

Abstract
Parkinson's disease (PD) is one of the most common neurodegenerative disorders that manifest various motor and nonmotor symptoms. Although currently available therapies can alleviate some of the symptoms, the disease continues to progress, leading eventually to severe motor and cognitive decline and reduced life expectancy. The past two decades have witnessed rapid progress in our understanding of the molecular and genetic pathogenesis of the disease, paving the way for the development of new therapeutic approaches to arrest or delay the neurodegenerative process. As a result of these advances, biomarker‐driven subtyping is making it possible to stratify PD patients into more homogeneous subgroups that may better respond to potential genetic‐molecular pathway targeted disease‐modifying therapies. Therapeutic nucleic acid oligomers can bind to target gene sequences with very high specificity in a base‐pairing manner and precisely modulate downstream molecular events. Recently, nucleic acid therapeutics have proven effective in the treatment of a number of severe neurological and neuromuscular disorders, drawing increasing attention to the possibility of developing novel molecular therapies for PD. In this review, we update the molecular pathogenesis of PD and discuss progress in the use of antisense oligonucleotides, small interfering RNAs, short hairpin RNAs, aptamers, and microRNA‐based therapeutics to target critical elements in the pathogenesis of PD that could have the potential to modify disease progression. In addition, recent advances in the delivery of nucleic acid compounds across the blood–brain barrier and challenges facing PD clinical trials are also reviewed.
Keywords: blood–brain barrier, molecular pathogenesis, nucleic acid therapeutics, Parkinson's disease, precision medicine
1. INTRODUCTION
Parkinson's disease (PD) is the second most prevalent neurodegenerative disease and it is estimated that by 2030 it will affect at least 8 million individuals worldwide. 1 It was first described by James Parkinson in his landmark 1817 publication: “An Essay on the Shaking Palsy”. In his original description, Parkinson drew attention to the cardinal motor features of the disease: “Involuntary tremulous motion, with lessened muscular power, in parts not in action and even when supported; with a propensity to bend the trunk forward and to pass from a walking to a running pace.” 2 It has since become apparent that the clinical phenotype is in fact much broader and encompasses a wide range of nonmotor manifestations that includes olfactory and gastrointestinal symptoms, autonomic dysfunction, sleep disorders, depression, and cognitive impairment. Some nonmotor symptoms may manifest in the prodromal stages of the disease and predate the initial motor presentation. 3 , 4 Systematic neuropathological studies have shown that the basic pathology and hallmark α‐synuclein containing Lewy body inclusions are not confined to the dopaminergic neurons of the midbrain, but appear to commence in the lower brainstem and olfactory bulb, before extending to more rostral brainstem centers and eventually to the cerebral cortex. It has been proposed that this progression may be due to a prion‐like cell‐to‐cell spread of α‐synuclein pathology. 5 Since the landmark discovery of the first mutation in the α‐synuclein (SNCA) gene in an autosomal dominant PD family, 6 a variety of mutations in other genes have been identified in autosomal dominant and recessive forms of familial PD that collectively account for around 5%–10% of cases of the disease. In addition, genome‐wide association studies have identified at least 90 common variants associated with risk of PD which are estimated to contribute to between 22% and 27% of the heritable risk of the disease. 7
Although symptomatic therapies for PD can improve the motor and nonmotor symptoms for patients, these drugs do not stop the ongoing neurodegeneration and progression of the disease, which eventually results in severe motor and cognitive disability and various secondary complications. Thus, there is an urgent need for the development of effective disease‐modifying therapies to slow or arrest the progression of PD. Unfortunately, many promising neuroprotective therapies in experimental animal models of PD have failed to demonstrate efficacy when tested in human clinical trials, and possible reasons for this failure have been discussed elsewhere. 8 One of the significant challenges is the heterogeneity of clinical phenotypes and genetic‐molecular pathogenesis across the spectrum of sporadic and familial forms of the disease, confounding treatment evaluation in clinical trials. There is thus increasing recognition of the importance of stratifying PD patients into more homogeneous groups according to the genes and/or molecular pathways involved and underlying pathophysiology, to better select patients who may respond to targeted therapies. 9 More importantly, improved understanding of the molecular pathogenesis is paving the way for novel targeted disease‐modifying approaches, including nucleic acid therapeutics. 10
Within the last few years, several nucleic acid therapeutic compounds have been approved by the US Food and Drug Administration (FDA) to modify gene expression in a number of inherited conditions including Duchenne muscular dystrophy, spinal muscular atrophy, familial amyloid neuropathy, and Batten disease. 11 , 12 , 13 , 14 Milasen®, an expedited personalized antisense oligomer, was approved by the FDA only 10 months after the genetic cause was identified. This study, by Professor Timothy Yu's group from Boston Children's Hospital, illustrates the efficiency and potential in the nucleic acid therapy discovery process. 14 The development of different types of nucleic acid therapeutics for PD using experimental in vitro and animal models has attracted increasing interest, with a few compounds under early‐stage clinical trials. Hence, in this review, we will first provide a general update on recent progress in understanding of the molecular pathogenesis of PD and will then focus on the development of precision nucleic acid therapeutics that target the key pathogenetic elements. Overall, it is encouraging that there is great enthusiasm for the development of precision and personalized therapies for PD, particularly for subgroups of patients with distinctive genetic and molecular characteristics, and for whom a more defined target pathway is evident.
2. MOLECULAR PATHOGENESIS OF PD
The aetiopathogenesis of PD has proven to be complex. The disease was for many years considered to be a sporadic disorder with no genetic associations until the first missense variant in SNCA (PARK1 and 4) was identified in 1997. 6 Since then, a large number of other genetic mutations have been determined to be responsible for familial forms of the disease (Table 1). As a result of these discoveries, several key molecular processes and pathways, including the ubiquitin–proteasomal system, the autophagy–lysosomal pathway, mitochondrial maintenance and integrity, oxidative stress, and neuroinflammation are now known to be involved in PD pathophysiology, as summarized in Figure 1. In addition, other pathways, including innate and adaptive immunity, have also been implicated. In this review, it is not our intent to discuss all of these PD pathways in detail, but rather to provide a general update on progress in some areas of the molecular pathogenesis, of relevance to drug development, and in particular to nucleic acid therapeutics.
Table 1.
Parkinson's disease related genes and phenotypes
| PARK symbol | Gene locus | Gene | Phenotype | Inheritance pattern | Penetrance | Presence of Lewy bodies | Clinical features |
|---|---|---|---|---|---|---|---|
| Confirmed causative genes | |||||||
| PARK1 and 4 | 4q21 | SNCA | EOPD | AD | 40% (duplication); 85% (A53T) | Yes | Rapid progression; high prevalence of dementia and psychiatric complications 15 |
| PARK2 | 6q26 | Parkin | EOPD | AR | 100% | No | Slow progression; frequent motor fluctuations; high prevalence of dystonia 16 |
| PARK6 | 1p36 | PINK1 | EOPD | AR | 100% | One case | Slow progression; good response to levodopa 17 |
| PARK7 | 1p36 | DJ1 | EOPD | AR | 100% | Unknown | Slow progression; good response to levodopa 15 |
| PARK8 | 12q12 | LRRK2 | LOPD | AD | 32%–75% (G2019S) | Yes | Slow progression; low prevalence of dementia and psychiatric disturbances 18 , 19 |
| PARK9 | 1p36 | ATP13A2 | EOPD | AR | Incomplete, age‐associated | Unknown | Kufor–Rakeb syndrome; rapid progression 15 |
| PARK14 | 22q13.1 | PLA2G6 | EOPD or adult‐onset | AR | Incomplete | Yes | Rapidly progressive parkinsonism with dystonia, and cognitive impairment 20 |
| PARK15 | 22q1 | FBXO7 | EOPD | AR | Unknown | Unknown | Variable phenotypes; classical PD ± pyramidal tracts signs 21 |
| PARK17 | 16q11.2 | VPS35 | Classical PD | AD | Incomplete, age‐associated | Yes | Parkinsonism; low prevalence of dyskinesia and dystonia 22 , 23 |
| PARK19 | 1p31.3 | DNAJC6 | EOPD | AR | Unknown | Unknown | Slowly progressive; classic PD with mental retardation and seizures 24 , 25 |
| PARK20 | 21q22.2 | SYNJ1 | EOPD | AR | 100% | Unknown | Progressive parkinsonism may include seizures, abnormal eye movements, and dystonia 26 |
| Susceptibility genes that need confirmation | |||||||
| PARK5 | 4p13 | UCHL1 | Classical PD | AD | Incomplete | Unknown | Typical idiopathic PD 15 |
| PARK11 | 2q37 | GIGYF2 | LOPD | AD | Incomplete, age‐associated | Unknown | Typical idiopathic PD with psychiatric symptoms 27 |
| PARK13 | 2p12 | HTRA2 | Classical PD | AD | Low penetrance | Unknown | Typical idiopathic PD; good response to levodopa 28 |
| PARK18 | 3q27.1 | EIF4G1 | Classical PD | AD | Incomplete, age‐associated | Yes | Parkinsonism; mild progression; preserved cognitive function 29 |
| PARK21 | 3q22 | TMEM230 | Classical PD | AD | Incomplete (heterozygous mutations) | Yes | Slowly progressive asymmetric parkinsonism 30 |
| PARK22 | 7p11.2 | CHCHD2 | Classical PD | AD | Incomplete | Unknown | Typical idiopathic PD; good response to levodopa 31 |
| Risk factor gene | |||||||
| NA | 1q21 | GBA | NA | AD/AR | Incomplete, age‐associated | Yes | Accelerated progression and high risk of cognitive impairment 32 , 33 |
Abbreviations: AD, autosomal dominant; AR, autosomal recessive; EOPD, early‐onset PD; LOPD, late‐onset PD; NA, not applicable; PD, Parkinson's disease.
Figure 1.
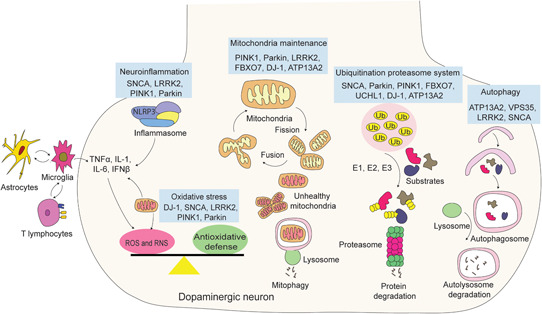
Confirmed causative Parkinson's disease genes and their roles in molecular pathogenesis pathways. E1, E1 ligase; E2, E2 ligase; E3, E3 ligase; NLRP3, nucleotide‐binding oligomerization domain‐like receptor protein 3; ROS, reactive oxygen species; RNS, reactive nitrogen species [Color figure can be viewed at wileyonlinelibrary.com]
2.1. Ubiquitin–proteasome system
The discovery that Lewy bodies are ubiquitin‐immunopositive provided an early indication that the ubiquitin–proteasome system (UPS) plays a role in PD pathogenesis. 34 As the main component of Lewy bodies, monomeric or α‐helically folded tetrameric α‐synuclein is actively degraded by the UPS under physiological conditions. 35 , 36 SNCA mutations, including duplication or triplication of the whole gene, or disease‐associated missense mutations, render α‐synuclein prone to misfolding and formation of toxic protein aggregates. These aggregates have prominent inhibitory effects on 20S/26S proteasomal protein cleavage in dopaminergic cells, 37 which in return further accumulates aggregates of toxic α‐synuclein. 38
Parkin is an auto‐inhibited RING‐between‐RING E3 ligase in the UPS which is deficient in autosomal recessive PD (PARK2). Upon activation by PINK1, Parkin undergoes a conformational change that facilitates its ubiquitin ligase activity. 39 Amongst the many Parkin substrates, aminoacyl‐tRNA synthetase complex interacting multifunctional protein‐2 accumulates when Parkin is deficient, which activates poly(ADP‐ribose) polymerase‐1 (PARP1) and causes selective loss of dopaminergic neurons. 40 Consequently, PARP1 inhibitors, which have been approved by the FDA for certain breast and ovarian cancers, are now being considered as repurposed drugs for the treatment of PD. 41 PINK1 has multiple functions, including modulating mitochondrial respiratory chain activity, regulating neuroinflammation, and promoting neuron survival. 42 In terms of its roles in the UPS, cytosolic PINK1 phosphorylates some Parkin substrates, primes Parkin‐mediated ubiquitination, and ultimately facilitates the degradation of pathological proteins. 43
The efficient and timely ubiquitination for substrate degradation requires the maintenance of cellular ubiquitin homeostasis. As a PD susceptibility gene, 44 the ubiquitin C‐terminal hydrolase L1 (UCHL1, PARK5) is one of the most abundant deubiquitinating enzymes that is predominantly expressed in the brain. UCHL1 is highly efficient in cleaving monoubiquitin from small peptides that are conjugated to the C‐terminus of a ubiquitinated protein. 45 , 46 UCHL1 also participates in other pathways including processing of proubiquitin, E3 ligase function, maintaining axonal function, and inhibiting autophagy. 47 , 48 , 49 , 50 Considering its multiple roles in the UPS and other cellular functions, UCHL1 might be considered as an attractive therapeutic target for PD and related disorders.
2.2. Autophagy–lysosomal pathway
Autophagy is a catabolic process that delivers dysfunctional organelles or misfolded proteins to the lysosome for degradation. With substantial evidence that proteins encoded by PD causative or risk genes directly or indirectly regulate the autophagy–lysosomal pathway, dysregulated autophagy is believed to play a major role in PD pathogenesis. Emerging studies are showing that multiple variants in lysosomal storage disorder genes can contribute to PD susceptibility. 51 , 52 Deficiency of the lysosomal hydrolase glucocerebrosidase, encoded by GBA, leads to the most common lysosomal storage disorder, Gaucher disease. Generally, around 5%–15% of PD patients are reported to carry heterozygous GBA mutations. However, the prevalence of GBA mutations varies in different populations, with the highest prevalence being in Ashkenazi Jewish PD patients (20%). 53 While, it is clear that GBA mutations cause autophagy–lysosomal dysfunction, the specific mechanisms involved remain unclear. 54
Mutations in ATP13A2, a transmembrane endo/lysosomal P‐type transport ATPase, cause Kufor–Rakeb syndrome (PARK9), a rare subtype of juvenile‐onset autosomal recessive parkinsonism. ATP13A2 controls lysosome homeostasis and regulates autophagosome–lysosome fusion through HDAC6 that is recruited to the lysosome and facilitate autophagy flux. 55 Moreover, ATP13A2 interacts with endocytic signaling lipids through its hydrophobic N‐terminal to enable endocytic cargo export. 56 Another PD‐related gene product that regulates endosomal–lysosomal trafficking is vacuolar protein sorting protein‐associated protein 35 (VPS35, PARK17), a member of the retromer complex. This complex is involved in intracellular trafficking of proteins including α‐synuclein and proteins that are important for autophagosome formation. 57 Reduced α‐synuclein degradation and profuse Lewy body pathology in the substantia nigra and other midbrain region were found in patients with VPS35 mutations. 58
The major pathogenic factor in both familial and sporadic PD is considered to be the formation of α‐synuclein aggregates, which is due in part to malfunction of the α‐synuclein degradation machinery. Normal monomeric α‐synuclein is usually degraded by chaperone‐mediated autophagy, while macroautophagy has been implicated in the clearance of α‐synuclein oligomers. 59 The burden of toxic α‐synuclein aggregates, may compromise the macroautophagy pathway through interfering with autophagosome formation or clearance. 60 On the other hand, when macroautophagy is affected or inhibited by gene mutations, intracellular α‐synuclein clearance is impeded and accumulation of the mutant protein is exacerbated. Mutations in LRRK2, which are the most common cause of familial PD (PARK8), have also been reported to dysregulate macroautophagy, although the findings in different studies have been somewhat contradictory. 61
2.3. Mitochondrial maintenance
The landmark observation that parkinsonism could result from the accidental intake of 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine, a potent inhibitor of mitochondrial complex I, was a critical finding that first implicated mitochondria in the pathogenesis of PD. 62 Since then, many aspects of mitochondrial dysfunction have been investigated, including impaired mitochondrial maintenance, defective mitophagy, calcium imbalance, oxidative stress, and effects of neuroinflammation. 63 LRRK2 is thought to act as a scaffold during mitochondrial fusion and fission, with the WD40 repeat‐containing domain and the dynamin‐related GTPase domain required for LRRK2 interacting with mitochondrial fission or fusion factors. 64 , 65 While, PINK1 and Parkin contribute to mitochondrial fission and fusion through ubiquitinating mitochondrial fusion regulators, including mitofusin 1/2. 66
Mitophagy is a cargo‐specific form of autophagy that removes damaged or excessive mitochondria to maintain an optimally functioning mitochondrial network. PINK1 is ubiquitously expressed in all mitochondria and maintained at a low level in healthy mitochondria. 67 Upon mitochondria damage or depolarization, PINK1 proteolysis is impeded. Subsequently, the overexpressed PINK1 recruits Parkin from the cytosol into mitochondria, 68 , 69 then mediates Lys63‐linked ubiquitination of outer mitochondrial membrane proteins and induces mitophagy. Loss‐of‐function mutations in the PINK1/Parkin pathway result in impaired mitophagy. The accumulation of dysfunctional mitochondria cause increased reactive oxygen species (ROS) generation, cytochrome c leakage, and mitochondrial DNA mutations, ultimately leading to dopaminergic neuron death. 70
Several other PD pathogenic genes are also reported to exert effects on the mitophagy pathway. A recent study found that LRRK2 mutations interfere with the interaction between dynamin‐related protein 1 and its mitochondrial receptor MiD51, which is required for the initiation of Parkin‐dependent mitophagy. 71 Under basal conditions, the outer mitochondrial membrane protein Miro is rapidly removed by LRRK2 to stop mitochondrial mobility and facilitate mitophagy, 72 while mutated LRRK2 disrupts this function, delays the arrest of damaged mitochondria and consequently hinders the induction of mitophagy. 73 Other factors such as the accumulation of α‐synuclein, ATP13A2 deficiency, and GBA mutations have also been shown to impair mitophagy. 74 , 75 , 76
In addition to their bioenergetic functions, mitochondria fine‐tune cytosolic Ca2+ levels by controlling Ca2+ uptake and efflux through several calcium transporters. 77 , 78 Mutations in SNCA, PINK1, and LRRK2 have been shown to dysregulate Ca2+ transporters, resulting in mitochondrial Ca2+ overload, increased ROS production, and ultimately neuronal cell death. 77 , 79 , 80 , 81 The tethering complexes connecting mitochondria and the endoplasmic reticulum are crucial for cellular calcium handling, 82 and mutations in SNCA, PARKIN, and DJ1 have been found to interfere with the coupling of the complexes and then impair Ca2+ dynamics. 82 , 83 , 84 , 85 Besides the role of PINK1, Parkin and DJ1 in mitophagy and Ca2+ dynamics, these proteins also modulate the function of the mitochondrial respiratory chain complex. 86 Mutations in these genes disrupt the assembly of the components of the respiratory chain complex, inhibit complexes I and III, and increase the generation of ROS and reactive nitrogen species (RNS).
2.4. Oxidative stress
Oxidative stress is a cascade reaction resulting from an imbalance between increasing free radicals, including ROS and RNS, and cellular antioxidant activity. The accumulation of free radicals causes DNA and protein oxidation, or lipid peroxidation that compromises cell integrity and leads to cell death. As neurons have a high demand for oxygen but relatively low levels of antioxidants, they are particularly susceptible to oxidative stress. 87 , 88 The impairment in mitochondrial homeostasis in PD could exacerbate the production of ROS that are the by‐products from the oxidative phosphorylation pathway. Some PD‐related gene products contribute to mitochondria‐related oxidative stress through regulating mitochondrial homeostasis or mitophagy as discussed above. In addition, α‐synuclein binds directly to mitochondria, impairing mitochondrial protein import and resulting in deficient mitochondrial respiration and enhanced ROS production. 89 Nonfibrillar phosphorylated α‐synuclein is a newly identified α‐synuclein neurotoxic isoform, and its aggregates have been shown to induce mitochondrial fragmentation via reduced lipoylation and aggravate oxidative stress. 90
The protein deglycase DJ1, encoded by PARK7, is ubiquitously expressed in the brain and acts as a sensor of oxidative stress. 91 DJ1 is involved in several signaling pathways that are protective against oxidative injury, including activating the extracellular signal‐regulated protein kinases 1 and 2 and Akt pathways, 92 , 93 , 94 inhibiting apoptosis signal‐regulating kinase 1, 95 and regulating transcription factors such as p53, nuclear factor kappa B, and nuclear factor erythroid 2–related factor 2. 91 , 96 , 97 , 98 The crosstalk between DJ1 and other major antioxidant systems, such as the thioredoxin system and the glutathione system 91 , 99 further supports the cellular antioxidant activity to provide an optimal response to oxidative stress. DJ1 mutants could produce selective mitochondrial pathologies that impair Ca2+ dynamics and free radical homeostasis through disrupting the interaction between DJ1 and mitochondrial accessory proteins. 84
2.5. Neuroinflammation
Since the first report of reactive microglia in postmortem PD brain tissue in 1988, 100 microglial activation and high levels of proinflammatory cytokines have been consistently detected in PD brains. 101 There is, therefore, convincing evidence that neuroinflammation is involved in dopaminergic neuron death and neurodegeneration. 102 As the primary central nervous system (CNS) resident immune cells, 103 microglia can be activated to yield two different phenotypes (M1/M2) to produce either proinflammatory or anti‐inflammatory cytokines to maintain CNS homeostasis. Chronic activation by misfolded proteins or environmental toxins results in M1 microglia that produce proinflammatory cytokines, including tumor necrosis factor‐α (TNF‐α), interleukin‐1β (IL‐1β), and interferon‐γ. The initial microglial activation and subsequent production of proinflammatory mediators may also result in activation of astrocytes that are considered to be integrative regulators of neuroinflammation in the nervous system. 104
Apart from the role of innate immunity in neuroinflammation, there is increasing evidence that adaptive immunity also plays a role in PD pathogenesis. 105 , 106 An increase in the number of cytotoxic and memory T cells, and changes in proportions of helper T cells and naive T lymphocytes in peripheral blood, indicates the activation of a cytotoxic immune response in PD. 106 , 107 , 108 Infiltration of T cells in the substantia nigra of PD mice was reported to affect neurodegeneration in other ways, including inducing microglial activation to the M1 phenotype. 109 , 110 Although the mechanism of T cell invasion into the CNS is still uncertain, microglia as the brain‐resident macrophages are believed to present CNS‐derived antigens to peripheral T lymphocytes, resulting in T cell activation and infiltration. 111 , 112
Along with the cellular mediators, several molecular mechanisms are involved in modulating neuroinflammation. Extracellular wild‐type or pathological α‐synuclein activates microglia through toll‐like receptor 2 and the Janus kinase–signal transducers and activators of transcription signaling pathways, which produces various proinflammatory cytokines and chemokines. 113 , 114 , 115 Blocking the toll‐like receptor 2 signaling alleviates α‐synuclein accumulation, neuroinflammation, and behavioral deficits in the α‐synuclein transgenic mouse model. 116 LRRK2 interacts with and phosphorylates nucleotide‐binding oligomerization domain‐like receptor C4, an inflammasome protein, and subsequently activates caspase‐1. 117 In addition, the LRRK2 G2019S mutation appears to drive microglia towards the reactive phenotype with enhanced inflammatory responses 118 and increases PD susceptibility. 119
PINK1‐ and Parkin‐mediated mitophagy can prevent mitochondrial‐induced inflammation. 120 Deficiency in either PINK1 or Parkin function results in elevated levels of cytosolic and circulating mitochondrial DNA, or double‐stranded mitochondrial RNA that promotes proinflammatory responses through different mechanisms. 121 , 122 Apart from roles in mitochondrial‐induced inflammation, PINK1 and Parkin also contribute to neuroinflammation via separate pathways. Through promoting proteasomal degradation of TNF‐α receptor‐associated factor 2/6 123 or regulating ubiquitin‐modifying enzyme A20 and NLRP3‐inflammasome, Parkin suppresses inflammation and cytokine‐induced neuron death. 124 , 125 PINK1 modulates neuroinflammation in a glial cell type‐specific manner, with studies showing that loss of PINK1 increases proinflammatory cytokines (TNF‐α, IL‐1β, and NO) in astrocytes. 126
The progress in the molecular findings is gradually leading to the clarification of PD pathogenesis and is providing justifications to develop molecular pathway targeted therapeutics. Although most of the pathways are closely related and/or overlapping, some pathways are proposed to result in a distinct disease process and resultant phenotypes. For example, mitochondrial damage is a particular problem in the substantia nigra, 127 and thus for patients with mitophagy failure, their clinical features are hypothesized to have a more dopaminergic nature. However, for those who have a dysregulated autophagy–lysosomal pathway, the clinical features might be more generalized. 128 , 129 The genes and molecular events involved are somewhat clearer in monogenic familial PD cases, which is facilitating the development of precision or personalized treatment strategies. Nucleic acid therapies are particularly attractive in this scenario since approaches including small interfering RNAs (siRNA), microRNAs (miRNA) and antisense oligonucleotides (ASO) can be applied to tackle some gain‐of‐function mutations, while splice‐switching antisense oligomers can be used to address selected loss‐of‐function mutations. Some common mutations of PD genes and potential nucleic acids‐based therapeutic strategies are summarized in Table 2.
Table 2.
Current status of nucleic acid therapeutic strategies for the common genetic forms of PD
| Genes | Common mutations | Mechanism | Potential therapeutic strategies | Current Status |
|---|---|---|---|---|
| SNCA | c.88G>A (p. A30P), c.152G>A (p. E46K), c.157G>A (p. A53T), increased copy number | Gain‐of‐function | Downregulation (ASO, siRNA, shRNA); Splice switching (ASO) | Under development (in vitro and in vivo) |
| Parkin | exon 3 deletion, exon 4 deletion, c.838G>A (p. A280A) | Loss‐of‐function | Exon skipping (ASO) | Under development (in vitro) |
| PINK1 | c.1231G>A (p. G411S), c.1311G>A (p. T437X) | Loss‐of‐function | Upregulation (ASO) | N/R |
| DJ1 | c.293G>A (p. A98G), c.310G>A (p. A104T) | Loss‐of‐function | Upregulation (ASO) | N/R |
| LRRK2 | c.6055G>A (p. G2019S), c.4322G>A (p. A1441H) | Gain‐of‐function | Knockdown (ASO, siRNA, miRNA) | Phase I clinical trial |
| ATP13A2 | No recurrent mutations | Loss‐of‐function | Exon skipping (ASO); Upregulation (ASO) | N/R |
| GBA | c.1226A>G (p. N370S), c.1448T>C (p. L444P) | Gain‐of‐function a | Downregulation (ASO, siRNA, miRNA, shRNA) | Under development |
Abbreviations: ASO, antisense oligonucleotides; miRNA, microRNA; N/R, no reports; PD, Parkinson's disease; shRNA, short hairpin RNA; siRNA, small interfering RNA.
Both gain‐of‐function and loss‐of‐function mechanisms are proposed for GBA‐associated Parkinsonism.
3. NUCLEIC ACID THERAPEUTICS AND ITS PROGRESS IN PD RESEARCH
Since the FDA first approved the nucleic acid drug, Vitravene in 1998 to treat cytomegalovirus retinitis in immunocompromised patients, nucleic acid therapeutics have gradually come‐of‐age. In excess of 200 nucleic acid drug clinical trials are registered at ClinicalTrials.gov and over 10, 000 patients have received nucleic acid drugs since 2016. 130 Nucleic acid therapeutics are emerging as a well‐established, validated class of drugs that could manipulate almost any gene with high specificity. 131 However, there are a number of hurdles that need to be overcome before successful widespread clinical application of nucleic acid compounds. The first and foremost is the instability of unmodified natural nucleic acids. As expected in normal RNA and DNA metabolism, the natural phosphodiester backbone is particularly sensitive to degradation by endonuclease and exonucleases. 132 , 133 , 134
3.1. Nucleic acid chemistries, modifications, and related toxicities
Chemical modifications (Figure 2) to the backbone and/or bases have been developed to enhance nuclease resistance and overall pharmacokinetic characteristics. The replacement of the nonbridging oxygen atom in the phosphodiester backbone with a sulfur atom creates a phosphorothioate linkage with enhanced resistance to degradation. 135 , 136 Furthermore, modifications at the 2′‐position include replacing the hydroxyl group with an O‐methyl (OMe), O‐methoxyethyl (MOE), fluoro (F), or amino (NH2) moiety, or linking the 2′ oxygen to the 4′ carbon to create locked nucleic acid (LNA). These chemical modifications not only increase the nuclease resistance but also the binding affinity of ASOs, siRNAs, aptamers, or other nucleic acids. 137 , 138 , 139 , 140 Modifications on both sugar and backbone such as peptide nucleic acid (PNA) 141 and phosphorodiamidate morpholino oligonucleotide (PMO) 142 have also been developed to enhance oligomer stability. Building on a backbone of morpholine rings connected by phosphorodiamidate linkages, the neutrally charged PMO has been shown to be a very safe chemistry in clinical trials, although a delivery system is needed to allow for better uptake of PMO. 143 Newer modifications including altritol nucleic acid, twisted intercalating nucleic acid, cyclohexeny nucleic acid, and hexitol nucleic acid are shown to have higher exon skipping efficiency compared with 2‐OMe ASO in vitro, 144 , 145 however, the toxicities and in vivo safety profile of these chemistries remain undetermined.
Figure 2.
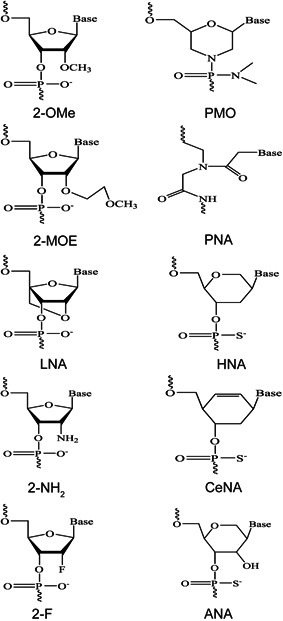
Examples of chemical modifications of nucleic acid analog oligomers. 2‐F, 2′‐fluoro; 2‐MOE, 2′‐O‐methoxyethyl; 2‐NH2, 2′‐amino; 2‐OMe, 2′‐O‐methyl; ANA, altritol nucleic acid; CeNA, cyclohexene nucleic acid; HNA, hexitol nucleic acid; LNA, locked nucleic acid; PMO, phosphorodiamidate morpholino oligomer; PNA, peptide nucleic acid
Actually, most chemical modifications incorporated into oligonucleotides result in drastic toxic consequences. 146 The phosphorothioate backbone has been demonstrated to confer innate immunostimulatory activity and activate the complement system. 147 , 148 , 149 Significant upregulation of immune system associated genes was observed in mouse brains after intracerebroventricular (ICV) administration of 2‐OMe ASO on the phosphorothioate backbone. 150 Chronic intravenous infusion of 2‐MOE modified oligomers on the phosphorothioate backbone in cynomolgus monkeys results in increased plasma concentration of the complement split product inducing Bb, C3a, and C5a, indicating activation of the alternative pathway of the complement system. 149 , 151 However, oligonucleotide‐induced complement activation was not observed in dog and human. 152
Due to their polyanionic nature, phosphorothioate backbone oligomers are known to bind to many serum proteins, including intracellular and extracellular receptors, causing renal or hepatic toxicities. For example, LNA‐related hepatoxicity is speculated to be caused by LNAs aptameric binding to hepatic intracellular proteins. 153 , 154 Specific oligonucleotide sequence motifs, such as TGC or TCC motifs of LNAs in a 3–8–3 gapmer design, tend to exhibit a high propensity to bind to mouse liver proteins and increase hepatotoxicity. 154 Binding with proteins in proximal tubule cells is also suspected of contributing to the LNA‐associated nephrotoxicity. However, the accumulation of the LNAs within proximal tubule lysosomes is more likely to cause renal toxicity, 155 as shown by tubular necrosis and oligomer accumulation in the kidney biopsy from the first human trial of an LNA ASO. 156
3.2. Off‐target effects of nucleic acid oligomers
Nucleic acid oligomers bind to targeted sequences in a highly specific base pairing manner, however, off‐target annealing to an unintended RNA that shows some homology is still one of the concerns over the safety of the oligonucleotide compounds. siRNAs inevitably cause unintended gene silencing due to miRNA‐like effects, because both exogenous siRNAs, including short hairpin RNAs (shRNAs) that eventually produce siRNAs, and endogenous miRNAs share the same downstream effector. 157 After loading with the effector and the Ago protein and then partial pairing to the 3′‐untranslated region (UTR), siRNAs intrinsically may suppress up to 1,000 unintended gene transcripts. 158 Such off‐target effects show the same dose‐response as on‐target effects, thus it is suggested that siRNA treatment should be titrated down to levels that maintain sufficient on‐target effects. 159 However, simply reducing concentration is not sufficient to eliminate off‐target effects. Since the hydroxyl group at siRNA nucleotide position 2 is essential to form a hydrogen bond with the asparagine residue of Ago, 160 chemical modifications on that 2′‐position could render steric constraints and interfere siRNA–Ago interaction, thus reducing siRNA off‐target effects. Inducing 2‐OMe at siRNA positions 1 and 2 is found to reduce around 80% of putative off‐target transcripts, resulting in a 66% decrease in unintended gene silencing. 157 Similar reduction in off‐target effects is observed when siRNA is chemically modified as LNA or unlocked nucleic acid, 161 however, the safety profile of these chemistries may hinder their further application. Novel strategies such as making circular siRNAs 162 or adding a biotin group at the 5′‐end of the sense strand 163 have been shown to improve siRNA specificity and reduce off‐target effects.
Off‐target effects of RNase H‐inducing ASO have also been known for decades, and increased oligonucleotide binding affinity by chemical modifications may potentially exaggerate the unwanted effects. 164 Reducing the overall binding affinity by limiting the number of LNAs in a gapmer ASO or increasing the length of ASO to make more specific binding to the target sequence have been shown as potential strategies to mitigate off‐target binding. 165 Splice‐switching ASOs were believed to have less off‐target effects because they must stringently bind to short splicing motifs such as exonic splicing enhancers or intronic splicing silencers. Moving one or a few nucleotides upstream or downstream of these splicing motifs may result in no splice‐switching activity. 166 , 167 However, in a recent study 17 missplicing events were detected by reverse transcription‐polymerase chain reaction after in vitro transfection of one uniformly modified 2‐MOE splice‐switching ASO, with that ASO predicted to have over 108 potential annealing sites in addition to its intended target. 168 Strategically placing mismatches within the ASO or combining two short ASOs were shown to reduce off‐target effects to some extent. Furthermore, delivering ASO through free uptake markedly reduced off‐target effects compared with delivering through lipid transfection, which may indicate that splice‐switching ASO off‐target effects are a greater concern in vitro than in vivo. 168 Since there are only a few reports on splice‐switching ASO induced missplicing activity, additional investigations are needed to assess global off‐target effects. Developing reliable in silico tools to predict off‐target effects 169 , 170 would allow for identifying and avoiding unwanted oligonucleotide actions before in vitro and in vivo evaluation.
As with any drug, effective delivery is of paramount importance to its therapeutic potential. Potent nucleic acid delivery systems are also needed to ensure effective cell uptake or even tissue‐specific delivery to allow for better assessment of therapeutic benefits. Various approaches, especially bioconjugates including cholesterol, cell‐penetrating peptides, nucleolipids, receptor ligands, and antibodies have been proposed to enhance nucleic acids delivery, 171 , 172 , 173 , 174 however even those approaches have limitations including immunogenic consequences and lack of efficiency and specificity. 175 To date, N‐acetylgalactosamine (GalNac) appears to be the most potent conjugate for siRNA delivery that specifically targets hepatocytes with limited off‐target effects. 176 , 177 Targeted ASO delivery was also achieved by conjugating ASO with GalNAc, resulting in selective ASO uptake by hepatocellular carcinoma cells. Systemically administered GalNAc‐conjugated ASO demonstrated enhanced antisense mediated antitumor activity both in vitro and in vivo. 178 However, up to now, cell‐penetrating peptides emerge as the most suitable vehicle for ASO systemic delivery which leads to two cell‐penetrating peptides conjugated splice‐switching ASOs, Eteplirsen 179 and Golodirsen 180 being approved by the FDA.
The development of nucleic acid therapeutics is rapidly evolving with many issues regarding drug stability, toxicity, off‐target effects, drug delivery being resolved to some extent, dramatically bringing these therapies closer to patients. The recent FDA approvals of Exondys 51 ®, Spinraza ®, Tegsedi ®, Onpattro ®, Vyondys 53 ®, Milasen ®, and Givlaari ® illustrate the broad therapeutic potential of nucleic acid drugs for the treatment of neurological and neuromuscular diseases. Consequently, an increasing number of researchers are actively investigating the therapeutic effects of different nucleic acid compounds for neurodegenerative diseases, including PD. Table 2 shows the current status of potential nucleic acid therapeutic strategies for PD precision or personalized medicines.
3.3. Progress in nucleic acid therapeutics for PD
3.3.1. Antisense oligonucleotides
ASOs are synthetic nucleic acid analogs that are designed to anneal to target RNA transcripts through Watson–Crick base pairing. Depending upon the base and backbone chemistries, ASOs can modify gene expression through inducing a variety of mechanisms including suppressing translation, altering splicing, modifying polyadenylation, or degrading RNA by activating RNase H or inducing RNA silencing (Figure 3i–iii). In situations where a gene lesion results in a misfolded toxic protein and the abnormal protein contributes to the disease pathway, RNA silencing or RNase H activating ASOs can be designed to bind to and degrade the corresponding messenger RNA (mRNA). The reduction in the targeted mRNA would subsequently reduce the protein levels and slow the disease course, as demonstrated by the development of Tegsedi ® for patients with hereditary transthyretin amyloidosis (HTA). 181
Figure 3.
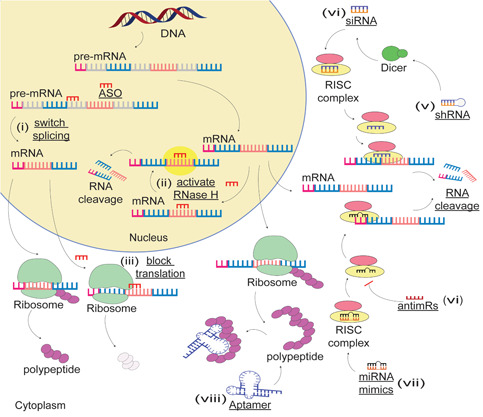
Examples of the mechanism of actions of nucleic acid compounds. (i) ASO‐mediated splice‐switching; ASOs bind to the acceptor or donor splice site, switch splicing, and induce alternative mRNA and protein isoforms; (ii) ASOs activate RNase H and cleave mRNA; (iii) ASOs inhibit mRNA translation by steric blockade of ribosomes; (iv) siRNA‐induced gene silencing; after being taken up by the RNA‐induced silencing complex and ejection of the passenger strand, the antisense strand of siRNA binds to mRNA and mediates mRNA cleavage; (v) shRNA‐mediated targeted gene silencing; (vi) miRNA‐induced RNA cleavage; (vii and viii) mechanism of actions of antimRs and aptamers. ASO, antisense oligonucleotides; miRNA, microRNA; mRNA, messenger RNA; RISC, RNA‐induced silencing complex; shRNA, short hairpin RNA; siRNA, small interfering RNA [Color figure can be viewed at wileyonlinelibrary.com]
Given that pharmacological inhibition of LRRK2 could abate α‐synuclein‐induced neurodegeneration, 182 a 2‐MOE ASO was designed to target LRRK2 mRNA for RNAse H‐mediated degradation to ameliorate pathologic α‐synuclein inclusion‐body formation. After the administration of the 2‐MOE ASO in a PD mouse model, reduced levels of LRRK2 mRNA and protein were evident, with diminished pathological aggregation of α‐synuclein in the substantia nigra of treated mice. 183 Since August 2019, a Phase I clinical trial (NCT03976349) has begun to evaluate this 2‐MOE ASO (BIIB094) through intrathecal administration in PD patients with or without a PD‐related LRRK2 mutation. This would be the first compound being tested and evaluated in PD patients with a defined genetic cause and some positive results were reported. 184
An oligonucleotide with amido‐bridged nucleic acid‐modified bases at each end and a DNA core to induce RNase H activity was designed to specifically degrade the human SNCA transcript. This compound reduced SNCA expression significantly and improved motor functions in a PD transgenic mouse model. 185 Due to the high prevalence of GBA mutations in PD patients, several therapeutic strategies including gene therapy and small molecules are under development for patients carrying this particular PD risk. 186 Chamishi Therapeutics was founded in September 2019 with a specific focus on developing ASO therapies for GBA‐associated PD.
It is well‐established that alternative splicing events are overrepresented in the brain, 187 with even subtle DNA variations able to catastrophically disrupt normal pre‐mRNA splicing. 188 Intense efforts are underway to develop ASO molecules capable of redirecting pre‐mRNA splicing by either promoting retention of a missing exon, excising a disease‐causing exon from the mature mRNA, or removing an exon to change the ratio between different transcripts. 189 , 190 α‐Synuclein has four isoforms that are generated by alternative splicing of SNCA exons 4, 6, or both. 191 The isoform lacking exon 6 has higher aggregation propensity than full‐length α‐synuclein, while the isoform lacking exon 4 confers a protective effect. 191 Thus, changing the ratio between the full‐length α‐synuclein and other isoforms by splice‐switching ASOs may initiate a new pathway for the development of disease‐modifying therapeutics for PD.
The recently reported one‐step strategy to convert astrocytes to functional neurons in situ by RNase H‐inducing ASOs is probably one of the most exciting advances in PD therapeutic research. Professor Xiangdong Fu's group designed ASOs on the phosphorothioate backbone to knockdown polypyrimidine tract‐binding protein 1, which successfully converted primary astrocytes to dopaminergic neurons in the mouse brain. 192 The converted dopaminergic neurons were shown to progressively mature and replenish lost dopaminergic neurons in a PD mouse model, thereby restoring striatal dopamine and reversing motor phenotypes. 192 This study could open up a completely novel avenue for development of treatments to “rebuild” damaged brains in PD, however, there are still some uncertainties about the effectiveness of this treatment in older individuals with reduced brain plasticity because of age. 193
3.3.2. Small interfering RNA
Twenty years after the first description of RNA interference (RNAi), the first RNAi drug Onpattro ® was approved by the FDA for the treatment of HTA in 2018. 194 , 195 The following year, the FDA approved the second siRNA drug Givlaari ® to treat patients with acute hepatic porphyria. 196 RNAi is a process that inhibits targeted gene expression with high specificity and is an exciting therapeutic strategy for precision medicine. siRNAs are generally synthetic, chemically modified short double‐stranded RNAs that are usually 21–23 nucleotides in length. siRNAs contain an antisense active strand and a sense mRNA sequence that is ejected when the double‐stranded siRNAs are taken up by the endogenous cytoplasmic RNA‐induced silencing complex. The active strand can bind to the target mRNA and initiate sequence‐specific mRNA degradation by the RNA‐induced silencing complex argonaute RNase (Figure 3iv). As with ASOs, chemical modifications of the 2′‐ribose and the phosphorothioate backbone help protect siRNAs from nuclease degradation and prolong its half‐life. 139 Incorporating siRNA into different lipid nanoparticles, especially second‐generation lipid nanoparticles, for example, Onpattro ®, substantially improves siRNA delivery and gene knockdown. 197 , 198 Other conjugates such as GalNac, lipoproteins, or exosomes are also showing improved siRNA delivery and increased knockdown of targeted genes.
SNCA overexpression and accumulation of α‐synuclein protein plays a crucial role in the pathogenesis of PD, particularly in families with SNCA copy number variations. 199 , 200 Thus, reducing SNCA mRNA levels and the resultant protein by siRNA is considered as a viable therapeutic strategy for PD. Rabies virus glycoprotein (RVG) decorated anionic liposomes loaded with siRNA‐protamine complex were used to silence SNCA expression. This complex was rapidly taken up by primary neuronal cells and efficiently reduced α‐synuclein protein in mouse primary cortical and hippocampal neurons. 201 In other preclinical studies, polyethylenimine‐siRNA and RVG‐exosome‐siRNA targeting α‐synuclein transcripts also demonstrated a robust reduction of both overexpressed human SNCA mRNA and α‐synuclein protein in the striatum of a PD mouse model and delayed the development of α‐synuclein pathology. 202 , 203 However, a delicate SNCA level must be maintained as it is obvious that too little α‐synuclein may also be harmful to the brain because of its essential biological functions at the synapse. Marked downregulation of SNCA expression by siRNA was associated with an increased risk of developing nigrostriatal degeneration. 204 Therefore, expression‐control siRNAs were generated by introducing nucleotide mismatches to control the levels of SNCA knockdown. Administration of various expression‐control siRNAs in PD transgenic fly models showed different levels of SNCA knockdown that correlated with motor function improvement. 205
3.3.3. Short hairpin RNAs
shRNAs are RNA transcripts that contain a loop structure and a stem that consists of a paired sense and antisense strand. As distinct from siRNAs, bacterial or viral vectors are needed to deliver shRNAs into cells, where they are processed into siRNAs and mediate targeted gene inhibition by the RNAi machinery (Figure 3v). Since host cells can continuously synthesize shRNAs, they have several advantages over siRNA including longer‐lasting effects, lower dose requirement, and less specific and nonspecific off‐target effects. 206 However, there are also some disadvantages to use viral vectors, including immunogenicity and potential risk of inducing mutations, making viral vectors potential safety hazards. 207 Adeno‐associated virus‐mediated delivery of shRNAs in normal rats resulted in a 35% knockdown of α‐synuclein without affecting motor function or causing degeneration of dopaminergic neurons. 208 The α‐synuclein knockdown by shRNAs in a PD rat model was observed to be neuroprotective, decreasing the degeneration of dopaminergic neurons and attenuating the progression of motor deficits. 208 shRNAs have also been used to knockdown polypyrimidine tract‐binding protein 1 and convert astrocytes to dopaminergic neurons in a PD mouse model, thereby replacing dopaminergic neuron loss and reversing motor deficits. 192 This approach has the potential to open up new therapeutic avenues for PD and other neurodegenerative disorders, but more in vivo and preclinical investigations will be required.
Since most viral vectors do not cross the blood–brain barrier, nanoparticles are being evaluated for delivery of shRNAs to the brain. N‐isopropylacrylamide is one type of nanopolymer material capable of controlled cargo targeting and release and is usually combined with acrylic acid as a drug delivery vehicle. This vehicle was further combined with photoactive nerve growth factor and oleic‐coated magnetic nanoparticles to deliver shRNAs targeting SNCA across the blood–brain barrier. Subsequently, the shRNAs reduced α‐synuclein expression and prevented dopamine neuron degeneration in the 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine PD mouse model. 209 Instead of directly targeting SNCA, other approaches, including reducing inflammation by shRNA‐mediated silencing of caspase‐1 210 or class II transactivator have also been attempted. 211 In addition, inhibition of Nurr1 212 or Shp‐2 213 by shRNAs was designed as a potential strategy for the management of levodopa‐induced dyskinesias. However, since Nurr1 has multiple functions including protection of dopaminergic neurons against neurotoxins and suppressing neuroinflammation, 214 there is still much research needed before the Nurr1‐based PD therapeutics can be considered for the treatment of PD.
3.3.4. miRNA therapeutics
miRNAs are short noncoding RNAs, generally, 20–25 nucleotides in length, that regulate gene expression through binding to the 3′‐UTR of a targeted mRNA. Understanding the critical roles of miRNAs in cellular and molecular biology, 215 and unveiling the mechanisms of dysregulation of miRNAs under disease conditions 216 , 217 , 218 has made these molecules very attractive propositions with respect to drug development. Since the first discovery of mammalian miRNAs in 2000, rapid progress in RNA chemistry and delivery technologies has enabled multiple miRNA‐based therapeutic compounds to move into clinical trials. 140 miRNA molecules can be synthesized with distinct chemical modifications that determine the mode of actions (Figure 3vi,vii). miR mimics are double‐stranded short RNA molecules and are often modified by O‐methylation of the passenger strand to increase stability. 140 When delivered to the targeted tissue, miR mimics can replenish the expression of downregulated miRNAs and silence targeted mRNA expression. 216 On the other hand, single‐stranded antimiRs (also called antagomiRs) either incorporating LNA or 2‐OME residues can inhibit specific miRNAs and upregulate targeted mRNA expression. 219 In conjunction with antimiRs, miRNA “sponges” or “decoys” are other strategies that can inhibit miRNA activity by providing multiple target sites complementary to specific miRNAs, and subsequently prevent miRNAs binding to endogenous target genes. 220 , 221
Studies have shown that miR‐7 regulates neuroinflammation and can also bind directly to specific regions of the α‐synuclein 3′‐UTR and repress α‐synuclein expression. 222 , 223 Injecting miR‐7 mimics directly into mouse striatum suppressed inflammasome activation and attenuated dopaminergic cell death. 224 Using a miR‐decoy stably reduced miR‐7 function and resulted in an increased expression of α‐synuclein and a rapid loss of dopaminergic neurons in vivo. 225 Other miRNAs that could downregulate α‐synuclein expression include miR‐153 and miR‐214, the latter also participating in brain repair in PD mouse models. 222 , 226 , 227 miR‐132 regulates embryonic stem cell differentiation into dopaminergic cells, while miR‐132 was upregulated in a PD mouse model and reduced dopamine neuron differentiation. The miRNA sponge mmu‐circRNA‐0003292 is hypothesized to sequester miR‐132 and affect PD pathogenesis, 228 however, further experimental verification is needed.
The miRNAs usually have a wide range of targets and a single miRNA can exert effects and influence multiple different cellular and molecular pathways. For example, transgenic mice overexpressing miR‐7 showed impaired insulin secretion and a decline in the expression level of transcriptional factors for pancreatic islet β‐cell differentiation and developed diabetes. 229 Over 1,000 genes were found to be dysregulated in an unbiased genome‐wide expression assay in a pancreatic β‐cell line after adenovirus delivered miR‐7 treatment. 229 Thus, before bringing miRNA therapeutics into clinical applications, potential perturbations to other body systems must be thoroughly investigated. In addition, unlike the application of miRNAs therapeutics in liver disease, where miRNA mimics or antimiRs may readily enter the target tissue and exert their effects, 230 difficulties in miRNA delivery across the blood–brain barrier is one of the factors that limit the progress in miRNA‐based therapeutics 231 and other therapeutic nucleic acids. Different miRNA profiles are found in PD and other neurodegenerative disorders, with increased expression of miR‐30a‐5p, miR‐153, and miR‐4639‐5p being identified in PD compared with unaffected healthy individuals. 232 , 233 , 234 Thus, it is likely that, in the near future, the application of miRNA panels on body‐fluids such as a serum, saliva, and cerebrospinal fluid may provide potential PD diagnostic biomarkers, and may also help stratify PD patients according to the genetic and molecular pathways involved.
3.3.5. Aptamers
Aptamers are single‐stranded DNA or RNA molecules that fold into defined three‐dimensional (3D) structures for specific target binding (Figure 3viii). Aptamers are generally selected from large DNA or RNA oligonucleotide libraries by systematic evolution of ligands by exponential enrichment. 235 The specific 3D configuration of these small molecules enables selected aptamers to bind to specific targets in a lock‐key manner. 236 Thus, aptamers have a wide range of potential applications including diagnostics, biomarker discovery, and targeted therapeutics where aptamers can be divided into three groups: antagonist, agonist, and carrier for other therapeutic compounds. Including the first FDA approved RNA aptamer drug Macugen ®, all aptamers currently in clinical trials were designed to function as antagonists.
The first DNA aptamer selected to bind to α‐synuclein initiated the exploration of these compounds in PD research, including investigating the pathogenesis and developing diagnostics and therapeutics for PD. 237 Since α‐synuclein oligomers are reported to be cytotoxic and are likely to play a major role in PD pathogenesis, 238 DNA aptamers were selected to bind to α‐synuclein oligomers specifically, rather than monomers or fibrils. 239 In light of additional evidence that antibodies targeting the C‐terminus of α‐synuclein reduced the extent of oligomerization and ameliorated nigral dopaminergic neuron loss, 240 , 241 two aptamers selected from 11, 019 sequences were found to bind to α‐synuclein with high affinity. 242 After entry into SK‐N‐SH cells and primary neurons, these peptide‐conjugated aptamers inhibited α‐synuclein aggregation, promoted α‐synuclein clearance, protected against α‐synuclein‐induced mitochondrial dysfunction, and rescued the cell defects. 242
Aptamers have also been applied to monitor dopamine concentration, with “Apta‐sensors” including the ultrasensitive and selective voltammetric apta‐sensor, 243 and gold nanoparticle enhanced surface plasmon resonance apta‐sensor being reported to quantitate dopamine levels down to 200 fM. 244 This superb level of sensitivity and specificity in detecting dopamine supports the development of apta‐sensors as tools for clinical diagnostics and monitoring disease progression in clinical trials. Although aptamer therapies have some disadvantages, including rapid clearance, metabolic instability, and poor translation in in vivo studies, 245 , 246 aptamers are believed to have considerable potential as novel therapeutics with advanced biological functions, once aptamer designs and modifications are optimized.
4. CHALLENGES IN NUCLEIC ACID THERAPEUTICS FOR PD
The first and foremost obstacle for the application of nucleic acid therapeutics for PD and other CNS disorders is the blood–brain barrier. Blood‐to‐brain transporters have been shown to facilitate transport of systemically‐delivered ASOs across the blood–brain barrier, and consequently bring a certain level of therapeutic benefit in animal models of Alzheimer's disease 247 and stroke. 248 However, the amount of ASOs that reached the brain in these studies was less than 1% of the dose administered intravenously. 249 Several strategies have been investigated to address the blood–brain barrier and increase drug delivery into the CNS, including direct CNS administration and the vehicle cargo drug delivery system, as illustrated in Figure 4.
Figure 4.
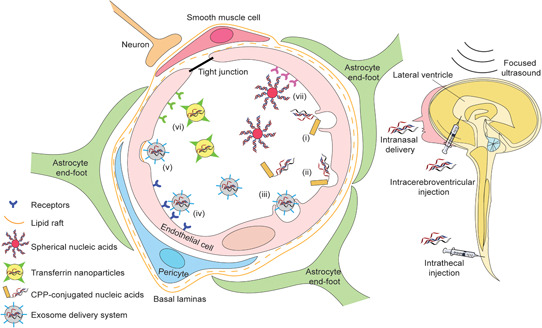
Strategies for the delivery of nucleic acid drugs across the blood–brain barrier into the central nervous system. The blood–brain barrier is formed by the cerebral endothelial cells with tight junctions at their margins, pericytes, basal lamina, and astrocytic end‐feet. Intracerebroventricular and intrathecal injection or infusion can directly administer compounds, including nucleic acids, into the central nervous system. Through intranasal administration, nucleic acid drugs can enter the central nervous system through the nose‐to‐brain route, mainly mediated by the olfactory and trigeminal nerve pathways. 250 Cell‐penetrating peptide‐conjugated nucleic acids are taken up through macropinocytosis (i) and endocytosis (ii) by the endothelial cells. The exosome system delivers nucleic acids across the blood–brain barrier mainly through receptor‐mediated endocytosis (iv), lipid‐raft mediated endocytosis (v), and macropinocytosis (iii). Receptor‐mediated endocytosis also contributes to the uptake of transferrin nanoparticles (vi) and spherical nucleic acids (vii) [Color figure can be viewed at wileyonlinelibrary.com]
ICV injection/infusion is considered to be a safe and long‐term route for CNS drug administration. 251 ASOs composed of 2‐OMe modified bases on a phosphorothioate backbone were shown to be evenly distributed in the parenchyma surrounding the ventricles in the mouse brain within a few hours after ICV infusion. 252 These ASOs were taken up preferentially by neurons in key brain structures such as the cerebellum, striatum, hippocampus, and dentate gyrus. 252 Although the pharmacokinetics and distribution pattern of other chemical modifications might be slightly different, ICV‐delivered ASOs using several different chemistries are generally showing therapeutic potential in preclinical studies. Continuous ICV infusion of a 2‐MOE phosphorothioate ASO was reported to mediate a sustained reversal of the Huntington's disease phenotype. 253 Furthermore, persisting allele‐specific mutant HTT gene silencing was achieved by ICV injection of constrained ethyl (cEt) ASOs. 254 However, the chance of adverse events, including infection and tissue damage induced by ICV cannot be ignored. The possibility of activation of the innate immune system, as was detected after ICV administration of 2‐OMe ASOs in mice, 255 is another factor that may eventually restrict the long term, repeated ICV ASO delivery in clinical studies.
Since the clinical trial (NCT01041222) using intrathecal delivery of an ASO to treat familial amyotrophic lateral sclerosis caused by SOD1 mutations, 256 most of the clinical trials for CNS indications are using intrathecal delivery. 130 Intrathecal injection of Nusinersen to infants and children with spinal muscular dystrophy was shown to be safe and tolerable. 257 However, as ICV and intrathecal drug delivery are both relatively invasive procedures, intranasal administration is an alternative approach that could deliver nucleic acid compounds to the CNS through endocytosis across the permeable olfactory epithelium. 250 , 258 Intranasally administered sertraline‐conjugated siRNA was internalized into 5‐hydroxytryptamine neurons and reduced serotonin transporter expression in raphe nuclei, evoking fast antidepressant‐like responses in mice. 258 In addition, intranasal delivery of LNA antagomirs targeting miR‐210 decreased brain miR‐210 levels and improved neurological function later in life, which outcomes were similar to those resulting from ICV‐mediated ASO treatment in a neonatal rat model. 259
In addition to selecting optimal delivery routes to achieve expected CNS therapeutic effects, multiple novel vehicle cargo drug delivery systems are being developed. One promising delivery approach is to conjugate cell‐penetrating peptides that penetrate the blood–brain barrier to the ASOs, thereby facilitating the uptake and actions of nucleic acid compounds. A feature of many of these peptides is the presence of tracts of arginine residues interrupted by short hydrophobic sequences. 260 PNA/PMO internalization peptide 6a (Pip6a), derived from the Penetratin peptide, consists of a central hydrophobic core flanked on either side by arginine‐rich sequences. 261 Pip6a‐PMO was shown to not only be taken up by muscle cells through endocytosis in vitro, 262 but also increased SMN expression in CNS tissues and resulted in profound phenotypic correction in SMA mice. 263 However, toxic effects, especially nephrotoxicity, remains as the main challenge to this category of peptide reaching clinical application. 264
Exosomes are endocytic nano‐vesicles that could enter the CNS through transcytosis or internalization by endothelial cells 265 and thus have been designed as drug vehicles. 266 Several preclinical studies are using exosomes to deliver siRNAs for neurological diseases, including Huntington's disease 267 and glioblastoma multiforme with few toxic effects or immunogenicity observed after repeated doses. 268 , 269 In one recent study, RVG peptide was fused to lysosome‐associated membrane glycoprotein (Lamp2b) on exosomes, and the RVG‐Lamp2b modified exosome was loaded with miR‐124 mimics. Systemic administration of the RVG‐Lamp2b‐miR‐124 localized to the infarct region in a focal cerebral ischemia model and ameliorated the ischemic injury by promoting neurogenesis. 269 Exosomes as the carriers for nucleic acid compounds are biocompatible, immunologically inert, and can efficiently reach the target. 270 However, efficiently and reliably isolating and characterizing exosomes is one of the obstacles that need to be addressed for future clinical applications.
Expanding research on the blood–brain barrier has changed the view on the blood–brain interface and transformed strategies to overcome its impermeability. 271 Blood–brain barrier‐penetrating peptides bearing the anti‐transferrin receptor antibody 272 and transferrin‐containing nanoparticles 273 promote the entry of therapeutic cargos into the CNS by absorptive transcytosis. Other nanoparticle‐nucleic acid delivery systems, such as spherical nucleic acids (gold nanoparticles) 274 and ASO‐liquid nanocapsules 275 are also being investigated to deliver nucleic acids across the blood–brain barrier (Figure 4). Spherical nucleic acids are generally composed of gold nanoparticles at the core and a dense layer of thiol‐modified oligonucleotide in the shell, which render spherical nucleic acids more resistant to degradation compared to linear nucleic acids. 276 More importantly, spherical nucleic acids can be rapidly internalized by any cell type and readily cross the blood–brain barrier when administered systemically. 277 This nanoparticle system has been given approval by the FDA for delivery of NU‐0129, a spherical nucleic acid gold nanoparticle containing siRNAs targeting the Bcl‐2‐like protein, for an early‐stage clinical trial in glioblastoma multiforme. In its Phase 0 first‐in‐human trial (NCT03020017), initial evidence of crossing blood–brain barrier, with no unexpected adverse effects, further validated this approach for CNS drug delivery. 278
These studies demonstrate the potential of vehicle cargo delivery systems to enhance the delivery of nucleic acid oligomers across the blood–brain barrier to their targets in the CNS. However, many outstanding challenges remain, particularly relating to the stability of the complex compound, possible adverse effects from off‐target distribution of peptides, toxicity, and efficacy of nanoparticles need to be addressed before these complex compounds can progress to clinical trials. Alternatively, physical methods such as MRI‐guided focused ultrasound‐induced blood–brain barrier opening has been approved as a novel means for CNS drug delivery. 279 Since enhanced delivery of ASOs to the target brain region was achieved by focused ultrasound, 280 many clinical trials are now applying MRI‐guided focused ultrasound, and showing safe and reversible opening of the blood–brain barrier. 281
5. CONCLUSIONS AND OUTLOOK
This review describes advances in the understanding of the complex molecular pathogenesis of PD, and the proteins and genes that play key roles in familial PD, as well as the more prevalent sporadic form of the disease. It also draws attention to the importance of biomarker‐driven subtyping of PD as a basis for the application of a precision‐medicine approach to the development of novel therapeutic strategies, including nucleic acid‐based therapeutics. Such an approach is an intuitive and logical way of addressing the problem of heterogeneity in PD and of identifying genetic‐molecular mechanisms that may be more specific to certain disease subtypes and could be targets for novel disease‐modifying therapies.
Nucleic acid therapeutics have demonstrated great diversity and potential for the treatment of various neurological disorders and have several important advantages, including high target specificity, stability, and low toxicity and immunogenicity. Other crucial advantages of nucleic acid therapeutics over other small molecule drugs, include the ease and predictable chemical synthesis of these compounds, efficient drug‐discovery process, and similarities in toxicity and pharmacokinetic profiling. 282 Within 10 months of identification of the genetic cause in a patient with Batten's disease, Milasen ®, a splice‐switching oligomer received FDA approval, demonstrating the potential for rapid design and development of nucleic acid therapeutics. Although Milasen ® is a patient‐customized drug development case, 14 it could serve as an exemplar to open up new avenues for clinical trials and regulation of individualized medicines for rare diseases. 283 The potential of nucleic acid therapeutics in PD is substantial, and studies to date have already shown that they have the ability to modify the expression of key PD proteins such as α‐synuclein and its specific isoforms. Although environmental factors such as pesticides or metals may participate in α‐synuclein aggregation, or the gastrointestinal microbiome could affect PD drug metabolism or clinical phenotypes, nucleic acid‐based therapeutics represent potentially new pathways for disease‐modifying therapies for PD that could be applicable to both familial and sporadic forms of the disease, and have enormous potential, warranting further investigation and development.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
ACKNOWLEDGMENTS
Dunhui Li receives a postgraduate scholarship from Muscular Dystrophy Western Australia. This study was supported by funding to Steve D. Wilton and Sue Fletcher from the National Health and Medical Research Council of Australia (Grant No. 1144791). The authors also acknowledge support from the Medical Health Research Infrastructure Fund of Western Australia.
Biographies
Dunhui Li graduated in Medicine from Binzhou Medical College in 2013 and obtained Masters' degree in Medicine in 2016. Under supervision of Professor Shengdi Chen, Dr. Li focused on exploring biomarkers especially neuroimage biomarkers including single‐photon emission computed tomography and transcranial sonography for the diagnosis and disease progression of Parkinson's disease. He was also involved in genetic studies for Parkinson's disease. In 2016, Dr. Li came to Australia and starts doing biomedical research as a PhD candidate at the Centre for Molecular Medicine and Innovative Therapeutics, Murdoch University, Australia, under the supervision of Professor Steve Wilton and Professor Sue Fletcher, aiming to explore novel nucleic acid therapies for Duchenne muscular dystrophy and Parkinson's disease. Dr. Li has published 12 papers so far including research articles and review papers and won a few awards at both local and international conferences.
Frank Mastaglia completed his MBBS from the University of Western Australia in 1964. He then went on to train at the Institute of Neurology in London and at the Regional Neurological Centre in Newcastle upon Tyne in the UK and the Massachusetts General Hospital in Boston, USA. He held the Chair of Experimental Neurology at the University of Newcastle. On his return to Australia, Professor Mastaglia was appointed Professor of Neurology and became Head of Neurology at the Sir Charles Gairdner Hospital and Head of the UWA Department of Medicine. He joined the Australian Neuromuscular Research Institute, now the Perron Institute for Neurological and Translational Science in 1990, and he is in a senior advisory role in this institute and the leader of the Parkinson's disease research group aiming to better understand the genetic and molecular pathogenesis, biomarkers for disease progression of Parkinson's disease.
Sue Fletcher is a Senior Principal Research Fellow at Murdoch University. Professor Fletcher obtained a BSc in Zoology and Biochemistry in 1976 and completed a PhD in Physiology at the University of Western Australia in 1988 and joined the UWA centre for Neuromuscular and Neurological Disorders as a molecular biologist in 1991. Together with Professor Steve Wilton, Professor Sue Fletcher has pioneered the development of antisense oligonucleotide therapies for Duchenne muscular dystrophy that lead to the accelerated approval of Exondys 51 ® and Vyondys 53 ® by the US FDA The translational impact of Professor Fletcher's research on Duchenne muscular dystrophy has been acknowledged through several awards, including the 2012 Western Australia Innovator of the Year Award and the 2013 Eureka Prize for Medical Research Translation jointly with Professor Wilton.
Steve Wilton completed his PhD at the University of Adelaide and joined the Perron Institute in 1991, working initially with Professor Nigel Laing developing molecular diagnostic tests for genes implicated in a number of neuromuscular conditions. Professor Wilton is now the Perron Institute's Director and holds the Foundation Chair in the Centre for Molecular Medicine and Innovative Therapeutics, Murdoch University focusing on molecular genetic therapies. Together with Professor Sue Fletcher, Steve Wilton developed the antisense oligonucleotide‐mediated exon skipping therapy for Duchenne muscular dystrophy, and the compound Exondys 51 ® is approved by the Food and Drug Administration USA for the treatment of Duchenne patient with amenable mutations in 2016. The translational impact of Professor Wilton's research has been acknowledged through several awards, including 2012 WA Innovator of the Year Award, the 2013 Eureka Prize for Medical Research Translation jointly with Professor Sue Fletcher, the 2014 Labgear Australia Discovery Science Award, and nominated Western Australian of the Year in the Professions category in 2016. His current research explores the use of nucleic acid therapies in other diseases, including spinal muscular atrophy, Spinocerebellar ataxia type 3, motor neuron disorders, and Parkinson's disease.
Li D, Mastaglia FL, Fletcher S, Wilton SD. Progress in the molecular pathogenesis and nucleic acid therapeutics for Parkinson's disease in the precision medicine era. Med Res Rev. 2020;40:2650‐2681. 10.1002/med.21718
REFERENCES
- 1. Dorsey ER, Constantinescu R, Thompson JP, et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007;68(5):384‐386. [DOI] [PubMed] [Google Scholar]
- 2. Parkinson J. An essay on the shaking palsy. J Neuropsychiatry Clin Neurosci. 2002;14(2):223‐236. [DOI] [PubMed] [Google Scholar]
- 3. Durcan R, Wiblin L, Lawson RA, et al. Prevalence and duration of non‐motor symptoms in prodromal Parkinson's disease. Eur J Neurol. 2019;26:979‐985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Poewe W, Seppi K, Tanner CM, et al. Parkinson disease. Nature Rev Dis Primers. 2017;3:17013. [DOI] [PubMed] [Google Scholar]
- 5. Visanji NP, Brooks PL, Hazrati LN, Lang AE. The prion hypothesis in Parkinson's disease: Braak to the future. Acta Neuropathol Commun. 2013;1:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha‐synuclein gene identified in families with Parkinson's disease. Science. 1997;276(5321):2045‐2047. [DOI] [PubMed] [Google Scholar]
- 7. Noyce A. GWAS of Parkinson's disease clinical biomarkers in 12 longitudinal patients' cohorts. Mov Disord. 2019;34:1839‐1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Athauda D, Foltynie T. Challenges in detecting disease modification in Parkinson's disease clinical trials. Parkinsonism Relat Disord. 2016;32:1‐11. [DOI] [PubMed] [Google Scholar]
- 9. Puschmann A. Monogenic Parkinson's disease and parkinsonism: clinical phenotypes and frequencies of known mutations. Parkinsonism Rel Disord. 2013;19(4):407‐415. [DOI] [PubMed] [Google Scholar]
- 10. Jankovic J. Pathogenesis‐targeted therapeutic strategies in Parkinson's disease. Mov Disord. 2019;34(1):41‐44. [DOI] [PubMed] [Google Scholar]
- 11. Charleston JS, Schnell FJ, Dworzak J, et al. Eteplirsen treatment for Duchenne muscular dystrophy: exon skipping and dystrophin production. Neurology. 2018;90(24):e2146‐e2154. [DOI] [PubMed] [Google Scholar]
- 12. Stein CA, Castanotto D. FDA‐approved oligonucleotide therapies in 2017. Mol Ther. 2017;25(5):1069‐1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sardone V, Zhou H, Muntoni F, Ferlini A, Falzarano M. Antisense oligonucleotide‐based therapy for neuromuscular disease. Molecules. 2017;22(4):563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim J, Hu C, Moufawad El Achkar C, et al. Patient‐customized oligonucleotide therapy for a rare genetic disease. N Engl J Med. 2019;381(17):1644‐1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schulte C, Gasser T. Genetic basis of Parkinson's disease: inheritance, penetrance, and expression. Appl Clin Genet. 2011;4:67‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Singleton AB, Farrer MJ, Bonifati V. The genetics of Parkinson's disease: progress and therapeutic implications. Mov Disord. 2013;28(1):14‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Truban D, Hou X, Caulfield TR, Fiesel FC, Springer W. PINK1, Parkin, and mitochondrial quality control: what can we learn about Parkinson's disease pathobiology? J Parkinsons Dis. 2017;7(1):13‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saunders‐Pullman R, Mirelman A, Alcalay RN, et al. Progression in the LRRK2‐asssociated Parkinson disease population. JAMA Neurol. 2018;75(3):312‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Trinh J, Guella I, Farrer MJ. Disease penetrance of late‐onset parkinsonism: a meta‐analysis. JAMA Neurol. 2014;71(12):1535‐1539. [DOI] [PubMed] [Google Scholar]
- 20. Paisan‐Ruiz C, Bhatia KP, Li A, et al. Characterization of PLA2G6 as a locus for dystonia‐parkinsonism. Ann Neurol. 2009;65(1):19‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Joseph S, Schulz JB, Stegmüller J. Mechanistic contributions of FBXO7 to Parkinson disease. J Neurochem. 2018;144(2):118‐127. [DOI] [PubMed] [Google Scholar]
- 22. Struhal W, Presslauer S, Spielberger S, et al. VPS35 Parkinson's disease phenotype resembles the sporadic disease. J Neural Transm. 2014;121(7):755‐759. [DOI] [PubMed] [Google Scholar]
- 23. Trinh J, Zeldenrust F, Huang J, et al. Genotype‐phenotype relations for the Parkinson's disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov Disord. 2018;33(12):1857‐1870. [DOI] [PubMed] [Google Scholar]
- 24. Elsayed LE, Drouet V, Usenko T, et al. A novel nonsense mutation in DNAJC6 expands the phenotype of autosomal‐recessive juvenile‐onset Parkinson's disease. Ann Neurol. 2016;79(2):335‐337. [DOI] [PubMed] [Google Scholar]
- 25. Olgiati S, Quadri M, Fang M, et al. DNAJC6 mutations associated with early‐onset Parkinson's disease. Ann Neurol. 2016;79(2):244‐256. [DOI] [PubMed] [Google Scholar]
- 26. Krebs CE, Karkheiran S, Powell JC, et al. The Sac1 domain of SYNJ1 identified mutated in a family with early‐onset progressive parkinsonism with generalized seizures. Hum Mutat. 2013;34(9):1200‐1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Necpál J, Vodička R, Kaňovský P. A novel mutation in the GIGYF2 gene in a patient with Parkinson's disease. CESKA A SLOVENSKA NEUROLOGIE A NEUROCHIRURGIE. 2017;80(6):719‐721. [Google Scholar]
- 28. Strauss KM, Martins LM, Plun‐Favreau H, et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson's disease. Hum Mol Genet. 2005;14(15):2099‐2111. [DOI] [PubMed] [Google Scholar]
- 29. Fujioka S, Sundal C, Strongosky AJ, et al. Sequence variants in eukaryotic translation initiation factor 4‐gamma (eIF4G1) are associated with Lewy body dementia. Acta Neuropathol. 2013;125(3):425‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Deng H‐X, Shi Y, Yang Y, et al. Identification of TMEM230 mutations in familial Parkinson's disease. Nature Genet. 2016;48:733‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Funayama M, Ohe K, Amo T, et al. CHCHD2 mutations in autosomal dominant late‐onset Parkinson's disease: a genome‐wide linkage and sequencing study. Lancet Neurol. 2015;14(3):274‐282. [DOI] [PubMed] [Google Scholar]
- 32. Robak LA, Jansen IE, Van Rooij J, et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson's disease. Brain. 2017;140(12):3191‐3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012;11(11):986‐998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kuzuhara S, Mori H, Izumiyama N, Yoshimura M, Ihara Y. Lewy bodies are ubiquitinated. A light and electron microscopic immunocytochemical study. Acta Neuropathol. 1988;75(4):345‐353. [DOI] [PubMed] [Google Scholar]
- 35. Bartels T, Choi JG, Selkoe DJ. α‐Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477(7362):107‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang W, Perovic I, Chittuluru J, et al. A soluble alpha‐synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci U S A. 2011;108(43):17797‐17802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zondler L, Kostka M, Garidel P, et al. Proteasome impairment by α‐synuclein. PLoS One. 2017;12(9):e0184040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Manecka D‐L, Vanderperre B, Fon EA, Durcan TM. The neuroprotective role of protein quality control in halting the development of alpha‐synuclein pathology. Front Mol Neurosci. 2017;10(311). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Walden H, Muqit MM. Ubiquitin and Parkinson's disease through the looking glass of genetics. Biochem J. 2017;474(9):1439‐1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee Y, Kang HC, Lee BD, Lee Y‐I, Kim YP, Shin J‐H. Poly (ADP‐ribose) in the pathogenesis of Parkinson's disease. BMB Rep. 2014;47(8):424‐432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Olsen AL, Feany MB. PARP inhibitors and Parkinson's disease. N Engl J Med. 2019;380(5):492‐494. [DOI] [PubMed] [Google Scholar]
- 42. Arena G, Valente EM. PINK1 in the limelight: multiple functions of an eclectic protein in human health and disease. J Pathol. 2017;241(2):251‐263. [DOI] [PubMed] [Google Scholar]
- 43. Lee Y, Stevens DA, Kang SU, et al. PINK1 primes parkin‐mediated ubiquitination of PARIS in dopaminergic neuronal survival. Cell Rep. 2017;18(4):918‐932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maraganore DM, Lesnick TG, Elbaz A, et al. UCHL1 is a Parkinson's disease susceptibility gene. Ann Neurol. 2004;55(4):512‐521. [DOI] [PubMed] [Google Scholar]
- 45. Bett JS, Ritorto MS, Ewan R, et al. Ubiquitin C‐terminal hydrolases cleave isopeptide‐ and peptide‐linked ubiquitin from structured proteins but do not edit ubiquitin homopolymers. Biochem J. 2015;466(3):489‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wilkinson KD, Lee KM, Deshpande S, Duerksen‐Hughes P, Boss JM, Pohl J. The neuron‐specific protein PGP 9.5 is a ubiquitin carboxyl‐terminal hydrolase. Science. 1989;246(4930):670‐673. [DOI] [PubMed] [Google Scholar]
- 47. Liu H, Povysheva N, Rose ME, et al. Role of UCHL1 in axonal injury and functional recovery after cerebral ischemia. Proc Natl Acad Sci U S A. 2019;116:4643‐4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yan C, Huo H, Yang C, Zhang T, Chu Y, Liu Y. Ubiquitin C‐terminal hydrolase L1 regulates autophagy by inhibiting autophagosome formation through its deubiquitinating enzyme activity. Biochem Biophys Res Commun. 2018;497(2):726‐733. [DOI] [PubMed] [Google Scholar]
- 49. Zheng Q, Huang T, Zhang L, et al. Dysregulation of ubiquitin‐proteasome system in neurodegenerative diseases. Front Aging Neurosci. 2016;8(303):303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Heride C, Urbé S, Clague MJ. Ubiquitin code assembly and disassembly. Curr Biol. 2014;24(6):R215‐R220. [DOI] [PubMed] [Google Scholar]
- 51. Robak LA, Jansen IE, van Rooij J, et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson's disease. Brain. 2017;140(12):3191‐3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ysselstein D, Shulman JM, Krainc D. Emerging links between pediatric lysosomal storage diseases and adult parkinsonism. Mov Disord. 2019;34(5):614‐624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med. 2009;361(17):1651‐1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stoker TB, Torsney KM, Barker RA. Pathological mechanisms and clinical aspects of GBA1 mutation‐associated Parkinson's disease In: Stoker TB, Greenland JC, eds. Parkinson's Disease: Pathogenesis and Clinical Aspects. Brisbane, Australia: Codon Publications; 2018:45–64. [PubMed] [Google Scholar]
- 55. Wang R, Tan J, Chen T, et al. ATP13A2 facilitates HDAC6 recruitment to lysosome to promote autophagosome–lysosome fusion. J Cell Biol. 2019;218(1):267‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Demirsoy S, Martin S, Motamedi S, et al. ATP13A2/PARK9 regulates endo‐/lysosomal cargo sorting and proteostasis through a novel PI(3, 5)P2‐mediated scaffolding function. Hum Mol Genet. 2017;26(9):1656‐1669. [DOI] [PubMed] [Google Scholar]
- 57. Rahman AA, Morrison BE. Contributions of VPS35 mutations to Parkinson's disease. Neuroscience. 2019;401:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Menšíková K, Tučková L, Kolařiková K, et al. Atypical parkinsonism of progressive supranuclear palsy–parkinsonism (PSP‐P) phenotype with rare variants in FBXO7 and VPS35 genes associated with Lewy body pathology. Acta Neuropathol. 2019;137(1):171‐173. [DOI] [PubMed] [Google Scholar]
- 59. Yu WH, Dorado B, Figueroa HY, et al. Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric α‐synuclein. Am J Pathol. 2009;175(2):736‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xilouri M, Brekk OR, Stefanis L. Autophagy and alpha‐synuclein: relevance to Parkinson's disease and related synucleopathies. Mov Disord. 2016;31(2):178‐192. [DOI] [PubMed] [Google Scholar]
- 61. Manzoni C. The LRRK2‐macroautophagy axis and its relevance to Parkinson's disease. Biochem Soc Trans. 2017;45(1):155‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine‐analog synthesis. Science. 1983;219(4587):979‐980. [DOI] [PubMed] [Google Scholar]
- 63. Park J‐S, Davis RL, Sue CM. Mitochondrial dysfunction in Parkinson's disease: new mechanistic insights and therapeutic perspectives. Curr Neurol Neurosci Rep. 2018;18(5):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Singh A, Zhi L, Zhang H. LRRK2 and mitochondria: recent advances and current views. Brain Res. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chen M‐L, Wu R‐M. LRRK 2 gene mutations in the pathophysiology of the ROCO domain and therapeutic targets for Parkinson's disease: a review. J Biomed Sci. 2018;25(1):52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gao F, Yang J, Wang D, et al. Mitophagy in Parkinson's disease: pathogenic and therapeutic implications. Front Neurol. 2017;8:527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nguyen TN, Padman BS, Lazarou M. Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol. 2016;26(10):733‐744. [DOI] [PubMed] [Google Scholar]
- 68. Narendra D, Tanaka A, Suen D‐F, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183(5):795‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Burchell VS, Nelson DE, Sanchez‐Martinez A, et al. The Parkinson's disease–linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nature Neurosci. 2013;16(9):1257‐1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ryan BJ, Hoek S, Fon EA, Wade‐Martins R. Mitochondrial dysfunction and mitophagy in Parkinson's: from familial to sporadic disease. Trends Biochem Sci. 2015;40(4):200‐210. [DOI] [PubMed] [Google Scholar]
- 71. Bonello F, Hassoun S‐M, Mouton‐Liger F, et al. LRRK2 impairs PINK1/Parkin‐dependent mitophagy via its kinase activity: pathologic insights into Parkinson's disease. Hum Mol Genet. 2019;28(10):1645‐1660. [DOI] [PubMed] [Google Scholar]
- 72. Lenka A, Pal PK. "Miro" in Parkinson's disease: here, there, everywhere! Mov Disord. 2017;32(6):839. [DOI] [PubMed] [Google Scholar]
- 73. Hsieh C‐H, Shaltouki A, Gonzalez AE, et al. Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson's disease. Cell Stem Cell. 2016;19(6):709‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shaltouki A, Hsieh C‐H, Kim MJ, Wang X. Alpha‐synuclein delays mitophagy and targeting miro rescues neuron loss in Parkinson's models. Acta Neuropathol. 2018;136(4):607‐620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Li H, Ham A, Ma TC, et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy. 2019;15(1):113‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang R, Tan J, Chen T, et al. ATP13A2 facilitates HDAC6 recruitment to lysosome to promote autophagosome‐lysosome fusion. J Cell Biol. 2019;218(1):267‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ludtmann MHR, Abramov AY. Mitochondrial calcium imbalance in Parkinson's disease. Neurosci Lett. 2018;663:86‐90. [DOI] [PubMed] [Google Scholar]
- 78. Doonan PJ, Chandramoorthy HC, Hoffman NE, et al. LETM1‐dependent mitochondrial Ca2+ flux modulates cellular bioenergetics and proliferation. FASEB J. 2014;28(11):4936‐4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Huang E, Qu D, Huang T, et al. PINK1‐mediated phosphorylation of LETM1 regulates mitochondrial calcium transport and protects neurons against mitochondrial stress. Nat Commun. 2017;8(1):1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Soman S, Keatinge M, Moein M, et al. Inhibition of the mitochondrial calcium uniporter rescues dopaminergic neurons in pink1−/− zebrafish. Eur J Neurosci. 2017;45(4):528‐535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Verma M, Callio J, Otero PA, Sekler I, Wills ZP, Chu CT. Mitochondrial calcium dysregulation contributes to dendrite degeneration mediated by PD/LBD‐associated LRRK2 mutants. J Neurosci. 2017;37(46):11151‐11165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gómez‐Suaga P, Bravo‐San Pedro JM, González‐Polo RA, Fuentes JM, Niso‐Santano M. ER‐mitochondria signaling in Parkinson's disease. Cell Death Dis. 2018;9(3):337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gautier CA, Erpapazoglou Z, Mouton‐Liger F, et al. The endoplasmic reticulum‐mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum Mol Genet. 2016;25(14):2972‐2984. [DOI] [PubMed] [Google Scholar]
- 84. Strobbe D, Robinson AA, Harvey K, et al. Distinct mechanisms of pathogenic DJ‐1 mutations in mitochondrial quality control. Front Mol Neurosci. 2018;11:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Liu Y, Ma X, Fujioka H, Liu J, Chen S, Zhu X. DJ‐1 regulates the integrity and function of ER‐mitochondria association through interaction with IP3R3‐Grp75‐VDAC1. Proc Natl Acad Sci U S A. 2019;116(50):25322‐25328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zanon A, Kalvakuri S, Rakovic A, et al. SLP‐2 interacts with Parkin in mitochondria and prevents mitochondrial dysfunction in Parkin‐deficient human iPSC‐derived neurons and Drosophila. Hum Mol Genet. 2017;26(13):2412‐2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Cho CH, Kim E‐A, Kim J, Choi SY, Yang S‐J, Cho S‐W. N‐Adamantyl‐4‐methylthiazol‐2‐amine suppresses amyloid β‐induced neuronal oxidative damage in cortical neurons. Free Radic Res. 2016;50(6):678‐690. [DOI] [PubMed] [Google Scholar]
- 88. Jiang T, Sun Q, Chen S. Oxidative stress: a major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson's disease and Alzheimer's disease. Prog Neurobiol. 2016;147:1‐19. [DOI] [PubMed] [Google Scholar]
- 89. Di Maio R, Barrett PJ, Hoffman EK, et al. α‐Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson's disease. Sci Transl Med. 2016;8(342):342ra378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Grassi D, Howard S, Zhou M, et al. Identification of a highly neurotoxic α‐synuclein species inducing mitochondrial damage and mitophagy in Parkinson's disease. Proc Natl Acad Sci U S A. 2018;115(11):E2634‐E2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Raninga PV, Di Trapani G, Tonissen KF. The multifaceted roles of DJ‐1 as an antioxidant. Adv Exp Med Biol. 2017;1037:67‐87. [DOI] [PubMed] [Google Scholar]
- 92. Lu L, Zhao S, Gao G, Sun X, Zhao H, Yang H. DJ‐1/PARK7, but not its L166P mutant linked to autosomal recessive Parkinsonism, modulates the transcriptional activity of the orphan nuclear receptor Nurr1 in vitro and in vivo. Mol Neurobiol. 2016;53(10):7363‐7374. [DOI] [PubMed] [Google Scholar]
- 93. Takahashi‐Niki K, Kato‐Ose I, Murata H, Maita H, Iguchi‐Ariga SM, Ariga H. Epidermal growth factor‐dependent activation of the extracellular signal‐regulated kinase pathway by DJ‐1 protein through its direct binding to c‐Raf protein. J Biol Chem. 2015;290(29):17838‐17847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Biosa A, Sandrelli F, Beltramini M, Greggio E, Bubacco L, Bisaglia M. Recent findings on the physiological function of DJ‐1: Beyond Parkinson's disease. Neurobiol Dis. 2017;108:65‐72. [DOI] [PubMed] [Google Scholar]
- 95. Oh SE, Mouradian MM. Cytoprotective mechanisms of DJ‐1 against oxidative stress through modulating ERK1/2 and ASK1 signal transduction. Redox Biol. 2018;14:211‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Fernandez‐Caggiano M, Schroder E, Cho H‐J, et al. Oxidant‐induced interprotein disulfide formation in cardiac DJ‐1 occurs via an interaction with peroxiredoxin 2. J Biol Chem. 2016;291(19):10399‐10410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Cuevas S, Yang Y, Konkalmatt P, et al. Role of nuclear factor erythroid 2–related factor 2 in the oxidative stress–dependent hypertension associated with the depletion of DJ‐1. Hypertension. 2015;65(6):1251‐1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Xu S, Yang X, Qian Y, Xiao Q. Parkinson's disease‐related DJ‐1 modulates the expression of uncoupling protein 4 against oxidative stress. J Neurochem. 2018;145(4):312‐322. [DOI] [PubMed] [Google Scholar]
- 99. Johnson WM, Golczak M, Choe K, et al. Regulation of DJ‐1 by glutaredoxin 1 in vivo: implications for Parkinson's disease. Biochemistry. 2016;55(32):4519‐4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA‐DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38(8):1285‐1291. [DOI] [PubMed] [Google Scholar]
- 101. Rocha NP, de Miranda AS, Teixeira AL. Insights into neuroinflammation in Parkinson's disease: from biomarkers to anti‐inflammatory based therapies. BioMed Res Int. 2015;2015:628192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Wang Q, Liu Y, Zhou J. Neuroinflammation in Parkinson's disease and its potential as therapeutic target. Transl Neurodegener. 2015;4:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lenz KM, Nelson LH. Microglia and beyond: innate immune cells as regulators of brain development and behavioral function. Front Immunol. 2018;9:698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Cekanaviciute E, Buckwalter MS. Astrocytes: integrative regulators of neuroinflammation in stroke and other neurological diseases. Neurotherapeutics. 2016;13(4):685‐701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Chen Z, Chen S, Liu J. The role of T cells in the pathogenesis of Parkinson's disease. Prog Neurobiol. 2018;169:1‐23. [DOI] [PubMed] [Google Scholar]
- 106. Sommer A, Winner B, Prots I. The Trojan horse—neuroinflammatory impact of T cells in neurodegenerative diseases. Mol Neurodegener. 2017;12:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Chen S, Liu Y, Niu Y, et al. Increased abundance of myeloid‐derived suppressor cells and Th17 cells in peripheral blood of newly‐diagnosed Parkinson's disease patients. Neurosci Lett. 2017;648:21‐25. [DOI] [PubMed] [Google Scholar]
- 108. Sommer A, Marxreiter F, Krach F, et al. Th17 lymphocytes induce neuronal cell death in a human iPSC‐based model of Parkinson's disease. Cell Stem Cell. 2018;23(1):123‐131.e6. [DOI] [PubMed] [Google Scholar]
- 109. Sommer A, Fadler T, Dorfmeister E, et al. Infiltrating T lymphocytes reduce myeloid phagocytosis activity in synucleinopathy model. J Neuroinflammation. 2016;13(1):174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Liu Z, Huang Y, Cao BB, Qiu YH, Peng YP. Th17 cells induce dopaminergic neuronal death via LFA‐1/ICAM‐1 interaction in a mouse model of Parkinson's disease. Mol Neurobiol. 2017;54(10):7762‐7776. [DOI] [PubMed] [Google Scholar]
- 111. Korn T, Kallies A. T cell responses in the central nervous system. Nat Rev Immunol. 2017;17(3):179‐194. [DOI] [PubMed] [Google Scholar]
- 112. Schetters STT, Gomez‐Nicola D, Garcia‐Vallejo JJ, Van Kooyk Y. Neuroinflammation: microglia and T cells get ready to tango. Front Immunol. 2017;8:1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Subramaniam SR, Federoff HJ. Targeting microglial activation states as a therapeutic avenue in Parkinson's disease. Front Aging Neurosci. 2017;9:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Zhang Q‐S, Heng Y, Yuan Y‐H, Chen N‐H. Pathological α‐synuclein exacerbates the progression of Parkinson's disease through microglial activation. Toxicol Lett. 2017;265:30‐37. [DOI] [PubMed] [Google Scholar]
- 115. Sanchez‐Guajardo V, Tentillier N, Romero‐Ramos M. The relation between α‐synuclein and microglia in Parkinson's disease: recent developments. Neuroscience. 2015;302:47‐58. [DOI] [PubMed] [Google Scholar]
- 116. Kim C, Spencer B, Rockenstein E, et al. Immunotherapy targeting toll‐like receptor 2 alleviates neurodegeneration in models of synucleinopathy by modulating alpha‐synuclein transmission and neuroinflammation. Mol Neurodegener. 2018;13(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Liu W, Liu X, Li Y, et al. LRRK2 promotes the activation of NLRC4 inflammasome during Salmonella typhimurium infection. J Exp Med. 2017;214(10):3051‐3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kim KS, Marcogliese PC, Yang J, et al. Regulation of myeloid cell phagocytosis by LRRK2 via WAVE2 complex stabilization is altered in Parkinson's disease. Proc Natl Acad Sci U S A. 2018;115(22):E5164‐E5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Alessi DR, Sammler E. LRRK2 kinase in Parkinson's disease. Science. 2018;360(6384):36‐37. [DOI] [PubMed] [Google Scholar]
- 120. Newman LE, Shadel GS. Pink1/Parkin link inflammation, mitochondrial stress, and neurodegeneration. J Cell Biol. 2018;217(10):3327‐3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Sliter DA, Martinez J, Hao L, et al. Parkin and PINK1 mitigate STING‐induced inflammation. Nature. 2018;561(7722):258‐262. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 122. Dhir A, Dhir S, Borowski LS, et al. Mitochondrial double‐stranded RNA triggers antiviral signalling in humans. Nature. 2018;560(7717):238‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Trudler D, Nash Y, Frenkel D. New insights on Parkinson's disease genes: the link between mitochondria impairment and neuroinflammation. J Neural Transm. 2015;122(10):1409‐1419. [DOI] [PubMed] [Google Scholar]
- 124. Afonina IS, Zhong Z, Karin M, Beyaert R. Limiting inflammation—the negative regulation of NF‐κB and the NLRP3 inflammasome. Nature Immunol. 2017;18(8):861‐869. [DOI] [PubMed] [Google Scholar]
- 125. Mouton‐Liger F, Jacoupy M, Corvol J‐C, Corti O. PINK1/Parkin‐dependent mitochondrial surveillance: from pleiotropy to Parkinson's disease. Front Mol Neurosci. 2017;10:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Sun L, Shen R, Agnihotri SK, Chen Y, Huang Z, Bueler H. Lack of PINK1 alters glia innate immune responses and enhances inflammation‐induced, nitric oxide‐mediated neuron death. Sci Rep. 2018;8(1):383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Reeve AK, Grady JP, Cosgrave EM, et al. Mitochondrial dysfunction within the synapses of substantia nigra neurons in Parkinson's disease. NPJ Parkinsons Dis. 2018;4(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Singleton A, Hardy J. Progress in the genetic analysis of Parkinson's disease. Hum Mol Genet. 2019;28:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson's disease. Lancet Neurol. 2019;19:170‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Khorkova O, Wahlestedt C. Oligonucleotide therapies for disorders of the nervous system. Nat Biotechnol. 2017;35(3):249‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Yin W, Rogge M. Targeting RNA: a transformative therapeutic strategy. Clin Transl Sci. 2019;12(2):98‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Park J, Park J, Pei Y, Xu J, Yeo Y. Pharmacokinetics and biodistribution of recently‐developed siRNA nanomedicines. Adv Drug Deliv Rev. 2016;104:93‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Geary RS, Norris D, Yu R, Bennett CF. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev. 2015;87:46‐51. [DOI] [PubMed] [Google Scholar]
- 134. Healy JM, Lewis SD, Kurz M, et al. Pharmacokinetics and biodistribution of novel aptamer compositions. Pharm Res. 2004;21(12):2234‐2246. [DOI] [PubMed] [Google Scholar]
- 135. Tang JY, Temsamani J, Agrawal S. Self‐stabilized antisense oligodeoxynucleotide phosphorothioates: properties and anti‐HIV activity. Nucleic Acids Res. 1993;21(11):2729‐2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Monia BP, Johnston JF, Sasmor H, Cummins LL. Nuclease resistance and antisense activity of modified oligonucleotides targeted to Ha‐ras. J Biol Chem. 1996;271(24):14533‐14540. [DOI] [PubMed] [Google Scholar]
- 137. Gustincich S, Zucchelli S, Mallamaci A. The Yin and Yang of nucleic acid‐based therapy in the brain. Prog Neurobiol. 2017;155:194‐211. [DOI] [PubMed] [Google Scholar]
- 138. Zhou J, Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat Rev Drug Discov. 2017;16(3):181‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Wittrup A, Lieberman J. Knocking down disease: a progress report on siRNA therapeutics. Nat Rev Genet. 2015;16(9):543‐552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16(3):203‐222. [DOI] [PubMed] [Google Scholar]
- 141. Schultz RG, Gryaznov SM. Oligo‐2′‐fluoro‐2′‐deoxynucleotide N3′→ P5′ phosphoramidates: synthesis and properties. Nucleic Acids Res. 1996;24(15):2966‐2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Amantana A, Iversen PL. Pharmacokinetics and biodistribution of phosphorodiamidate morpholino antisense oligomers. Curr Opin Pharmacol. 2005;5(5):550‐555. [DOI] [PubMed] [Google Scholar]
- 143. Iversen PL, Aird KM, Wu R, Morse MM, Devi GR. Cellular uptake of neutral phosphorodiamidate morpholino oligomers. Curr Pharm Biotechnol. 2009;10(6):579‐588. [DOI] [PubMed] [Google Scholar]
- 144. Le BT, Chen S, Abramov M, Herdewijn P, Veedu RN. Evaluation of anhydrohexitol nucleic acid, cyclohexenyl nucleic acid and d‐altritol nucleic acid‐modified 2′‐O‐methyl RNA mixmer antisense oligonucleotides for exon skipping in vitro. Chem Commun. 2016;52(92):13467‐13470. [DOI] [PubMed] [Google Scholar]
- 145. Le BT, Filichev VV, Veedu RN. Investigation of twisted intercalating nucleic acid (TINA)‐modified antisense oligonucleotides for splice modulation by induced exon‐skipping in vitro. RSC Adv. 2016;6(97):95169‐95172. [Google Scholar]
- 146. Stanton R, Sciabola S, Salatto C, et al. Chemical modification study of antisense gapmers. Nucleic Acid Ther. 2012;22(5):344‐359. [DOI] [PubMed] [Google Scholar]
- 147. Hartmann G, Krug A, Waller‐Fontaine K, Endres S. Oligodeoxynucleotides enhance lipopolysaccharide‐stimulated synthesis of tumor necrosis factor: dependence on phosphorothioate modification and reversal by heparin. Mol Med. 1996;2(4):429‐438. [PMC free article] [PubMed] [Google Scholar]
- 148. Henry SP, Beattie G, Yeh G, et al. Complement activation is responsible for acute toxicities in rhesus monkeys treated with a phosphorothioate oligodeoxynucleotide. Int Immunopharmacol. 2002;2(12):1657‐1666. [DOI] [PubMed] [Google Scholar]
- 149. Henry SP, Giclas PC, Leeds J, et al. Activation of the alternative pathway of complement by a phosphorothioate oligonucleotide: potential mechanism of action. J Pharmacol Exp Ther. 1997;281(2):810‐816. [PubMed] [Google Scholar]
- 150. Toonen L, Casaca‐Carreira J, Pellisé‐Tintoré M, et al. Intracerebroventricular administration of a 2′‐O‐methyl phosphorothioate antisense oligonucleotide results in activation of the innate immune system in mouse brain. Nucleic Acid Ther. 2018;28(2):63‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Shen L, Engelhardt JA, Hung G, et al. Effects of repeated complement activation associated with chronic treatment of cynomolgus monkeys with 2′‐o‐methoxyethyl modified antisense oligonucleotide. Nucleic Acid Ther. 2016;26(4):236‐249. [DOI] [PubMed] [Google Scholar]
- 152. Henry SP, Jagels MA, Hugli TE, et al. Mechanism of alternative complement pathway dysregulation by a phosphorothioate oligonucleotide in monkey and human serum. Nucleic Acid Ther. 2014;24(5):326‐335. [DOI] [PubMed] [Google Scholar]
- 153. Kakiuchi‐Kiyota S, Koza‐Taylor PH, Mantena SR, et al. Comparison of hepatic transcription profiles of locked ribonucleic acid antisense oligonucleotides: evidence of distinct pathways contributing to non‐target mediated toxicity in mice. Toxicol Sci. 2014;138(1):234‐248. [DOI] [PubMed] [Google Scholar]
- 154. Burdick AD, Sciabola S, Mantena SR, et al. Sequence motifs associated with hepatotoxicity of locked nucleic acid—modified antisense oligonucleotides. Nucleic Acids Res. 2014;42(8):4882‐4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Henry SP, Kim T‐W, Kramer‐Stickland K, Zanardi TA, Fey RA, Levin AA. Toxicologic properties of 2′‐O‐methoxyethyl chimeric antisense inhibitors in animals and man In: Crooke ST, ed. Antisense Drug Technology: Principles, Strategies and Applications. 2nd ed New York, NY: CRC Press; 2007:327‐363. [Google Scholar]
- 156. van Poelgeest EP, Swart RM, Betjes MG, et al. Acute kidney injury during therapy with an antisense oligonucleotide directed against PCSK9. Am J Kidney Dis. 2013;62(4):796‐800. [DOI] [PubMed] [Google Scholar]
- 157. Seok H, Lee H, Jang E‐S, Chi SW. Evaluation and control of miRNA‐like off‐target repression for RNA interference. Cell Mol Life Sci. 2018;75(5):797‐814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Persengiev SP, Zhu X, Green MR. Nonspecific, concentration‐dependent stimulation and repression of mammalian gene expression by small interfering RNAs (siRNAs). RNA. 2004;10(1):12‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Caffrey DR, Zhao J, Song Z, et al. siRNA off‐target effects can be reduced at concentrations that match their individual potency. PLoS One. 2011;6(7):e21503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Jackson AL, Burchard J, Leake D, et al. Position‐specific chemical modification of siRNAs reduces “off‐target” transcript silencing. RNA. 2006;12(7):1197‐1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Mook O, Vreijling J, Wengel SL, et al. In vivo efficacy and off‐target effects of locked nucleic acid (LNA) and unlocked nucleic acid (UNA) modified siRNA and small internally segmented interfering RNA (sisiRNA) in mice bearing human tumor xenografts. Artif DNA PNA XNA. 2010;1(1):36‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Zhang L, Liang D, Chen C, et al. Circular siRNAs for reducing off‐target effects and enhancing long‐term gene silencing in cells and mice. Mol Ther Nucleic Acids. 2018;10:237‐244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Mobergslien A, Sioud M. Exploring 5′‐biotinylation of the sense strand to improve siRNA specificity and potency In: Sioud M, ed. RNA Interference and CRISPR Technologies. New York, NY: Springer; 2020:163‐170. [DOI] [PubMed] [Google Scholar]
- 164. Hagedorn PH, Hansen BR, Koch T, Lindow M. Managing the sequence‐specificity of antisense oligonucleotides in drug discovery. Nucleic Acids Res. 2017;45(5):2262‐2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Hagedorn PH, Pontoppidan M, Bisgaard TS, et al. Identifying and avoiding off‐target effects of RNase H‐dependent antisense oligonucleotides in mice. Nucleic Acids Res. 2018;46(11):5366‐5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Hua Y, Vickers TA, Baker BF, Bennett CF, Krainer AR. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007;5(4):73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Wang Z, Jeon HY, Rigo F, Bennett CF, Krainer AR. Manipulation of PK‐M mutually exclusive alternative splicing by antisense oligonucleotides. Open Biol. 2012;2(10):120133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Scharner J, Ma WK, Zhang Q, et al. Hybridization‐mediated off‐target effects of splice‐switching antisense oligonucleotides. Nucleic Acids Res. 2020;48(2):802‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Liu W, Wang X. Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol. 2019;20(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Lück S, Kreszies T, Strickert M, Schweizer P, Kuhlmann M, Douchkov D. siRNA‐finder (si‐Fi) software for RNAi‐target design and off‐target prediction. Front Plant Sci. 2019;10(1023):1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Zhou X, Wang S, Zhu Y, Pan Y, Zhang L, Yang Z. Overcoming the delivery barrier of oligonucleotide drugs and enhancing nucleoside drug efficiency: the use of nucleolipids. Med Res Rev. 2019;40(4):1178‐1199. [DOI] [PubMed] [Google Scholar]
- 172. Ndeboko B, Ramamurthy N, Lemamy GJ, Jamard C, Nielsen PE, Cova L. Role of cell‐penetrating peptides in intracellular delivery of peptide nucleic acids targeting hepadnaviral replication. Mol Ther Nucleic Acids. 2017;9:162‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Catuogno S, Esposito CL, Condorelli G, de Franciscis V. Nucleic acids delivering nucleic acids. Adv Drug Deliv Rev. 2018;134:79‐93. [DOI] [PubMed] [Google Scholar]
- 174. Betker JL, Xu L, Zhang Y, Anchordoquy TJ. Utilizing cholesterol nanodomains for nucleic acid delivery In: Ilies MA, ed. Control of Amphiphile Self‐Assembling at the Molecular Level: Supra‐Molecular Assemblies with Tuned Physicochemical Properties for Delivery Applications. Washington, DC: ACS Publications; 2017:71‐93. [Google Scholar]
- 175. Chernikov IV, Vlassov VV, Chernolovskaya EL. Current development of siRNA bioconjugates: from research to the clinic. Front Pharmacol. 2019;10:444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Janas MM, Schlegel MK, Harbison CE, et al. Selection of GalNAc‐conjugated siRNAs with limited off‐target‐driven rat hepatotoxicity. Nat Commun. 2018;9(1):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Springer AD, Dowdy SF. GalNAc‐siRNA conjugates: leading the way for delivery of RNAi therapeutics. Nucleic Acid Ther. 2018;28(3):109‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Kim Y, Jo M, Schmidt J, et al. Enhanced potency of GalNAc‐conjugated antisense oligonucleotides in hepatocellular cancer models. Mol Ther. 2019;27(9):1547‐1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.DRUG USF. FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. 2016; https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-drug-duchenne-muscular-dystrophy. Accessed 19 September 2016.
- 180.DRUG USF. FDA grants accelerated approval to first targeted treatment for rare Duchenne muscular dystrophy mutation. 2019; https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation. Accessed 12 December 2019.
- 181. Benson MD, Waddington‐Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Daher JP, Abdelmotilib HA, Hu X, et al. Leucine‐rich repeat Kinase 2 (LRRK2) pharmacological inhibition abates alpha‐synuclein gene‐induced neurodegeneration. J Biol Chem. 2015;290(32):19433‐19444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Zhao HT, John N, Delic V, et al. LRRK2 antisense oligonucleotides ameliorate alpha‐synuclein inclusion formation in a Parkinson's disease mouse model. Mol Ther Nucleic Acids. 2017;8:508‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. The Michael J. Fox Foundation. Positive results from Parkinson's LRRK2 drug trial. 2018; https://www.michaeljfox.org/news/positive-results-parkinsons-lrrk2-drug-trial. Accessed 2 August 2018.
- 185. Uehara T, Choong C‐J, Nakamori M, et al. Amido‐bridged nucleic acid (AmNA)‐modified antisense oligonucleotides targeting α‐synuclein as a novel therapy for Parkinson's disease. Sci Rep. 2019;9(1):7567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Schneider SA, Alcalay RN. Precision medicine in Parkinson's disease: emerging treatments for genetic Parkinson's disease. J Neurol. 2020;267(3):860‐869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Mills JD, Janitz M. Alternative splicing of mRNA in the molecular pathology of neurodegenerative diseases. Neurobiol Aging. 2012;33(5):1012.e11‐1012.e24. [DOI] [PubMed] [Google Scholar]
- 188. Sterne‐Weiler T, Sanford JR. Exon identity crisis: disease‐causing mutations that disrupt the splicing code. Genome Biol. 2014;15(1):201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Flynn LL, Mitrpant C, Pitout IL, Fletcher S, Wilton SD. Antisense oligonucleotide‐mediated terminal intron retention of the SMN2 transcript. Mol Ther Nucleic Acids. 2018;11:91‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Li D, Mastaglia FL, Fletcher S, Wilton SD. Precision medicine through antisense oligonucleotide‐mediated exon skipping. Trends Pharmacol Sci. 2018;39:982‐994. [DOI] [PubMed] [Google Scholar]
- 191. Beyer K, Ariza A. Alpha‐synuclein posttranslational modification and alternative splicing as a trigger for neurodegeneration. Mol Neurobiol. 2013;47(2):509‐524. [DOI] [PubMed] [Google Scholar]
- 192. Qian H, Kang X, Hu J, et al. Reversing a model of Parkinson's disease with in situ converted nigral neurons. Nature. 2020;582(7813):550‐556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Mahncke HW, Bronstone A, Merzenich MM. Brain plasticity and functional losses in the aged: scientific bases for a novel intervention. Prog Brain Res. 2006;157:81‐109. [DOI] [PubMed] [Google Scholar]
- 194. Ledford H. Gene‐silencing technology gets first drug approval after 20‐year wait. Nature. 2018;560(7718):291‐292. [DOI] [PubMed] [Google Scholar]
- 195. Adams D, Gonzalez‐Duarte A, O'Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11‐21. [DOI] [PubMed] [Google Scholar]
- 196.FDA. FDA approves givosiran for acute hepatic porphyria. 2019; https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-givosiran-acute-hepatic-porphyria. Accessed 20 November 2019.
- 197. Fitzgerald K, Frank‐Kamenetsky M, Shulga‐Morskaya S, et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single‐blind, placebo‐controlled, phase 1 trial. Lancet. 2014;383(9911):60‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819‐829. [DOI] [PubMed] [Google Scholar]
- 199. Ahn TB, Kim SY, Kim JY, et al. Alpha‐synuclein gene duplication is present in sporadic Parkinson disease. Neurology. 2008;70(1):43‐49. [DOI] [PubMed] [Google Scholar]
- 200. Singleton AB, Farrer M, Johnson J, et al. Alpha‐synuclein locus triplication causes Parkinson's disease. Science. 2003;302(5646):841. [DOI] [PubMed] [Google Scholar]
- 201. Schlich M, Longhena F, Faustini G, et al. Anionic liposomes for small interfering ribonucleic acid (siRNA) delivery to primary neuronal cells: evaluation of alpha‐synuclein knockdown efficacy. Nano Res. 2017;10(10):3496‐3508. [Google Scholar]
- 202. Helmschrodt C, Höbel S, Schöniger S, et al. Polyethylenimine nanoparticle‐mediated siRNA delivery to reduce α‐synuclein expression in a model of Parkinson's disease. Mol Ther Nucleic Acids. 2017;9:57‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Cooper JM, Wiklander PB, Nordin JZ, et al. Systemic exosomal siRNA delivery reduced alpha‐synuclein aggregates in brains of transgenic mice. Mov Disord. 2014;29(12):1476‐1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Gorbatyuk OS, Li S, Nash K, et al. In vivo RNAi‐mediated alpha‐synuclein silencing induces nigrostriatal degeneration. Mol Ther. 2010;18(8):1450‐1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Takahashi M, Suzuki M, Fukuoka M, et al. Normalization of overexpressed alpha‐synuclein causing Parkinson's disease by a moderate gene silencing with RNA interference. Mol Ther Nucleic Acids. 2015;4:e241. [DOI] [PubMed] [Google Scholar]
- 206. Rao DD, Vorhies JS, Senzer N, Nemunaitis J. siRNA vs. shRNA: similarities and differences. Adv Drug Deliv Rev. 2009;61(9):746‐759. [DOI] [PubMed] [Google Scholar]
- 207. Jiao Y, Xia ZL, Ze LJ, Jing H, Xin B, Fu S. Research progress of nucleic acid delivery vectors for gene therapy. Biomed Microdevices. 2020;22(1):16. [DOI] [PubMed] [Google Scholar]
- 208. Zharikov AD, Cannon JR, Tapias V, et al. shRNA targeting alpha‐synuclein prevents neurodegeneration in a Parkinson's disease model. J Clin Invest. 2015;125(7):2721‐2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Niu S, Zhang LK, Zhang L, et al. Inhibition by multifunctional magnetic nanoparticles loaded with alpha‐synuclein RNAi plasmid in a Parkinson's disease model. Theranostics. 2017;7(2):344‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Wang W, Nguyen LT, Burlak C, et al. Caspase‐1 causes truncation and aggregation of the Parkinson's disease‐associated protein alpha‐synuclein. Proc Natl Acad Sci U S A. 2016;113(34):9587‐9592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Williams GP, Schonhoff AM, Jurkuvenaite A, Thome AD, Standaert DG, Harms AS. Targeting of the class II transactivator attenuates inflammation and neurodegeneration in an alpha‐synuclein model of Parkinson's disease. J Neuroinflammation. 2018;15(1):244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Sellnow RC, Steece‐Collier K, Kanaan NM, et al. 709. rAAV‐mediated regulation of striatal Nurr1 expression alters development and severity of levodopa‐induced dyskinesias in the 6‐OHDA rat model of Parkinson's disease. Mol Ther. 2015;23:S282‐S283. [Google Scholar]
- 213. Fiorentini C, Savoia P, Savoldi D, et al. Shp‐2 knockdown prevents l‐dopa‐induced dyskinesia in a rat model of Parkinson's disease. Mov Disord. 2016;31(4):512‐520. [DOI] [PubMed] [Google Scholar]
- 214. Dong J, Li S, Mo JL, Cai HB, Le WD. Nurr1‐based therapies for Parkinson's disease. CNS Neurosci Ther. 2016;22(5):351‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281‐297. [DOI] [PubMed] [Google Scholar]
- 216. Chakraborty C, Sharma AR, Sharma G, Doss CGP, Lee S‐S. Therapeutic miRNA and siRNA: moving from bench to clinic as next generation medicine. Mol Ther Nucleic Acids. 2017;8:132‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Greenberg DS, Soreq H. MicroRNA therapeutics in neurological disease. Curr Pharm Des. 2014;20(38):6022‐6027. [DOI] [PubMed] [Google Scholar]
- 218. Leggio L, Vivarelli S, L'Episcopo F, et al. MicroRNAs in Parkinson's disease: from pathogenesis to novel diagnostic and therapeutic approaches. Int J Mol Sci. 2017;18(12):2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Cheng CJ, Bahal R, Babar IA, et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature. 2014;518:107‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4(9):721‐726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Haraguchi T, Ozaki Y, Iba H. Vectors expressing efficient RNA decoys achieve the long‐term suppression of specific microRNA activity in mammalian cells. Nucleic Acids Res. 2009;37(6):e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Doxakis E. Post‐transcriptional regulation of alpha‐synuclein expression by mir‐7 and mir‐153. J Biol Chem. 2010;285:12726‐12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Junn E, Lee KW, Jeong BS, Chan TW, Im JY, Mouradian MM. Repression of alpha‐synuclein expression and toxicity by microRNA‐7. Proc Natl Acad Sci U S A. 2009;106(31):13052‐13057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Zhou Y, Lu M, Du RH, et al. MicroRNA‐7 targets Nod‐like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson's disease. Mol Neurodegener. 2016;11:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. McMillan KJ, Murray TK, Bengoa‐Vergniory N, et al. Loss of microRNA‐7 regulation leads to α‐synuclein accumulation and dopaminergic neuronal loss in vivo. Mol Ther. 2017;25(10):2404‐2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Wang ZH, Zhang JL, Duan YL, Zhang QS, Li GF, Zheng DL. MicroRNA‐214 participates in the neuroprotective effect of Resveratrol via inhibiting α‐synuclein expression in MPTP‐induced Parkinson's disease mouse. Biomed Pharmacother. 2015;74:252‐256. [DOI] [PubMed] [Google Scholar]
- 227. Saraiva C, Paiva J, Santos T, Ferreira L, Bernardino L. MicroRNA‐124 loaded nanoparticles enhance brain repair in Parkinson's disease. J Control. Release. 2016;235:291‐305. [DOI] [PubMed] [Google Scholar]
- 228. Jia E, Zhou Y, Liu Z, et al. Transcriptomic profiling of circular RNA in different brain regions of Parkinson's disease in a mouse model. Int J Mol Sci. 2020;21(8):3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Latreille M, Hausser J, Stützer I, et al. MicroRNA‐7a regulates pancreatic beta cell function. J Clin Invest. 2014;124(6):2722‐2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Fu Y, Chen J, Huang Z. Recent progress in microRNA‐based delivery systems for the treatment of human disease. ExRNA. 2019;1(1):1‐14. [Google Scholar]
- 231. Petrescu GE, Sabo AA, Torsin LI, Calin GA, Dragomir MP. MicroRNA based theranostics for brain cancer: basic principles. J Exp Clin Cancer Res. 2019;38(1):231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Chen Y, Gao C, Sun Q, et al. MicroRNA‐4639 is a regulator of DJ‐1 expression and a potential early diagnostic marker for Parkinson's disease. Front Aging Neurosci. 2017;9:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Schwienbacher C, Foco L, Picard A, et al. Plasma and white blood cells show different miRNA expression profiles in Parkinson's disease. J Mol Neurosci. 2017;62(2):244‐254. [DOI] [PubMed] [Google Scholar]
- 234. Cressatti M, Juwara L, Galindez JM, et al. Salivary microR‐153 and microR‐223 levels as potential diagnostic biomarkers of idiopathic Parkinson's disease. Mov Disord. 2020;35(3):468‐477. [DOI] [PubMed] [Google Scholar]
- 235. Wang T, Chen C, Larcher LM, Barrero RA, Veedu RN. Three decades of nucleic acid aptamer technologies: lessons learned, progress and opportunities on aptamer development. Biotech Adv. 2019;37(1):28‐50. [DOI] [PubMed] [Google Scholar]
- 236. Chen L, Rashid F, Shah A, et al. The isolation of an RNA aptamer targeting to p53 protein with single amino acid mutation. Proc Natl Acad Sci U S A. 2015;112(32):10002‐10007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Tsukakoshi K, Harada R, Sode K, Ikebukuro K. Screening of DNA aptamer which binds to α‐synuclein. Biotechnol Lett. 2010;32(5):643‐648. [DOI] [PubMed] [Google Scholar]
- 238. Kalia LV, Kalia SK, McLean PJ, Lozano AM, Lang AE. α‐Synuclein oligomers and clinical implications for Parkinson disease. Ann Neurol. 2013;73(2):155‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Tsukakoshi K, Abe K, Sode K, Ikebukuro K. Selection of DNA aptamers that recognize alpha‐synuclein oligomers using a competitive screening method. Anal Chem. 2012;84(13):5542‐5547. [DOI] [PubMed] [Google Scholar]
- 240. Tran HT, Chung CH, Iba M, et al. Alpha‐synuclein immunotherapy blocks uptake and templated propagation of misfolded alpha‐synuclein and neurodegeneration. Cell Rep. 2014;7(6):2054‐2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Näsström T, Gonçalves S, Sahlin C, et al. Antibodies against alpha‐synuclein reduce oligomerization in living cells. PLoS One. 2011;6(10):e27230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Zheng Y, Qu J, Xue F, et al. Novel DNA aptamers for Parkinson's disease treatment inhibit alpha‐synuclein aggregation and facilitate its degradation. Mol Ther Nucleic Acids. 2018;11:228‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Wang W, Wang W, Davis JJ, Luo X. Ultrasensitive and selective voltammetric aptasensor for dopamine based on a conducting polymer nanocomposite doped with graphene oxide. Microchim Acta. 2015;182(5):1123‐1129. [Google Scholar]
- 244. Cao Y, McDermott MT. Femtomolar and selective dopamine detection by a gold nanoparticle enhanced surface plasmon resonance aptasensor. bioRxiv. 2018. 10.1101/273078 [DOI] [Google Scholar]
- 245. Kanwar JR, Shankaranarayanan JS, Gurudevan S, Kanwar RK. Aptamer‐based therapeutics of the past, present and future: from the perspective of eye‐related diseases. Drug Discov Today. 2014;19(9):1309‐1321. [DOI] [PubMed] [Google Scholar]
- 246. Qu J, Yu S, Zheng Y, Zheng Y, Yang H, Zhang J. Aptamer and its applications in neurodegenerative diseases. Cell Mol Life Sci. 2017;74(4):683‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Erickson MA, Niehoff ML, Farr SA, et al. Peripheral administration of antisense oligonucleotides targeting the amyloid‐β protein precursor reverses AβPP and LRP‐1 overexpression in the aged SAMP8 mouse brain. J Alzheimers Dis. 2012;28(4):951‐960. [DOI] [PubMed] [Google Scholar]
- 248. Dogrukol‐Ak D, Kumar VB, Ryerse JS, et al. Isolation of peptide transport system‐6 from brain endothelial cells: therapeutic effects with antisense inhibition in Alzheimer and stroke models. J Cereb Blood Flow Metab. 2009;29(2):411‐422. [DOI] [PubMed] [Google Scholar]
- 249. Banks WA, Farr SA, Butt W, Kumar VB, Franko MW, Morley JE. Delivery across the blood‐brain barrier of antisense directed against amyloid beta: reversal of learning and memory deficits in mice overexpressing amyloid precursor protein. J Pharmacol Exp Ther. 2001;297(3):1113‐1121. [PubMed] [Google Scholar]
- 250. Alarcón‐Arís D, Recasens A, Galofré M, et al. Selective α‐synuclein knockdown in monoamine neurons by intranasal oligonucleotide delivery: potential therapy for Parkinson's disease. Mol Ther. 2018;26(2):550‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Cohen‐Pfeffer JL, Gururangan S, Lester T, et al. Intracerebroventricular delivery as a safe, long‐term route of drug administration. Pediatr Neurol. 2017;67:23‐35. [DOI] [PubMed] [Google Scholar]
- 252. Casaca‐Carreira J, Temel Y, Larrakoetxea I, Jahanshahi A. Distribution and penetration of intracerebroventricularly administered 2′OMePS oligonucleotide in the mouse brain. Nucleic Acid Ther. 2017;27(1):4‐10. [DOI] [PubMed] [Google Scholar]
- 253. Kordasiewicz HB, Stanek LM, Wancewicz EV, et al. Sustained therapeutic reversal of Huntington's disease by transient repression of huntingtin synthesis. Neuron. 2012;74(6):1031‐1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Southwell AL, Skotte NH, Kordasiewicz HB, et al. In vivo evaluation of candidate allele‐specific mutant huntingtin gene silencing antisense oligonucleotides. Mol Ther. 2014;22(12):2093‐2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Toonen LJA, Casaca‐Carreira J, Pellisé‐Tintoré M, et al. Intracerebroventricular administration of a 2′‐O‐methyl phosphorothioate antisense oligonucleotide results in activation of the innate immune system in mouse brain. Nucleic Acid Ther. 2018;28(2):63‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Miller TM, Pestronk A, David W, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first‐in‐man study. Lancet Neurol. 2013;12(5):435‐442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Haché M, Swoboda KJ, Sethna N, et al. Intrathecal injections in children with spinal muscular atrophy: nusinersen clinical trial experience. J Child Neurol. 2016;31(7):899‐906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Ferrés‐Coy A, Galofré M, Pilar‐Cuéllar F, et al. Therapeutic antidepressant potential of a conjugated siRNA silencing the serotonin transporter after intranasal administration. Mol Psychiatry. 2016;21(3):328‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Ma Q, Dasgupta C, Li Y, et al. Inhibition of microRNA‐210 provides neuroprotection in hypoxic‐ischemic brain injury in neonatal rats. Neurobiol Dis. 2016;89:202‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Juliano RL. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016;44(14):6518‐6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Boisguérin P, Deshayes S, Gait MJ, et al. Delivery of therapeutic oligonucleotides with cell penetrating peptides. Adv Drug Deliv Rev. 2015;87:52‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Lehto T, Castillo Alvarez A, Gauck S, et al. Cellular trafficking determines the exon skipping activity of Pip6a‐PMO in mdx skeletal and cardiac muscle cells. Nucleic Acids Res. 2014;42(5):3207‐3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Hammond SM, Hazell G, Shabanpoor F, et al. Systemic peptide‐mediated oligonucleotide therapy improves long‐term survival in spinal muscular atrophy. Proc Natl Acad Sci U S A. 2016;113(39):10962‐10967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Gait MJ, Arzumanov AA, McClorey G, et al. Cell‐penetrating peptide conjugates of steric blocking oligonucleotides as therapeutics for neuromuscular diseases from a historical perspective to current prospects of treatment. Nucleic Acid Ther. 2019;29(1):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Jarmalaviciute A, Pivoriunas A. Exosomes as a potential novel therapeutic tools against neurodegenerative diseases. Pharmacol Res. 2016;113(pt B):816‐822. [DOI] [PubMed] [Google Scholar]
- 266. Ohno S, Takanashi M, Sudo K, et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther. 2013;21(1):185‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Lee ST, Im W, Ban JJ, et al. Exosome‐based delivery of miR‐124 in a Huntington's disease model. J Mov Disord. 2017;10(1):45‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Yang T, Fogarty B, LaForge B, et al. Delivery of small interfering RNA to inhibit vascular endothelial growth factor in zebrafish using natural brain endothelia cell‐secreted exosome nanovesicles for the treatment of brain cancer. AAPS J. 2017;19(2):475‐486. [DOI] [PubMed] [Google Scholar]
- 269. Yang J, Zhang X, Chen X, Wang L, Yang G. Exosome mediated delivery of miR‐124 promotes neurogenesis after ischemia. Mol Ther Nucleic Acids. 2017;7:278‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Zhou Y, Zhou G, Tian C, et al. Exosome‐mediated small RNA delivery for gene therapy. Wiley Interdiscip Rev: RNA. 2016;7(6):758‐771. [DOI] [PubMed] [Google Scholar]
- 271. Banks WA. From blood–brain barrier to blood–brain interface: new opportunities for CNS drug delivery. Nat Rev Drug Discov. 2016;15(4):275‐292. [DOI] [PubMed] [Google Scholar]
- 272. Sonoda H, Morimoto H, Yoden E, et al. A blood‐brain‐barrier‐penetrating anti‐human transferrin receptor antibody fusion protein for neuronopathic mucopolysaccharidosis II. Mol Ther. 2018;26(5):1366‐1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Clark AJ, Davis ME. Increased brain uptake of targeted nanoparticles by adding an acid‐cleavable linkage between transferrin and the nanoparticle core. Proc Natl Acad Sci U S A. 2015;112(40):12486‐12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Graczyk A, Pawlowska R, Jedrzejczyk D, Chworos A. Gold nanoparticles in conjunction with nucleic acids as a modern molecular system for cellular delivery. Molecules. 2020;25(1):204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Griveau A, Bejaud J, Anthiya S, Avril S, Autret D, Garcion E. Silencing of miR‐21 by locked nucleic acid‐lipid nanocapsule complexes sensitize human glioblastoma cells to radiation‐induced cell death. Int J Pharm. 2013;454(2):765‐774. [DOI] [PubMed] [Google Scholar]
- 276. Mokhtarzadeh A, Vahidnezhad H, Youssefian L, Mosafer J, Baradaran B, Uitto J. Applications of spherical nucleic acid nanoparticles as delivery systems. Trends Mol Med. 2019;25(12):1066‐1079. [DOI] [PubMed] [Google Scholar]
- 277. Choi CHJ, Hao L, Narayan SP, Auyeung E, Mirkin CA. Mechanism for the endocytosis of spherical nucleic acid nanoparticle conjugates. Proc Natl Acad Sci U S A. 2013;110(19):7625‐7630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Kumthekar P, Rademaker A, Ko C, et al. A phase 0 first‐in‐human study using NU‐0129: a gold base spherical nucleic acid (SNA) nanoconjugate targeting BCL2L12 in recurrent glioblastoma patients. J Clin Oncol. 2019;37(15_suppl):3012. [Google Scholar]
- 279. Chen K‐T, Wei K‐C, Liu H‐L. Theranostic strategy of focused ultrasound induced blood‐brain barrier opening for CNS disease treatment. Front Pharmacol. 2019;10:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Negishi Y, Yamane M, Kurihara N, et al. Enhancement of blood‐brain barrier permeability and delivery of antisense oligonucleotides or plasmid DNA to the brain by the combination of bubble liposomes and high‐intensity focused ultrasound. Pharmaceutics. 2015;7(3):344‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Lipsman N, Meng Y, Bethune AJ, et al. Blood–brain barrier opening in Alzheimer's disease using MR‐guided focused ultrasound. Nat Commun. 2018;9(1):2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Levin AA. Treating disease at the RNA level with oligonucleotides. N Engl J Med. 2019;380(1):57‐70. [DOI] [PubMed] [Google Scholar]
- 283. Woodcock J, Marks P. Drug regulation in the era of individualized therapies. N Engl J Med. 2019;381(17):1678‐1680. [DOI] [PubMed] [Google Scholar]