

Abstract
Objective
To evaluate the efficacy and safety of PF‐06651600 (ritlecitinib), an irreversible inhibitor of JAK3 and the tyrosine kinase expressed in hepatocellular carcinoma (TEC) kinase family, in comparison with placebo in patients with rheumatoid arthritis (RA).
Methods
An 8‐week, phase II, double‐blind, parallel‐group study was conducted. Seventy patients who were seropositive for anti–citrullinated protein antibodies and/or rheumatoid factor were randomized 3:2 to receive oral PF‐06651600 (200 mg once daily) or placebo for 8 weeks. Eligible patients had an inadequate response to methotrexate, and the study design allowed up to 50% of patients to have previously received 1 tumor necrosis factor inhibitor that was inadequately effective and/or not tolerated. The primary end point was change from baseline in the Simplified Disease Activity Index (SDAI) score at week 8, assessed by Bayesian analysis using an informative prior distribution for placebo response.
Results
Mean change from baseline in the SDAI score at week 8 was greater in the PF‐06651600 group (−26.1 [95% credible interval −29.7, −22.4]) than in the placebo group (−16.8 [95% credible interval −20.9, −12.7]; P < 0.001). Most adverse events (AEs) were mild in severity, and no treatment‐related serious AEs, severe AEs, or deaths were reported. The most common classes of AE were infections and infestations as well as skin and subcutaneous tissue disorders; there was 1 mild case of herpes simplex in the PF‐06651600 group that was considered to be treatment related, which resolved within 3 days without study treatment discontinuation or antiviral therapy.
Conclusion
Treatment with the oral JAK3/TEC inhibitor PF‐06651600 (200 mg once daily) was associated with significant improvements in RA disease activity and was generally well‐tolerated in this small 8‐week study.
INTRODUCTION
Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by joint inflammation and destruction, progressive disability, and adverse physiologic effects. Epidemiologic data from 2010 indicate that RA affects ~0.24% of the global population, with numbers expected to increase as populations age and mortality rates decrease (1). Anti–citrullinated protein antibodies (ACPAs) have been observed in patients with severe RA experiencing rapidly progressing disease and nonresponse to treatment (2).
As there is no curative treatment for RA, European and US guidelines recommend a treat‐to‐target approach, with the aim of achieving sustained clinical remission or low disease activity, minimizing joint damage, and enhancing physical function and quality of life (3, 4). Disease‐modifying antirheumatic drugs (DMARDs) are standard RA treatments (5). In patients with inadequate response to conventional synthetic DMARDs, such as methotrexate, treatment may be switched or added (3, 4); other treatments include biologic DMARDs (including tumor necrosis factor inhibitors [TNFi] and non‐TNFi) and JAK inhibitors (5, 6, 7, 8). Despite these treatments, remission rates remain low (9, 10), and pain and fatigue may persist.
Tofacitinib is an oral JAK1/3 inhibitor for the treatment of RA (11). Recently, data have emerged from preclinical and clinical studies of JAK inhibitors with varying selectivity profiles. These inhibitors include the JAK1/2 inhibitor baricitinib and the JAK1 inhibitor upadacitinib, both recently approved for the treatment of moderate‐to‐severe RA (12, 13, 14, 15, 16, 17), and the JAK1 inhibitor filgotinib and the moderately selective JAK3 inhibitor peficitinib, both under investigation as potential treatments for RA (18, 19, 20, 21, 22, 23).
PF‐06651600 (now known as ritlecitinib) is an orally bioavailable small molecule that inhibits JAK3 by irreversibly blocking the ATP binding site and the tyrosine kinase expressed in hepatocellular carcinoma (TEC) family of kinases (Bruton’s tyrosine kinase [BTK], bone marrow tyrosine kinase on chromosome X [BMX], interleukin‐2–inducible T cell kinase [ITK], resting lymphocyte kinase, and TEC), with high selectivity over the other JAK isoforms, JAK1, JAK2, and Tyk2, and the broader human kinome (24, 25). TEC kinases are involved in immunologic regulation. BTK and ITK are being explored as treatments for inflammatory diseases as they contribute to signal transduction from antigen receptors on B and T cells, respectively (26, 27).
JAK3 inhibition leads to modulation of γ–common chain cytokines, including interleukin‐2 (IL‐2), IL‐4, IL‐7, IL‐9, IL‐15, and IL‐21 (24, 25, 28), some of which have been implicated in RA pathophysiology (11). PF‐06651600 is expected to spare inhibition of immunoregulatory cytokines that may play protective roles, including IL‐10 and IL‐27 (11, 24), by JAK1 inhibition. Furthermore, sparing inhibition of JAK2 cytokine signaling, which is associated with hematologic adverse effects including neutropenia, thrombocytopenia, and anemia (28), makes JAK3 inhibitors an attractive therapeutic modality for inflammatory diseases including RA.
This phase IIa, single‐dose, randomized, placebo‐controlled, proof‐of‐concept study evaluated the efficacy and safety of PF‐06651600 in patients with moderate‐to‐severe RA who were seropositive for ACPAs and/or rheumatoid factor (RF), and had an inadequate response to methotrexate. The study design allowed for up to 50% of the patients to have received 1 prior TNFi.
PATIENTS AND METHODS
Patients
Eligible patients were ages 18–75 years and were diagnosed as having RA according to the American College of Rheumatology (ACR)/European League Against Rheumatism 2010 classification criteria (score ≥6/10) (29). Patients had active disease at screening and baseline (defined by ≥6 tender or painful joints, ≥6 swollen joints, and high‐sensitivity C‐reactive protein [hsCRP] ≥7 mg/liter), and were seropositive for ACPAs and/or RF at screening.
Exclusion criteria included preexisting chronic autoimmune disease other than RA; active or historic recurrent bacterial, viral, fungal, mycobacterial, or other infections; evidence of untreated latent or active tuberculosis, or residing with or in close contact with individuals with active tuberculosis; history of any lymphoproliferative disorder; current or history of malignancy (except patients with adequately treated or excised nonmetastatic basal cell cancer, squamous cell cancer, or in situ cervical cancer); and other illnesses, clinical conditions, or laboratory anomalies that would have increased risk to the patient and/or affected study interpretation.
Before participating in the study, patients must have taken oral methotrexate at doses of 15–25 mg weekly for ≥3 months. In cases where patients experienced toxicity or intolerance of methotrexate, a dose between 10 and <15 mg was allowed. Patients must have had an inadequate response to methotrexate, defined as a lack of adequate benefit from methotrexate as determined by the patient and investigator, plus residual disease activity that met the study entry criteria. Patients continued their prestudy dose of methotrexate, which must have been stable for ≥4 weeks before the first dose of study drug and remained stable throughout the study. Patients were required to take a stable dose of supplemental folic acid ≥5 mg weekly, 24 hours after taking methotrexate.
The study design allowed up to 50% of patients to have taken 1 approved TNFi that was inadequately effective and/or not tolerated. TNFi were discontinued during a washout period for ≥4 weeks (etanercept) or ≥10 weeks (infliximab, adalimumab, golimumab, or certolizumab). A 3‐month washout period was required for previous tofacitinib treatment. Patients treated with other non‐TNFi biologic DMARDs were excluded.
Concomitant medications were permitted at stable doses, including antimalarials, nonsteroidal antiinflammatory drugs, selective cyclooxygenase 2 inhibitors, acetaminophen or paracetamol at doses ≤2.6 gm/day, opioids at doses with a potency equivalent to ≤30 mg of oral morphine, and oral glucocorticoids ≤10 mg prednisone or equivalent daily, beginning ≥4 weeks before the first study dose. For all other permitted medications, including chloroquine phosphate and hydroxychloroquine, patients must have been on a stable regimen before and during the study. DMARDs, including leflunomide, tofacitinib, and TNFi, were prohibited.
Patients provided written informed consent. This study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice Guidelines, and approved by the institutional review boards and/or ethics committees at each investigational center.
Study design and treatment
This phase IIa, randomized, double‐blind, placebo‐controlled, parallel‐group study was conducted at 32 centers across 9 countries (Bulgaria, Czech Republic, Georgia, Germany, Hungary, Poland, Serbia, Slovakia, and the US) and comprised a 5‐week screening period, an 8‐week treatment period, and a 4‐week follow‐up period. Using an interactive web‐based response system, patients were randomized 3:2 to receive oral PF‐06651600 (200 mg once daily supplied as 50‐mg tablets in blister packs) or oral matching placebo (once daily supplied in blister packs matching the PF‐06651600 tablets) for 8 weeks. Study staff entered the protocol and patient numbers to generate the treatment assignment, randomization number, and dispensable unit or container number for each patient. The sponsor, study investigators, and participants were blinded with regard to treatment allocation, and no one accessed the randomization scheme or study results until study completion.
For this first study of PF‐06651600 in patients with RA, a dose of 200 mg once daily was chosen because doses ≤400 mg once daily and single doses ≤800 mg were well‐tolerated in a phase I trial (ClinicalTrials.gov identifier: NCT02309827). Furthermore, pharmacokinetic (PK) modeling, assuming similar PKs in healthy subjects in the same trial (ClinicalTrials.gov identifier: NCT02309827) and patients with RA, predicted that treatment with PF‐06651600 at a dose of 200 mg once daily for 8 weeks would yield a median peak plasma maximum concentration (Cmax) of 1,254 ng/ml and an area under the concentration–time curve of 3,712 ng·hour/ml; modeling predicted these PK levels would inhibit in vitro signaling by 81% and 71% for IL‐15 and IL‐21, respectively. This degree of cytokine modulation indicated that the 200 mg once daily dose of PF‐06651600 would provide adequate pharmacology in the 8‐week treatment period to permit preliminary testing of the mechanism in this phase IIa study. Treatment duration beyond 8 weeks was limited by duration of nonclinical toxicity studies; however, 8 weeks was considered an adequate timeframe to conduct this study based on the rapid onset of action of JAK inhibition pharmacology (30).
Efficacy and safety end points
The primary efficacy end point was change from baseline in the Simplified Disease Activity Index (SDAI) score at week 8. SDAI has been demonstrated to be a valid and sensitive measure of RA disease activity (31).
Secondary efficacy end points included change from baseline in the SDAI score and components of the SDAI score (tender joint count [TJC], swollen joint count [SJC], physician global assessment of arthritis, patient global assessment of arthritis [PtGA], and hsCRP level) at weeks 1, 2, 4, 6, and 8; the proportion of patients who had low disease activity according to the SDAI (score ≤11) and disease in remission according to the SDAI (score ≤3.3) at weeks 4, 6, and 8 (week 8 data shown); change from baseline in the 4‐variable Disease Activity Score in 28 joints using the erythrocyte sedimentation rate (DAS‐ESR) and the 4‐variable DAS28 using the C‐reactive protein level (DAS28‐CRP) at weeks 1, 2, 4, 6, and 8; and the proportion of patients with 4‐variable DAS28‐ESR or 4‐variable DAS28‐CRP scores <2.6 at weeks 4, 6, and 8 (week 8 data shown). Exploratory efficacy end points included the ACR criteria for 20% improvement (ACR20), ACR50, and ACR70 response rates at weeks 4, 6, and 8 (week 8 data shown).
Safety and tolerability of PF‐06651600 were assessed by monitoring treatment‐emergent adverse events (TEAEs), and physical features, vital signs, and laboratory findings. Severity of AEs was classified by a medically qualified investigator as mild (the AE does not interfere with the patient’s usual function), moderate (the AE interferes to some extent with the patient’s usual function), or severe (the AE interferes significantly with the patient’s usual function). Grading was based on the Medical Dictionary for Regulatory Activities v20.1 coding. Absolute lymphocyte counts were graded post hoc using the Common Terminology Criteria for Adverse Events (CTCAE) version 5.0 as follows: grade 1 (less than the lower limit of normal [LLN] to 800/mm3); grade 2 (<800–500/mm3); grade 3 (<500–200/mm3); and grade 4 (<200/mm3). Patient‐reported outcomes were assessed as change from baseline in the disability index (DI) of the Health Assessment Questionnaire (HAQ) at weeks 1, 2, 4, 6, and 8.
Evaluation of PK (PF‐06651600 plasma concentration) was an exploratory objective. Blood plasma samples were analyzed using validated, sensitive, and specific high‐performance liquid chromatography–tandem mass spectrometry (Covance Bioanalytical Services). The lower limit of quantification for PF‐06651600 was 1.00 ng/ml.
Statistical analysis
The primary population for the efficacy analyses was the intent‐to‐treat population, which included patients who were randomized and received ≥1 dose of the randomized treatment. The safety population included patients who received ≥1 dose of the randomized treatment.
The study planned to enroll 60 patients, expecting a 15% dropout rate to yield ~50 completers: 30 completers in the PF‐06651600 group and 20 completers in the placebo group. The study had >90% power to detect a difference from placebo, assuming a 13‐point difference in response between the PF‐06651600 and placebo groups (the assumed SD was 14 points for both groups) and controlling the Type I error rate at 5%.
The primary efficacy end point analysis was conducted using a Bayesian analysis of covariance (ANCOVA) model with baseline SDAI score as a covariate. This model used an informative prior distribution for the placebo group (i.e., placebo data from recent tofacitinib trials with a similar patient population, study design, and efficacy/safety end points) after adjustment for baseline SDAI score and TNFi status from the present study. Utilizing historic placebo data from previous RA trials allowed for a decreased placebo sample size, thereby limiting the exposure of patients to placebo in this trial. The prior model for placebo assumed that the mean SDAI response in the placebo group followed a normal distribution with a mean of −13.7 and variance of 25. Noninformative prior distribution was used for the mean SDAI response in the PF‐06651600 group and for all other model parameters. The P values in the primary analysis were derived using posterior distribution for placebo‐adjusted treatment effects, which represents the likelihood that the treatment effect is ≤0.
Multiple sensitivity analyses were considered for the prior mean of the placebo distribution. The main sensitivity analysis was conducted using noninformative prior distribution (i.e., without historic placebo information) for a normally distributed placebo response with a mean of 0 and variance of 1,000.
Change from baseline in the SDAI, the components of the SDAI score, 4‐variable DAS28‐ESR, 4‐variable DAS28‐CRP, and HAQ DI were analyzed via a mixed‐effects repeated‐measures model (with treatment, visit, and treatment‐by‐visit as fixed effects, baseline value as a covariate, and patient as a random effect) with an unstructured covariance matrix assumed and no imputation for missing data. Due to the exploratory nature of the study, no statistical multiplicity adjustment was made.
The proportion of patients who had low disease activity or disease in remission, according to the SDAI scores or a 4‐variable DAS28‐ESR or 4‐variable DAS28‐CRP score of <2.6, and who met ACR20/50/70 criteria, were summarized descriptively, with missing values imputed using nonresponder imputation. Safety and clinical laboratory data were summarized descriptively. Statistical analysis was performed using SAS software version 9.4.
RESULTS
Patient characteristics
Seventy patients (15 from Bulgaria, 1 from Czech Republic, 20 from Georgia, 2 from Germany, 7 from Poland, 14 from Serbia, 6 from Slovakia, and 5 from the US) were randomized to receive PF‐06651600 (n = 42) or placebo (n = 28) between December 20, 2016 (first patient, first visit) and December 12, 2017 (last patient, last visit). No patients from Hungary were randomized. (Figure 1).
Figure 1.
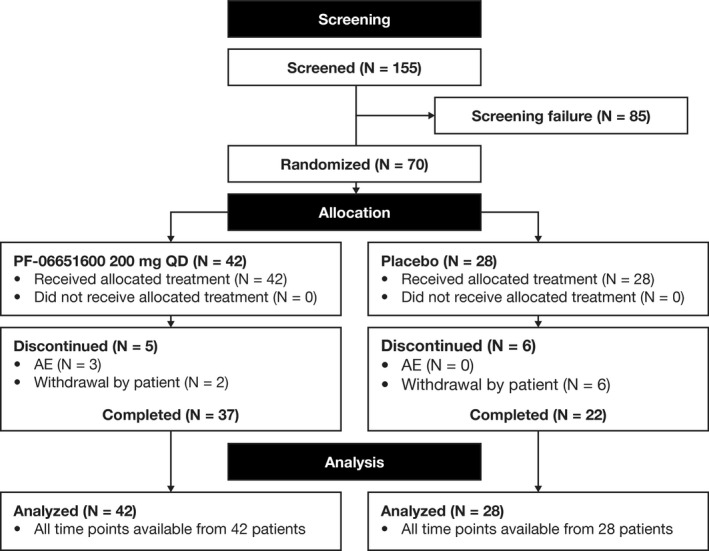
Disposition of the study patients. QD = once daily; AE = adverse event.
Patient demographic and baseline disease characteristics were generally balanced between treatment groups (Table 1). At baseline, patients had been receiving methotrexate at a mean ± SD dose of 15.0 ± 3.4 mg/week in the PF‐06651600 group (duration of treatment ranging 72–7,361 days) and 16.0 ± 4.4 mg/week in the placebo group (duration of treatment ranging 115–4,752 days); patients continued this treatment throughout the study. Overall, 24.3% of patients were previously treated with 1 TNFi, which was balanced across treatment groups. Prior and concomitant treatments are summarized in Table 1.
Table 1.
Baseline disease and demographic characteristics of the patients with RA treated with PF‐06651600 or placebo*
|
PF‐06651600 200 mg once daily (n = 42) |
Placebo (n = 28) |
|
|---|---|---|
| Age, years | 55.4 ± 11.7 | 54.2 ± 11.8 |
| Female, no. (%) | 33 (78.6) | 24 (85.7) |
| Race, no. (%) | ||
| White | 41 (97.6) | 28 (100.0) |
| Asian | 1 (2.4) | 0 (0.0) |
| Weight, mean kg | 77.0 | 74.5 |
| Duration since onset, years | 7.1 ± 7.1 | 8.4 ± 7.2 |
| Baseline disease characteristics | ||
| SDAI | 45.2 ± 13.2 | 44.9 ± 14.0 |
| SJC | 13.0 ± 5.4 | 12.1 ± 6.2 |
| TJC | 16.7 ± 6.8 | 16.8 ± 6.7 |
| PtGA, cm | 6.8 ± 1.6 | 6.9 ± 1.6 |
| PhGA, cm | 6.6 ± 1.4 | 7.4 ± 1.3 |
| 4‐variable DAS28‐CRP | 6.1 ± 0.9 | 6.0 ± 0.9 |
| 4‐variable DAS28‐ESR | 6.8 ± 0.8 | 6.7 ± 0.9 |
| hsCRP, mg/dl | 2.0 ± 1.8 | 1.7 ± 2.9 |
| ESR, mm/hour | 46.8 ± 20.7 | 42.0 ± 19.3 |
| HAQ DI | 1.8 ± 0.6 | 1.7 ± 0.6 |
| Prior treatment, no. (%)† | ||
| Methotrexate | 42 (100.0) | 28 (100.0) |
| Chloroquine phosphate | 2 (4.8) | 2 (7.1) |
| Hydroxychloroquine | 1 (2.4) | 0 |
| Leflunomide | 0 | 2 (7.1) |
| Tofacitinib | 2 (4.8) | 0 |
| TNFi | 10 (23.8) | 7 (25) |
| Glucocorticoids | 24 (57.1) | 16 (38.1) |
| Concomitant treatment† | ||
| Methotrexate | ||
| No. (%) | 42 (100.0) | 28 (100.0) |
| Dose, mg/week | 15.0 ± 3.4 | 16.0 ± 4.4 |
| Prednisone‡ | ||
| No. (%) | 4 (9.5) | 1 (3.6) |
| Dose, mg/day | 8.1 ± 2.4 | 2.5 ± – |
| Prednisolone | ||
| No. (%) | 7 (16.7) | 5 (17.9) |
| Dose, mg/day | 9.3 ± 1.9 | 8.5 ± 2.2 |
| Methylprednisolone | ||
| No. (%) | 10 (23.8) | 9 (32.1) |
| Dose, mg/day | 5.2 ± 1.9 | 4.7 ± 2.0 |
Except where indicated otherwise, values are the mean ± SD. RA = rheumatoid arthritis; SDAI = Simplified Disease Activity Index; SJC = swollen joint count; TJC = tender joint count; PtGA = patient global assessment of arthritis; PhGA = physician global assessment of arthritis; DAS28‐CRP = Disease Activity Score in 28 joints using the C‐reactive protein level; DAS28‐ESR = DAS28 using the erythrocyte sedimentation rate; hsCRP = high‐sensitivity CRP; HAQ DI = Health Assessment Questionnaire disability index; TNFi = tumor necrosis factor inhibitor.
World Health Organization Drug Dictionary Enhanced v201703 coding was applied.
One patient was excluded from this analysis because the patient started prednisone on day 78 of the study.
Efficacy outcomes
The primary efficacy end point, mean change from baseline in SDAI score at week 8, was greater with PF‐06651600 (−26.1 [95% credible interval −29.7, −22.4]) than placebo (−16.8 [95% credible interval −20.9, −12.7]; P < 0.001) using a Bayesian informative prior distribution for placebo response.
A Bayesian ANCOVA with a noninformative prior distribution for placebo response confirmed the significant treatment effect based on the change from baseline in SDAI scores observed in the primary analysis (P = 0.0023). A parametric ANCOVA (using the week 8 end point) and a mixed‐effects repeated‐measures model (including all SDAI time points) were consistent with the primary analysis. Two modifications to the primary analysis population (excluding 5 patients after applying the inclusion/exclusion criteria and 4 patients who did not complete the week 8 visit) produced similar estimates of the significant treatment effect.
Least squares mean (LSM) change from baseline in SDAI score showed statistical separation of the PF‐06651600 and placebo groups at week 6 (P = 0.011) and week 8 (P = 0.005) (Figure 2). LSM change from baseline in SDAI components showed a trend for improvement with PF‐06651600 versus placebo throughout the 8‐week treatment period (Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.41316/abstract); statistical separation of PF‐06651600 from placebo was shown at week 8 for the TJC (P = 0.008), week 6 for the PhGA score (P = 0.017), and weeks 2, 4, 6, and 8 for the PtGA score (all P < 0.05); there were no differences for these components at other time points, or for the SJC at any time point. To account for variability, hsCRP was expressed as median change (interquartile range). No differences in hsCRP were observed between the groups at any time point (Supplementary Figure 1, http://onlinelibrary.wiley.com/doi/10.1002/art.41316/abstract).
Figure 2.
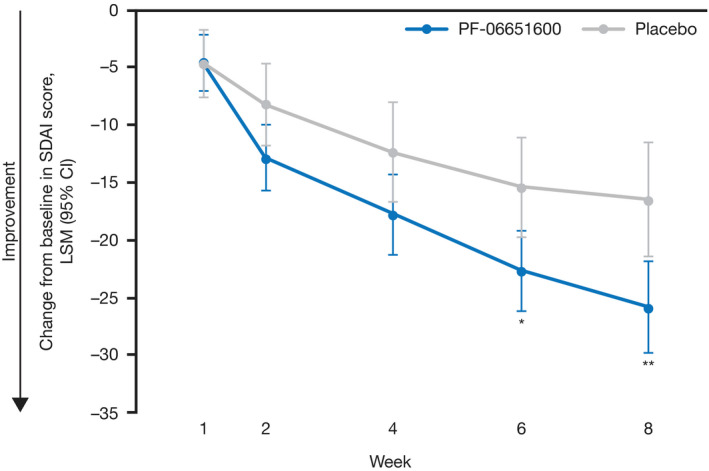
Least squares mean (LSM) change from baseline in Simplified Disease Activity Index (SDAI) at weeks 1, 2, 4, 6, and 8 in patients with rheumatoid arthritis receiving PF‐06651600 or placebo. A mixed‐effects repeated‐measures model was used for these analyses, with treatment, visit, and treatment‐by‐visit interaction as fixed effects, baseline value as a covariate, and patient as random effect (an unstructured covariance matrix assumed). Baseline was defined as the last nonmissing value from pretreatment period. Statistical multiplicity adjustment was not made due to the exploratory nature of this study. Values are the LSM change from baseline in SDAI score (95% credible interval [95% CI]). * = P < 0.05; ** = P < 0.01 versus placebo.
Low disease activity according to the SDAI was achieved in a higher proportion of patients receiving PF‐06651600 (23.8%) than those receiving placebo (7.1%) at week 8 (P = 0.042) (Table 2). No differences in rates of remission according to the SDAI were observed between the groups.
Table 2.
Proportion of patients in the ITT population who met study efficacy end points at week 8*
|
PF‐06651600 200 mg once daily (n = 42) |
Placebo (n = 28) |
P versus placebo | |
|---|---|---|---|
| Low disease activity according to SDAI (SDAI ≤11) | |||
| No. | 10 | 2 | 0.042 |
| % (SE) | 23.8 (6.6) | 7.1 (4.9) | |
| Remission according to SDAI (SDAI ≤3.3) | |||
| No. | 3 | 0 | 0.072 |
| % (SE) | 7.1 (4.0) | 0 (0) | |
| 4‐variable DAS28‐ESR <2.6 | |||
| No. | 3 | 0 | 0.072 |
| % (SE) | 7.1 (4.0) | 0 (0) | |
| 4‐variable DAS28‐CRP <2.6 | |||
| No. | 4 | 0 | 0.036 |
| % (SE) | 9.5 (4.5) | 0 (0) | |
| ACR response | |||
| ACR20 | |||
| No. | 30 | 10 | 0.002 |
| % (SE) | 71.4 (7.0) | 35.7 (9.1) | |
| ACR50 | |||
| No. | 15 | 3 | 0.008 |
| % (SE) | 35.7 (7.4) | 10.7 (5.9) | |
| ACR70 | |||
| No. | 5 | 0 | 0.017 |
| % (SE) | 11.9 (5.0) | 0 (0) |
The SE was calculated based on normal approximation of binomial proportions. Missing values were imputed using the nonresponder imputation method. Statistical multiplicity adjustment was not made due to the exploratory nature of this study. ITT = intent‐to‐treat; ACR20 = American College of Rheumatology 20% improvement criteria (see Table 1 for other definitions).
LSM change from baseline in the 4‐variable DAS28‐ESR and 4‐variable DAS28‐CRP showed a trend toward improvement with PF‐06651600 versus placebo during the 8‐week treatment period; statistical separation of PF‐06651600 from placebo was shown at weeks 2, 6, and 8 (all P < 0.05) for the 4‐variable DAS28‐ESR and at weeks 2, 4, 6, and 8 (all P < 0.05) for the 4‐variable DAS28‐CRP (Supplementary Figure 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.41316/abstract). The proportion of patients with a 4‐variable DAS28‐ESR and 4‐variable DAS28‐CRP score of <2.6 is shown in Table 2.
Exploratory efficacy analyses were also performed to determine the proportion of patients who met the ACR20/50/70 response criteria at week 8 (Table 2). At week 8, an ACR20 response was achieved in 30 patients (71.4%) who received PF‐06651600 versus 10 patients (35.7%) who received placebo (P = 0.002). An ACR50 response was achieved in 15 patients (35.7%) who received PF‐06651600 versus 3 patients (10.7%) who received placebo (P = 0.008). An ACR70 response was achieved in 5 patients (11.9%) who received PF‐06651600 versus no patients who received placebo (P = 0.017).
Safety outcomes
Median treatment duration was 57 days in both groups. Most patients (54 of 70 patients [77.1%]) received treatment for ≥57 days. All‐cause TEAEs, the number of patients with TEAEs, discontinuations due to TEAEs, and temporary discontinuations or dose reductions due to TEAEs were numerically higher in the PF‐06651600 group than in the placebo group (Table 3). No serious TEAEs, severe TEAEs, or deaths were reported. Overall, 25 patients (35.7%) experienced 36 TEAEs. Twelve events reported by 10 patients (14.3%) were considered treatment‐related, including 9 patients (21.4%) in the PF‐06651600 group and 1 patient (3.6%) in the placebo group. Most TEAEs (77.8%) were mild in severity (23 of 28 patients [82.1%] who received PF‐06651600 and 5 of 8 patients [62.5%] who received placebo). In the PF‐06651600 group, 3 patients (7.1%) discontinued due to TEAEs, including 1 patient due to suicidal ideation, 1 patient due to mild lymphopenia, and 1 patient due to mild hepatotoxicity. Suicidal ideation and hepatotoxicity were considered treatment‐related; suicidal ideation resolved upon withdrawal. The lymphopenia was not considered treatment‐related and resolved without intervention. Three patients (7.1%) in the PF‐06651600 group temporarily discontinued use due to AEs, including 1 patient due to arthralgia, 1 patient due to peripheral edema, and 1 patient due to pruritus, which were considered TEAEs, and 1 patient due to a urinary tract infection that started on day 1 before the first dose of PF‐06651600 and thus was considered an AE. The AE of pruritus was considered treatment‐related. All AEs that led to temporary discontinuation were mild in severity and resolved. No patients in the placebo group discontinued, experienced dose reduction, or temporarily discontinued due to TEAEs.
Table 3.
Summary of all‐cause TEAEs*
|
PF‐06651600 200 mg once daily (n = 42) |
Placebo (n = 28) |
Total (n = 70) |
|
|---|---|---|---|
| No. of TEAEs | 28 | 8 | 36 |
| No. (%) with TEAEs | 20 (47.6) | 5 (17.9) | 25 (35.7) |
| No. (%) with serious TEAEs | 0 (0) | 0 (0) | 0 (0) |
| No. (%) with severe TEAEs | 0 (0) | 0 (0) | 0 (0) |
| No. (%) who discontinued due to TEAE | 3 (7.1) | 0 (0) | 3 (4.3) |
| No. (%) with dose reduction or temporary discontinuation due to TEAEs | 2 (4.8) | 0 (0) | 2 (2.9) |
| No. (%) with mild/moderate/severe TEAEs by system organ class and preferred term† | |||
| Infections and infestations | 5 (11.9)/0 (0)/0 (0) | 1 (3.6)/0 (0)/0 (0) | 6 (8.6)/0 (0)/0 (0) |
| Influenza | 3 (7.1)/0 (0)/0 (0) | 0 (0)/0 (0)/0 (0) | 3 (4.3)/0 (0)/0 (0) |
| Asymptomatic bacteriuria | 1 (2.4)/0 (0)/0 (0) | 0 (0)/0 (0)/0 (0) | 1 (1.4)/0 (0)/0 (0) |
| Fungal skin infection | 1 (2.4)/0 (0)/0 (0) | 0 (0)/0 (0)/0 (0) | 1 (1.4)/0 (0)/0 (0) |
| Oral herpes | 1 (2.4)/0 (0)/0 (0) | 0 (0)/0 (0)/0 (0) | 1 (1.4)/0 (0)/0 (0) |
| Upper respiratory tract infection | 0 (0)/0 (0)/0 (0) | 1 (3.6)/0 (0)/0 (0) | 1 (1.4)/0 (0)/0 (0) |
| Skin and subcutaneous tissue disorders | 5 (11.9)/0 (0)/0 (0) | 1 (3.6)/0 (0)/0 (0) | 6 (8.6)/0 (0)/0 (0) |
| Pruritus | 2 (4.8)/0 (0)/0 (0) | 1 (3.6)/0 (0)/0 (0) | 3 (4.3)/0 (0)/0 (0) |
| Acne | 1 (2.4)/0 (0)/0 (0) | 0 (0)/0 (0)/0 (0) | 1 (1.4)/0 (0)/0 (0) |
| Dermatitis | 1 (2.4)/0 (0)/0 (0) | 0 (0)/0 (0)/0 (0) | 1 (1.4)/0 (0)/0 (0) |
| Maculopapular rash | 1 (2.4)/0 (0)/0 (0) | 0 (0)/0 (0)/0 (0) | 1 (1.4)/0 (0)/0 (0) |
| Blood and lymphatic system disorders | 2 (4.8)/1 (2.4)/0 (0) | 0 (0)/0 (0)/0 (0) | 2 (2.9)/1 (1.4)/0 (0) |
| Lymphopenia | 2 (4.8)/1 (2.4)/0 (0) | 0 (0)/0 (0)/0 (0) | 2 (2.9)/1 (1.4)/0 (0) |
| Gastrointestinal disorders | 2 (4.8)/1 (2.4)/0 (0) | 0 (0)/0 (0)/0 (0) | 2 (2.9)/1 (1.4)/0 (0) |
| Abdominal distension | 0 (0)/1 (2.4)/0 (0) | 0 (0)/0 (0)/0 (0) | 0 (0)/1 (1.4)/0 (0) |
| Glossitis | 1 (2.4)/0 (0)/0 (0) | 0 (0)/0 (0)/0 (0) | 1 (1.4)/0 (0)/0 (0) |
| Nausea | 1 (2.4)/0 (0)/0 (0) | 0 (0)/0 (0)/0 (0) | 1 (1.4)/0 (0)/0 (0) |
| Vomiting | 1 (2.4)/0 (0)/0 (0) | 0 (0)/0 (0)/0 (0) | 1 (1.4)/0 (0)/0 (0) |
| Abnormal laboratory findings | 2 (4.8)/0 (0)/0 (0) | 0 (0)/1 (3.6)/0 (0) | 2 (2.9)/1 (1.4)/0 (0) |
| Increased alanine aminotransferase | 0 (0)/0 (0)/0 (0) | 0 (0)/1 (3.6)/0 (0) | 0 (0)/1 (1.4)/0 (0) |
| Increased aspartate aminotransferase | 0 (0)/0 (0)/0 (0) | 0 (0)/1 (3.6)/0 (0) | 0 (0)/1 (1.4)/0 (0) |
| Increased blood creatine phosphokinase | 1 (2.4)/0 (0)/0 (0) | 0 (0)/0 (0)/0 (0) | 1 (1.4)/0 (0)/0 (0) |
| Positive cytomegalovirus test | 1 (2.4)/0 (0)/0 (0) | 0 (0)/0 (0)/0 (0) | 1 (1.4)/0 (0)/0 (0) |
| Musculoskeletal and connective tissue disorders | 2 (4.8)/0 (0)/0 (0) | 1 (3.6)/0 (0)/0 (0) | 3 (4.3)/0 (0)/0 (0) |
| Arthralgia | 1 (2.4)/0 (0)/0 (0) | 0 (0)/0 (0)/0 (0) | 1 (1.4)/0 (0)/0 (0) |
| Spinal pain | 0 (0)/0 (0)/0 (0) | 1 (3.6)/0 (0)/0 (0) | 1 (1.4)/0 (0)/0 (0) |
| Synovitis | 1 (2.4)/0 (0)/0 (0) | 0 (0)/0 (0)/0 (0) | 1 (1.4)/0 (0)/0 (0) |
| Nervous system disorders | 0 (0)/0 (0)/0 (0) | 2 (7.1)/1 (3.6)/0 (0) | 2 (2.9)/1 (1.4)/0 (0) |
| Headache | 0 (0)/0 (0)/0 (0) | 2 (7.1)/1 (3.6)/0 (0) | 2 (2.9)/1 (1.4)/0 (0) |
Data were collected beginning at the time of the first dose of study drug. For treatment‐emergent adverse events (TEAEs), patients were counted once per treatment, except for the total number of TEAEs. The severity of a TEAE was determined by the investigator. A serious TEAE is defined as any untoward medical occurrence at any dose that is life‐threatening, requires inpatient hospitalization or prolongation of existing hospitalization, or results in persistent or significant disability/incapacity, congenital anomaly/birth defect, or death, or is considered to be an important medical event. A severe TEAE is defined as one that interferes significantly with a patient’s usual function. A moderate TEAE is defined as one that interferes to some extent with a patient’s usual function. A mild TEAE is defined as one that does not interfere with a patient’s usual function. If the same patient in a treatment group had >1 occurrence in a preferred term category, the most severe occurrence was counted. Medical Dictionary for Regulatory Activities v20.1 coding was applied.
TEAEs that occurred in ≥3% of patients across both treatment arms. The number of patients in the system organ class (i.e., infections and infestations) is not necessarily the sum of the numbers of patients in the subcategories, since a patient may have reported ≥2 different TEAEs in the system organ class.
The most common TEAEs (all mild) were infections and infestations and skin and subcutaneous tissue disorders (11.9% in the PF‐06651600 group and 3.6% in the placebo group for both classes). The most frequently reported infection and infestation and skin and subcutaneous TEAEs were influenza (7.1% in the PF‐06651600 group and 0% in the placebo group), and pruritus (4.8% in the PF‐06651600 group and 3.6% in the placebo group), respectively. There was 1 mild case of herpes simplex in the PF‐06651600 group considered to be treatment‐related; this resolved within 3 days without study treatment discontinuation or antiviral therapy.
Mean absolute values for hematology tests, liver function tests, and other laboratory tests are shown in Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.41316/abstract. Median counts of hemoglobin (3.0%), platelets (25.0%), lymphocytes (21.0%), and neutrophils (24.0%) were decreased from baseline by week 8 only in the PF‐06651600 group. These values were not considered clinically relevant and returned to near baseline levels by follow‐up at week 12. No bleeding events, reductions in platelet counts <100 × 103/mm3, or clinically significant symptoms of anemia or hemodynamic compromise were reported. Three patients in the PF‐06651600 group met the protocol‐specified discontinuation criteria for hemoglobin decline from baseline (>2 gm/dl). Two patients had already completed the 8‐week treatment period and continued with follow‐up. The third patient met the discontinuation criteria at week 1, but hemoglobin levels were so robust that it was deemed not clinically significant. PF‐06651600 treatment continued and hemoglobin returned to baseline levels by week 12. In the PF‐06651600 group, 1 patient with a low normal baseline absolute lymphocyte count (890/mm3; normal range 800–3,000/mm3), had an absolute lymphocyte count of 480/mm3, met the discontinuation criteria for absolute lymphocyte count (<500/mm3), and was diagnosed as having mild lymphopenia by a medically qualified investigator. The lymphopenia was not considered treatment‐related and resolved without medication within 5 days after study discontinuation. Two patients in the PF‐06651600 group were diagnosed as having lymphopenia (1 mild case and 1 moderate case) by a medically qualified investigator; both were considered treatment‐related. The mild case of lymphopenia, with an absolute lymphocyte count of 450/mm3 (normal range 800–3,000/mm3), resolved without intervention and PF‐06651600 treatment continued. The moderate case of lymphopenia, with an absolute lymphocyte count of 450/mm3 (normal range 910–4,280/mm3), resolved within 3 days following 1 dose of the supplement oral transfer factor 10 mg once daily and PF‐06651600 treatment continued. All 3 lymphopenia cases were in the CTCAE grade 3 range (absolute lymphocyte count <500–200/mm3). No patients met the discontinuation criteria (<0.75 x 109/liter) for neutropenia.
In the placebo group, 3 cases (1 per test) met the abnormality criteria for alanine aminotransferase, aspartate aminotransferase (both >3.0 times the upper limit of normal [ULN]), or high‐density lipoprotein cholesterol (<0.5 times the LLN). No patients met the abnormality criteria for bilirubin (>1.5 times the ULN), total cholesterol (>1.3 times the ULN), low‐density lipoprotein cholesterol (>1.2 times the ULN), creatine kinase (>2.0 times the ULN), or serum creatinine (>1.3 times the ULN).
Patient‐reported outcomes
No significant differences in LSM change from baseline for HAQ DI were observed between the treatment groups at any time point (Supplementary Figure 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.41316/abstract).
Pharmacokinetics
On day 57, median plasma concentrations of PF‐06651600 at the nominal time points of 0.5, 1, and 2 hours after dosing were 1,660 ng/ml, 1,910 ng/ml, and 1,400 ng/ml, respectively (predicted Cmax 1,254 ng/ml), indicating that adequate exposure was achieved to produce pharmacologic activity.
DISCUSSION
This phase IIa proof‐of‐concept study was the first to evaluate the efficacy and safety of the oral JAK3/TEC inhibitor PF‐06651600 in patients with RA and an inadequate response to methotrexate. Approximately 25% of patients were previously treated with 1 TNFi, which was balanced across treatment groups. PF‐06651600 treatment was associated with significant improvements in RA disease scores (as measured by primary and secondary end points) and was generally well‐tolerated across the 8‐week trial.
Common γ‐chain cytokine signaling, via JAK3, has been implicated in RA pathophysiology and other inflammatory diseases, including alopecia areata, psoriasis, ulcerative colitis, and Crohn’s disease (32, 33) and provided the basis for the development of JAK3 inhibitors. The moderately selective JAK3 inhibitor decernotinib demonstrated efficacy, as monotherapy (34) and in combination with a DMARD (35, 36), for the treatment of RA. However, decernotinib has shown some activity against JAK1 in in vitro assays (37, 38), and neutropenia was observed in clinical trials (34, 36). Treatment with the moderately selective JAK3 inhibitor peficitinib demonstrated efficacy as monotherapy (39) and in combination with methotrexate (22) in patients with RA, and as monotherapy in patients with plaque psoriasis (40).
PF‐06651600 has a high degree of selectivity toward JAK3 due to a covalent interaction with a cysteine residue (Cys‐909) in the JAK3 catalytic domain that is replaced by a serine residue in other JAK isoforms (25). Furthermore, PF‐06651600 irreversibly inhibits the TEC kinases due to the presence of a cysteine residue in the same position as Cys‐909 for JAK3 (25). Clinical consequences of JAK1 inhibition include elevation of cholesterol and liver enzyme levels (33, 41), and rapid reduction of CRP levels (32), which may be associated with IL‐6 signaling (42, 43). Since JAK3‐selective inhibition only inhibits γ–common chain receptor signaling, it spares IL‐6 signaling and other JAK1‐dependent immunoregulatory cytokines, including IL‐10 and IL‐27 (11, 24). This is one of several characteristics that may contribute to the overall benefit/risk profile of PF‐06651600. There were no clinically significant cases of anemia or neutropenia, findings sometimes observed with JAK1/2 inhibition. Although there were no cases of clinically relevant thrombocytopenia, reductions in platelet levels were observed in some patients who received PF‐06651600. The mechanism for this reduction is unknown but may result from TEC kinase inhibition, which plays a role in platelet aggregation (44). This study provides preliminary data that PF‐06651600 may have therapeutic potential in RA. Further preclinical, pharmacokinetic, and clinical studies are required to fully characterize the PF‐06651600 selectivity profile and determine the associated clinical consequences and optimal dose.
PF‐06651600 was generally well‐tolerated when administered for 8 weeks in this small trial. Most TEAEs were mild in severity, and no serious AEs or deaths were reported. There were no events of herpes zoster, but there was 1 mild case of herpes simplex in the PF‐06651600 group that was considered treatment‐related, which resolved within 3 days without intervention.
The absence of any clinically relevant changes in HAQ DI between the groups may be due to the short study duration or the small number of patients included. A longer trial with a larger patient population should be conducted to explore HAQ DI changes further.
Results from this trial support further exploration of PF‐06651600 in patients with RA and other inflammatory conditions. PF‐06651600 is being evaluated for treatment of alopecia areata (ClinicalTrials.gov identifiers: NCT02974868 and NCT03732807), vitiligo (ClinicalTrials.gov identifier: NCT03715829), ulcerative colitis (ClinicalTrials.gov identifier: NCT02958865), and Crohn’s disease (ClinicalTrials.gov identifier: NCT03395184). Recently, PF‐06651600 received breakthrough therapy designation by the US Food and Drug Administration for treatment of patients with alopecia areata, which was supported by positive results from a phase IIa study (ClinicalTrials.gov identifier: NCT02974868). Long‐term studies are warranted to evaluate the efficacy and safety of PF‐06651600 in patients with RA to understand the lifelong impact of PF‐06651600 on patients’ health and quality of life.
Study limitations are similar to those in other phase IIa studies and include a small sample size (n = 70) and a limited treatment period (8 weeks). In this preliminary proof‐of‐concept study, we evaluated a single dose of PF‐06651600 to verify the therapeutic potential of PF‐06651600 in RA. A phase IIb dose‐ranging study with a larger sample size would be required to probe optimal dosing and confirm the treatment effect. Study inclusion required patients to be seropositive for ACPAs and/or RF; thus, results cannot be generalized to patients with RA who are not seropositive. Most patients were European; therefore, a confirmatory study with a wider cohort will be required to determine whether the findings are generalizable to a broader population. Finally, P values were not controlled for multiple comparisons due to the exploratory nature of this study.
In this phase IIa study of patients with moderate‐to‐severe RA, treatment with the oral JAK3/TEC inhibitor PF‐06651600 reduced RA disease activity and was generally well‐tolerated over 8 weeks in this limited study. Additional trials will be required to understand the safety and efficacy of PF‐06651600 with a longer treatment duration.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content and all authors approved the final version to be published. Dr. Robinson had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Banfield, Peeva, Beebe.
Acquisition of data
Robinson, Damjanov, Stamenkovic, Radunovic, Kivitz, Peeva, Beebe.
Analysis and interpretation of the data
Robinson, Damjanov, Stamenkovic, Radunovic, Kivitz, Cox, Manukyan, Banfield, Saunders, Chandra, Vincent, Mancuso, Peeva, Beebe.
ROLE OF THE STUDY SPONSOR
This study was sponsored by Pfizer Inc. Medical writing support under the guidance of the authors was provided by Jennifer Arnold, PhD, CMC Connect, McCann Health Medical Communications and was funded by Pfizer Inc. in accordance with Good Publication Practice guidelines (Ann Intern Med 2015;163:461–464).
Supporting information
Supplementary Material
Acknowledgments
We would like to thank all of the patients, study investigators, and staff who were involved with this study.
ClinicalTrials.gov identifier: NCT02969044.
Supported by Pfizer, Inc.
Dr. Damjanov has received consulting fees, speaking fees, and/or honoraria from AbbVie, Boehringer Ingelheim, Gedeon Richter, Merck, Novartis, Pfizer, and Roche (less than $10,000 each). Dr. Kivitz has received consulting fees, speaking fees, and/or honoraria from AbbVie, Boehringer Ingelheim, Celgene, Flexion, Genzyme, Janssen, Merck, Novartis, Regeneron, Sanofi, Sun Pharma Advanced Research, and UCB (less than $10,000 each) and from Pfizer (more than $10,000), and owns stock or stock options in Amgen, Gilead, GlaxoSmithKline, Novartis, Pfizer, and Sanofi. Drs. Cox, Manukyan, Banfield, Saunders, Vincent, Mancuso, Peeva, and Beebe and Ms Chandra own stock or stock options in Pfizer, Inc. No other disclosures relevant to this article were reported.
Upon request, and subject to certain criteria, conditions, and exceptions (see https ://www.pfizer.com/science/clinical‐trials/trial‐data‐and‐results for more information), Pfizer will provide access to individual de‐identified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (1) for indications that have been approved in the US and/or EU or (2) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de‐identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
References
- 1. Cross M, Smith E, Hoy D, Carmona L, Wolfe F, Vos T, et al. The global burden of rheumatoid arthritis: estimates from the global burden of disease 2010 study. Ann Rheum Dis 2014;73:1316–22. [DOI] [PubMed] [Google Scholar]
- 2. Willemze A, Trouw LA, Toes RE, Huizinga TW. The influence of ACPA status and characteristics on the course of RA [review]. Nat Rev Rheumatol 2012;8:144–52. [DOI] [PubMed] [Google Scholar]
- 3. Smolen JS, Breedveld FC, Burmester GR, Bykerk V, Dougados M, Emery P, et al. Treating rheumatoid arthritis to target: 2014 update of the recommendations of an international task force. Ann Rheum Dis 2016;75:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Singh JA, Saag KG, Bridges SL Jr, Akl EA, Bannuru RR, Sullivan MC, et al. 2015 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol 2016;68:1–26. [DOI] [PubMed] [Google Scholar]
- 5. Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet 2010;376:1094–108. [DOI] [PubMed] [Google Scholar]
- 6. Smolen JS, Maini RN. Interleukin‐6: a new therapeutic target. Arthritis Res Ther 2006;8 Suppl 2:S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cohen MD, Keystone E. Rituximab for rheumatoid arthritis. Rheumatol Ther 2015;2:99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Korhonen R, Moilanen E. Abatacept, a novel CD80/86-CD28 T cell costimulation modulator, in the treatment of rheumatoid arthritis. Basic Clin Pharmacol Toxicol 2009;104:276–84. [DOI] [PubMed] [Google Scholar]
- 9. Sokka T, Hetland ML, Makinen H, Kautiainen H, Horslev‐Petersen K, Luukkainen RK, et al. Remission and rheumatoid arthritis: data on patients receiving usual care in twenty‐four countries. Arthritis Rheum 2008;58:2642–51. [DOI] [PubMed] [Google Scholar]
- 10. Felson DT, Smolen JS, Wells G, Zhang B, van Tuyl LH, Funovits J, et al. American College of Rheumatology/European League Against Rheumatism provisional definition of remission in rheumatoid arthritis for clinical trials. Arthritis Rheum 2011;63:573–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hodge JA, Kawabata TT, Krishnaswami S, Clark JD, Telliez JB, Dowty ME, et al. The mechanism of action of tofacitinib: an oral janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin Exp Rheumatol 2016;34:318–28. [PubMed] [Google Scholar]
- 12. Fridman JS, Scherle PA, Collins R, Burn TC, Li Y, Li J, et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol 2010;184:5298–307. [DOI] [PubMed] [Google Scholar]
- 13. Genovese MC, Kremer J, Zamani O, Ludivico C, Krogulec M, Xie L, et al. Baricitinib in patients with refractory rheumatoid arthritis. N Engl J Med 2016;374:1243–52. [DOI] [PubMed] [Google Scholar]
- 14. Taylor PC, Keystone EC, van der Heijde D, Weinblatt ME, Morales LD, Gonzaga JR, et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N Engl J Med 2017;376:652–62. [DOI] [PubMed] [Google Scholar]
- 15. Parmentier JM, Voss J, Graff C, Schwartz A, Argiriadi M, Friedman M, et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT‐494). BMC Rheumatol 2018;2:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Burmester GR, Kremer JM, van den Bosch F, Kivitz A, Bessette L, Li Y, et al. Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease‐modifying antirheumatic drugs (SELECT‐NEXT): a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet 2018;391:2503–12. [DOI] [PubMed] [Google Scholar]
- 17. Fleischmann R, Pangan AL, Song IH, Mysler E, Bessette L, Peterfy C, et al. Upadacitinib versus placebo or adalimumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase III, double‐blind, randomized controlled trial. Arthritis Rheumatol 2019;71:1788–800. [DOI] [PubMed] [Google Scholar]
- 18. Van Rompaey L, Galien R, van der Aar EM, Clement‐Lacroix P, Nelles L, Smets B, et al. Preclinical characterization of GLPG0634, a selective inhibitor of JAK1, for the treatment of inflammatory diseases. J Immunol 2013;191:3568–77. [DOI] [PubMed] [Google Scholar]
- 19. Westhovens R, Taylor PC, Alten R, Pavlova D, Enriquez‐Sosa F, Mazur M, et al. Filgotinib (GLPG0634/GS-6034), an oral JAK1 selective inhibitor, is effective in combination with methotrexate (MTX) in patients with active rheumatoid arthritis and insufficient response to MTX: results from a randomised, dose‐finding study (DARWIN 1). Ann Rheum Dis 2017;76:998–1008. [DOI] [PubMed] [Google Scholar]
- 20. Genovese MC, Kalunian K, Gottenberg JE, Mozaffarian N, Bartok B, Matzkies F, et al. Effect of filgotinib vs placebo on clinical response in patients with moderate to severe rheumatoid arthritis refractory to disease‐modifying antirheumatic drug therapy: the FINCH 2 randomized clinical trial. JAMA 2019;322:315–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hamaguchi H, Amano Y, Moritomo A, Shirakami S, Nakajima Y, Nakai K, et al. Discovery and structural characterization of peficitinib (ASP015K) as a novel and potent JAK inhibitor. Bioorg Med Chem 2018;26:4971–83. [DOI] [PubMed] [Google Scholar]
- 22. Kivitz AJ, Gutierrez‐Urena SR, Poiley J, Genovese MC, Kristy R, Shay K, et al. Peficitinib, a JAK inhibitor, in the treatment of moderate‐to‐severe rheumatoid arthritis in patients with an inadequate response to methotrexate. Arthritis Rheumatol 2017;69:709–19. [DOI] [PubMed] [Google Scholar]
- 23. Tanaka Y, Takeuchi T, Tanaka S, Kawakami A, Iwasaki M, Song YW, et al. Efficacy and safety of peficitinib (ASP015K) in patients with rheumatoid arthritis and an inadequate response to conventional DMARDs: a randomised, double‐blind, placebo‐controlled phase III trial (RAJ3). Ann Rheum Dis 2019;78:1320–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Telliez JB, Dowty ME, Wang L, Jussif J, Lin T, Li L, et al. Discovery of a JAK3‐selective inhibitor: functional differentiation of JAK3‐selective inhibition over pan‐JAK or JAK1‐selective inhibition. ACS Chem Biol 2016;11:3442–51. [DOI] [PubMed] [Google Scholar]
- 25. Xu H, Jesson MI, Seneviratne UI, Lin TH, Sharif MN, Xue L, et al. PF‐06651600, a dual JAK3/TEC family kinase inhibitor. ACS Chem Biol 2019;14:1235–42. [DOI] [PubMed] [Google Scholar]
- 26. Andreotti AH, Schwartzberg PL, Joseph RE, Berg LJ. T cell signaling regulated by the TEC family kinase. ITK. Cold Spring Harb Perspect Biol 2010;2:a002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Corneth OB, Wolterink RG, Hendriks RW. BTK signaling in B cell differentiation and autoimmunity. Curr Top Microbiol Immunol 2016;393:67–105. [DOI] [PubMed] [Google Scholar]
- 28. O'Shea JJ, Kontzias A, Yamaoka K, Tanaka Y, Laurence A. Janus kinase inhibitors in autoimmune diseases. Ann Rheum Dis 2013;72 Suppl 2:ii111–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO III, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010;62:2569–81. [DOI] [PubMed] [Google Scholar]
- 30. Fleischmann R, Kremer J, Tanaka Y, Gruben D, Kanik K, Koncz T, et al. Efficacy and safety of tofacitinib in patients with active rheumatoid arthritis: review of key Phase 2 studies. Int J Rheum Dis 2016;19:1216–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Machold KP, Smolen JS. Adalimumab: a new TNF‐ɑ antibody for treatment of inflammatory joint disease. Expert Opin Biol Ther 2003;3:351–60. [DOI] [PubMed] [Google Scholar]
- 32. Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol 2017;76:736–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. De Vries LC, Wildenberg ME, de Jonge WJ, D'Haens GR. The future of janus kinase inhibitors in inflammatory bowel disease. J Crohns Colitis 2017;11:885–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fleischmann RM, Damjanov NS, Kivitz AJ, Legedza A, Hoock T, Kinnman N. A randomized, double‐blind, placebo‐controlled, twelve‐week, dose‐ranging study of decernotinib, an oral selective JAK‐3 inhibitor, as monotherapy in patients with active rheumatoid arthritis. Arthritis Rheumatol 2015;67:334–43. [DOI] [PubMed] [Google Scholar]
- 35. Genovese MC, Yang F, Ostergaard M, Kinnman N. Efficacy of VX‐509 (decernotinib) in combination with a disease‐modifying antirheumatic drug in patients with rheumatoid arthritis: clinical and MRI findings. Ann Rheum Dis 2016;75:1979–83. [DOI] [PubMed] [Google Scholar]
- 36. Genovese MC, van Vollenhoven RF, Pacheco‐Tena C, Zhang Y, Kinnman N. VX‐509 (decernotinib), an oral selective JAK‐3 inhibitor, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheumatol 2016;68:46–55. [DOI] [PubMed] [Google Scholar]
- 37. Clark JD, Flanagan ME, Telliez JB. Discovery and development of janus kinase (JAK) inhibitors for inflammatory diseases. J Med Chem 2014;57:5023–38. [DOI] [PubMed] [Google Scholar]
- 38. Farmer LJ, Ledeboer MW, Hoock T, Arnost MJ, Bethiel RS, Bennani YL, et al. Discovery of VX‐509 (decernotinib): a potent and selective janus kinase 3 inhibitor for the treatment of autoimmune diseases. J Med Chem 2015;58:7195–216. [DOI] [PubMed] [Google Scholar]
- 39. Takeuchi T, Tanaka Y, Iwasaki M, Ishikura H, Saeki S, Kaneko Y. Efficacy and safety of the oral janus kinase inhibitor peficitinib (ASP015K) monotherapy in patients with moderate to severe rheumatoid arthritis in Japan: a 12‐week, randomised, double‐blind, placebo‐controlled phase IIb study. Ann Rheum Dis 2016;75:1057–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Papp K, Pariser D, Catlin M, Wierz G, Ball G, Akinlade B, et al. A phase 2a randomized, double‐blind, placebo‐controlled, sequential dose‐escalation study to evaluate the efficacy and safety of ASP015K, a novel janus kinase inhibitor, in patients with moderate‐to‐severe psoriasis. Br J Dermatol 2015;173:767–76. [DOI] [PubMed] [Google Scholar]
- 41. Genovese MC, Smolen JS, Weinblatt ME, Burmester GR, Meerwein S, Camp HS, et al. Efficacy and safety of ABT‐494, a selective JAK‐1 inhibitor, in a phase IIb study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Rheumatol 2016;68:2857–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nishimoto N, Yoshizaki K, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, et al. Treatment of rheumatoid arthritis with humanized anti–interleukin‐6 receptor antibody: a multicenter, double‐blind, placebo‐controlled trial. Arthritis Rheum 2004;50:1761–9. [DOI] [PubMed] [Google Scholar]
- 43. Smolen JS, Beaulieu A, Rubbert‐Roth A, Ramos‐Remus C, Rovensky J, Alecock E, et al. Effect of interleukin‐6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double‐blind, placebo‐controlled, randomised trial. Lancet 2008;371:987–97. [DOI] [PubMed] [Google Scholar]
- 44. Chen J, Kinoshita T, Gururaja T, Sukbuntherng J, James D, Lu D, et al. The effect of Bruton's tyrosine kinase (BTK) inhibitors on collagen‐induced platelet aggregation, BTK, and tyrosine kinase expressed in hepatocellular carcinoma (TEC). Eur J Haematol 2018;101:604–12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material