

Abstract
Evolution of eukaryotic species and their genomes has been traditionally understood as a vertical process in which genetic material is transmitted from parents to offspring along a lineage, and in which genetic exchange is restricted within species boundaries. However, mounting evidence from comparative genomics indicates that this paradigm is often violated. Horizontal gene transfer and mating between diverged lineages blur species boundaries and challenge the reconstruction of evolutionary histories of species and their genomes. Nonvertical evolution might be more restricted in eukaryotes than in prokaryotes, yet it is not negligible and can be common in certain groups. Recognition of such processes brings about the need to incorporate this complexity into our models, as well as to conceptually reframe eukaryotic diversity and evolution. Here, I review the recent work from genomics studies that supports the effects of nonvertical modes of evolution including introgression, hybridization, and horizontal gene transfer in different eukaryotic groups. I then discuss emerging patterns and effects, illustrated by specific examples, that support the conclusion that nonvertical processes are often at the root of important evolutionary transitions and adaptations. I will argue that a paradigm shift is needed to naturally accommodate nonvertical processes in eukaryotic evolution.
Keywords: reticulated evolution, introgression, genetic exchange, horizontal gene transfer, hybridization
Here, I review recent work from genomics studies that supports the significant effect of nonvertical modes of evolution, including introgression, hybridization, and horizontal gene transfer in different eukaryotic groups. I then discuss emerging patterns and impacts, illustrated by specific examples, that support the conclusion that nonvertical processes are often at the root of important evolutionary transitions and adaptations. I will argue that a paradigm shift is needed to naturally accommodate nonvertical processes in eukaryotic evolution.

Introduction
Reticulated evolution—also known as network evolution or nonvertical inheritance—refers to the total or partial merging of genetic material between two diverged lineages, leading to evolutionary histories that are better depicted by a phylogenetic network than by a steadily bifurcating tree. 1 Such processes have long been acknowledged as an important factor in the evolution of prokaryotes, where horizontal gene transfer (HGT)—also known as lateral gene transfer—has been shown to be rampant and is considered a major force that shapes genomes, to the point that the concept of a tree of life for prokaryotes has been challenged. 2 HGT refers to the transfer of material between different lineages without specifying what possible mechanisms may underlie the transfer. Generally, mechanisms other than sexual reproduction are considered for HGT, while transfer of genetic material between different species through sexual reproduction is considered within the framework of interspecies hybridization (see below). Many known mechanisms can result in HGT, including bacterial conjugation, but also the transmission of genetic material via viruses or direct uptake of genetic material from the environment and subsequent integration in the genome. These processes have been well documented in bacteria. In eukaryotes, processes of HGT have been generally considered to be oddities, that is, only of relevance to few major events such as the transfer of genes from organelles or endosymbionts to certain taxons, such as phagotrophic microbes. 3 Similarly, hybridization and genetic introgression, the latter being the result of reproduction among divergent lineages, have been long acknowledged in plants and animals but were considered to be anecdotal, only occurring within a short range of evolutionary distances. Hybridization in particular was considered to lead to evolutionary dead ends, given the inability of most hybrids to undergo sexual reproduction. 4
Recent genomic research, however, has brought reticular evolution to the forefront of eukaryotic genome evolution. Focused studies have shown that reticulated processes can occur between eukaryotic lineages with varying degrees of divergence and reproductive isolation, and that they can be mediated by a variety of mechanisms, ranging from virus‐ or symbiont‐mediated transference of genetic material to the fusion of nuclei from different species. In addition, depending on the mechanism, nonvertical inheritance can involve small fractions of the genome, such as in the HGT of single genes, or larger regions, including complete chromosomal sets, such as in interspecies hybridization. Both HGT and interspecies hybridization are considered potential sources for the acquisition of “transgressive” phenotypic traits in a lineage, and for the origin of new species. 5 , 6 , 7 Finally, reticulated evolution can have not only notable ecological and evolutionary consequences for the species involved but also a significant effect on genome evolution.
Importantly, processes of nonvertical evolution can be the source of incongruence among phylogenetic trees constructed from different genes in a genome. HGT of genes can cross large phylogenetic boundaries, which results in clear‐cut phylogenetic incongruence between gene trees and species trees, so that sequences from unrelated species appear close. By contrast, hybridization affects closely related species, with the viability of hybrids diminishing dramatically with phylogenetic distance. As a result, hybridization can also result in phylogenetic incongruence, which is generally limited to closely related lineages but can affect a larger fraction of the genome. Because patterns of nonvertical evolution leave a footprint in the form of phylogenetic incongruence, phylogenetic approaches can be used to uncover past reticulation events that are otherwise difficult to detect. However, the inference of past reticulated events through phylogenetic approaches is not simple, as phylogenetic incongruence can also arise from analytical factors. 8 As a consequence, phylogenetic evidence supporting nonvertical evolution in eukaryotes has been the focus of criticism, 9 despite the same approach being trusted for the detection of reticulated patterns among bacteria.
In recent years, accumulating evidence from genomic and phylogenomic studies has facilitated the recognition that HGT and interspecies hybridization are more widespread and have more complex consequences in eukaryotes than previously anticipated. In addition, while historical studies on reticulated evolution in eukaryotes have focused on phenotypic or ecological consequences in a few model plant and animal organisms, recent advances in genomic technologies have allowed tracing of the effects at the genomic level on virtually any organism of interest. Similarly, an emerging generation of phylogenetic analysis tools are being developed that allow going beyond bifurcating evolutionary histories by accounting for nonvertical inheritance and network‐like structures. 10 , 11 Despite recent progress, however, we still have a very limited understanding of the overall effects of reticulated evolution across eukaryotes. The emerging picture is complex and fragmented, and there is a need to assess global patterns that shed light on what factors modulate nonvertical inheritance across the diversity of eukaryotes. In the discussion below, I survey emerging trends and impacts of nonvertical evolution across eukaryotes and discuss current challenges and opportunities.
Phylogenetic incongruence in eukaryotes
Evidence of nonvertical evolution in prokaryotes is often derived from the observation of phylogenetic incongruence, in which the evolutionary signal of a given gene is largely dissimilar to that of most genes of the same species, so that HGT between different organisms is the most plausible explanation of the incongruence. 12 The plausibility of HGT events in prokaryotes has been a natural notion for a long time, given that specific mechanisms driving the acquisition of foreign DNA, such as transformation or conjugation, were known for this group of organisms. 12 Phylogenetic incongruence in gene trees of eukaryotic species was also recognized early on, 13 and it has been further encountered when a certain number of gene trees was compared. 14 , 15 , 16 , 17 , 18 Traditionally, as processes of nonvertical evolution were not naturally considered for eukaryotes, incongruences were generally attributed to analytical factors, such as the lack of sufficient phylogenetic signal or the effect of analytical artifacts. Many analytical factors can generate errors in a phylogenetic reconstruction. These generally involve underlying causes emerging from the data, such as biases in sequence composition, unbalanced divergence rates, or insufficient phylogenetic signal mostly affecting short ancient internodes and short sequences. 19 In addition, several data acquisition strategies can be a source of phylogenetic incongruence. For instance, incomplete or uncurated database annotations can lead to incorrect inference of gene absence, or blast cutoffs can enrich for sampling of divergent sequences. The consideration that analytical factors, including noisy and poor signal, were the main source of phylogenetic incongruence sustained efforts directed to maximize the signal by combining genes and to resolve unique, fully bifurcating, species trees. 17 , 20
As genome sequencing technology progressed and data sets of relatively closely related species became available, it was evident that analytical factors could not be the sole source of phylogenetic incongruence. Closely related species that have similar lifestyles and biology are likely to have similar evolutionary rates, and with fully sequenced genomes one should expect to find sufficiently “well‐behaved” sequences. In addition, the use of synteny conservation analysis served to either discard or confirm possible sequence contamination by, for example, inspecting the assembly context of incongruent genes, assessing the congruence in the phylogenetic signal of neighboring genes, and detecting breakpoints consistent with insertion by, or association with, transposable elements. Yet, some internodes of the eukaryotic species trees seemed to present robust tree incongruence. For instance, the branching patterns of human, chimpanzee, and gorilla were later shown to be variable across genomic regions. 21 Such incongruences were attributed to incomplete lineage sorting (ILS), a process by which alleles present in an ancestral species population are assorted differentially in two consecutive speciations (Fig. 1), leading to genome segments that are more closely related between two more distant species (i.e., human and gorilla) than a closer species pair (i.e., human and chimp). ILS results in predictable patterns of gene tree incongruence. 22 For the relative speciation of three lineages, ILS is expected to result in one dominant topology and two minor alternatives present in similar proportions: 50%, 25%, and 25% being the expected proportions of the dominant and the two minor topologies in the most extreme case. Subsequent gene loss can, of course, alter these relative proportions, but this expectation serves as a test for the likelihood of ILS, compared with other potential processes. Other purely vertical evolutionary processes that result in tree incongruence include duplication followed by differential gene loss (Fig. 1), which can lead to the well‐known phylogenetic artifact of hidden paralogy. 23 Both ILS and hidden paralogy are the result of vertical processes of evolution; in principle, given sufficient genetic information, they could theoretically be overcome by sufficient gene and taxon sampling in a gene concatenation approach, resulting in a single bifurcating species phylogeny. In the face of disturbing gene incongruence, the assumption of analytical or vertical‐evolutionary causes seemed satisfactory, and the quest for phylogenetic relationships among eukaryotes kept an idealized, fully bifurcating tree as a paradigm. As we will see, nonvertical processes have emerged as plausible and as a common source of tree incongruence at a growing number of eukaryotic clades. Different processes of nonvertical evolution leave different footprints in terms of incongruence among species trees and gene trees that can be detected in phylogenetic analysis (Fig. 2). Although the type and extent of nonvertical processes seem to differ among eukaryotic clades, the emerging picture is that these processes appear throughout the entire eukaryotic tree of life.
Figure 1.
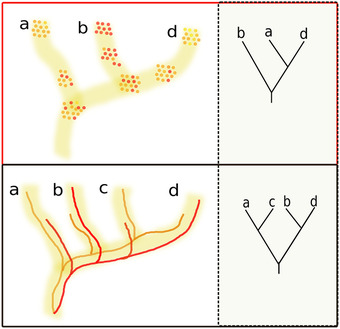
Vertical evolutionary processes leading to gene tree incongruence. Incomplete lineage sorting (top) and duplication followed by differential gene loss (bottom) are vertical evolutionary processes that can result in topologies that are incongruent with the underlying species tree. In incomplete lineage sorting (top), two consecutive speciation events lead to different assortment of alleles present in a population (small colored circles) that can be fixed differentially in the three resulting lineages (top left). As a result, some gene trees (top right) will result in closer relationships for the two most distant lineages of the trio, simply reflecting the histories of the alleles that were fixed in each lineage. In duplication followed by differential gene loss (bottom), an ancestor carries two paralogs resulting from a gene duplication; in subsequent speciations, different paralogs are lost in the different lineages (bottom left), resulting in gene tree topologies that are incongruent with the species tree (bottom right).
Figure 2.
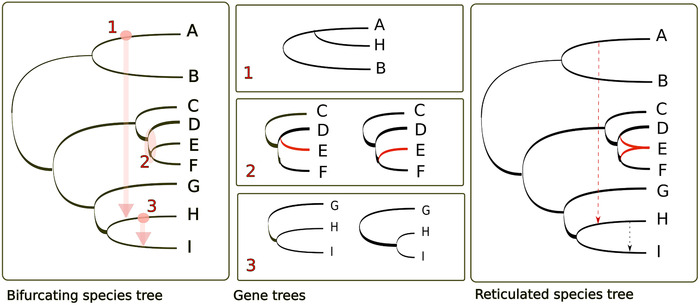
Reticulated patterns. Different processes can result in reticulated patterns of evolution. The left panel shows an idealized, fully bifurcating species tree representing the evolutionary relationships among species A to I. Three types of events are marked with circles, ellipses, and arrows: (1) an HGT event from species A to H; (2) a hybridation event between species D and F, originating the hybrid species E; and (3) a hybridization between species H and I resulting in introgression from species H into the genome of species I. The central panels shows gene phylogenies presenting incongruences or altered branch patterns resulting from these events: (1) a gene from species H clusters with homologs from the phylogenetically distant species A and B; (2) conflicting patterns found among gene trees in D and E form a clade to the exclusion of F; in others E and F form a clade to the exclusion of D; and (3) genes from the introgressed regions in species I show shorter distances with homologs of species H (right), as compared with nonintrogressed regions (left). The right panel shows a reticulated tree for the same species, here including information on the past reticulation events.
HGT in eukaryotes: a taboo no more
Traditionally, HGT has been considered extremely rare in eukaryotes, limited to only a few microbial taxa with particular characteristics such as bearing endosymbionts or phagocitizing other microbes. 24 The study of HGT in eukaryotes went through a dark phase after initial claims of the presence of bacterial‐derived genes in the human genome were proven to be an artifact. 25 This early error cast a shadow of skepticism and even criticism of subsequent attempts to identify HGT in eukaryotes, leading the issue to be considered almost taboo. 9 , 26 , 27 As a result, over a long period the search for HGT events was not considered among standard analyses of newly sequenced genomes. This unfortunately limited comprehension of evolutionary processes in eukaryotes for too long, as the assumption of fully vertical evolution was nearly always applied when interpreting genomic differences.
In recent years, however, the number of described HGT events involving eukaryotes has increased exponentially, suggesting that HGT is more widespread in eukaryotes than previously anticipated. 3 HGT has been observed in groups previously thought to be refractory to the acquisition of foreign genes. For instance, fungi possess a thick cell wall, are osmotrophs, and lack the ability to phagocitize, but all analyzed fungal genomes contain horizontally acquired genes, even from very distantly related organisms such as bacteria. 28 Similarly, there is accumulating evidence that HGT can occur in multicellular organisms, including plants and animals. 12 , 29 A growing number of surveys report events of HGT in eukaryotes, but these are limited to a few clades and use different methodologies, which makes it difficult to deduce global patterns. In addition, a great diversity of microbial eukaryotes remains poorly explored in this respect. It is not currently well understood, for example, what traits may be related to an increased propensity for HGT in certain groups of eukaryotes or what functional classes of genes are more prone to be mobile in different eukaryotic clades. For instance, current hypotheses posit that both lowly expressed genes and enzymes having a function in the periphery of metabolism are more prone to HGT in prokaryotes. 30 , 31 Such hypotheses have been tested using empirical data in prokaryotes; but studies on eukaryotes are much more limited, and sufficient evidence is lacking on whether additional patterns operate in eukaryotes. Although a complete understanding of how HGT specifically affects the major eukaryotic groups has not been achieved, some patterns can be discerned from a growing body of studies. An obvious factor influencing the difficulty for a lineage to stably integrate foreign DNA is whether somatic and germ lines are separated, as occurs in multicellular lineages of eukaryotes. The following two sections summarize recent findings regarding the occurrence of HGT in multicellular and unicellular eukaryotes.
HGT in multicellular organisms: rare but a potential source of disruptive phenotypes
To be permanently incorporated into a lineage, horizontally transferred genes need to be incorporated into the germline. In multicellular organisms there is generally strong separation of somatic tissue and the germ line, which undoubtedly poses a significant barrier to HGT. This reasoning has served as the basis to neglect any type of HGT in eukaryotic groups such as animals and plants. One might ask, however, whether such barriers are fully impenetrable or whether, under certain circumstances, the germ line can be exposed to even a small chance of acquiring a foreign gene. The study of evolution has shown that the probability of an event is only one of the factors in the interplay of chance and necessity that governs the emergence of new traits. 32 Extant organisms are rippled with examples of improbable events that eventually were subsequently selected and fixed. Although still a limited set, careful analyses have uncovered several clear‐cut cases of HGT of functional genes in animals and plants (Fig. 3). One of the best studied is the transfer of genes from endosymbionts of the genus Wolbachia to their multicellular hosts. Wolbachia sp. are intracellular bacteria that are vertically transmitted in arthropods, where they can manipulate host reproduction. In addition, other species of Wolbachia are obligate symbionts of several filarial nematode species. Several genome analyses have revealed instances where large sections of the Wolbachia genome have been transferred to the host chromosomes; 33 however, most cases of large‐scale transfers are recent, and most genes are thought to be inactive and in the process of pseudogeneization. With time, some genes may become fully integrated and functional, as it is the case in some well‐studied cases. 34 Other cases of HGT in multicellular organisms include, among others, the acquisition of fungal carotenoid pigment pathways in aphids 35 and spider mites, 36 and the acquisition of bacterial genes in mealybug, 37 plants, 38 bdelloid rotifers, 39 and nematodes. 40 Most of these examples comprise transference from microorganisms to multicellular organisms, which probably reflects the fact that multicellular organisms are constantly exposed to microbial organisms that function as parasites or endosymbionts (as in the case of Wolbachia) or are part of the multicellular organism's natural microbiota; however, this fact can also be concerning because it represents a challenge for identifying bona fide HGT, as traces of contaminant sequences are common in large‐scale sequencing projects. 41
Figure 3.

HGT in multicellular eukaryotes. Examples of multicellular eukaryotes (animals and plants) in which HGTs have been robustly identified (these are mentioned in the main text). Depicted are black bean aphids Aphis fabae (picture by Gaspar Alves), unidentified bdelloid rotifer (picture by Bob Blaylock), the mealybug Planococcus citri (picture by Jeffrey W. Lotz), Amborella trichopoda (picture by Scott Zona), red spider mite Tetranychus urticae (Charles Lam), the moss Physcomitrella patens (Hermann Schachner), the grass Alloteropsis cimicina (J.M. Garg), the fern Polypodium vulgare (André Karwath), and the mouth of a lamprey Petromyzon marinus (public domain). All pictures were taken from wikimedia commons and are shared under the Creative Commons Attribution‐Share Alike 3.0 license (https://creativecommons.org/licenses/by/3.0/), the GNU Free Documentation License, version 1.2 (https://commons.wikimedia.org/wiki/Commons:GNU_Free_Documentation_License,_version_1.2), or the public domain (CC0).
There are also some noteworthy examples of horizontal transfer of genetic material between multicellular organisms, the majority of which includes virus‐mediated transference of mobile elements or transfer of organelle genomes. 12 , 42 , 43 The flowering plant Amborella trichopoda has acquired several mitochondrial genes from other plants, 44 and sea lampreys and sturgeons—the former known to feed on the latter—share a transposable element. 45 There are other clear‐cut examples of gene transfer from multicellular organisms to other multicellular organisms, including parasitic plants that acquired genes from their hosts or genes that were likely shared among plant parasites that alternate hosts. 46 Additionally, HGT has facilitated switches to C4 photosynthesis in Alloteropsis grassess 47 and adaptation to low light conditions in ferns. 48 Other examples include possible ancient transfers, such as the actinoporin gene (Cjtox I) present in mosses, proposed to be the result of HGT from aquatic animals before the adaptation of mosses to land. 49
The above studies show that the barriers to gene transfer in multicellular eukaryotes are not unbreakable, and that, even if rare, the acquisition of foreign genes can have a significant impact on the adaptation to new niches. Given the widespread resistance to the idea of HGT in multicellular organisms, the reported cases may represent the tip of the iceberg. Although incomplete, the emerging picture serves to identify some trends, such as the larger representation of metabolic genes among transferred genes. In addition, some of the observations seem to be in line with the earlier proposed “weak link” model, 50 which states that unicellular stages and early developmental stages are the key entry points for HGT. This model makes several testable predictions: (1) multicellular eukaryotes with fully exposed zygotes, spores, or early developmental stages are more prone to HGT than are other multicellular eukaryotes; (2) multicellular eukaryotes with external fertilization will be more exposed to HGT than those with internal modes of fertilization; (3) eukaryotes with asexual modes of reproduction will be more prone to HGT; and (4) stable association with symbionts or pathogens will facilitate HGT among these organisms.
HGT in unicellular eukaryotes: anything goes?
The majority of eukaryotic diversity is found among unicellular organisms, and most eukaryotic supergroups derive from unicellular lineages; for the remaining supergroups, unicellular organisms constitute a majority of the lineages. It thus seems reasonable to consider typical eukaryotic features as naturally following on the consideration of the diversity of unicellular lineages. However, in the case of HGT, observations from multicellular organisms have often dominated the conceptual realm of what is to be expected from a typical eukaryote; thus, even among unicellular eukaryotes, it was long understood that they only rarely undergo HGT. On the one hand, unicellular and multicellular eukaryotes may share barriers to HGT, such as the confinement of DNA within a nucleus or a complex gene regulatory system; on the other hand, given their lack of separation of germ and somatic lines, unicellular eukaryotes are more exposed to HGT than multicellular organisms. Early ideas, such as the “you are what you eat” hypothesis, suggested that phagotrophic eukaryotes inevitably accumulate genes acquired from their bacterial prey. 24 Although initially formulated to explain the presence of nuclear genes of bacterial descent (other than for the mitochondrion in early eukaryotes), this idea was extended to extant phagotrophic microorganisms, as the same ratchet process would be expected to occur. As genomic sequences of microbial eukaryotes began to accumulate, however, the evidence for widespread and nonnegligible occurrence of HGT in microbial eukaryotes became overwhelmingly strong. 3 , 12 , 29 , 51 Examples of HGT in unicellular eukaryotes are many and range from the acquisition of bacterial enzymes for degrading complex carbohydrates by rumen ciliates, 52 to the acquisition of fungal genes related to plant pathogenesis in filamentous oomycetes, 53 to the acquisition of bacterial and archaeal genes in the red alga Galdieria that facilitated its adaptation to extreme environment, 54 to the acquisition of gene clusters for synthesizing alkaloids toxic to insects in plant‐associated fungi. 55 The list could be extended to include hundreds of robustly supported examples from every main lineage that has been carefully inspected. These HGT events often relate to the acquisition of important functions related to the physiology or ecology of the recipient lineage. Fungi are a particularly illustrating example. This group of unicellular eukaryotes has relatively broad taxonomic representation and several well‐sampled groups that allow accurate phylogenetic reconstruction and detection of phylogenetic incongruence; in addition, most genomes of fungal species have been obtained from axenic cultures, minimizing the risk of contaminating sequences. These factors provide a relatively firm founding on which to make claims about the existence of HGT. As mentioned above, fungi might be expected to be particularly refractory to HGT, as their cells are surrounded by a thick cell wall and lack phagotrophic capabilities. However, studies have shown that HGT is common within fungi, with clear‐cut cases found in almost every genome that has been inspected. 28 , 56 , 57 , 58
The above examples are among the current evidence indicating that HGT in microbial eukaryotic lineages is common and often underlies the acquisition of important traits and adaptation. The evolutionary distances involved in these transfers can be large, even involving interdomain transfers (though concluding this may rely on ascertainment bias, as transfers between related organisms are more difficult to detect and establish robustly). Although no broad systematic study has been performed, an overview of the published literature and of a few large‐scale studies suggests a predominance of metabolic genes among those transferred, with some surprising examples of enzymatic functions having been recurrently transferred in multiple lineages independently. 59 Thus, similar to what has been found in prokaryotes, gene transfers in eukaryotes seem to support the complexity hypothesis, which posits that new members can be added more easily to the boundaries of existing interaction networks. 31
Hybridization and introgression at close evolutionary distance: the norm rather than the exception?
Hybridization between different species can be considered a form of massive HGT, and yet some authors treat it as indistinct from HGT. However, since it involves a specific mechanism (i.e., cell fusion and combination of two complete genomes) and has a number of known particularities, I will here treat hybridization separately, restricting HGT to the transfer of genes by means other than mating mechanisms. According to the degree of divergence and the amount of gene flow between the parental species, I have previously defined three types of hybrid genetic zones 60 that may lead to different outcomes (see Fig. 4 for details). At short evolutionary distances, hybridization followed by backcrossing with the parental lineages (i.e., introgression) is a common mechanism that drives genetic exchange between related incipient species. 61 Although the process was known from the study of genetic markers, access to complete genome sequences from closely related species has revealed a broader picture, and instances of genomic introgression have been found in many lineages. In vertebrates, for almost every group that has been carefully inspected, and those for which sufficient genomic sampling exists, evidence of relatively recent introgression events has been found. This includes cases, for example, in felids, bears, fishes, and birds. 62 , 63 , 64 , 65 In humans, access to ancient genomes from Neanderthals and Denisovans has provided evidence that ancient hybridization resulted in introgressed regions in different populations of modern humans, some of which might be related to certain adaptive traits. 66 , 67 Of note, that this became evident only after access to ancient genomes of extinct hominid lineages suggests that such past hybridization events likely remain invisible for most organisms. A study of a more recent introgression in house mice (Mus musculus) provides a compelling example of how hybridization, followed by backcrossing and selection, can result in introgressed regions with adaptative value in the recipient population. 68 In this case, recent hybridization with the Algerian mouse (Mus spretus)—estimated to have occurred 20–30 years ago—led to the introgression of a genomic region carrying an allele of the vitamin K epoxide reductase complex, subunit 1 gene (Vkorc1), which confers resistance to warfarin, an anticoagulant drug used as a rodenticide. Interestingly, this gene acquired important changes in the Algerian mouse as an adaptation to a vitamin K–deficient diet; in other words, warfarin resistance was a pleiotropic effect that acquired adaptive value in the recipient species. This example highlights how fast hybridization can result in specific introgressed regions when they include a gene with a selective advantage. It also shows that the selective advantage of the introgressed gene(s) might be different in donor and recipient species. Comparative genomics research in plants has also underscored the relevance of introgression in adaptive processes. 69 In microbial eukaryotes, the lack of sufficient sampling of genomes for closely related species hampers the detection of introgression. Again, fungi stand out as an exception, with multiple clades of closely related species having been sequenced, 70 revealing that introgression is common in fungi and that, contrary to plants and animals, is not limited to closely related species. 71 , 72
Figure 4.
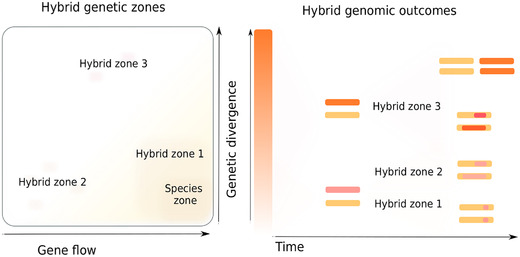
Hybridization at different genetic distances and their possible genomic outcomes. Left panel represents the three possible hybrid zones, as described in Ref. 60. The square indicates a space of connectivity (x‐axis, the amount of gene flow) and genetic relatedness (y‐axis, genetic divergence) between putative lineages. Three different hybrid zones (1, 2, and 3) and a species zone are represented. The species zone corresponds to an area of low genetic divergence and high gene flow between populations. The hybrid zone 1 is defined in an area where either gene flow or genetic relatedness is beyond the boundaries that usually define a species, so that those populations rarely cross and often present some genetic incompatibilities. Hybrid zone 2 corresponds to hybrids between lineages that abruptly separated relatively recently, so that genetic divergence is still low, but the absence of gene flow between the lineages may have resulted in the appearance of incompatibilities. Hybrid zone 3 defines hybrids between very divergent lineages. The right panel depicts typical genomic evolution of the different types of hybrids. Here, the x‐axis represents time (i.e., the amount of generations after the hybridization) and the y‐axis indicates genetic divergence. For simplicity, chromosomes of diploid hybrids with only two homologous chromosomes are represented (center). Hybrids of zone 1 and 2 have initially low sequence divergence (bottom). Given the high gene flow between hybridizing lineages and the ability to backcross with the parentals, hybrids of zone 1 generally result in introgressed genomic regions that are reduced as time progresses. Introgressed regions comprising genes that confer a selective advantage are selectively retained. Given the lack of backcrossing, hybrids of zone 2 evolve as a new lineage and genomes are shaped by chromosomal recombination, leading to the loss of heterozygosity. Hybrids of zone 2 present high divergence among homologous chromosomes from the start (top); they can either evolve through recombination and loss of heterozygosity or, in some circumstances, undergo WGD.
These processes of genetic admixture blur the phylogenetic signal of fast speciation events that occur in the presence of gene flow, resulting in apparent shorter phylogenetic distances between species, as well as greater heterogeneity in divergence times between genes. An important extrapolation from this observation is that similar processes in the past may have also affected ancient speciations, which now represent relatively deep nodes in a species phylogeny. It is currently unknown to what extent this has happened and what could be the contribution to current phylogenetic conflict between gene and species trees in eukaryotes. As discussed earlier, such incongruences have been historically attributed to phylogenetic noise or other processes, such as differential gene duplication and either loss or ILS. The above observations suggest that gene flux between closely related lineages is likely the norm rather than the exception, and that we need to incorporate this in our interpretation of past events.
Finally, it is unclear what role hybridization may have in spreading previously acquired genes across a given clade. The mode of HGT in eukaryotes remains controversial, and it is unclear whether transferred genes are acquired individually or are the consequence of past introgression events. Such a process, for instance, may explain intriguing observations of large clusters of transferred genes 56 and of the apparent recursive transfers of the same genes among relatively distant lineages. 28 , 59 Indeed, in a comprehensive phylogenetic analysis of transferred d‐amino acid metabolism genes, at least one well‐supported nested transfer event was detected involving, first, a transfer from bacteria to fungi and then a subsequent transfer between two distantly related fungal genera. 59 In this regard, given the possibility of nested transfers and that some intermediate organisms are extinct or not sampled, one must always consider reconstructed scenarios with caution.
Hybridization between diverged lineages: some can really do it
The above examples refer to hybridization between closely related lineages (i.e., those that entail close relatives), including lineages at the incipient stages of speciation, in which geographical or behavioral barriers moved in and out before robust genetic barriers were established to prevent successful mating between them (Fig. 4). But how far can interspecies hybridization go? Is there a limit to genetic divergence that prevents the formation of chimeric organisms? These questions necessarily have multiple answers, depending on the specific eukaryotic group of focus. Hybrids of mammalian organisms are generally only formed between closely related species typically belonging to the same genus, whereas plants hybridization can occur between somewhat more distant species, sometimes involving different related genera. 73 Although species and genus levels refer to different levels of genetic divergence in different clades, most described hybridizations in multicellular organisms involve parental lineages with less than 1–3% average sequence divergence at the nucleotide level. In microbial eukaryotes, however, hybridization can sometimes involve truly highly diverged lineages. In yeasts, for example, natural hybrids between yeast species that are more than 25% divergent at the nucleotide level have been found, and artificial hybrids have also been successfully formed in a laboratory setting between species of two distantly related lineages within the Ascomycota, Saccharomyces and Schizosaccharomyces. 71 Although researchers are far from understanding all constraints that act on hybrid formation, differences between groups of eukaryotes reflect underlying differences in barriers to hybridization. For interspecies hybrids to occur, individuals from the different species must meet, reproduce, and form viable zygotes that result in individuals sufficiently fit to their environment and that survive and successfully compete with nonhybrid individuals. Importantly, for a new hybrid lineage to persist it must be able to reproduce. Differences in dispersion ability, reproductive modes, and genomic plasticity, among others, will influence the ability of different groups to form successful hybrids; take mammals, which must necessarily reproduce sexually in each generation to leave progeny. This imposes the need for a new chimeric chromosomal set to successfully undergo meiosis, otherwise, the hybrid would be sterile. In lineages that can reproduce asexually, this constraint is relaxed, and hybrids can form populations that reproduce clonally.
The formation of hybrids between divergent organisms has many implications that relate, in the first place, to how such chimeric organisms can survive and, second, to how they evolve subsequently. Hybrids have two divergent sets of chromosomes within a single nucleus, and most theoretical frameworks, such as the Bateson, Dobzhansky, and Muller model, 74 , 75 , 76 would predict that such extreme chimerism will result in numerous incompatibilities at the genetic, transcriptomic, or proteomic level. These incompatibilities are expected because combinations of alleles that have evolved independently in different populations (here species) are expected to be less fit than combinations of alleles in the same population, simply because the later are not exposed to the filter of selection. As a result, mutations that tend to result in a loss of heterozygosity will be favored by selection. In this regard, hybrid genomes would be quickly shaped by many processes that ultimately lead to the overall reduction of both heterozygosity and stabilization of the genome. These include, among others, gene conversion, gene loss, the duplication or the loss of entire chromosomes or large chromosomal regions leading to chromosomal aneuploidies, or even whole genome duplication (WGD; discussed in more detail below).
At the transcriptome level, hybridization results in “genomic shock,” 77 involving massive disregulation of genes. The effects of hybridization at the transcriptional level have been the focus of intense research in plants and animals, with most studies supporting the existence of large transcriptional shocks. 78 , 79 , 80 , 81 The study of such process in microbial eukaryotes lags behind, but the few reports in fungi suggest a much lower effect. For instance, the impact at the transcriptional level of a hybridization between Saccharomyces cerevisiae and Saccharomyces uvarum results in a limited number of significantly altered genes (1–2%), five‐fold smaller than the transcriptional changes resulting from a thermal shock. 82 Although preliminary, such results may suggest that transcriptional incompatibilities between divergent species in yeasts (and perhaps other microbial eukaryotes) might be comparatively smaller than in multicellular organisms. Additionally, the view of genomic shocks in multicellular animals might be biased from observations made in model species. Overall, the implications of hybridization at the genetic, physiological, and evolutionary levels, and how they vary across different eukaryotic groups, are still poorly understood.
The genomic aftermath of hybridization is signaled by a change in chromosome number (ploidy). This form of WGD is referred to as allopolyploidization, in contrast to autopolyploidization in which the replication of the genetic material is not followed by cell division. 83 If the genomes of the two hybridizing lineages are sufficiently divergent, imperfect pairing of chromosomes may result in the inability to undergo meiosis. Proper pairing of chromosomes can be restored upon further doubling of the genetic complement: polyploid with respect to the two haploid genomes that form the zygote but behaving as a pseudohaploid genome, as it cannot reduce ploidy through meiosis. This process, common among plant hybrids, 84 , 85 has been proposed as the mechanism leading to yeast WGD. 86 Although it remains unclear whether other WGDs result from hybridization, processes of allopolyploidization better explain the origin of a new lineage sexually isolated from the parental populations and having unique traits. Autopolyploidization, by contrast, implies scenarios where the initial selective advantage is conferred solely by a change in ploidy. Thus, I argue that hybridization is likely to be the most common process underlying the origin of WGDs.
An emerging paradigm
The above sections provide an overview of the growing body of evidence for the existence of a significant impact of nonvertical evolution in eukaryotes (Fig. 3). As previous views gradually disappear, more and more genomic studies will undertake the search for footprints of past HGT or hybridization. So far, the emerging picture is already revealing.
Far from being an anecdotal process, the evidence for a widespread role of reticulated processes in eukaryotes is now difficult to question. Although not as common as in prokaryotes, HGT has been found in every major eukaryotic group that has been carefully investigated, with some groups of microbial eukaryotes showing a relatively high propensity to exchange genes. The literature now contains many examples showing not only transfer genes across distant lineages and even across different domains but also that transferred genes played essential roles in the acquisition of ecologically relevant traits or of the ability to colonize new niches. HGT in multicellular eukaryotes is rarer though nonnegligible, with some multicellular lineages showing a surprisingly high history of acquiring alien genes. The examples show that, among multicellular organisms too, transferred genes seem to have provided important selective advantages to the recipient species. Overall, the picture remains opaque of the impact of HGT across eukaryotic lineages and how variability of HGT relates to differences in physiological barriers or ecological constraints across the different taxa. Moreover, although the complexity hypothesis seems to hold for eukaryotes, 3 and metabolic genes are more subjected to HGT, 28 , 58 little is known about the idiosyncrasies of HGT in eukaryotes: it is easy to detect when occurring across large genetic distances, yet such occurrences may be under‐representative of how widespread it is. Although a strong methodological framework is lacking to robustly infer the occurrence of HGT between closely related lineages, increased taxonomic sampling of genomic datasets and algorithmic developments are likely to provide better solutions in the near future. Other open issues include the difficulty of inferring the directionality of the transfer event, that is, identifying donor from recipient. Although several phylogenetic approaches have been developed to infer this from gene tree topologies, 59 , 87 unspoken assumptions, such as transfer goes from small to large organism, still dominate interpretations. Finally, as illustrated by the case of gene transfer from Wolbachia endosymbionts to their hosts, transfer of single genes may have initiated from transfer of larger genomic regions; and yet the process of gene acclimation following the transfer remains little understood.
Regarding hybridization, the emerging picture is that genetic flux between incipient species may be the rule rather than the exception. This process may underlie a large part of the pervasive incongruence among gene trees that have long been noted. Vertical and nonvertical sources of phylogenetic incongruence are expected to result in different genome‐wide patterns, and thus, at least theoretically, it should be possible to distinguish them by careful analyses. Again, increased taxonomic sampling and novel algorithmic solutions are likely to help improve the awareness of the differential patterns and impacts of hybridization across different eukaryotic lineages. Hybridization occurs along a continuous gradient of genetic divergence, with outcomes varying according to different factors. Hybridization at close distances is usually followed by backcrossing with one (or two) of the parental lineages, diluting the chimeric nature of the hybrid lineages and resulting in genetic introgression. At larger distances, the hybrids might be sexually isolated, and a new lineage can be established only if nonsexual reproduction is a possibility. In some cases, these hybrids might undergo WGD, restore fertility, and thus settle a new lineage capable of sexual reproduction. The study of the factors that influence one or the other outcome, or how they vary across lineages, is still in its infancy. Similarly, little is known about the genomic, transcriptomic, and ecological aftermath of hybridization, beyond a few well‐studied groups.
That nonvertical processes of evolution are widespread and common across the entirety of eukaryotic diversity, I argue, should bring fundamental shifts to some current paradigms of eukaryotic evolution. First, nonvertical evolution should have important consequences on how adaptive processes in eukaryotic species are understood. In classical frameworks, populations are depicted as “navigating” genotypic and fitness landscapes, with driving forces being only vertical processes of inheritance, mutation, and selection. Nonvertical evolution, by contrast, introduces the possibility of more radical, nongradual changes in genotypes, resulting in large jumps at both the genotypic and fitness landscapes. For this reason, the role of nonvertical processes of evolution should be particularly scrutinized in the context of radical adaptation or when innovations appear in a relatively short evolutionary time.
Another area of reconsideration is the concept of species. Although there are clear boundaries between lineages, these do not always correspond to previously defined species; moreover, the boundaries should not be assumed to be impenetrable. Species complexes—comprising species with different degrees of introgression among them—and their hybrids are increasingly being recognized, best represented by reticulate trees. At a general level, the idea that blurred boundaries exist between species, particularly in microbial lineages but also in multicellular ones, should become less foreign. Gene flow between species, particularly closely related ones, should always be considered a possibility. Acquisition of genes from distantly related organisms should also be considered a possible scenario, and never assumed to be impossible. Whereas caution should always be taken and possible artifacts discarded, we should openly look at the possibilities with as much awareness as possible of negative preconceptions.
Finally, the interpretation of how species trees and gene trees relate to each other should be reframed. The classical paradigm of vertical‐only evolution and fully bifurcating species trees has been dominant in conceptual interpretation of how eukaryotic species evolve and adapt. In most cases, the alternative hypothesis that reticulation could have affected the observed patterns is not considered. I argue that this has severely limited the advancement of the field. Indeed, controversies regarding nodes in the eukaryotic tree of life may reflect underlying processes of deep reticulation. Recognizing this may be difficult, but it may also be eye‐opening and pave the way for reconstructing more realistic scenarios of how ancient lineages originated and diverged.
The problem is not limited to whether the idealized fully bifurcating nature of a species tree is right or wrong. I would argue that in many cases a fully bifurcating tree is a reasonably good approximation of how eukaryotic species diverged from one another, despite an underlying diversity of gene evolutionary histories. A more important consequence of the vertical‐only paradigm extends to the realm of understanding how species have shaped their genetic constituents and adapted to different niches, even in scenarios where the reconstruction of fully‐bifurcating trees is not problematic. Imposing the strong assumption that only vertical processes are in place will, I believe, slow down reconstruction of ancestral genomes and how they evolved. However, embracing nonvertical processes in models of eukaryotic evolution is not a simple task. For this to happen, a first step is to study these processes thoroughly, to know their real extent, and to clarify their patterns and effects across diverse eukaryotic lineages.
Competing interests
The author declares no competing interests.
Acknowledgments
The author acknowledges support from the Spanish Ministry of Science and Innovation for the Grant PGC2018‐099921‐B‐I00, cofounded by European Regional Development Fund (ERDF); from the CERCA Programme/Generalitat de Catalunya; from the Catalan Research Agency (AGAUR) SGR423, and Grants from the European Union's Horizon 2020 research and innovation programme under the Grant agreement ERC‐2016‐724173, and the Marie Sklodowska‐Curie Grant agreement H2020‐MSCA‐IF‐2017‐793699. The author also receives support from an INB Grant (PT17/0009/0023 – ISCIII‐SGEFI/ERDF).
References
- 1. Mallet, J. , Besansky N. & Hahn M.W.. 2016. How reticulated are species? BioEssays 38: 140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Doolittle, W.F. 1999. Phylogenetic classification and the universal tree. Science 284: 2124–2128. [DOI] [PubMed] [Google Scholar]
- 3. Keeling, P.J. & Palmer J.D.. 2008. Horizontal gene transfer in eukaryotic evolution. Nat. Rev. Genet. 9: 605–618. [DOI] [PubMed] [Google Scholar]
- 4. Neiman, M. , Sharbel T.F. & Schwander T.. 2014. Genetic causes of transitions from sexual reproduction to asexuality in plants and animals. J. Evol. Biol. 27: 1346–1359. [DOI] [PubMed] [Google Scholar]
- 5. Rieseberg, L.H. , Archer M.A. & Wayne R.K.. 1999. Transgressive segregation, adaptation and speciation. Heredity (Edinb.) 83: 363–372. [DOI] [PubMed] [Google Scholar]
- 6. Abbott, R. , Albach D., Ansell S., et al 2013. Hybridization and speciation. J. Evol. Biol. 26: 229–246. [DOI] [PubMed] [Google Scholar]
- 7. Mallet, J. 2007. Hybrid speciation. Nature 446: 279–283. [DOI] [PubMed] [Google Scholar]
- 8. Huelsenbeck, J.P. , Rannala B. & Masly J.P.. 2000. Accommodating phylogenetic uncertainty in evolutionary studies. Science 288: 2349–2350. [DOI] [PubMed] [Google Scholar]
- 9. Martin, W.F. 2017. Too much eukaryote LGT. BioEssays 39: 1700115. [DOI] [PubMed] [Google Scholar]
- 10. Bapteste, E. , van Iersel L., Janke A., et al 2013. Networks: expanding evolutionary thinking. Trends Genet. 29: 439–441. [DOI] [PubMed] [Google Scholar]
- 11. Szöllösi, G.J. , Tannier E., Daubin V., et al 2015. The inference of gene trees with species trees. Syst. Biol. 64: e42–e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Soucy, S.M. , Huang J. & Gogarten J.P.. 2015. Horizontal gene transfer: building the web of life. Nat. Rev. Genet. 16: 472–482. [DOI] [PubMed] [Google Scholar]
- 13. Penny, D. , Foulds L.R. & Hendy M.D.. 1982. Testing the theory of evolution by comparing phylogenetic trees constructed from five different protein sequences. Nature 297: 197–200. [DOI] [PubMed] [Google Scholar]
- 14. Huerta‐Cepas, J. , Dopazo H., Dopazo J., et al 2007. The human phylome. Genome Biol. 8: R109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jarvis, E.D. , Mirarab S., Aberer A.J., et al 2014. Whole‐genome analyses resolve early branches in the tree of life of modern birds. Science 346: 1320–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marcet‐Houben, M. & Gabaldón T.. 2009. The tree versus the forest: the fungal tree of life and the topological diversity within the yeast phylome. PLoS One 4: e4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Salichos, L. & Rokas A.. 2013. Inferring ancient divergences requires genes with strong phylogenetic signals. Nature 497: 327–331. [DOI] [PubMed] [Google Scholar]
- 18. Hahn, M.W. , Han M.V. & Han S.G.. 2007. Gene family evolution across 12 Drosophila genomes. PLoS Genet. 3: 2135–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Philippe, H. , Brinkmann H., Lavrov D.V., et al 2011. Resolving difficult phylogenetic questions: why more sequences are not enough. PLoS Biol. 9: e1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gee, H. 2003. Ending incongruence. Nature 425: 782. [DOI] [PubMed] [Google Scholar]
- 21. Chen, F.C. & Li W.H.. 2001. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am. J. Hum. Genet. 68: 444–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pollard, D.A. , Iyer V.N., Moses A.M., et al 2006. Widespread discordance of gene trees with species tree in Drosophila: evidence for incomplete lineage sorting. PLoS Genet. 2: 1634–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gribaldo, S. & Philippe H.. 2002. Ancient phylogenetic relationships. Theor. Popul. Biol. 61: 391–408. [DOI] [PubMed] [Google Scholar]
- 24. Doolittle, W.F. 1998. You are what you eat: a gene transfer ratchet could account for bacterial genes in eukaryotic nuclear genomes. Trends Genet. 14: 307–311. [DOI] [PubMed] [Google Scholar]
- 25. Stanhope, M.J. , Lupas A., Italia M.J., et al 2001. Phylogenetic analyses do not support horizontal gene transfers from bacteria to vertebrates. Nature 411: 940–944. [DOI] [PubMed] [Google Scholar]
- 26. Kurland, C.G. , Canback B. & Berg O.G.. 2003. Horizontal gene transfer: a critical view. Proc. Natl. Acad. Sci. USA 100: 9658–9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Martin, W.F. 2018. Eukaryote lateral gene transfer is Lamarckian. Nat. Ecol. Evol. 2: 754. [DOI] [PubMed] [Google Scholar]
- 28. Marcet‐Houben, M. & Gabaldón T.. 2010. Acquisition of prokaryotic genes by fungal genomes. Trends Genet. 26: 5–8. [DOI] [PubMed] [Google Scholar]
- 29. Husnik, F. & McCutcheon J.P.. 2018. Functional horizontal gene transfer from bacteria to eukaryotes. Nat. Rev. Microbiol. 16: 67–79. [DOI] [PubMed] [Google Scholar]
- 30. Cohen, O. , Gophna U. & Pupko T.. 2010. The complexity hypothesis revisited: connectivity rather than function constitutes a barrier to horizontal gene transfer. Mol. Biol. Evol. 28: 1481–1489. [DOI] [PubMed] [Google Scholar]
- 31. Jain, R. , Rivera M.C. & Lake J.A.. 1999. Horizontal gene transfer among genomes: the complexity hypothesis. Proc. Natl. Acad. Sci. USA 96: 3801–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Monod, J. 1974. On chance and necessity In Studies in the Philosophy of Biology. Ayala F.J. & Dobzhansky T., Eds.: 357–375. Macmillan Education UK. [Google Scholar]
- 33. Dunning Hotopp, J.C. , Clark M.E., Oliveira D.C.S.G., et al 2007. Widespread lateral gene transfer from intracellular bacteria to multicellular eukaryotes. Science 317: 1753–1756. [DOI] [PubMed] [Google Scholar]
- 34. Cordaux, R. & Gilbert C.. 2017. Evolutionary significance of Wolbachia‐to‐animal horizontal gene transfer: female sex determination and the f element in the isopod Armadillidium vulgare . Genes (Basel) 8: 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moran, N.A. & Jarvik T.. 2010. Lateral transfer of genes from fungi underlies carotenoid production in aphids. Science 328: 624–627. [DOI] [PubMed] [Google Scholar]
- 36. Altincicek, B. , Kovacs J.L. & Gerardo N.M.. 2012. Horizontally transferred fungal carotenoid genes in the two‐spotted spider mite Tetranychus urticae . Biol. Lett. 8: 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Husnik, F. , Nikoh N., Koga R., et al 2013. Horizontal gene transfer from diverse bacteria to an insect genome enables a tripartite nested mealybug symbiosis. Cell 153: 1567–15678. [DOI] [PubMed] [Google Scholar]
- 38. White, F.F. , Garfinkel D.J., Huffman G.A., et al 1983. Sequences homologous to Agrobacterium rhizogenes T‐DNA in the genomes of uninfected plants. Nature 301: 348–350. [Google Scholar]
- 39. Gladyshev, E.A. , Meselson M. & Arkhipova I.R.. 2008. Massive horizontal gene transfer in bdelloid rotifers. Science 320: 1210–1213. [DOI] [PubMed] [Google Scholar]
- 40. Noon, J.B. & Baum T.J.. 2016. Horizontal gene transfer of acetyltransferases, invertases and chorismate mutases from different bacteria to diverse recipients. BMC Evol. Biol. 16: 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Richards, T.A. & Monier A.. 2016. A tale of two tardigrades. Proc. Natl. Acad. Sci. USA 113: 4892–4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wallau, G.L. , Vieira C. & Loreto É.L.S.. 2018. Genetic exchange in eukaryotes through horizontal transfer: connected by the mobilome. Mob. DNA 9: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schaack, S. , Gilbert C. & Feschotte C.. 2010. Promiscuous DNA: horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol. Evol. 25: 537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bergthorsson, U. , Richardson A.O., Young G.J., et al 2004. Massive horizontal transfer of mitochondrial genes from diverse land plant donors to the basal angiosperm Amborella . Proc. Natl. Acad. Sci. USA 101: 17747–17752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang, H.‐H. , Feschotte C. & Han M.‐J., et al 2014. Recurrent horizontal transfers of Chapaev transposons in diverse invertebrate and vertebrate animals. Genome Biol. Evol. 6: 1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wickell, D.A. & Li F.. 2020. On the evolutionary significance of horizontal gene transfers in plants. New Phytol. 225: 113–117. [DOI] [PubMed] [Google Scholar]
- 47. Dunning, L.T. , Olofsson J.K., Parisod C., et al 2019. Lateral transfers of large DNA fragments spread functional genes among grasses. Proc. Natl. Acad. Sci. USA 116: 4416–4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li, F.W. , Villarreal J.C., Kelly S., et al 2014. Horizontal transfer of an adaptive chimeric photoreceptor from bryophytes to ferns. Proc. Natl. Acad. Sci. USA 111: 6672–6677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hoang, Q.T. , Cho S.H., McDaniel S.F., et al 2009. An actinoporin plays a key role in water stress in the moss Physcomitrella patens . New Phytol. 184: 502–510. [DOI] [PubMed] [Google Scholar]
- 50. Huang, J. 2013. Horizontal gene transfer in eukaryotes: the weak‐link model. Bioessays 35: 868–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Andersson, J.O. 2009. Gene transfer and diversification of microbial eukaryotes. Annu. Rev. Microbiol. 63: 177–193. [DOI] [PubMed] [Google Scholar]
- 52. Ricard, G. , McEwan N.R., Dutilh B.E., et al 2006. Horizontal gene transfer from bacteria to rumen ciliates indicates adaptation to their anaerobic, carbohydrates‐rich environment. BMC Genomics 7: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Richards, T.A. , Dacks J.B., Jenkinson J.M., et al 2006. Evolution of filamentous plant pathogens: gene exchange across eukaryotic kingdoms. Curr. Biol. 16: 1857–1864. [DOI] [PubMed] [Google Scholar]
- 54. Schonknecht, G. , Chen W.‐H., Ternes C.M., et al 2013. Gene transfer from bacteria and archaea facilitated evolution of an extremophilic eukaryote. Science 339: 1207–1210. [DOI] [PubMed] [Google Scholar]
- 55. Marcet‐Houben, M. & Gabaldón T.. 2016. Horizontal acquisition of toxic alkaloid synthesis in a clade of plant associated fungi. Fungal Genet. Biol. 86: 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wisecaver, J.H. & Rokas A.. 2015. Fungal metabolic gene clusters—caravans traveling across genomes and environments. Front. Microbiol. 6: 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gluck‐Thaler, E. & Slot J.C.. 2015. Dimensions of horizontal gene transfer in eukaryotic microbial pathogens. PLoS Pathog. 11: e1005156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Naranjo‐Ortiz, M.A. & Gabaldón T.. 2020. Fungal evolution: cellular, genomic and metabolic complexity. Biol. Rev. Camb. Philos. Soc. 95: 1198–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Naranjo‐Ortíz, M.A. , Brock M., Brunke S., et al 2016. Widespread inter‐ and intra‐domain horizontal gene transfer of d‐amino acid metabolism enzymes in eukaryotes. Front. Microbiol. 7 10.3389/fmicb.2016.02001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gabaldón, T. 2020. Hybridization and the origin of new yeast lineages. FEMS Yeast Res. 20: foaa040 10.1093/femsyr/foaa040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Harrison, R.G. & Larson E.L.. 2014. Hybridization, introgression, and the nature of species boundaries. J. Hered. 105(Suppl.): 795–809. [DOI] [PubMed] [Google Scholar]
- 62. Figueiró, H.V. , Li G., Trindade F.J., et al 2017. Genome‐wide signatures of complex introgression and adaptive evolution in the big cats. Sci. Adv. 3: e1700299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cahill, J.A. , Stirling I., Kistler L., et al 2015. Genomic evidence of geographically widespread effect of gene flow from polar bears into brown bears. Mol. Ecol. 24: 1205–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Souissi, A. , Bonhomme F., Manchado M., et al 2018. Genomic and geographic footprints of differential introgression between two divergent fish species (Solea spp.). Heredity (Edinb.) 121: 579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ottenburghs, J. , Kraus R.H.S., van Hooft P., et al 2017. Avian introgression in the genomic era. Avian Res. 8: 1–11. [Google Scholar]
- 66. Gokcumen, O. 2020. Archaic hominin introgression into modern human genomes. Am. J. Phys. Anthropol. 171(Suppl. 70): 60–73. [DOI] [PubMed] [Google Scholar]
- 67. Simonti, C.N. , Vernot B., Bastarache L., et al 2016. The phenotypic legacy of admixture between modern humans and Neandertals. Science 351: 737–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Song, Y. , Endepols S., Klemann N., et al 2011. Adaptive introgression of anticoagulant rodent poison resistance by hybridization between old world mice. Curr. Biol. 21: 1296–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Suarez‐Gonzalez, A. , Lexer C. & Cronk Q.C.B.. 2018. Adaptive introgression: a plant perspective. Biol. Lett. 14 10.1098/rsbl.2017.0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Naranjo‐Ortiz, M.A. & Gabaldón T.. 2019. Fungal evolution: diversity, taxonomy and phylogeny of the fungi. Biol. Rev. 94: 2101–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Morales, L. & Dujon B.. 2012. Evolutionary role of interspecies hybridization and genetic exchanges in yeasts. Microbiol. Mol. Biol. Rev. 76: 721–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gladieux, P. , Ropars J., Badouin H., et al 2014. Fungal evolutionary genomics provides insight into the mechanisms of adaptive divergence in eukaryotes. Mol. Ecol. 23: 753–773. [DOI] [PubMed] [Google Scholar]
- 73. Goulet, B.E. , Roda F. & Hopkins R.. 2017. Hybridization in plants: old ideas, new techniques [OPEN]. Plant Physiol. 173: 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bateson, W. 2009. Heredity and variation in modern lights In Darwin and Modern Science. Seward A.C., Ed.: 85–101. Cambridge: Cambridge University Press. [Google Scholar]
- 75. Dobzhansky, T. 1934. Studies on hybrid sterility ‐ I. Spermatogenesis in pure and hybrid Drosophila pseudoobscura . Z. Zellforsch. Mikrosk. Anat. 21: 169–223. [Google Scholar]
- 76. Muller, H.J. 1942. Isolating mechanisms, evolution, and temperature In Biological Symposia: A Series of Volumes to Current Symposia in the Field of Biology. Vol. 6 Dobzhansky T., Ed.: 71–125. Lancaster, PA: Jaques Cattell Press. [Google Scholar]
- 77. McClintock, B. 1984. The significance of responses of the genome to challenge. Science 226: 792–801. [DOI] [PubMed] [Google Scholar]
- 78. McManus, C.J. , Coolon J.D., Duff M.O., et al 2010. Regulatory divergence in Drosophila revealed by mRNA‐seq. Genome Res. 20: 816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Maheshwari, S. & Barbash D.A.. 2011. The genetics of hybrid incompatibilities. Annu. Rev. Genet. 45: 331–355. [DOI] [PubMed] [Google Scholar]
- 80. Ranz, J.M. , Namgyal K., Gibson G., et al 2004. Anomalies in the expression profile of interspecific hybrids of Drosophila melanogaster and Drosophila simulans . Genome Res. 14: 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Malone, J.H. , Chrzanowski T.H. & Michalak P.. 2007. Sterility and gene expression in hybrid males of Xenopus laevis and X. muelleri . PLoS One 2: e781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hovhannisyan, H. , Saus E., Ksiezopolska E., et al 2020. Integrative omics analysis reveals a limited transcriptional shock after yeast inter‐species hybridization. Front. Genet. 11: 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ohno, S. 1970. Evolution by Gene Duplication. Heidelberg: Springer‐Verlag. [Google Scholar]
- 84. Soltis, P.S. , Marchant D.B., Van de Peer Y., et al 2015. Polyploidy and genome evolution in plants. Curr. Opin. Genet. Dev. 35: 119–125. [DOI] [PubMed] [Google Scholar]
- 85. Soltis, P.S. & Soltis D.E.. 2009. The role of hybridization in plant speciation. Annu. Rev. Plant Biol. 60: 561–588. [DOI] [PubMed] [Google Scholar]
- 86. Marcet‐Houben, M. & Gabaldón T.. 2015. Beyond the whole‐genome duplication: phylogenetic evidence for an ancient interspecies hybridization in the baker's yeast lineage. PLoS Biol. 13: e1002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Boc, A. , Philippe H. & Makarenkov V.. 2010. Inferring and validating horizontal gene transfer events using bipartition dissimilarity. Syst. Biol. 59: 195–211. [DOI] [PubMed] [Google Scholar]