the vehicle group.
3.7. Effect of arctigenin on microglial activation and expression of inflammatory mediators in HMGB1‐ or TNF‐α‐stimulated microglia
The primary cultures of microglia were incubated with or without HMGB1 (100 ng·ml−1) or TNF‐α (10 ng·ml−1) in the presence of various concentrations of arctigenin (0–40 μM) and sertraline (1 μM) for 24 h, before microglia viability was evaluated by MTS assay. The result showed that arctigenin (0–10 μM) and sertraline (1 μM) did not exert significant cytotoxicity towards microglia, with or without HMGB1 or TNF‐α (Figure 7b,c). Therefore, the concentrations of arctigenin (5 or 10 μM) and sertraline (1 μM) that resulted in cell viability of more than 90% were chosen for the following experiments.
We analysed the expression of Iba‐1 and inflammatory mediators in microglia after activation with HMGB1 or TNF‐α. As shown in Figure 7d,f, the protein levels of Iba‐1, TNF‐α, IL‐1β, and iNOS increased in the HMGB1‐ or TNF‐α‐stimulated microglia. Pretreatment with arctigenin (5 or 10 μM) or sertraline (1 μM) significantly decreased these levels of inflammatory proteins in the HMGB1‐ or TNF‐α‐stimulated microglia, in a dose‐dependent manner. Interestingly, the inhibition of the expression of TNF‐α in HMGB1‐ or TNF‐α‐stimulated microglia by arctigenin (10 μM) was greater than that by sertraline (1 μM).
We used immunofluorescence staining to assess the effects of arctigenin (10 μM) or sertraline (1 μM) on the cellular morphology of HMGB1‐ or TNF‐α‐stimulated microglia (Figure 7e,g). Microglia, after challenge by HMGB1 or TNF‐α, transformed from a resting state to an active state, presenting the shape of phagocytic amoeba, with a larger cell body and shorter protuberant. Arctigenin or sertraline pretreatment normalized the morphology of microglia.
In addition, immunofluorescence staining was used to observe changes in the location of HMGB1 in the microglia (Figure 7h). HMGB1 is found mainly in the nucleus of normal microglia. TNF‐α stimulation induces translocation of HMGB1 from the nucleus to the cytoplasm and extracellular matrix. In contrast, treatment with arctigenin (10 μM) or sertraline (1 μM) resulted in the retention of HMGB1 in the nucleus. Furthermore, elisa showed that the production of HMGB1 was significantly elevated in the TNF‐α‐stimulated microglia. However, pretreatment with arctigenin (5 or 10 μM) or sertraline (1 μM) restricted TNF‐α‐stimulated HMGB1 production in a dose‐dependent manner (Figure 7i).
3.8. Effect of arctigenin on the interaction between HMGB1 and TLR4 or TNF‐α and TNFR1
To analyse the interaction between arctigenin or sertraline and TLR4, we first investigated their binding mode through a molecular docking study. Arctigenin or sertraline adopted a compact conformation to bind inside the pocket of the TLR4 (Figures 8a(i) and S1B(i)). One of the aryl groups of arctigenin or sertraline was located at the hydrophobic pocket, surrounded by the residues Tyr292, Leu293, and Tyr296, forming a stable hydrophobic binding (Figures 8a(ii) and S1B(ii)). Detailed analysis showed that the phenyl groups of arctigenin formed cation–π interactions with residues Arg264 and Lys362. Importantly, one key hydrogen bond was observed between arctigenin and Arg264 (bond length: 2.3 Å), which was the main interaction between the arctigenin and the TLR4 (Figure 8a(ii)). Meanwhile, detailed analysis showed that the phenyl groups of sertraline formed cation–π interactions with residues Arg264, Lys341 and Lys362 (Figure S1B(ii)). All these interactions enable arctigenin or sertraline to anchor in the binding site of the TLR4.
FIGURE 8.
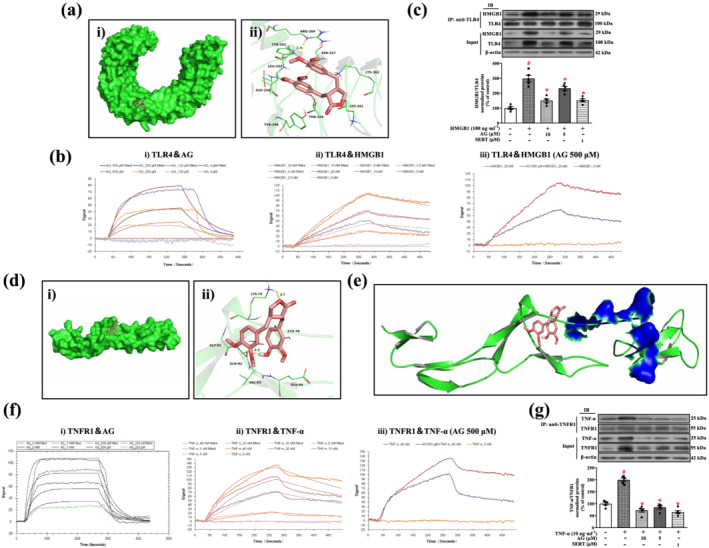
Arctigenin disrupts the interaction between HMGB1 and TLR4 or TNF‐α and TNFR1. (a) Arctigenin docked into the binding pocket of TLR4, the total view of arctigenin (i), the detailed view of arctigenin (ii). (b) Binding affinities of arctigenin for HMGB1 and TLR4. Arctigenin (AG) directly interacts with TLR4 in a dose‐dependent manner by LSPR (i), HMGB1 directly interacts with TLR4 in a dose‐dependent manner by LSPR (ii), and arctigenin blocked the binding capacity between HMGB1 and TLR4 by LSPR (iii). (c) Primary microglia were pretreated with arctigenin (AG; 5 or 10 μM) and sertraline (SERT; 1 μM) for 12 h, followed by HMGB1 (100 ng·ml−1) stimulation for 24 h; then cell lysates were immunoprecipitated with anti‐TLR4 antibody. HMGB1 and TLR4 were detected on the western blot with anti‐HMGB1 and anti‐TLR4 antibodies after immunoprecipitation with anti‐TLR4 antibody. Western blot analysis with anti‐HMGB1 and anti‐TLR4 antibodies was performed. IP, immunoprecipitation; IB, immunoblot (n = 5). (d) Arctigenin docked into the binding pocket of the TNFR1, the total view of arctigenin (i), the detailed view of arctigenin (ii). (e) Total view of the binding mode between arctigenin and TNFR1. TNFR1 is shown as green cartoon mode. Arctigenin is shown as salmon sticks; the TNF‐α binding sites are shown in blue surface mode. (f) Binding affinities of arctigenin for TNF‐α and TNFR1. Arctigenin directly interacts with TNFR1 in a dose‐dependent manner by LSPR (i), TNF‐α directly interacts with TNFR1 in a dose‐dependent manner by LSPR (ii), and arctigenin blocked the binding capacity between TNF‐α and TNFR1, as assayed by LSPR (iii). (g) Primary microglia were pretreated with arctigenin (AG; 5 or 10 μM) and sertraline (SERT; 1 μM) for 12 h, followed by TNF‐α (10 ng·ml−1) stimulation for 24 h; then cell lysates were immunoprecipitated with anti‐TNFR1 antibody. TNF‐α and TNFR1 were detected by western blot with anti‐TNF‐α and anti‐TNFR1 antibodies after immunoprecipitation with anti‐TNFR1 antibody. Western blot analysis with anti‐TNF‐α and anti‐TNFR1 antibodies was performed. IP, immunoprecipitation; IB, immunoblot (n = 5). The data are expressed as means ± SEM. # P < 0.05, significantly different from control group; *P < 0.05, significantly different from vehicle group; △ P < 0.05, significantly different from sertraline group; one‐way ANOVA followed by post hoc Tukey's test
In the following experiment, we performed LSPR assays to explore whether arctigenin or sertraline directly interact with TLR4 and affect the interaction between TLR4 and its ligand HMGB1. The K D values for the binding of arctigenin or sertraline to TLR4 were 786 and 7.68 μM, respectively (Figures 8b(i) and S1C(i)), indicating that arctigenin or sertraline binds directly to TLR4. The K D value for the binding of HMGB1 to TLR4 was 8.65 nM (Figure 8b(ii)), indicating a strong affinity between them. Notably, arctigenin or sertraline could interfere with the binding between HMGB1 and TLR4 (Figures 8b(iii) and S1C(ii)). To further confirm the effect of arctigenin or sertraline on the interaction between HMGB1 and TLR4 in HMGB1‐stimulated microglia, we performed co‐immunoprecipitation (Co‐IP) assay and western blotting (Figure 8c). After treating with arctigenin (5 or 10 μM) or sertraline (1 μM), TLR4 protein in microglia was immunoprecipitated with specific antibodies against TLR4 and analysed by western blot using antibodies against HMGB1. As expected, the expression of HMGB1 in the vehicle group was significantly increased, compared with the control group. The elevated expression of HMGB1 after treatment with arctigenin (5 or 10 μM) or sertraline (1 μM) decreased in a dose‐dependent manner. Taken together, these results showed that arctigenin or sertraline might effectively inhibit the binding of HMGB1 to TLR4, thereby inhibiting the activation of HMGB1/TLR4 signalling cascades.
Our previous studies demonstrated that TNFR1 also plays a key role in the development of stress‐induced depression‐like behaviours (Lu et al., 2019). Therefore, we tested if arctigenin or sertraline could also dock into the binding site of the TNFR1. Arctigenin or sertraline adopted a compact conformation to bind inside the pocket of the TNFR1 (Figures 8d(i) and S1B(iii)). The compound arctigenin formed van der Waals interactions with residues Cys76, Lys78, Gly81, Gln82, Val83 and Glu84 (Figure 8d(ii)). Detailed analysis showed that anion–π interaction was observed between arctigenin and the residue Glu84. Importantly, two hydrogen bonds were observed between the arctigenin and residues Lys78 and Gln82, with bond lengths of 2.7 and 2.3 Å, respectively. Meanwhile, sertraline formed van der Waals interactions with the residues Lys75, Cys76, Lys78, Gln82, Val83, Glu84 and Arg99 (Figure S1B(iv)). Detailed analysis showed that sertraline formed cation–π interactions with the residues Lys75, Lys78, and Arg99. In addition, anion–π interaction was observed between sertraline and the residue Glu84. All these interactions enable arctigenin or sertraline to anchor in the binding site of TNFR1.
Arctigenin could inhibit the binding of TNF‐α by direct competition at the binding site for TNF‐α (orthosteric) or by binding to an allosteric site, potentially changing conformation of TNFR1. We compared the binding site on TNFR1 for TNF‐α with the arctigenin or sertraline binding site. As shown in Figures 8e and S1B(v), the TNF‐α binding residues are Glu56, His69, and Ser72 of TNFR1 (Mukai et al., 2009), and none of these residues is involved in the binding of arctigenin or sertraline. It seems that arctigenin or sertraline do not directly compete with TNF‐α for binding to TNFR1 but, rather, these compounds bind to an allosteric site in TNFR1, potentially changing its conformation and thus interfering with the binding between TNF‐α and TNFR1. Our previous studies have shown that sertraline has a good affinity for TNFR1 (Lu et al., 2019). We used LSPR assay to detect the affinity of arctigenin and TNFR1. The K D value for the binding of arctigenin to TNFR1 was 884 μM (Figure 8f(i)). It suggests that arctigenin has direct binding effect on TNFR1. We further tested the effect of arctigenin or sertraline on the binding between TNF‐α and TNFR1. The K D value for the binding of TNF‐α to TNFR1 was 11.7 nM, which indicated the potential strong affinity (Figure 8f(ii)). However, arctigenin or sertraline could interfere with their binding (Figures 8f(iii) and S1C(iii)). This action of arctigenin or sertraline was further confirmed by Co‐IP assay and western blotting (Figure 8g). TNFR1 protein in microglia was immunoprecipitated with specific antibodies against TNFR1 and analysed by western blot using antibodies against TNF‐α. Interestingly, the level of TNF‐α in TNF‐α‐stimulated microglia controls was high; treatment with arctigenin (5 or 10 μM) or sertraline (1 μM) markedly decreased the expression of TNF‐α in a dose‐dependent manner. Taken together, these results suggest that arctigenin or sertraline may effectively inhibit the binding between TNF‐α and TNFR1, thereby inhibiting the activation of the TNF‐α/TNFR1 signalling cascade.
3.9. Effect of arctigenin on HMGB1/TLR4/NF‐κB or TNF‐α/TNFR1/NF‐κB signalling pathway in HMGB1‐ or TNF‐α‐stimulated microglia
In the following experiments, we analysed the expression of TLR4 and MyD88 in microglia after activation with HMGB1. As shown in Figure 9a, HMGB1 significantly induced the TLR4 and MyD88 expression in microglia, and their expressions were significantly decreased by arctigenin (5 or 10 μM) or sertraline (1 μM). Arctigenin had a greater effect than sertraline on TLR4 protein expression in HMGB1‐stimulated microglia. Similar effects of arctigenin and sertraline on TLR4 expression in HMGB1‐stimulated microglia were confirmed by immunofluorescence staining (Figure 9b).
FIGURE 9.
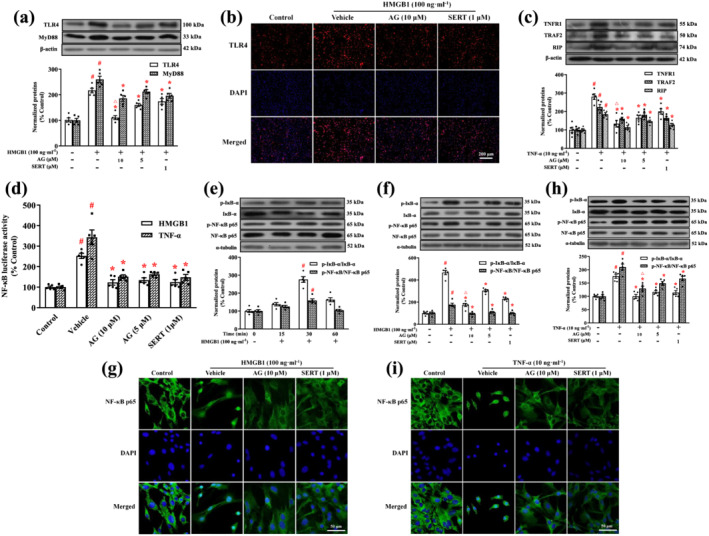
Arctigenin inhibited HMGB1/TLR4/NF‐κB or TNF‐α/TNFR1/NF‐κB signalling pathway in HMGB1‐ or TNF‐α‐stimulated microglia. (a) Expression of TLR4 and MyD88 in HMGB1‐stimulated microglia was assessed by western blot. The protein levels were normalized to the level of β‐actin. (b) Representative immunofluorescence microscopic images showed TLR4 (red) and DAPI (blue) in HMGB1‐stimulated microglia. (c) Expression of TNFR1, TRAF2, and RIP in TNF‐α‐stimulated microglia was assessed by western blot. The protein levels were normalized to the level of β‐actin. (d) Primary microglia were transfected with an NF‐κB luciferase reporter plasmid. After 24 h of transfection, the cells were pretreated with arctigenin (AG; 10 μM) and sertraline (SERT; 1 μM) for 12 h, followed by HMGB1 (100 ng·ml−1) or TNF‐α (10 ng·ml−1) stimulation for 12 h. Luciferase activity was determined by using the Luciferase Assay System. (e) The total p‐IκB‐α and p‐NF‐κB p65 protein levels in HMGB1‐stimulated microglia were assessed for the indicated time by western blot. Each immunoreactive band of the phosphorylated protein was normalized against unphosphorylated protein. (f, h) The total p‐IκB‐α and p‐NF‐κB p65 protein levels in HMGB1‐ or TNF‐α‐stimulated microglia were assessed by western blot. Each immunoreactive band of the phosphorylated protein was normalized against unphosphorylated protein. (g, i) Representative immunofluorescence microscopic images showed NF‐κB p65 (green) and DAPI (blue) in HMGB1‐ or TNF‐α‐stimulated microglia. The data are expressed as means ± SEM (n = 5). # P < 0.05, significantly different from control group; *P < 0.05, significantly different from vehicle group; △ P < 0.05, significantly different from sertraline group; one‐way ANOVA followed by post hoc Tukey's test
Next, we assessed the effects of arctigenin or sertraline on TNFR1, TRAF2, and RIP protein expression in TNF‐α‐stimulated microglia. Expression of TRAF2 and RIP was strongly enhanced in TNF‐α‐stimulated microglia. Treatment with arctigenin (5 or 10 μM) or sertraline (1 μM) also reversed these alterations in the TNF‐α‐stimulated microglia. Strikingly, arctigenin was more effective than sertraline on TNFR1 protein expression in TNF‐α‐stimulated microglia (Figure 9c).
NF‐κB signalling pathways are activated in response to extracellular stimuli, including HMGB1 (Li, Bao, Wang, Sun, & Zhang, 2017) and TNF‐α (Lu et al., 2019), leading to inflammatory response in microglia (Li et al., 2017; Lu et al., 2019). We examined the effect of arctigenin or sertraline on the activation of NF‐κB by using an NF‐κB transcription reporter vector and luciferase reporter assay. As shown in Figure 9d, HMGB1 (100 ng·ml−1) or TNF‐α (10 ng·ml−1) significantly increased the NF‐κB‐dependent luciferase activity in microglia, and this activity was attenuated by treatment with arctigenin (5 or 10 μM) or sertraline (1 μM) in a dose‐dependent manner.
To determine whether the inhibition of HMGB1‐induced NF‐κB p65 activation was caused by the inhibition of IκB‐α phosphorylation, we stimulated the microglia with HMGB1 for different times (Figure 9e). The HMGB1‐induced phosphorylation and degradation of IκB‐α in the microglia occurred within 30 min. This was consistent with our previous study in which TNF‐α induced the phosphorylation and degradation of IκB‐α in BV2 cells, also in 30 min (Lu et al., 2019).
The effects of arctigenin and sertraline treatments on IκB‐α and NF‐κB p65 phosphorylation were evaluated in the microglia by western blot (Figure 9f,h). Stimulation of microglia with HMGB1 or TNF‐α led to increases in the expression of p‐IκB‐α and p‐NF‐κB. In contrast, pretreatment with arctigenin (5 or 10 μM) and sertraline (1 μM) significantly decreased their expression. Notably, treatment with arctigenin inhibited the expression of p‐IκB‐α in the HMGB1‐stimulated microglia and the expression of p‐NF‐κB in the TNF‐α‐stimulated microglia more effectively than sertraline. Meanwhile, the immunofluorescence staining showed that stimulation of microglia with HMGB1 or TNF‐α led to NF‐κB p65 translocation, and the application of arctigenin (10 μM) or sertraline (1 μM) inhibited the NF‐κB p65 translocation in the HMGB1‐ or TNF‐α‐stimulated microglia (Figure 9g,i).
4. DISCUSSION
This study has demonstrated that arctigenin has clear antidepressant effects in mouse models of depression and attenuated microglial activation and neuroinflammation through HMGB1/TLR4/NF‐κB and TNF‐α/TNFR1/NF‐κB signalling pathways.
Arctigenin exerted antidepressant‐ and anxiolytic‐like effects when administered i.p. (Du et al., 2019). However, patients with depression need to take their medication over a long time, and administration by injection is not conducive to improving patients' compliance. In our previous study, arctigenin improved Toxoplasma gondii infection‐induced depressive behaviour in mice, after oral administration. In microglia, arctigenin attenuated excessive activation and neuroinflammatory responses by down‐regulating TLR4/NF‐κB and TNFR1/NF‐κB signalling pathways (Cheng et al., 2020). Thus, we used oral administration of arctigenin to study its antidepressant effects and its potential, underlying mechanisms in models of depression, using CUMS‐exposed mice.
The pathogenesis of depression has been related to the neuroinflammation caused by the activation of microglia in the brain (Brites & Fernandes, 2015; Yirmiya, Rimmerman, & Reshef, 2015) and excessive neuroinflammatory reactions have been shown to lead to neuronal damage in the PFC of CUMS‐induced depression model in rats (Fan et al., 2018a, 2018b). The PFC is critical for the translation of emotional information into stress action responses and participates in the neural mechanisms underlying stress adaptation and pathology (Pan, Chen, Zhang, & Kong, 2014). In the present study, we have focused on the PFC to study the activation of microglia that contributes to overproduction of neuroinflammation and neuronal damage.
Microglia are important immune cells in the CNS and account for 10–15% of all the cells of the CNS (Xavier, Menezes, Goldman, & Nedergaard, 2014). Other studies revealed that microglia were highly activated in the PFC of mice (Geng, Liu, Yuan, Liu, & Guo, 2019) and rats (Pan et al., 2014) exposed to CUMS, by promoting high expression of inflammatory mediators. These inflammatory mediators can cause inflammation‐related neuronal degeneration that is associated with depression (Fan et al., 2018a). Our results also showed overexpression of Iba‐1 and proinflammatory cytokines (TNF‐α and IL‐1β) and enzyme (iNOS) in the PFC of CUMS‐exposed mice and HMGB1‐ or TNF‐α‐stimulated primary cultured microglia, whereas arctigenin inhibited microglia activation and inflammatory responses and reduced the production of these proinflammatory cytokines and enzyme. Arctigenin also ameliorated neuronal damage and apoptosis in the PFC of CUMS‐exposed mice. Earlier studies showed that HMGB1, a core component of the late inflammatory response, was released from macrophages after stimulation with endotoxins, TNF or IL‐1β (Wang et al., 1999). Our results indicate that arctigenin inhibits the translocation and overproduction of HMGB1 in primary cultures of microglia, upon TNF‐α stimulation. Thus, our study has confirmed that arctigenin attenuates excessive neuroinflammation and relieves neuron damage by inhibiting the activation of microglia.
Metabolic disorders of monoamine neurotransmitters such as 5‐HT and dopamine are closely related to the development of neuropsychiatric disorders including depression (Chen et al., 2019). CUMS‐exposed rats have a significant reduction in 5‐HT and dopamine production in the PFC (Shen et al., 2019). In addition, proinflammatory cytokines (IFN‐γ, TNF‐α, and IL‐1β) (Chaves Filho et al., 2018) and HMGB1 (Wang, Lian, Dong, et al., 2018; Wang, Lian, Su, et al., 2018) can increase the activity of IDO. This is a rate‐limiting enzyme that catalyses the degradation of tryptophan through the kynurenine pathway, causing an increase in tryptophan catabolism and a decrease in 5‐HT biosynthesis, and leading to depression‐like behaviour (Myint & Kim, 2014). Moreover, studies have shown that NO inhibited levels of 5‐HT and dopamine in the hippocampus of rats (Wegener, Volke, & Rosenberg, 2000), and the biosynthesis of NO is regulated by iNOS (Lee, Ryu, Ferrante, Morris, & Ratan, 2003). Thus, increasing iNOS contributes to the biosynthesis of NO, thereby inhibiting 5‐HT and dopamine levels and leading to depression‐like behaviour. In the present study, we found that arctigenin inhibited the increase of IDO and the decrease of 5‐HT and dopamine in the PFC and serum of CUMS‐exposed mice. Interestingly, the effects of arctigenin were similar to those of sertraline. These findings suggest that arctigenin can alleviate depression‐like behaviours by reducing the depletion of monoamine neurotransmitters.
TLR4 is one of the receptors for HMGB1 which is expressed on microglia and mediates neuroinflammatory diseases (Le et al., 2019). Cheng et al. (2016) reported that TLR4−/− mice were less susceptible to depression‐like behaviours induced by learned helplessness. Similar results were found in our study, where TLR4−/− mice were less susceptible to depression‐like behaviours induced by CUMS, suggesting that TLR4 plays an important role in depression. MyD88 is essential for inflammatory cytokine production in response to TLR4 agonists and can activate and regulate NF‐κB signalling (Akira & Sato, 2003). NF‐κB is a key transcription factor in the regulation of inflammatory responses (Zhang et al., 2018). NF‐κB is usually retained in the cytoplasm by interacting with its inhibitor IκB, and when activated by the stimulus, the phosphorylation and degradation of IκBα is required for NF‐κB nuclear translocation (Ma, Mi, Wang, Lee, & Jin, 2016). TLR4 (but not RAGE or TLR2) is required for HMGB1‐dependent activation of macrophage TNF release (Yang et al., 2010). HMGB1 can activate TLR4 receptors leading to NF‐κB activation to induce the cytokine release in microglia (Yang et al., 2011). Studies in the mouse neonatal hypoxic‐ischaemic brain injury model have revealed that microglia can actively release HMGB1, which in turn activates the TLR4/MyD88/NF‐κB signalling pathway in microglia to initiate neuroinflammation (Le et al., 2019). Moreover, HMGB1 is highly expressed in the cerebral cortex (Lian et al., 2017) and hippocampus (Liu et al., 2019; Wang, Lian, Dong, et al., 2018; Wang, Lian, Su, et al., 2018) of CUMS mice. Stress can induce depression‐like behaviours through the HMGB1/TLR4/NF‐κB signalling pathway in the hippocampus (Liu et al., 2019) and PFC (Xu et al., 2020).
In the present study, the molecular docking study showed that arctigenin or sertraline directly interacts with TLR4. Subsequently, we confirmed that arctigenin or sertraline can bind to TLR4 with high affinity using the LSPR assay. Meanwhile, we also found that arctigenin or sertraline effectively blocks HMGB1 binding to TLR4 in the LSPR and Co‐IP assays. In addition, we have demonstrated, using double immunohistochemical staining, that arctigenin inhibits the increased expression of HMGB1, TLR4, and p‐NF‐κB p65 in the microglia of PFC in CUMS‐exposed mice. Western blot results showed that arctigenin or sertraline inhibited the high expression of TLR4, MyD88, p‐IκB‐α, and p‐NF‐κB p65 in the PFC of CUMS‐exposed mice. The in vitro experiment used HMGB1‐induced microglia activation and confirmed that high expressions of neuroinflammatory mediators were produced through TLR4/MyD88/NF‐κB signalling pathway in the primary cultured microglia. These were successfully inhibited by arctigenin or sertraline.
Binding of the TNF‐α to TNFR1 and TNFR2 induces the recruitment of several signalling proteins to the cytoplasmic domains of the receptors such as RIP1 and TRAF2, thereby activating the NF‐κB signalling pathway (Baud & Karin, 2001). Among them, TNFR1 plays an important role in the TNF‐induced cell activation and proinflammatory effects (Probert, 2015). Furthermore, the immobility time in TST and FST was found to be significantly prolonged when TNF‐α was injected into the brain of WT mice, while the immobility time in TST and FST of TNFR1−/− mice was shorter than that of normal mice (Kaster, Gadotti, Calixto, Santos, & Rodrigues, 2012). This suggests that TNFR1 is involved in the development of depression‐like behaviour. Importantly, our previous study had demonstrated that CUMS induced depression‐like behaviours in mice by activating the microglia TNF‐α/TNFR1 signalling pathway, increasing phosphorylation and nuclear translocation of NF‐κB, and inducing inflammation. Sertraline directly binds to TNF‐α and TNFR1, effectively inhibits TNF‐α‐induced inflammation, blocks TNFR1‐triggered NF‐κB signalling activity, and reduces depression‐like behaviours caused by inflammation (Lu et al., 2019; Xu et al., 2020). In the present study, the molecular docking experiment showed that arctigenin or sertraline directly interacted with TNFR1, potentially changing its conformation and thereby interfering with the binding between TNF‐α and TNFR1. Moreover, LSPR experiments showed that arctigenin or sertraline not only had high affinity binding with TNFR1 but also effectively blocked the binding of TNF‐α to TNFR1. This result was further confirmed by Co‐IP using anti‐TNFR1 antibody experiment. In addition, western blot results showed that arctigenin or sertraline down‐regulates the expression of TNFR1 downstream proteins, such as TRAF2, RIP, p‐NF‐κB p65, and p‐IκB‐α in the PFC of CUMS‐exposed mice or in the primary cultures of microglia. Arctigenin or sertraline could block the physical interaction between TNF‐α and TNFR1, and TNFR1‐mediated signalling, thereby suppressing IκB‐α/NF‐κB activation.
5. CONCLUSION
In conclusion, our data showed that arctigenin reduced depression by attenuating the activation of microglia and neuroinflammation, through the HMGB1/TLR4/NF‐κB and the TNF‐α/TNFR1/NF‐κB signalling pathways, as illustrated in Figure 10. Our findings indicate that these two signalling pathways in the microglia are potential pharmacological targets for the analysis of depression mechanisms. Moreover, it is noticeable that the antidepressant efficiency of arctigenin resembles that of sertraline, currently the primary drug of choice for depression. However, arctigenin was superior to sertraline in the measure of weight gain and some anti‐neuroinflammatory actions in depressed mice. Thus, arctigenin is expected to become as a new drug candidate, suitable for clinical trials to treat depression.
FIGURE 10.
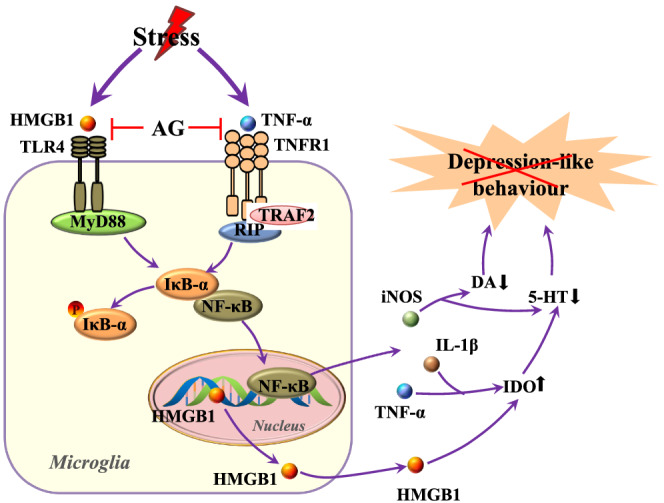
Arctigenin acts on signalling mechanisms of stress‐induced depression‐like behaviours. Stress activates TLR4 and TNFR1, possibly through HMGB1 and TNF‐α, resulting in the phosphorylation and degradation of IκB‐α, which leads to the nuclear translocation of NF‐κB and the expression of inflammatory mediators. This results in perturbed neurotransmission and neuronal damage, ultimately leading to depression‐like behaviours. Arctigenin (AG) inhibits microglial activation and protects against CUMS‐induced depression‐like behaviours by inhibiting the HMGB1/TLR4/NF‐κB pathway as well as the TNF‐α/TNFR1/NF‐κB signalling pathway. DA, dopamine
AUTHOR CONTRIBUTIONS
L.‐X.P. and H.‐N.P. designed the whole study. X.X. performed all the experiments. X.‐Y.Z. performed parts of in vivo experiments. X.X. and J.‐H.C. wrote the manuscript. F.A., H.‐N.P., and X.J. participated in the discussion and gave important suggestions. Y.‐X.C., J.‐L., H.‐R.P., and J.M. contributed reagents/materials/analysis tools. Y.‐X.C. and J.‐L. participated in part of statistical analysis. L.‐X.P. and F.A. reviewed and edited the manuscript. All authors read and approved the manuscript.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry and Animal Experimentation, and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
Supporting information
Figure S1 SERT disrupts the interaction between TLR4 and HMGB1 or TNFR1 and TNF‐α. (A) Schematic timeline of in vitro experimental procedures. (B) SERT docked into the binding pocket of the TLR4 or TNFR1, the total view of SERT in TLR4 (i), the detailed view of SERT in TLR4 (ii), the total view of SERT in TNFR1 (iii), the detailed view of SERT in TNFR1 (iv). Total view of the binding mode between the SERT and the TNFR1. The TNFR1 was represented with green cartoon mode. The SERT was represented with rose red sticks. The TNF‐α binding sites were shown in blue surface mode (v). (C) Binding affinities of SERT for HMGB1 and TLR4 or TNFR1 and TNF‐α. SERT directly interacts with TLR4 in a dose‐dependent manner by LSPR (i), SERT blocked the binding capacity between TLR4 and HMGB1 by LSPR (ii), SERT blocked the binding capacity between TNFR1 and TNF‐α by LSPR (iii)
{kind=link}
ACKNOWLEDGEMENTS
This study was supported by a grant from the National Natural Science Foundation of China (81960375, 81660344, and 81260251) and partially by Science and Technology Planning Projects from the Science and Technology Department of Jilin Province (20190201149JC).
Xu X, Piao H‐N, Aosai F, et al. Arctigenin protects against depression by inhibiting microglial activation and neuroinflammation via HMGB1/TLR4/NF‐κB and TNF‐α/TNFR1/NF‐κB pathways. Br J Pharmacol. 2020;177:5224–5245. 10.1111/bph.15261
Xiang Xu and Hu‐Nan Piao contributed equally to this study.
Contributor Information
Xuejun Jin, Email: xjjin@ybu.edu.cn.
Lian‐Xun Piao, Email: lxpiao@ybu.edu.cn.
REFERENCES
- Akira, S. , & Sato, S. (2003). Toll‐like receptors and their signaling mechanisms. Scandinavian Journal of Infectious Diseases, 35(9), 555–562. 10.1080/00365540310015683 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , … CGTP Collaborators . (2019). The Concise Guide to PHARMACOLOGY 2019/20: Introduction and other protein targets. British Journal of Pharmacology, 176(Suppl 1), S1–S20. 10.1111/bph.14747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology . British Journal of Pharmacology, 175, 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aosai, F. , Chen, M. , Kang, H. K. , Mun, H. S. , Norose, K. , Piao, L. X. , … Yano, A. (2002). Toxoplasma gondii‐derived heat shock protein HSP70 functions as a B cell mitogen. Cell Stress & Chaperones, 7(4), 357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud, V. , & Karin, M. (2001). Signal transduction by tumor necrosis factor and its relatives. Trends in Cell Biology, 11(9), 372–377. 10.1016/s0962-8924(01)02064-5 [DOI] [PubMed] [Google Scholar]
- Brites, D. , & Fernandes, A. (2015). Neuroinflammation and depression: Microglia activation, extracellular microvesicles and microRNA dysregulation. Frontiers in Cellular Neuroscience, 9, 476 10.3389/fncel.2015.00476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekhar, Y. , Ramya, E. M. , Navya, K. , Phani Kumar, G. , & Anilakumar, K. R. (2017). Antidepressant like effects of hydrolysable tannins of Terminalia catappa leaf extract via modulation of hippocampal plasticity and regulation of monoamine neurotransmitters subjected to chronic mild stress (CMS). Biomedicine & Pharmacotherapy, 86, 414–425. 10.1016/j.biopha.2016.12.031 [DOI] [PubMed] [Google Scholar]
- Chaves Filho, A. , Lima, C. , Vasconcelos, S. , de Lucena, D. F. , Maes, M. , & Macedo, D. (2018). IDO chronic immune activation and tryptophan metabolic pathway: A potential pathophysiological link between depression and obesity. Progress in Neuro‐Psychopharmacology & Biological Psychiatry, 80(Pt C), 234–249. 10.1016/j.pnpbp.2017.04.035 [DOI] [PubMed] [Google Scholar]
- Chen, B. , Li, J. , Xie, Y. , Ming, X. , Li, G. , Wang, J. , … Xiong, L. (2019). Cang‐ai volatile oil improves depressive‐like behaviors and regulates DA and 5‐HT metabolism in the brains of CUMS‐induced rats. Journal of Ethnopharmacology, 244, 112088 10.1016/j.jep.2019.112088 [DOI] [PubMed] [Google Scholar]
- Cheng, J. H. , Xu, X. , Li, Y. B. , Zhao, X. D. , Aosai, F. , Shi, S. Y. , … Piao, L. X. (2020). Arctigenin ameliorates depression‐like behaviors in Toxoplasma gondii‐infected intermediate hosts via the TLR4/NF‐κB and TNFR1/NF‐κB signaling pathways. International Immunopharmacology, 82, 106302 Advance online publication. 10.1016/j.intimp.2020.106302 [DOI] [PubMed] [Google Scholar]
- Cheng, Y. , Desse, S. , Martinez, A. , Worthen, R. J. , Jope, R. S. , & Beurel, E. (2018). TNFα disrupts blood brain barrier integrity to maintain prolonged depressive‐like behavior in mice. Brain, Behavior, and Immunity, 69, 556–567. 10.1016/j.bbi.2018.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, Y. , Pardo, M. , Armini, R. S. , Martinez, A. , Mouhsine, H. , Zagury, J. F. , … Beurel, E. (2016). Stress‐induced neuroinflammation is mediated by GSK3‐dependent TLR4 signaling that promotes susceptibility to depression‐like behavior. Brain, Behavior, and Immunity, 53, 207–222. 10.1016/j.bbi.2015.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellarole, A. , Morton, P. , Brambilla, R. , Walters, W. , Summers, S. , Bernardes, D. , … Bethea, J. R. (2014). Neuropathic pain‐induced depressive‐like behavior and hippocampal neurogenesis and plasticity are dependent on TNFR1 signaling. Brain, Behavior, and Immunity, 41, 65–81. 10.1016/j.bbi.2014.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou, F. , Chu, X. , Zhang, B. , Liang, L. , Lu, G. , Ding, J. , & Chen, S. (2018). EriB targeted inhibition of microglia activity attenuates MPP+ induced DA neuron injury through the NF‐κB signaling pathway. Molecular Brain, 11(1), 75 10.1186/s13041-018-0418-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, Y. , Li, W. , Li, Y. , Yang, J. , Wang, X. , Yin, S. , … Shi, H. (2019). Repeated arctigenin treatment produces antidepressant‐ and anxiolytic‐like effects in mice. Brain Research Bulletin, 146, 79–86. 10.1016/j.brainresbull.2018.12.005 [DOI] [PubMed] [Google Scholar]
- Duan, C. M. , Zhang, J. R. , Wan, T. F. , Wang, Y. , Chen, H. S. , & Liu, L. (2020). SRT2104 attenuates chronic unpredictable mild stress‐induced depressive‐like behaviors and imbalance between microglial M1 and M2 phenotypes in the mice. Behavioural Brain Research, 378, 112296 10.1016/j.bbr.2019.112296 [DOI] [PubMed] [Google Scholar]
- Fan, C. , Song, Q. , Wang, P. , Li, Y. , Yang, M. , Liu, B. , & Yu, S. Y. (2018a). Curcumin protects against chronic stress‐induced dysregulation of neuroplasticity and depression‐like behaviors via suppressing IL‐1β pathway in rats. Neuroscience, 392, 92–106. 10.1016/j.neuroscience.2018.09.028 [DOI] [PubMed] [Google Scholar]
- Fan, C. , Song, Q. , Wang, P. , Li, Y. , Yang, M. , & Yu, S. Y. (2018b). Neuroprotective effects of ginsenoside‐Rg1 against depression‐like behaviors via suppressing glial activation, synaptic deficits, and neuronal apoptosis in rats. Frontiers in Immunology, 9, 2889 10.3389/fimmu.2018.02889 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gao, M. , Hu, P. , Cai, Z. , Wu, Y. , Wang, D. , Hu, W. , … Huang, C. (2019). Identification of a microglial activation‐dependent antidepressant effect of amphotericin B liposome. Neuropharmacology, 151, 33–44. 10.1016/j.neuropharm.2019.04.005 [DOI] [PubMed] [Google Scholar]
- Geng, J. , Liu, J. , Yuan, X. , Liu, W. , & Guo, W. (2019). Andrographolide triggers autophagy‐mediated inflammation inhibition and attenuates chronic unpredictable mild stress (CUMS)‐induced depressive‐like behavior in mice. Toxicology and Applied Pharmacology, 379, 114688 10.1016/j.taap.2019.114688 [DOI] [PubMed] [Google Scholar]
- Givens, C. J. (2016). Adverse drug reactions associated with antipsychotics, antidepressants, mood stabilizers, and stimulants. The Nursing Clinics of North America, 51(2), 309–321. 10.1016/j.cnur.2016.01.013 [DOI] [PubMed] [Google Scholar]
- Grohmann, U. , Fallarino, F. , & Puccetti, P. (2003). Tolerance, DCs and tryptophan: Much ado about IDO. Trends in Immunology, 24(5), 242–248. 10.1016/s1471-4906(03)00072-3 [DOI] [PubMed] [Google Scholar]
- Gu, L. , Xie, J. , Long, J. , Chen, Q. , Chen, Q. , Pan, R. , … Su, L. (2013). Epidemiology of major depressive disorder in mainland china: A systematic review. PLoS ONE, 8(6), e65356 10.1371/journal.pone.0065356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, L. T. , Wang, S. Q. , Su, J. , Xu, L. X. , Ji, Z. Y. , Zhang, R. Y. , … Ma, S. P. (2019). Baicalin ameliorates neuroinflammation‐induced depressive‐like behavior through inhibition of toll‐like receptor 4 expression via the PI3K/AKT/FoxO1 pathway. Journal of Neuroinflammation, 16(1), 95 10.1186/s12974-019-1474-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, X. , Shi, Y. , Du, P. , Wang, J. , Han, Y. , Sun, B. , & Feng, J. (2019). HMGB1/TLR4 promotes apoptosis and reduces autophagy of hippocampal neurons in diabetes combined with OSA. Life Sciences, 239, 117020 10.1016/j.lfs.2019.117020 [DOI] [PubMed] [Google Scholar]
- Guo, Z. , Zhao, F. , Wang, Y. , Wang, Y. , Geng, M. , Zhang, Y. , … Xu, X. (2019). Sevoflurane exerts an anti‐depressive action by blocking the HMGB1/TLR4 pathway in unpredictable chronic mild stress rats. Journal of Molecular Neuroscience: MN, 69(4), 546–556. 10.1007/s12031-019-01380-2 [DOI] [PubMed] [Google Scholar]
- Habib, P. , & Beyer, C. (2015). Regulation of brain microglia by female gonadal steroids. The Journal of Steroid Biochemistry and Molecular Biology, 146, 3–14. 10.1016/j.jsbmb.2014.02.018 [DOI] [PubMed] [Google Scholar]
- Jiang, B. , Wang, Y. J. , Wang, H. , Song, L. , Huang, C. , Zhu, Q. , … Zhang, W. (2017). Antidepressant‐like effects of fenofibrate in mice via the hippocampal brain‐derived neurotrophic factor signalling pathway. British Journal of Pharmacology, 174(2), 177–194. 10.1111/bph.13668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, B. , Xiong, Z. , Yang, J. , Wang, W. , Wang, Y. , Hu, Z. L. , … Chen, J. G. (2012). Antidepressant‐like effects of ginsenoside Rg1 are due to activation of the BDNF signalling pathway and neurogenesis in the hippocampus. British Journal of Pharmacology, 166(6), 1872–1887. 10.1111/j.1476-5381.2012.01902.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, R. , Chen, R. , Zhang, Q. , Hou, W. , Wu, S. , Cao, L. , … Tang, D. (2014). HMGB1 in health and disease. Molecular Aspects of Medicine, 40, 1–116. 10.1016/j.mam.2014.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaster, M. P. , Gadotti, V. M. , Calixto, J. B. , Santos, A. R. , & Rodrigues, A. (2012). Depressive‐like behavior induced by tumor necrosis factor‐α in mice. Neuropharmacology, 62(1), 419–426. 10.1016/j.neuropharm.2011.08.018 [DOI] [PubMed] [Google Scholar]
- Kessler, R. C. , Berglund, P. , Demler, O. , Jin, R. , Koretz, D. , Merikangas, K. R. , … Wang, P. S. (2003). The epidemiology of major depressive disorder: Results from the National Comorbidity Survey Replication (NCS‐R). JAMA, 289(23), 3095–3105. 10.1001/jama.289.23.3095 [DOI] [PubMed] [Google Scholar]
- Le, K. , Chibaatar Daliv, E. , Wu, S. , Qian, F. , Ali, A. I. , Yu, D. , & Guo, Y. (2019). SIRT1‐regulated HMGB1 release is partially involved in TLR4 signal transduction: A possible anti‐neuroinflammatory mechanism of resveratrol in neonatal hypoxic‐ischemic brain injury. International Immunopharmacology, 75, 105779 10.1016/j.intimp.2019.105779 [DOI] [PubMed] [Google Scholar]
- Lee, J. , Ryu, H. , Ferrante, R. J. , Morris, S. M. Jr. , & Ratan, R. R. (2003). Translational control of inducible nitric oxide synthase expression by arginine can explain the arginine paradox. Proceedings of the National Academy of Sciences of the United States of America, 100(8), 4843–4848. 10.1073/pnas.0735876100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard, B. E. (2014). Impact of inflammation on neurotransmitter changes in major depression: An insight into the action of antidepressants. Progress in Neuro‐Psychopharmacology & Biological Psychiatry, 48, 261–267. 10.1016/j.pnpbp.2013.10.018 [DOI] [PubMed] [Google Scholar]
- Li, D. C. , Bao, X. Q. , Wang, X. L. , Sun, H. , & Zhang, D. (2017). A novel synthetic derivative of squamosamide FLZ inhibits the high mobility group box 1 protein‐mediated neuroinflammatory responses in murine BV2 microglial cells. Naunyn‐Schmiedeberg's Archives of Pharmacology, 390(6), 643–650. 10.1007/s00210-017-1363-6 [DOI] [PubMed] [Google Scholar]
- Li, M. Y. , Zhang, Z. H. , Wang, Z. , Zuo, H. X. , Wang, J. Y. , Xing, Y. , … Jin, X. (2019). Convallatoxin protects against dextran sulfate sodium‐induced experimental colitis in mice by inhibiting NF‐κB signaling through activation of PPARγ. Pharmacological Research, 147, 104355 10.1016/j.phrs.2019.104355 [DOI] [PubMed] [Google Scholar]
- Lian, Y. J. , Gong, H. , Wu, T. Y. , Su, W. J. , Zhang, Y. , Yang, Y. Y. , … Wang, Y. X. (2017). Ds‐HMGB1 and fr‐HMGB induce depressive behavior through neuroinflammation in contrast to nonoxid‐HMGB1. Brain, Behavior, and Immunity, 59, 322–332. 10.1016/j.bbi.2016.09.017 [DOI] [PubMed] [Google Scholar]
- Lilley, E. , Stanford, S. C. , Kendall, D. E. , Alexander, S. P. , Cirino, G. , Docherty, J. R. , … Ahluwalia, A. (2020). ARRIVE 2.0 and the British Journal of Pharmacology: Updated guidance for 2020. British Journal of Pharmacology. 10.1111/bph.15178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L. , Dong, Y. , Shan, X. , Li, L. , Xia, B. , & Wang, H. (2019). Anti‐depressive effectiveness of baicalin in vitro and in vivo . Molecules (Basel, Switzerland), 24(2), 326 10.3390/molecules24020326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou, D. , Wang, J. , & Wang, X. (2019). miR‐124 ameliorates depressive‐like behavior by targeting STAT3 to regulate microglial activation. Molecular and Cellular Probes, 48, 101470 10.1016/j.mcp.2019.101470 [DOI] [PubMed] [Google Scholar]
- Lu, Y. , Xu, X. , Jiang, T. , Jin, L. , Zhao, X. D. , Cheng, J. H. , … Piao, L. X. (2019). Sertraline ameliorates inflammation in CUMS mice and inhibits TNF‐α‐induced inflammation in microglia cells. International Immunopharmacology, 67, 119–128. 10.1016/j.intimp.2018.12.011 [DOI] [PubMed] [Google Scholar]
- Lu, Y. N. , Zhao, X. D. , Xu, X. , Piao, J. , Aosai, F. , Li, Y. B. , … Piao, L. X. (2020). Arctigenin exhibits hepatoprotective activity in Toxoplasma gondii‐infected host through HMGB1/TLR4/NF‐κB pathway. International Immunopharmacology, 84, 106539 10.1016/j.intimp.2020.106539 [DOI] [PubMed] [Google Scholar]
- Ma, J. , Mi, C. , Wang, K. S. , Lee, J. J. , & Jin, X. (2016). 4′,6‐Dihydroxy‐4‐methoxyisoaurone inhibits TNF‐α‐induced NF‐κB activation and expressions of NF‐κB‐regulated target gene products. Journal of Pharmacological Sciences, 130(2), 43–50. 10.1016/j.jphs.2015.10.002 [DOI] [PubMed] [Google Scholar]
- Morris, G. M. , Huey, R. , Lindstrom, W. , Sanner, M. F. , Belew, R. K. , Goodsell, D. S. , & Olson, A. J. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry, 30(16), 2785–2791. 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai, Y. , Shibata, H. , Nakamura, T. , Yoshioka, Y. , Abe, Y. , Nomura, T. , … Tsutsumi, Y. (2009). Structure‐function relationship of tumor necrosis factor (TNF) and its receptor interaction based on 3D structural analysis of a fully active TNFR1‐selective TNF mutant. Journal of Molecular Biology, 385(4), 1221–1229. 10.1016/j.jmb.2008.11.053 [DOI] [PubMed] [Google Scholar]
- Myint, A. M. , & Kim, Y. K. (2014). Network beyond IDO in psychiatric disorders: Revisiting neurodegeneration hypothesis. Progress in Neuro‐Psychopharmacology & Biological Psychiatry, 48, 304–313. 10.1016/j.pnpbp.2013.08.008 [DOI] [PubMed] [Google Scholar]
- Pan, Y. , Chen, X. Y. , Zhang, Q. Y. , & Kong, L. D. (2014). Microglial NLRP3 inflammasome activation mediates IL‐1β‐related inflammation in prefrontal cortex of depressive rats. Brain, Behavior, and Immunity, 41, 90–100. 10.1016/j.bbi.2014.04.007 [DOI] [PubMed] [Google Scholar]
- Percie du Sert, N. , Hurst, V. , Ahluwalia, A. , Alam, S. , Avey, M. T. , Baker, M. , … Würbel, H. (2020). The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biology, 18(7), e3000410 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probert, L. (2015). TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience, 302, 2–22. 10.1016/j.neuroscience.2015.06.038 [DOI] [PubMed] [Google Scholar]
- Qian, Z. , Wang, S. L. , Tao, W. W. , Long, H. Y. , & Wang, J. W. (2017). Effects of Jiaotaiwan on depressive‐like behavior in mice after lipopolysaccharide administration. Metabolic Brain Disease, 32(2), 415–426. 10.1007/s11011-016-9925-8 [DOI] [PubMed] [Google Scholar]
- Ribeiro, Â. , Ribeiro, J. P. , & von Doellinger, O. (2018). Depression and psychodynamic. Psychotherapy, 40(1), 105–109. 10.1590/1516-4446-2016-2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanner, M. F. (1999). Python: A programming language for software integration and development. Journal of Molecular Graphics & Modelling, 17(1), 57–61. [PubMed] [Google Scholar]
- Shen, M. , Yang, Y. , Wu, Y. , Zhang, B. , Wu, H. , Wang, L. , … Chen, J. (2019). l‐theanine ameliorate depressive‐like behavior in a chronic unpredictable mild stress rat model via modulating the monoamine levels in limbic–cortical–striatal–pallidal–thalamic‐circuit related brain regions. Phytotherapy Research: PTR, 33(2), 412–421. 10.1002/ptr.6237 [DOI] [PubMed] [Google Scholar]
- Song, T. , Yang, Y. , Wei, H. , Xie, X. , Lu, J. , Zeng, Q. , … Peng, J. (2019). Zfp217 mediates m6A mRNA methylation to orchestrate transcriptional and post‐transcriptional regulation to promote adipogenic differentiation. Nucleic Acids Research, 47(12), 6130–6144. 10.1093/nar/gkz312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, W. J. , Zhang, Y. , Chen, Y. , Gong, H. , Lian, Y. J. , Peng, W. , … Jiang, C. L. (2017). NLRP3 gene knockout blocks NF‐κB and MAPK signaling pathway in CUMS‐induced depression mouse model. Behavioural Brain Research, 322(Pt A), 1–8. 10.1016/j.bbr.2017.01.018 [DOI] [PubMed] [Google Scholar]
- Trott, O. , & Olson, A. J. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455–461. 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, B. , Lian, Y. J. , Dong, X. , Peng, W. , Liu, L. L. , Su, W. J. , … Wang, Y. X. (2018). Glycyrrhizic acid ameliorates the kynurenine pathway in association with its antidepressant effect. Behavioural Brain Research, 353, 250–257. 10.1016/j.bbr.2018.01.024 [DOI] [PubMed] [Google Scholar]
- Wang, B. , Lian, Y. J. , Su, W. J. , Peng, W. , Dong, X. , Liu, L. L. , … Wang, Y. X. (2018). HMGB1 mediates depressive behavior induced by chronic stress through activating the kynurenine pathway. Brain, Behavior, and Immunity, 72, 51–60. 10.1016/j.bbi.2017.11.017 [DOI] [PubMed] [Google Scholar]
- Wang, H. , Bloom, O. , Zhang, M. , Vishnubhakat, J. M. , Ombrellino, M. , Che, J. , … Tracey, K. J. (1999). HMG‐1 as a late mediator of endotoxin lethality in mice. Science (New York, N.Y.), 285(5425), 248–251. 10.1126/science.285.5425.248 [DOI] [PubMed] [Google Scholar]
- Wang, W. , Pan, Q. , Han, X. Y. , Wang, J. , Tan, R. Q. , He, F. , … Kang, T. G. (2013). Simultaneous determination of arctiin and its metabolites in rat urine and feces by HPLC. Fitoterapia, 86, 6–12. 10.1016/j.fitote.2013.01.016 [DOI] [PubMed] [Google Scholar]
- Wegener, G. , Volke, V. , & Rosenberg, R. (2000). Endogenous nitric oxide decreases hippocampal levels of serotonin and dopamine in vivo . British Journal of Pharmacology, 130(3), 575–580. 10.1038/sj.bjp.0703349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willner, P. , Towell, A. , Sampson, D. , Sophokleous, S. , & Muscat, R. (1987). Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology, 93(3), 358–364. 10.1007/BF00187257 [DOI] [PubMed] [Google Scholar]
- Xavier, A. L. , Menezes, J. R. , Goldman, S. A. , & Nedergaard, M. (2014). Fine‐tuning the central nervous system: Microglial modelling of cells and synapses. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 369(1654), 20130593 10.1098/rstb.2013.0593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing, Y. , Mi, C. , Wang, Z. , Zhang, Z. H. , Li, M. Y. , Zuo, H. X. , … Ma, J. (2018). Fraxinellone has anticancer activity in vivo by inhibiting programmed cell death‐ligand 1 expression by reducing hypoxia‐inducible factor‐1α and STAT3. Pharmacological Research, 135, 166–180. 10.1016/j.phrs.2018.08.004 [DOI] [PubMed] [Google Scholar]
- Xu, X. , Zeng, X. Y. , Cui, Y. X. , Li, Y. B. , Cheng, J. H. , Zhao, X. D. , … Piao, L. X. (2020). Anti‐depressive effect of arctiin by attenuating neuroinflammation via HMGB1/TLR4‐ and TNF‐α/TNFR1‐mediated NF‐κB activation. ACS Chemical Neuroscience, 11(15), 2214–2230. 10.1021/acschemneuro.0c00120 [DOI] [PubMed] [Google Scholar]
- Yang, H. , Hreggvidsdottir, H. S. , Palmblad, K. , Wang, H. , Ochani, M. , Li, J. , … Tracey, K. J. (2010). A critical cysteine is required for HMGB1 binding to Toll‐like receptor 4 and activation of macrophage cytokine release. Proceedings of the National Academy of Sciences of the United States of America, 107(26), 11942–11947. 10.1073/pnas.1003893107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Q. W. , Lu, F. L. , Zhou, Y. , Wang, L. , Zhong, Q. , Lin, S. , … Wang, J. Z. (2011). HMBG1 mediates ischemia‐reperfusion injury by TRIF‐adaptor independent Toll‐like receptor 4 signaling. Journal of Cerebral Blood Flow and Metabolism, 31(2), 593–605. 10.1038/jcbfm.2010.129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yirmiya, R. , Rimmerman, N. , & Reshef, R. (2015). Depression as a microglial disease. Trends in Neurosciences, 38(10), 637–658. 10.1016/j.tins.2015.08.001 [DOI] [PubMed] [Google Scholar]
- Zhang, Z. H. , Mi, C. , Wang, K. S. , Wang, Z. , Li, M. Y. , Zuo, H. X. , … Jin, X. (2018). Chelidonine inhibits TNF‐α‐induced inflammation by suppressing the NF‐κB pathways in HCT116 cells. Phytotherapy Research: PTR, 32(1), 65–75. 10.1002/ptr.5948 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 SERT disrupts the interaction between TLR4 and HMGB1 or TNFR1 and TNF‐α. (A) Schematic timeline of in vitro experimental procedures. (B) SERT docked into the binding pocket of the TLR4 or TNFR1, the total view of SERT in TLR4 (i), the detailed view of SERT in TLR4 (ii), the total view of SERT in TNFR1 (iii), the detailed view of SERT in TNFR1 (iv). Total view of the binding mode between the SERT and the TNFR1. The TNFR1 was represented with green cartoon mode. The SERT was represented with rose red sticks. The TNF‐α binding sites were shown in blue surface mode (v). (C) Binding affinities of SERT for HMGB1 and TLR4 or TNFR1 and TNF‐α. SERT directly interacts with TLR4 in a dose‐dependent manner by LSPR (i), SERT blocked the binding capacity between TLR4 and HMGB1 by LSPR (ii), SERT blocked the binding capacity between TNFR1 and TNF‐α by LSPR (iii)