

Abstract
A bifunctional 3d‐metal catalyst for the cascade synthesis of diverse pyrroles from nitroarenes is presented. The optimal catalytic system Co/NGr‐C@SiO2‐L is obtained by pyrolysis of a cobalt‐impregnated composite followed by subsequent selective leaching. In the presence of this material, (transfer) hydrogenation of easily available nitroarenes and subsequent Paal–Knorr/Clauson‐Kass condensation provides >40 pyrroles in good to high yields using dihydrogen, formic acid, or a CO/H2O mixture (WGSR conditions) as reductant. In addition to the favorable step economy, this straightforward domino process does not require any solvents or external co‐catalysts. The general synthetic utility of this methodology was demonstrated on a variety of functionalized substrates including the preparation of biologically active and pharmaceutically relevant compounds, for example, (+)‐Isamoltane.
Keywords: cobalt, heterogeneous catalysis, hydrogenation, nitroarene, pyrrole
A general cascade synthesis of pyrroles from nitroarenes is reported. The process is catalyzed by a heterogeneous cobalt catalyst using either dihydrogen, formic acid, or a CO/H2O mixture as reducing agents. This strategy was applied to the synthesis of biologically active compounds, including (+)‐Isamoltane.

Introduction
Synthetic methodologies, such as tandem, domino, or cascade reactions, involve multiple reaction steps in a single pot. They are true power tools to increase the molecular complexity of the final products in a straightforward manner.1, 2 Following this general concept, the amount of time and energy consumed for a given product is significantly reduced compared to the corresponding stepwise procedures. In fact, the avoidance of separation steps and isolation of intermediates offers significant economic and environmental benefits. The main challenge in developing such methodologies involving several catalytic events is the design of the appropriate catalysts compatible with each other, as well as the reagents and intermediates. Due to the intrinsic advantages, interesting developments applying homogeneous organometallic complexes or organo‐catalysts have been achieved.3, 4 On the other hand, heterogeneous catalytic materials are scarcely known in this area despite their advantageous recyclability, separation, and stability.5, 6
In the past decade, significant progress in base‐metal catalysis for redox transformations has been observed.7 In particular, metal‐nitrogen‐carbon (3d‐M/N/C) catalysts prepared by pyrolysis of supported molecularly‐defined complexes or immobilized MOFs have been widely applied for organic transformations and energy storage applications. An interesting feature of most of these catalysts is the presence of 3d‐metal/3d‐metal oxide core–shell nanoparticles embedded in N‐doped carbonaceous material.8, 9, 10 This specific architecture is believed to be crucial for the activation of hydrogen and related processes. Thus, (transfer) hydrogenations of alkenes, ketones, nitriles, and nitroarenes can be performed under comparably mild conditions. In addition, reductive aminations to prepare a wide variety of amines were realized.11 Apart from such basic C−N bond forming reactions, the synthesis of important N‐heterocycles has been explored to a lesser extent. In 2016 Kempe and co‐workers reported a cobalt‐based nanocatalyst for the preparation of benzimidazoles from nitroarenes and aldehydes.12 This pioneering work sparked the development of other 3d‐metal catalysts for the synthesis of various N‐heterocycles from nitroarenes.13 Among the different heterocycles, pyrroles are an important class of compounds with useful biological and physical properties. Hence, a variety of methods for their synthesis exists,14, 15, 16, 17 including traditional, but still popular, Hantzsch and Paal–Knorr synthesis. Since its discovery in 1884,18, 19 the latter reaction (1,4‐diketones and primary amines) remains a reliable transformation that continues to be used, for example, in the total synthesis of natural products. As a prominent example, the pyrrole ring of the drug Lipitor is produced on an industrial scale using Paal–Knorr synthesis.20
For the preparation of N‐aryl pyrroles anilines must be used; these are typically obtained via hydrogenation of the corresponding nitroarenes. Obviously, the direct use of nitroarenes in Paal–Knorr type reactions has an attractive step‐economic advantage over anilines. Known synthetic methodologies for this transformation typically involve the use of stoichiometric amounts of a reducing agent in the presence of a strong acid (e.g. In/HCl, In/AcOH),21, 22 which is an evident drawback. Surprisingly, only few catalytic systems using more environmentally friendly reducing agents are known for such processes. While Chung demonstrated the principal feasibility of using CO/H2O as a hydrogen source in a single example using a Co2Rh2/C catalyst,23 Guerrero‐Ríos and co‐workers used formic acid as a reductant in the presence of stabilized rhodium nanoparticles in ionic liquids (two examples).24 In fact, only the Zhang group developed a general catalyst for the reaction of nitroarenes with formic acid to give pyrroles.25 To the best of our knowledge, only one report (one example) for such transformations using H2 is known, which is based on Pt and Pd as noble metals.26
Inspired by the recent success of M/N/C materials to promote selective hydrogenations of nitroarenes in the presence of sensitive functional groups, we became interested in the ability of these materials for pyrrole synthesis from nitroarenes. Ideally, a suitable catalyst system should not only be able to catalyze the initial hydrogenation step, but also to drive the subsequent condensation without an external acid. Thus, in the desired catalytic material, metal nanoparticles should be located on an acidic support. Following this idea, herein we report cobalt/cobalt oxide core–shell particles surrounded by a nitrogen‐doped graphene shell supported on fumed silica (Co/NGr‐C@SiO2‐L), which allow for the preparation of diverse N‐aryl pyrroles from nitroarenes and different 1,4‐dicarbonyl compounds and their surrogates. This process allows the use of different reductants, including hydrogen, and affords the desired heterocycles in good to high yield under solvent‐free conditions with only environmentally benign by‐products (Scheme 1 A).
Scheme 1.

A) Co‐catalyzed synthesis of pyrroles from nitroarenes. B) Preparation and structural representation of the catalysts.
Results and Discussion
Identification of the Active Catalyst
We started our catalytic investigations using model substrate 1, 40 bar dihydrogen as a reductant under solvent‐free conditions for the synthesis of N‐phenylpyrrole 2 via a Clauson‐Kaas modification27 of classical Paal–Knorr synthesis (Table 1). In this approach, 2,5‐dimethoxytetrahydrofuran (DMTHF) is utilized as a masked succinaldehyde giving unsubstituted pyrroles.
Table 1.
Synthesis of 2 from nitrobenzene 1 with heterogeneous catalysts.[a]
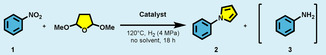
|
Entry |
Catalyst |
mol % (mg) |
Conv. [%][b] |
2, (3) [%][b] |
|---|---|---|---|---|
|
1 |
Co/NGr‐C@SiO2 |
4.3 (40) |
100 |
82 (0) |
|
2 |
Co/NGr‐C@SiO2‐L |
1.8 (40) |
100 |
83 (0) |
|
3 |
Co/NGr‐C@SiO2 |
2.1 (20) |
91 |
20 (63) |
|
4 |
Co‐Co3O4/NGr@C |
4.1 (40) |
100 |
83 (0) |
|
5 |
Ni‐NiO/NGr@C |
4.3 (40) |
0 |
0 (0) |
|
6 |
Fe2O3/NGr@C |
4.2 (40) |
7 |
5 (0) |
|
7 |
Ni‐Si/NiO‐SiO2@SiO2 |
4.8 (40) |
9 |
5 (0) |
[a] Reaction conditions: 0.5 mmol 1, 20–40 mg catalyst, 40 bar H2, 120 °C, 18 hours; 0.8 mL DMTHF; [b] Determined by gas chromatography‐flame ionization detector (GC‐FID) using n‐hexadecane as an internal standard.
For an initial catalyst testing, different M/N/C nanoparticles on silica and carbon (M=Co, Fe, Ni, Mn) were selected and used for 18 h at 120 °C. In general, the M/NGr‐C@support catalysts were prepared starting from simple metal salts and 1,10‐phenanthroline was used as a nitrogen‐containing ligand and graphene precursor (Scheme 1 B). As an example, the in situ generated cobalt phenanthroline complex was supported on fumed silica, then dried and pyrolyzed at 800 °C for 2 h to give Co/NGr‐C@SiO2. Recently, our group demonstrated that this latter material is active in the semihydrogenation of alkynes to alkenes,28 while He et al. used it in oxidative syntheses of 2‐arylbenzoxazoles.29
Indeed, Co/NGr‐C@SiO2 (40 mg; 4.3 mol % Co) allowed for full conversion of 1 giving N‐phenylpyrrole 2 in 82 % yield, with no aniline 3 detected (Table 1, entry 1). Previous detailed characterizations of this catalyst revealed the presence of a variety of different types of Co species: metallic Co particles, Co oxide particles and core–shell type particles, where a metal center was covered by small Co oxide crystallites.28 In particular, the individual cobalt oxide particles were not covered by graphitic shells, whereas some metallic particles had graphitic structures attached to them. To test a more well‐defined catalyst material, Co/NGr‐C@SiO2 was washed with 2 m hydrochloric acid to remove the cobalt oxide phase and large unprotected cobalt particles, leaving only small cobalt‐containing particles protected by graphitic structures (Figures S1, S2).28 This modified material Co/NGr‐C@SiO2‐L (L=leached), obtained after selective leaching, was more active despite the lower catalyst loading (1.8 mol % Co) (entry 2). When a similar amount (20 mg, 2.1 mol % Co) of Co/NGr‐C@SiO2 was used, only 20 % of the desired pyrrole 2 was obtained (entry 3), demonstrating the advantage of the Co/NGr‐C@SiO2‐L.
In addition, a related cobalt‐based catalyst supported on carbon (Co‐Co3O4/NGr@C) which was previously demonstrated to reduce nitroarenes to anilines10 worked well in this transformation (entry 4). Although, Co‐Co3O4/NGr@C showed high catalytic activity using nitrobenzene 1 as a starting material, later on we found that this catalyst has much lower functional group tolerance (Table S1). Testing other supported metal nanoparticles under similar reaction conditions, for example, Ni‐NiO/NGr@C30 and Fe2O3/NGr@C,8 poor activity was observed (entries 5 and 6). Similarly, a recently reported nickel silicide9 catalyst (Ni‐Si/NiO‐SiO2@SiO2) did not give the desired pyrrole 2 with any reasonable yield (entry 7). Notably, the role of cobalt nanoparticles in Co/NGr‐C@SiO2‐L in the second step of the cascade reaction is diminished and the Clauson‐Kaas condensation of anilines to form pyrroles is mainly driven by the acidic nature of the SiO2 support (Table S3).
Scope and Limitations
Next, we explored the applicability of Co/NGr‐C@SiO2‐L in the synthesis of pyrroles from various nitroarenes, including substrates with sensitive functional groups (Scheme 2). Hydrogenation of halogen‐containing nitroarenes occurred smoothly and the resulting anilines were converted into their corresponding unsubstituted pyrroles 4–6 in the presence of DMTHF. Notably, 3‐iodonitrobenzene bearing a labile iodine substituent gave 6 with no dehalogenation side‐reaction. Nitroarenes bearing electron‐rich substituents are well‐tolerated under standard reaction conditions, as shown with pyrroles 7 and 8. Even the presence of sulfur atoms did not affect the rate of the reaction; thus, thioether‐containing nitroarenes reacted smoothly, giving 9 and 10. To our delight, pyrroles containing nitriles and ketones sensitive towards hydrogenation 11–13 could be obtained from the corresponding nitroarenes using the Co/NGr‐C@SiO2‐L catalyst. Similarly, ethyl cinnamate containing pyrrole 14 was obtained without any products of C=C bond hydrogenation. In addition, heterocyclic compounds were successfully employed including quinolines and benzothiazole 15–17. Under standard reaction conditions 2‐nitroaniline was converted to the corresponding pyrrole 18, albeit at slower reaction rate and giving 5 % of dipyrrolyl benzene byproduct.
Scheme 2.

Synthesis of pyrroles from nitroarenes in the presence of Co/NGr‐C@SiO2‐L and H2. Reaction conditions: 0.5 mmol nitroarene, 40 mg catalyst, 40 bar H2, 120 °C, 24 hours; 0.8 mL DMTHF or 0.8 mL 2,5‐hexanedione. [a] after 36 h; [b] newly reported compound; [c] corresponding anilines were obtained as the only products.
When 2,5‐hexanedione was used as a coupling partner, a variety of 2,5‐dimethylpyrroles were obtained. Notably, this process can be achieved pressureless (1 bar H2; Table S2). Although this novel pyrrole synthesis proved to be chemoselective in the presence of sensitive functional groups—such as halogens (20, 25), olefins (24), carbonyl compounds (23, 26) and N‐heterocycles (27)—certain limitations could be identified. Specifically, substrates containing basic nitrogen atoms (28–30) were not suitable for this protocol using H2 as a reductant. More specifically, these nitroarenes were fully converted into the corresponding anilines, however, due to the basicity of the reaction media, condensation with dicarbonyl compounds and DMTHF was inefficient.
Then, we explored the possibility to access more diverse substituted pyrroles using commercially available 2,5‐diketones (Scheme 3). Due to practical considerations 1.2 equiv of the corresponding diketone was used and the reaction was performed in THF. Again, reducible groups such as nitriles (32), ketones (33), esters (34) were compatible with the Co/NGr‐C@SiO2‐L catalyst. However, condensation of in situ generated aniline 3 with 2,5‐diphenylhexanedione under the studied reaction conditions proved to be difficult, providing 35 in low yield.
Scheme 3.

Synthesis of 2,5‐disubstituted pyrroles from nitroarenes. Reaction conditions: 0.5 mmol nitroarene, 0.6 mmol 2,5‐diketone, 40 mg catalyst, 40 bar H2, 120 °C, 24 hours; 0.8 mL THF; [a] newly reported compound; [b] aniline 3 80 % yield.
Recycling and reusability are most important features of heterogeneous catalysts. In this respect, we tested the reusability of Co/NGr‐C@SiO2‐L for the synthesis of 19. To our delight, hydrogenation with H2 and subsequent Paal–Knorr synthesis of nitrobenzene proceeded smoothly in similar yields after ten times (Figure 1).
Figure 1.

Catalyst recycling test: synthesis of 19.
Transfer Hydrogenation with HCOOH: Improved Scope and Milder Conditions
Addressing the limitations of the methodology described above, we developed an improved N‐aryl pyrrole synthesis from nitroarenes using HCOOH‐Et3N as an alternative to molecular hydrogen (Scheme 4). This alternative protocol uses formic acid, which is readily available from bio‐waste and does not require any special experimental setup, such as high‐pressure autoclaves. Interestingly, formic acid not only serves as a hydrogen donor, but also has a positive impact on the rate of the Paal–Knorr pyrrole synthesis. Using cobalt‐catalyzed transfer hydrogenation, various pyrroles were obtained under milder reaction conditions vide supra (Scheme 3). In the presence of 1.8 mol % Co/NGr‐C@SiO2‐L under solvent‐free conditions, the starting nitroarenes were converted to their corresponding N‐phenylpyrroles. In addition to chemical building blocks, this cascade reaction can be easily used to prepare biologically active compounds such as 37 and 38 which are known for their anti‐HIV and antifungal activities.31 Notably, the acidic nature of formic acid allows for expansion of the reaction scope to include previously inaccessible substrates with basic N‐atoms, including pyridines (40), benzimidazoles (39), pyrazoles (41) and 1,10‐phenanthroline (42). When piperazine substrate 43 was tested under the optimized reaction conditions, the hydrogenation of nitro group and subsequent pyrrole formation proceeded smoothly. However, the secondary amine was formylated with HCOOH giving product 44 in 54 % yield.
Scheme 4.

Synthesis of pyrroles from nitroarenes using formic acid as a hydrogen source. Reaction conditions: 0.5 mmol nitroarene, HCOOH/TEA 5:2 mixture (1.75 mmol HCOOH, 0.7 mmol TEA), 40 mg catalyst, 100 °C, 0.8 mL 2,5‐hexanedione, 24 hours. [a] newly reported compound.
Preparation of Pyrroles under WGSR Conditions
The water‐gas shift reaction (WGSR) of CO and H2O to give H2 and CO2 is an industrially relevant transformation which found its application in the production of methanol, as well as in the Haber‐Bosch and Fischer–Tropsch processes. Despite the importance of this reaction, its application for hydrogenation of organic compounds remains less established compared to hydrogenations using H2 and transfer hydrogenations.32 Recently, our research group has developed the first heterogeneous non‐noble metal‐based catalysts for the reduction of nitroarenes to anilines using CO/H2O as a hydrogen donor.33 Encouraged by these results, we tested the ability of Co/NGr‐C@SiO2‐L to operate under WGSR conditions (Table 2). When hydrogen was replaced with 5 bar CO, 100 μL H2O and the standard amount of catalyst (1.8 mol %), after 24 h at 120 °C the desired N‐pyrrole 19 was observed in 30 % yield. Interestingly, here the addition of catalytic amounts of triethylamine (TEA) significantly improved the reaction rate: with just 5 mol % TEA, 95 % conversion was achieved (entry 2). A further increase of TEA to 10 mol % resulted in complete conversion and gave 19 as the only product (entry 3).
Table 2.
Synthesis of pyrroles under WGSR conditions.[a]

|
Entry |
TEA, mol % |
CO [bar] |
notes |
Conv. [%][b] |
19 [%][b] |
|---|---|---|---|---|---|
|
1 |
0 |
5 |
– |
30 |
30 |
|
2 |
5 |
5 |
– |
95 |
89 |
|
3 |
10 |
5 |
– |
100 |
93 |
|
4 |
10 |
0 |
– |
0 |
0 |
|
5 |
0 |
5 |
No H2O |
1 |
1 |
|
6 |
10 |
1 |
– |
91 |
60[c] |
[a] Reaction conditions: 0.5 mmol 1, 40 mg catalyst, 0–5 bar CO, 120 °C, 100 μL H2O, 24 hours; 0.8 mL DMTHF; [b] Yields were determined by GC‐FID using n‐hexadecane as an internal standard; [c] 14 % of aniline 3 by‐product.
To better understand the mechanism of the cobalt‐catalyzed pyrrole formation under WGSR conditions, control experiments have been performed. As expected, the reaction does not occur in the absence of either water or carbon monoxide (entries 4 and 5), however with just 1 atm of CO, 60 % of 19 was obtained (entry 6). In the absence of the nitroarene substrate, Co/NGr‐C@SiO2‐L at 120 °C proved to be a poor WGSR catalyst, giving only trace amounts of CO2 and H2 (Figure S4). In contrast, using substrate 1 undergoing full conversion to the desired product 19, the amount of CO2 increased to 0.93 vol % (p =0.047 bar), with no H2 being present (Figure S5).
Since the activity of Co/NGr‐C@SiO2‐L as a pure WGSR catalyst is low, the participation of H2 as an intermediate in the reduction process is unlikely; rather the active cobalt hydride species are formed with CO/H2O. The observed rate acceleration with TEA is quite intriguing. A similar effect on Co‐ and Fe‐catalyzed hydrogenation of nitroarenes with H2 has been observed and we postulated that base promotes heterolytic cleavage of molecular dihydrogen, generating the metal hydride species.34 As the formation of H2 as an intermediate is excluded under the reaction conditions studied, TEA must operate via a different mechanistic pathway. Participation of TEA as a pure reductant for nitroarenes is highly unlikely since the base is not required for the reaction and a significant rate acceleration is observed already with only catalytic amounts of TEA.
This acceleration effect, however, is not specific to cobalt metal. In fact an iron‐based catalyst was shown to operate better in the presence of TEA.35 Thus, the likely scenario for the observed effect does not involve the metal directly; instead we believe that under WGSR conditions TEA facilitates the metal‐hydride formation via base‐assisted activation of H2O to attack the CO.
Application: Synthesis of Bioactive Compounds
To showcase the synthetic utility of this general cobalt‐catalyzed pyrrole synthesis from nitroarenes, we performed a multistep synthesis of several biologically active and pharmaceutically important lead compounds. We aimed to demonstrate the utility of our catalyst for gram scale synthesis of pyrrole intermediates using all three reducing systems (H2, HCOOH, CO/H2O).
At first we prepared 46′ which belongs to a family of substituted pyrroles with antitubercular properties (Scheme 5).36 The cobalt‐catalyzed pyrrole formation was utilized as a key step converting p‐fluoronitrobenzene 45 to pyrrole 4 in 85 % yield (1.10 g) in a single step using H2 as the external reductant. Next, following a reported procedure we performed a double Mannich reaction between pyrrole and a phenylpiperazine/formaldehyde mixture to obtain the desired product. Although the NMR data of the compound matched the data from the report by Castagnolo, X‐ray analysis37 revealed that the original structure 46′ was improperly assigned.38 The corrected structure 46 is in agreement with the expected C‐2 nucleophilic properties of pyrroles.
Scheme 5.
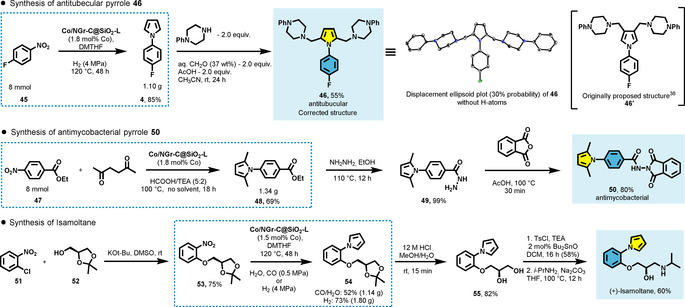
Synthesis of biologically active pyrroles on gram scale.
Our second target was pyrrole 50 with antimycobacterial properties (Scheme 5).39 Starting from ethyl 4‐nitrobenzoate 47 the key pyrrole 48 has been obtained in 69 % yield (1.34 g) via cobalt‐catalyzed transfer hydrogenation with formic acid. Next, following established procedures, the target compound 50 was obtained in two steps. Treatment of 48 with hydrazine hydrate in boiling ethanol gave 49 in quantitative yield. A subsequent acid‐catalyzed reaction with phthalic anhydride produced pyrrole 50 in 80 % yield.
Finally, we prepared (+)‐Isamoltane (CGP 361A) in four steps starting from commercially available materials (Scheme 5). Isamoltane is an antagonist of β‐adrenergic receptors, 5‐HT 1A receptors and 5‐HT 1B receptors.40 The synthesis was commenced with a nucleophilic aromatic substitution of o‐chloronitrobenzene 51 with the protected alcohol 52, giving ether 53 in 75 % yield. Next, 53 was converted into the corresponding pyrrole 54 under WGSR conditions using our cobalt catalyst, providing 1.14 g 54 in 52 % yield. Utilization of hydrogen as an alternative reductant gave better results (73 %). Next, acid‐catalyzed acetonide deprotection of 54 give the unprotected diol 55 in 82 % isolated yield. We found that treatment of 54 with HCl in aqueous methanol for 15 minutes was sufficient and prolonged exposure of 54 in acidic solution resulted in product decomposition. Finally, the diol 55 was selectively sulfonylated with tosyl chloride in the presence of catalytic amounts of dibutyltin oxide and subsequent substitution with isopropylamine giving the desired (+)‐Isamoltane.41
Conclusion
We have developed a heterogeneous Co/NGr‐C@SiO2‐L catalyst for an efficient cascade synthesis of pyrroles starting from the corresponding nitroarenes using either hydrogen or formic acid or CO/H2O mixtures as reducing agents. Our robust catalyst can be used up to ten times without significant loss of activity. The key transformation in these cobalt‐catalyzed pyrrole syntheses is the selective reduction of nitroarenes with active cobalt hydride species. These active species can be commonly generated in the presence of various benign reducing agents (Figure 2). The presented methodology shows a good functional group tolerance and has been successfully applied in the synthesis of pharmaceutical lead compounds. As an example, the enantioselective synthesis of (+)‐Isamoltane is reported.
Figure 2.

Generation of cobalt hydride species using H2, HCOOH and CO/H2O.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The Federal Ministry of Education and Research (BMBF), the State of Mecklenburg‐Vorpommern, and EU (ERC Advanced grant NoNaCat) are gratefully acknowledged for their financial support. We also thank Susann Buchholz, Dr. Christine Fischer, Susanne Schareina and Gudrun Wenzel for their excellent analytical service; Dr. Nils Rockstroh for the TEM analysis of cobalt catalysts (LIKAT analytical department). Dr. Dario Formenti, Johannes Fessler for valuable discussions and David K. Leonard (all at LIKAT) for his help in the preparation of this article. Open access funding enabled and organized by Projekt DEAL.
P. Ryabchuk, T. Leischner, C. Kreyenschulte, A. Spannenberg, K. Junge, M. Beller, Angew. Chem. Int. Ed. 2020, 59, 18679.
References
- 1. Ardkhean R., Caputo D. F. J., Morrow S. M., Shi H., Xiong Y., Anderson E. A., Chem. Soc. Rev. 2016, 45, 1557–1569. [DOI] [PubMed] [Google Scholar]
- 2. Nicolaou K. C., Edmonds D. J., Bulger P. G., Angew. Chem. Int. Ed. 2006, 45, 7134–7186; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 7292–7344. [Google Scholar]
- 3. Huang H.-M., Garduño-Castro M. H., Morrill C., Procter D. J., Chem. Soc. Rev. 2019, 48, 4626–4638. [DOI] [PubMed] [Google Scholar]
- 4. Zhou J., Chem. Asian J. 2010, 5, 422–434. [DOI] [PubMed] [Google Scholar]
- 5. Climent M. J., Corma A., Iborra S., Sabater M. J., ACS Catal. 2014, 4, 870–891. [Google Scholar]
- 6. Huang Y.-B., Liang J., Wang X.-S., Cao R., Chem. Soc. Rev. 2017, 46, 126–157. [DOI] [PubMed] [Google Scholar]
- 7. Beller M., Chem. Rev. 2019, 119, 2089–2089. [DOI] [PubMed] [Google Scholar]
- 8. Jagadeesh R. V., Surkus A.-E., Junge H., Pohl M.-M., Radnik J., Rabeah J., Huan H., Schünemann V., Brückner A., Beller M., Science 2013, 342, 1073–1076. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Ryabchuk P., Agostini G., Pohl M.-M., Lund H., Agapova A., Junge H., Junge K., Beller M., Sci. Adv. 2018, 4, eaat0761; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Ryabchuk P., Agapova A., Kreyenschulte C., Lund H., Junge H., Junge K., Beller M., Chem. Commun. 2019, 55, 4969–4972. [DOI] [PubMed] [Google Scholar]
- 10. Westerhaus F. A., Jagadeesh R. V., Wienhöfer G., Pohl M.-M., Radnik J., Surkus A.-E., Rabeah J., Junge K., Junge H., Nielsen M., Brückner A., Beller M., Nat. Chem. 2013, 5, 537–543. [DOI] [PubMed] [Google Scholar]
- 11. Jagadeesh R. V., Murugesan K., Alshammari A. S., Neumann H., Pohl M.-M., Radnik J., Beller M., Science 2017, 358, 326–332. [DOI] [PubMed] [Google Scholar]
- 12. Schwob T., Kempe R., Angew. Chem. Int. Ed. 2016, 55, 15175–15179; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15400–15404. [Google Scholar]
- 13.
- 13a. Bäumler C., Kempe R., Chem. Eur. J. 2018, 24, 8989–8993; [DOI] [PubMed] [Google Scholar]
- 13b. Schwob T., Ade M., Kempe R., ChemSusChem 2019, 12, 3013–3017. [DOI] [PubMed] [Google Scholar]
- 14. Michlik S., Kempe R., Nat. Chem. 2013, 5, 140–144. [DOI] [PubMed] [Google Scholar]
- 15. Lu Y., Arndtsen B. A., Angew. Chem. Int. Ed. 2008, 47, 5430–5433; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 5510–5513. [Google Scholar]
- 16. St. Cyr D. J., Arndtsen B. A., J. Am. Chem. Soc. 2007, 129, 12366–12367. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Zhang M., Neumann H., Beller M., Angew. Chem. Int. Ed. 2013, 52, 597–601; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 625–629; [Google Scholar]
- 17b. Wozniak B., Li Y., Hinze S., Tin S., de Vries J. G., Eur. J. Org. Chem. 2018, 2009–2012. [Google Scholar]
- 18. Knorr L., Chem. Ber. 1884, 17, 2863–2870. [Google Scholar]
- 19. Paal C., Chem. Ber. 1884, 17, 2756–2767. [Google Scholar]
- 20. Roth B. D. in Progress in Medicinal Chemistry, Vol. 40 (Eds.: F. D. King, A. W. Oxford, A. B. Reitz, S. L. Dax), Elsevier, Amsterdam, 2002, pp. 1–22. [Google Scholar]
- 21.
- 21a. Kim B. H., Bae S., Go A., Lee H., Gong C., Lee B. M., Org. Biomol. Chem. 2016, 14, 265–276; [DOI] [PubMed] [Google Scholar]
- 21b. Lee H., Kim B. H., Tetrahedron 2013, 69, 6698–6708. [Google Scholar]
- 22. Kim E., Jeong M., Lee H., Kim B. H., Heterocycles 2019, 98, 1105–1118. [Google Scholar]
- 23. Park J. W., Chung Y. K., ACS Catal. 2015, 5, 4846–4850. [Google Scholar]
- 24. Serrano-Maldonado A., Martin E., Guerrero-Ríos I., Eur. J. Inorg. Chem. 2019, 2863–2870. [Google Scholar]
- 25. Gong Z., Lei Y., Zhou P., Zhang Z., New J. Chem. 2017, 41, 10613–10618. [Google Scholar]
- 26. Cirujano F. G., Leyva-Pérez A., Corma A., Xamena F. X. L. I., ChemCatChem 2013, 5, 538–549. [Google Scholar]
- 27. Clauson-Kaas N., Tyle Z., Acta Chem. Scand. 1952, 6, 667–670. [Google Scholar]
- 28. Chen F., Kreyenschulte C., Radnik J., Lund H., Surkus A.-E., Junge K., Beller M., ACS Catal. 2017, 7, 1526–1532. [Google Scholar]
- 29. He J., Lin F., Yang X., Wang D., Tan X., Zhang S., Org. Process Res. Dev. 2016, 20, 1093–1096. [Google Scholar]
- 30. Pisiewicz S., Formenti D., Surkus A.-E., Pohl M.-M., Radnik J., Junge K., Topf C., Bachmann S., Scalone M., Beller M., ChemCatChem 2016, 8, 129–134. [Google Scholar]
- 31. Brites N. P., Dilelio M. C., Martins G. M., do Carmo G., Morel A. F., Kaufman T. S., Silveira C. C., ChemistrySelect 2019, 4, 5398–5406. [Google Scholar]
- 32. Ambrosi A., Denmark S. E., Angew. Chem. Int. Ed. 2016, 55, 12164–12189; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12348–12374. [Google Scholar]
- 33. Westerhaus F. A., Sorribes I., Wienhöfer G., Junge K., Beller M., Synlett 2015, 26, 313–317. [Google Scholar]
- 34. Formenti D., Topf C., Junge K., Ragaini F., Ragaini M., Catal. Sci. Technol. 2016, 6, 4473–4477. [Google Scholar]
- 35. Ryabchuk P., Junge K., Beller M., Synthesis 2018, 50, 4369–4376. [Google Scholar]
- 36. Bhakta S., Scalacci N., Maitra A., Brown A. K., Dasugari S., Evangelopoulos D., McHugh T. D., Mortazavi P. N., Twist A., Petricci E., Manetti F., Castagnolo D., J. Med. Chem. 2016, 59, 2780–2793. [DOI] [PubMed] [Google Scholar]
- 37. Deposition Number 2005593 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
- 38. Scalacci N., Black G. W., Mattedi G., Brown N. L., Turner N. J., Castagnolo D., ACS Catal. 2017, 7, 1295–1300. [Google Scholar]
- 39. Joshi S. D., More U. A., Dixit S. R., Korat H. H., Aminabhavi T. M., Badiger A. M., Med. Chem. Res. 2014, 23, 1123–1147. [Google Scholar]
- 40.
- 40a. Rényi L., Larsson L.-G., Berg S., Svensson B. E., Thorell G., Ross S. B., Naunyn-Schmiedeberg′s Arch. Pharmacol. 1991, 343, 1–6; [DOI] [PubMed] [Google Scholar]
- 40b. Langlois M., Brémont B., Rousselle D., Gaudy F., Eur. J. Pharmacol. 1993, 244, 77–87. [DOI] [PubMed] [Google Scholar]
- 41. Martinelli M. J., Nayyar N. K., Moher E. D., Dhokte U. P., Pawlak J. M., Vaidyanathan R., Org. Lett. 1999, 1, 447. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary