

Abstract
A series of WIV alkyne complexes with the sulfur‐rich ligand hydridotris(2‐mercapto‐1‐methylimidazolyl) borate) (TmMe) are presented as bio‐inspired models to elucidate the mechanism of the tungstoenzyme acetylene hydratase (AH). The mono‐ and/or bis‐alkyne precursors were reacted with NaTmMe and the resulting complexes [W(CO)(C2R2)(TmMe)Br] (R=H 1, Me 2) oxidized to the target [WE(C2R2)(TmMe)Br] (E=O, R=H 4, Me 5; E=S, R=H 6, Me 7) using pyridine‐N‐oxide and methylthiirane. Halide abstraction with TlOTf in MeCN gave the cationic complexes [WE(C2R2)(MeCN)(TmMe)](OTf) (E=CO, R=H 10, Me 11; E=O, R=H 12, Me 13; E=S, R=H 14, Me 15). Without MeCN, dinuclear complexes [W2O(μ‐O)(C2Me2)2(TmMe)2](OTf)2 (8) and [W2(μ‐S)2(C2Me2)(TmMe)2](OTf)2 (9) could be isolated showing distinct differences between the oxido and sulfido system with the latter exhibiting only one molecule of C2Me2. This provides evidence that a fine balance of the softness at W is important for acetylene coordination. Upon dissolving complex 8 in acetonitrile complex 13 is reconstituted in contrast to 9. All complexes exhibit the desired stability toward water and the observed effective coordination of the scorpionate ligand avoids decomposition to disulfide, an often‐occurring reaction in sulfur ligand chemistry. Hence, the data presented here point toward a mechanism with a direct coordination of acetylene in the active site and provide the basis for further model chemistry for acetylene hydratase.
Keywords: acetylene hydratase, bioinorganic chemistry, soft scorpionate, tungsten, tungsten sulfide
Reduction to essential: A series of new structural models for the tungstoenzyme acetylene hydratase are presented bearing the soft scorpionate ligand TmMe. The introduction of a sulfido group gave the biologically relevant WIV complex, leading to higher electrophilic acetylene relative to the oxido system. The second substrate water did not coordinate upon bromide abstraction, pointing toward a W(C2H2) adduct in the ongoing debate on the mechanism of this enzyme.
Introduction
Molybdoenzymes are ubiquitous in nature and virtually all forms of life depend on it.1 However some organisms exchanged the molybdenum in the active site of these enzymes with the congener tungsten.2 Among these scarce tungstoenzymes acetylene hydratase (AH) stands out as the only one being able to convert acetylene to acetaldehyde.3 In this net hydration reaction the metal does not change its +IV oxidation state during catalysis which is contrary to all other molybdo‐ and tungstoenzymes.4 The WIV in the active site is coordinated by five sulfur atoms of two molybdopterin co‐factors and a cysteine residue. Its distorted octahedral geometry is completed by a water molecule as the sixth ligand.4 As to date no crystal structure of the enzyme containing the substrate could be derived, the mechanism of acetylene hydratase is still under debate.5 Two principle mechanisms are discussed with either the acetylene molecule directly coordinated to tungsten or the water molecule (Figure 1).6 In the suggested first‐shell mechanism the observed water ligand is substituted with the incoming substrate acetylene in the initial step.4 This water then engages in a nucleophilic attack of the now W‐activated acetylene. The formed vinyl anion is attacked by the water molecule and subsequent tautomerization yields acetaldehyde.7 On the other hand, a second‐shell mechanism is discussed in which the coordinated water molecule is activated by hydrogen bonding to the nearby Asp13. The thereby generated oxonium protonates the acetylene substrate which is presumably located in a hydrophobic pocket right above the active site.4 Theoretical evaluation based on DFT QM/MM calculations revealed a more realistic energy barrier of 16.7 kcal mol−1[8] for the mechanism involving a W‐C2H2 adduct and for calculation on the model complex [W(C2H2)(SCH3)(mnt)2] (mnt=malonitrile) an even lower barrier of 15.2 kcal mol−1 was obtained.9 Nevertheless the second‐shell mechanism cannot be ruled out yet as for the only structural‐functional model of AH, [Et4N]2[WO(mnt)2], no acetylene‐adduct could be spectroscopically observed.10 However, an adduct with the activated dimethylacetylenedicarboxylate could be detected and latest efforts to reproduce the catalytic activity of the functional model failed, thus supporting the first‐shell proposal.10, 11
Figure 1.
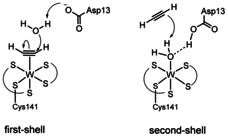
Two proposals for the disputed reactions mechanism of the enzyme acetylene hydratase.
Beside the above mentioned putative functional model, the three monomeric complexes in Figure 2 qualify as structural models. These complexes have a biological relevant WIV center with octahedral geometry and the substrate acetylene coordinated to it. The dithiocarbamate complexes [WO(C2H2)(S2CNR2)2] (R=Me, Et) (Figure 2 A) were synthesized 26 years prior to AH crystallization to study alkynes as four electron donors. They may be considered the closest active site model for a first‐shell mechanism, due to the sulfur‐rich surrounding.12 The recent complex [WO(C2H2)(S‐Phoz)2] (S‐Phoz=2‐(4′4′‐dimethyloxazoline‐2′‐yl)thiophenolate) (Figure 2 B) is bio‐inspired, featuring a non‐natural S,N‐donor motif and was specifically synthesized to resemble AH. Additionally to the structural similarity, it was able to release and re‐coordinate acetylene upon exposure to light.13 Complex [WO(C2H2)(Tp′)I] (Figure 2 C), also synthesized prior to AH crystallization, is coordinated by the N3‐ligand hydridotris(3,5‐dimethyl‐1‐pyrazolyl)borate (Tp′) and thus provides no sulfur in the first coordination sphere of W. Despite this difference to the native enzyme, [WO(C2H2)(H2O)(Tp′)](OTf) could be obtained, containing both substrates of acetylene hydratase on a WIV center, upon iodide abstraction.14
Figure 2.
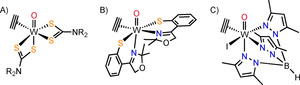
Complexes which qualify as structural models of AH.
With respect to AH, a more suitable sulfur analogue to the well‐established Tp“ ligand was introduced by Reglinsky in 1996.15 This hydridotris(2‐mercapto‐1‐methylimidazolyl) borate) ligand (TmMe) was employed in the complex [MoO2(TmMe)Cl] which successfully mimicked the catalytic reactivity of sulfite oxidase, a molybdoenzyme.16 Hill and co‐workers used this ligand as they were generally interested in alkyne binding to Mo and W complexes with a sulfur‐rich coordination sphere.17 In addition, the only monomeric tungsten complex [W(CO)(C2Me2)(TmMe)I] (Figure 3 A) containing this soft scorpionate ligand and a substituted alkyne (2‐butyne) was reported, yet its full characterization is lacking.17 In contrast, the C2H2 analogue remained elusive. In general, tungsten complexes with κ3‐S,S,S ligands as well as a coordinated alkyne are extremely rare. Besides TmMe, only the ligands bis(2‐(methylthio)ethyl)sulfane (Figure 3 B) and 2,5,8‐trithia[9]orthocyclophane (Figure 3 C) are established in the cationic complexes of the type [W(CO)(C2R2)(S,S,S)X]X, both with a substituted alkyne only.18 Herein, we have turned to the synthesis of tungsten complexes coordinated by the soft scorpionate ligand TmMe, in order to better mimic the enzyme's active site and to evaluate the influence of a sulfur‐rich W center on the coordination capability of AH′s substrates acetylene and water. As the terminal alkyne moiety of acetylene is known to be involved in side reactions like polymerization19 and was found to impose different properties and reactivity also in our hands20 a series of biologically relevant WIV complexes bearing the natural substrate acetylene as well as the more well‐behaved surrogate substrate 2‐butyne (dimethylacetylene) were prepared. This is achieved by the introduction of an oxido or a sulfido moiety at tungsten, with the latter paying respect to the five sulfur donors in the natural active site of AH. The structural design of the complexes allows for halide abstraction leading to cationic and thus presumably more electrophilic compounds which should facilitate nucleophilic attack on the coordinated acetylene. Moreover it also permits the investigation toward the direct coordination of the second substrate water. Their comparison with the analogous nitrogen‐based Tp’ system permits conclusions on the debated mechanism of acetylene hydratase.
Figure 3.
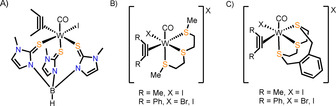
Alkyne complexes employing a S3‐donor motif.
Results and Discussion
Synthesis of [W(CO)(C2R2)(TmMe)Br]
Complexes of the type [W(CO)(C2R2)(TmMe)Br] (R=H 1, Me 2, Ph 3) were synthesized in a salt metathesis reaction starting from mono‐ and/or bis‐alkyne precursors and the NaTmMe ligand (Scheme 1).17, 20, 21 For the substituted alkynes, the precursors [W(CO)(C2R2)2(MeCN)Br2] (R=Me, Ph)22 are well established while for the respective acetylene precursor only a mixture of the bis‐ and mono‐acetylene complexes [W(CO)(C2H2)2(MeCN)Br2]/[W(CO)(C2H2)(MeCN)2Br2] is available as previously described.20 The latter was used for the preparation of [W(CO)(C2H2)(SPy)2], where low temperatures (−60 °C) and immediate work‐up at low temperatures was necessary.20 With the soft scorpionate ligand investigated here the reaction proceeds smoothly at room temperature and we found that both species in the precursor mixture reacted with the ligand to the product leading to a significantly higher yield. Also the precursors with substituted alkynes reacted selectively to the mono‐alkyne products under elimination of the second alkyne. This is in contrast to our previously investigated soft scorpionate system phenyltris((methylthio)methyl)borate (PhTt) which only allowed isolation of bis‐alkyne complexes of the type [W(CO)(C2R2)2(PhTt‐S,S′)Br] (R=Me, Ph).23 Thus, complexes 1–3 were synthesized by stirring the respective precursor and NaTmMe for 1–12 h at room temperature. After work‐up as described in the experimental section they were isolated in high yields. The yield of the acetylene complex 1 was calculated by using a mass of 472.29 g mol−1 (average of the two alkyne precursors 479.80 and 464.78 g mol−1) as the composition of the starting material mixture [W(CO)(C2H2)2(MeCN)Br2]/[W(CO)(C2H2)(MeCN)2Br2] was not determined due to its very poor solubility.
Scheme 1.
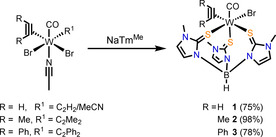
Synthetic approaches of complexes of the type [W(CO)(C2R2)(TmMe)Br].
Single crystals suitable for X‐ray diffraction analysis were obtained from CH2Cl2/heptane at −35 °C which unambiguously confirmed their molecular structures (Figures S68–S71).
The complexes show good solubility in chlorinated solvents such as CH2Cl2 and CHCl3 but also in MeCN. Complex 2 and 3 are also soluble in THF but insoluble in hydrocarbons or diethyl ether. Best solubility is observed for the pale blue 2‐butyne complex 2, followed by the grass green phenyl substituted alkyne complex 3. Green–turquoise complex 1 displays significantly lower solubility in all solvents than the substituted alkyne complexes.
IR spectroscopy shows one shifted carbonyl frequency for each complex at 1903 (1), 1882 (2) and 1913 (3) cm−1 compared with the respective precursor and virtually identical B−H stretchings between 2428–2432 cm−1. The CO stretching frequencies are in accordance with the electronic influences of the different alkyne substituents where donors result in more efficient π‐back‐donation into the CO and thus lower frequencies. The 1H NMR spectra of [W(CO)(C2R2)(TmMe)Br] (R=H 1, Me 2, Ph 3) are consistent with the asymmetric structure as they reveal three sets of methimazole signals, giving six doublets for the CH=CH group ranging from 6.79–7.04 ppm and three singlets between 3.05 and 4.00 ppm for the N‐CH3 groups. Also the alkynes give rise of two separate signals which are particularly distinct in complex 1 (acetylenic protons at 12.13 and 12.71 ppm) and in 2 (methyl protons of C2Me2 at 3.0 and 3.05 ppm). In contrast, the 1H NMR spectrum of [W(CO)3(TmMe)I]24 displays a single set of signals due to the three typical fluxional CO ligands which create an average chemical environment on the NMR time scale.25 Nevertheless, the alkynes in complexes 1–3 seem to be also involved in a fluxional behavior as their alkyne proton resonances are broadened relative to that of methimazole N‐CH3.
Oxidation to W≡O and W≡S
To oxidize the formal WII complexes [W(CO)(C2R2)(TmMe)Br] (R=H 1, Me 2, Ph 3) to the biologically relevant WIV compounds, two different oxidation agents were used (Scheme 2), namely pyridine‐N‐oxide (PyNOx) to obtain, according to literature,13 complexes of the type [WO(C2R2)(TmMe)Br] and methylthiirane for the preparation of the respective sulfido complexes [WS(C2R2)(TmMe)Br]. The latter renders the metal environment even more sulfur‐rich which is relevant to the native enzyme. The oxidation reactions were performed in MeCN at 40–45 °C with 1.1–1.3 equiv of oxidizing agent to yield the complexes [WE(C2R2)(TmMe)Br] (E=O, H 4, Me 5; E=S, H 6, Me 7). All oxidation attempts of the diphenylacetylene complex 3 turned out to be futile and either showed no conversion or led to decomposition. The oxidation reactions of 1 and 2 using PyNOx led to the desired oxido complexes. However, the reactions were accompanied by various side reactions including formation of polyoxometalates (POM) which led to lowered yields. Generally, the 2‐butyne complex 2 was found to be more reactive toward oxidation than the corresponding acetylene complex, as no full conversion of [W(CO)(C2H2)(TmMe)Br] (1) to 4 or 6 was observed which may possibly be attributed to the significant lower solubility of the starting complex 1 relative to the oxidation products.
Scheme 2.
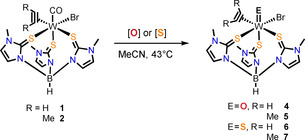
Oxidation with pyridine‐N‐oxide (O) and methylthiirane (S) to complexes of the type [WE(C2R2)(TmMe)Br].
The reaction of the acetylene compound [W(CO)(C2H2)(TmMe)Br] (1) with PyNOx allowed the isolation of [WO(C2H2)(TmMe)Br] (4) after fractionated recrystallization from chlorinated solvents combined with toluene/heptane. With this, 19 % of yellow microcrystalline 4 could be obtained as a mixture containing approximately equimolar amounts of pyridinium hydrobromide regarding elemental analysis (Figure S11). Due to similar solubility properties, only milligram amounts of PyHBr‐free [WO(C2H2)(TmMe)Br] (4) could be obtained after repeated recrystallization from CH2Cl2/THF. The origin of HBr remains unclear but presumably arises from decomposition during synthesis and purification. The respective reaction with the 2‐butyne compound 2 gave [WO(C2Me2)(TmMe)Br] (5) in 24 % yield where we were able to remove any pyridine‐containing side products by recrystallization. Compound 5 proved to crystallize well in CH2Cl2 which supported purification, however, it was isolated as a mixture containing also the analogous chlorinated complex [WO(C2Me2)(TmMe)Cl]. We assume the latter to arise from halogen exchange reaction with the chlorinated solvent as repeated crystallization from CH2Cl2/heptane leads to an increase of chlorinated complex as shown by 1H NMR spectroscopy (Figure S14). High‐resolution mass spectrometry supports the occurrence of [WO(C2Me2)(TmMe)Cl] revealing a Cl‐fragment besides the expected Br analogue (Figure S47–S50). In CDCl3 even further chlorination to paramagnetic [W(Cl)3(TmMe)] occurred, evidenced by X‐ray diffraction analysis of single crystals which formed in the NMR tube (Figures S78 and S79). To avoid such exchange reactions the non‐chlorinated solvent acetonitrile was considered but purification by recrystallization in this alternative solvent was unsuccessful. Furthermore, [WO(C2Me2)(TmMe)Br] (5) could be obtained by oxidation of [W(CO)(C2Me2)(TmMe)Br] (2) with O2 over two days at 42 °C.
The oxidation reactions using methylthiirane led to the corresponding sulfido complexes of the type [WS(C2Me2)(TmMe)Br] (R=H 6; Me 7). While the 2‐butyne complex [WS(C2Me2)(TmMe)Br] (7) was isolated in pure form in 54 % yield as a microcrystalline powder, the acetylene complex [WS(C2H2)(TmMe)Br] (6) was again more challenging to isolate and could be obtaining as a 9:1 mixture of 6 and 1 in 31 % yield.
Complexes 4–6 are highly soluble, whereas complex 7 is moderately soluble in CH2Cl2 and CHCl3. They are also soluble in acetonitrile with the W≡O compounds 4 and 5 being significantly better than [WS(C2R2)(TmMe)Br] (R=H 6, Me 7). The latter were found to be sensitive toward sulfido substitution with O2 and H2O to form the W≡O complexes [WO(C2R2)(TmMe)Br] (R=H 4, Me 5) beside decomposition.
The solid‐state structures of all four complexes 4–7 were determined by single crystal X‐ray diffraction analysis, confirming the coordination of an alkyne ligand and a terminal oxygen or sulfur atom (Figures 4 and 5). Characterization by ATR‐IR spectroscopy reveals the W≡O stretching frequencies at 933 (4) and 919 cm−1 (5) which is within the expected range.12, 26 The W≡S band at 479 cm−1 in both sulfido complexes 6 and 7 is also in accordance with the only other known WS‐alkyne complexes [WS(C2Ph2)(S2CNMe2)2] and [WS(C2Ph2)(SCNMe2)(S2CNMe2)] (485 and 492 cm−1).27 In W≡S complexes of the type [WS(CO)(Tp′)(L)] (L=I, Br, Cl, S S), bearing a π‐acidic CO instead of a π‐acidic alkyne ligand, the sulfur‐tungsten stretch is found between 502–512 cm−1 indicating a stronger bond.28 The weaker W≡S stretch in our scorpionate complexes is in line with the observed ease of exchange with oxygen.
Figure 4.
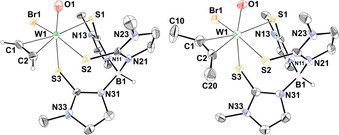
Molecular structure of [WO(C2H2)(TmMe)Br] (4, left) and [WO(C2Me2)(TmMe)Br] (5, right) showing the atomic numbering scheme. The probability ellipsoids are drawn at the 50 % probability level. The hydrogen atom at boron and acetylene are drawn with an arbitrary radius, the other H atoms and the disordered molecules were omitted for clarity. For 4 only one of the two molecules in the unit cell is depicted.
Figure 5.
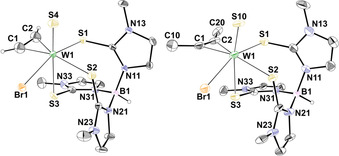
Molecular structure of [WS(C2H2)(TmMe)Br] (6, left) and [WS(C2Me2)(TmMe)Br] (7, right) showing the atomic numbering scheme. The probability ellipsoids are drawn at the 50 % probability level. The hydrogen atom at boron and acetylene are drawn with an arbitrary radius, the other H atoms and the solvent molecules were omitted for clarity.
Characterization of complexes 4–7 in solution by 1H and 13C NMR spectroscopy confirms the asymmetric coordination environment giving rise of three sets of methimidazole signals of the scorpionate ligand, similar to the CO compounds 1–3. Comparison of the 1H NMR acetylene proton resonances in our three acetylene complexes [W(CO)(C2H2)(TmMe)Br] (1), [WO(C2H2)(TmMe)Br] (4) and [WS(C2H2)(TmMe)Br] (6) reveals the carbonyl compound 1 exhibiting the most low‐field shifted signals (12.13/12.71 ppm), followed by those of the sulfido compound 6 (11.4/11.96 ppm) and last those of the oxido species 4 (10.17/10.77 ppm). Those shifts are consistent with a π‐accepting carbonyl ligand in 1 rendering the coordinated acetylene more electropositive than in the higher oxidized complexes 4 and 6 with π‐donor ligands. It is furthermore consistent with the higher π‐donor capability of the oxido relative to the sulfido moiety. Similar trends are apparent in the 13C NMR spectra as well as within the 2‐butyne complexes 2, 5 and 7. In contrast to our expectations complex 7 shows higher acetylene shifts than the acetylene complex 6 (Table 2). It was assumed that the ‐I effect of the methyl substituents increases the electron density on the C≡C atoms. However, this contradiction can already be observed in the uncoordinated alkynes which show higher carbon shifts for 2‐butyne comparted to acetylene (74.83 ppm vs. 72.24 ppm).
Table 2.
Overview of data of complexes of the type [WE(C2R2)(L)(TmMe)] in CD3CN.
|
Compound |
1H NMR[a] |
13C NMR[a] |
IR[b] |
X‐ray[c] |
||||
|---|---|---|---|---|---|---|---|---|
|
|
δ η2‐RC2R |
δ (C≡C) |
|
C≡C |
W‐C2R2 |
W‐R |
||
|
1 |
[W(CO)(C2H2)(TmMe)Br] |
12.13 12.72 |
192.06 197.47 |
C≡O |
1903 |
1.279(7)[d] |
2.0541(5)[d] 2.041(18)[d] |
1.174(11)[d] C≡O |
|
2 |
[W(CO)(C2Me2)(TmMe)Br] |
3.00 3.05 |
196.79 203.43 |
1882 |
1.314(7) |
2.050(6) 2.050(6) |
1.134(7) C≡O |
|
|
4 |
[WO(C2H2)(TmMe)Br] |
10.22 10.70 |
145.83 155.82 |
W≡O |
933 |
1.252(12)[d] |
2.096(8)[d] 2.103(8)[d] |
1.725(6)[d] W≡O |
|
5 |
[WO(C2Me2)(TmMe)Br] |
2.64 3.06 |
148.84 158.95 |
919 |
1.255(8) |
2.077(6) 2.114(5) |
1.791(4) W≡O |
|
|
6 |
[WS(C2H2)(TmMe)Br] |
11.48 11.91 |
161.97 173.61 |
W≡S |
479 |
1.2605(13) |
2.1035(10)[e] 2.1175(10)[e] |
2.1529(13)[e] W≡S |
|
7 |
[WS(C2Me2)(TmMe)Br] |
2.84 3.32 |
173.88 185.56 |
479 |
1.240(9) |
2.069(7) 2.118(7) |
2.1730(18) W≡S |
|
|
10 |
[W(CO)(C2H2)(MeCN)(TmMe)](OTf) |
12.19 12.97 |
192.66 196.29 |
C≡O |
1921 |
– |
||
|
11 |
[W(CO)(C2Me2)(MeCN)(TmMe)](OTf) |
3.09 |
198.70 203.16 |
1907 |
– |
|||
|
12 |
[WO(C2H2)(MeCN)(TmMe)](OTf) |
10.30 11.03 |
142.72 147.69 |
W≡O |
935 |
– |
||
|
13 |
[WO(C2Me2)(MeCN)(TmMe)](OTf) |
2.71 2.97 |
145.23 152.64 |
925 |
– |
|||
|
14 |
[WS(C2H2)(MeCN)(TmMe)](OTf) |
11.35 12.10 |
158.77 164.55 |
W≡S |
515 |
|
||
|
15 |
[WS(C2Me2)(MeCN)(TmMe)](OTf) |
2.92 3.12 |
167.55 175.20 |
484 |
– |
|||
[a] In ppm. [b] Neat in cm−1. [c] In Å. [d] Average length from two crystals or two molecules in the unit cell. [e] Values from disordered bonds.
In view of reactivity of the coordinated alkyne toward a nucleophilic attack, the carbonyl compounds seem to be the desired ones despite their non‐biological formal oxidation state of +II. Nevertheless, the introduction of the sulfido ligand to yield the biological WIV center lead to more electropositive carbons relative to the established W≡O complexes, although the sulfido moiety turned out to be labile under aqueous conditions if not embedded in a protein environment. The additional S‐donor in the native enzyme could further increase the electrophilicity of acetylene and thus be responsible for its unique reactivity.
Comparison of oxido acetylene WIV complexes [WO(C2H2)L2] described to date (Figure 2), reveal very similar acetylene resonances in the 1H NMR spectra irrespective of the nature of the two additional ligands L (Table 1) which is relevant for bio‐inspired modelling using mixed‐ or non‐sulfur‐containing ligands. The pure N‐donor ligand Tp“ shows the highest acetylene shift but only for one acetylene proton and carbon and it also displays the highest W≡O stretching frequency. Also our N‐containing S‐Phoz system displays slightly higher C≡C shifts. Probably the sulfur donor atoms increase the electron density at the metal center relative to the nitrogen donors. As a consequence, a lone pair of the terminal oxido group might perform more π‐donation to the metal center and thus increase the W≡O frequency.12
Table 1.
Overview of complexes of the type [WO(C2H2)L2] in CD2Cl2.
|
Complex |
1H NMR[a] |
13C NMR[a] |
W≡O[b] |
X‐ray[c] |
Ref. |
||
|---|---|---|---|---|---|---|---|
|
|
δ η2‐HC2H |
δ C≡C |
|
C≡C |
W‐C2R2 |
W≡O |
|
|
[WO(C2H2)(TmMe)Br] (4) |
10.22 10.70 |
145.5 155.5 |
933 |
1.252(12) |
2.096(8) 2.103(8) |
1.725(6) |
this work |
|
[WO(C2H2)(S‐Phoz)2] |
10.50 10.55 |
151.6 152.9 |
939 |
1.268(6) |
2.094(4) 2.109(4) |
1.724(3) |
|
|
[WO(C2H2)(Tp′)I] |
10.49 11.82 |
145.3 156.8 |
953 |
– |
– |
– |
|
|
[WO(C2H2)(S2CNEt2)2] |
10.56 10.69 |
146.9 154.1 |
930 |
1.282(3) |
2.092(2) 2.112(2) |
1.7137(16) |
12 and this work[d] |
[a] In ppm. [b] Neat in cm−1. [c] In Å. [d] See SI Figure S85 and Table S25.
Syntheses of cationic complexes: dinuclear
To adjust the electrophilic nature, the biologically relevant WIV complexes were investigated toward halide abstraction forming cationic species, both in the coordinating solvent acetonitrile as well as in dichloromethane. Addition of one equiv of thallium triflate (TlOTf) to [WE(C2Me2)(TmMe)Br] (E=O 5; E=S 7) in CH2Cl2 allowed the isolation of two cationic dinuclear complexes namely [W2O(μ‐O)(C2Me2)2(TmMe)2](OTf)2 (8) and [W2(μ‐S)2(C2Me2)(TmMe)2](OTf)2 (9) after work‐up as described in the experimental section. Surprisingly, the two compounds feature very different colors with 8 being yellow and 9 being purple pointing to different structures. In fact, while both compounds are dinuclear, their bridging moiety is different as evidenced by X‐ray diffraction data (see below, Figure 6). In the oxido compound 8 the two tungsten atoms are connected via a single μ‐oxido and in the sulfido compound 9 by two μ‐sulfido groups. In both compounds one scorpionate ligand is fully coordinated to one tungsten atom in κ3‐S,S,S fashion and the other one features a κ3‐S,S,H coordination to the second tungsten with its third S‐donor arm bridging to the first tungsten atom (Scheme 3). To sustain hexa‐coordination at each tungsten atom, in the (μ‐S)2 compound one alkyne molecule is displaced leading to the bis‐ cationic, mono‐alkyne complex 9. This is reflected by the reaction times for the preparation of the two compounds which were monitored by 1H NMR spectroscopy. The formation of [W2O(μ‐O)(C2Me2)2(TmMe)2](OTf)2 (8) was found to occur immediately whereas the (μ‐S)2 dimer 9 is formed over 40 h via rearrangement and loss of a 2‐butyne molecule (Figure S26). Compounds 8 and 9 display quite different solubility properties in chlorinated solvents with 8 being moderately and 9 being very good soluble. It is interesting to note that in acetonitrile the oxido dimer undergoes splitting to a mononuclear compound with a coordinated solvent molecule of the type [WO(C2Me2)(MeCN)(TmMe)](OTf) (13) whereas upon dissolving 9 the two μ‐sulfido groups persist in solution (Figure S25). This is furthermore observed by high resolution mass spectrometry where the spectra show the [M]2+ cation at m/z 594.012 for the dimer [W2(μ‐S)2(C2Me2)(TmMe)2](OTf)2 (9) and the [M]+ cation at m/z 605.058 for the monomeric [WO(C2Me2)(TmMe)]+ fragment of [W2O(μ‐O)(C2Me2)2(TmMe)2](OTf)2 (8).
Figure 6.
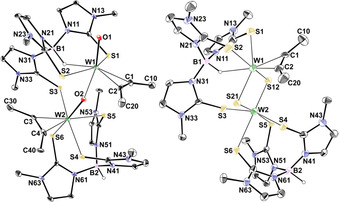
Molecular structure of [W2O(μ‐O)(C2Me2)2(TmMe)2](OTf)2 (8, left) and [W2(μ‐S)2(C2Me2)(TmMe)2](OTf)2 (9, right) showing the atomic numbering scheme. The probability ellipsoids are drawn at the 30 % (8) and 20 % (9) probability level. The hydrido atom is drawn with an arbitrary radius, the other H atoms, the OTf‐Anions and the solvent molecules were omitted for clarity.
Scheme 3.
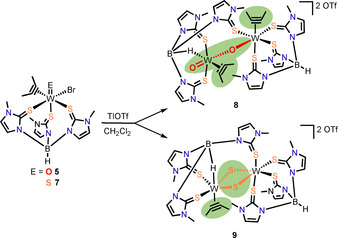
Dinuclear complex formation upon halide abstraction in the non‐coordinating solvent CH2Cl2.
The analogous reactions with the acetylene complexes 4 and 6 were futile as they only resulted in an undefined mixture according to 1H NMR spectroscopy. Possibly, the more labile nature of acetylene compared to 2‐butyne and the above demonstrated ease of dimerization leads finally to the formation of polymeric materials which is largely prevented in the halide compounds and also by a coordinated solvent molecule (see below).
Due to the asymmetric nature of [W2O(μ‐O)(C2Me2)2(TmMe)2](OTf)2 (8) and [W2(μ‐S)2(C2Me2)(TmMe)2](OTf)2 (9) their 1H NMR spectra are quite complex showing distinct signals for the 12 inequivalent protons at the heterocycles and singlets for the six N‐CH3 groups. The methyl groups of the coordinated 2‐butynes appear in 8 as four singlets and in 9 as one broad singlet for two methyl groups. The 2‐butyne signals were identified by their carbon shifts in the HSQC spectrum. Nevertheless, the resonances of 9 exhibit much narrower line widths than 8, indicating dynamic behavior of the latter which is in accordance with the reversible monomer formation upon addition of MeCN. The less dynamic behavior of 9 allows the detection of a broad quadruplet at −3.01 ppm which can be assigned to the B−H proton involved in an agostic‐like interaction with the W center. This interaction even persists when the complex is dissolved in the coordinating solvent CD3CN. A similar observation was made in the κ3‐S,S,H complex [W(CO)(BmMe)2] in [D6]DMSO.29 The occurrence of this interaction in both compounds 8 and 9 is confirmed by X‐ray diffraction analysis (Figure 6). However, the dynamic nature of 8 prevents its detection by 1H NMR spectroscopy.
Syntheses of cationic complexes: mononuclear
For the preparation of the desired mononuclear cations, halide abstraction was performed in the coordinating solvent acetonitrile. Therefore, [WE(C2R2)(TmMe)Br] (E=CO, H 1, Me 2; E=O; R=H 4, Me 5; E=S, R=H 6, Me 7) in MeCN were reacted with one equiv TlOTf allowing the isolation of the respective monomeric cations [WE(C2R2)(MeCN)(TmMe)](OTf) (E=CO, H 10, Me 11; E=O, R=H 12, Me 13; E=S, R=H 14, Me 15) (Scheme 4). In contrast to the reaction in CH2Cl2, in the coordinating solvent also the acetylene complexes [WE(C2H2)(MeCN)(TmMe)](OTf) (E=O 12; E=S 14) were accessible. However, because the respective starting materials for the abstraction of [WS(C2H2)(TmMe)Br] (6) contained residual 1 as described above, also the products 14 contained the respective cation as we were not able to remove it. However, NMR and MS data convincingly support the formation of the mononuclear cationic acetylene complex 14.
Scheme 4.
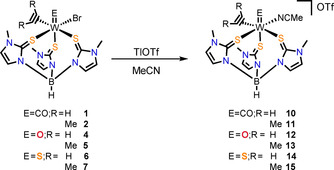
Formation of the cationic complexes of the type [WE(C2R2)(MeCN)(TmMe)](OTf) upon bromide abstraction in coordinating solvent MeCN.
In general, the oxido complexes were found to be stable in MeCN solutions whereas the sulfido complexes are prone to sulfur substitution with water, oxygen, dimerization and/or decomposition over time. This is consistent with the sulfido lone pairs being a stronger σ‐donor than MeCN and with the observed stability of the (μ‐S)2 species persisting in acetonitrile solution as described above. Furthermore, all cationic compounds were found to be extremely soluble preventing crystallization of single crystal suitable for X‐ray diffraction analysis. However, high resolution mass spectra for all six complexes 10–15 display the expected [WE(C2R2)(TmMe)]+ fragments with correct isotopic pattern (Figures S55–S67). The spectra were recorded under ambient conditions under which the oxygen sensitive sulfido compounds 14 and 15 extensively converted into the oxido complexes.
NMR spectroscopic data of the cations 10–15 support their structure as they reveal one set of ligand signals for an asymmetric scorpionate complex (Table 2 and Figures S27–S40). The resonance for coordinated acetonitrile could not be detected indicating fast exchange with excess CD3CN. The data of complex 13 are identical to those of the dinuclear complex [W2O(μ‐O)(C2Me2)2(TmMe)2](OTf)2 (8) in coordinating CD3CN confirming the conversion of the dimer into the solvent stabilized monomer in the coordinating solvent.
Interestingly, the amount of MeCN observed in the 1H NMR spectra of dried samples of the cationic complexes was <1 equivalent, indicating a loosely bound solvent molecule which can be removed in vacuo. This behavior is also reflected by the formation of dinuclear complex [W2O(μ‐O)(C2Me2)2(TmMe)2](OTf)2 (8) upon dissolving complex [WO(C2Me2)(MeCN)(TmMe)](OTf) (11) in the non‐coordinating NMR solvent CD2Cl2, revealing full reversibility of this system. Dimerization was also observed when re‐dissolving complex 15 in CD2Cl2 but it was accompanied by side product formation due to the higher sensitivity of the sulfido compounds. The hard triflate anion seems to be unable to stabilize the complexes as observed in the nitrogen‐based scorpionate system in which [WO(C2H2)(H2O)(Tp′)](OTf) converts into [WO(C2H2)(Tp′)(OTf)] and [WO(C2H2)(MeCN)(Tp′)](OTf) upon addition of MeCN.14 Our complexes rather stabilize themselves with their W≡S or W≡O moieties which is possibly due to the generally softer coordination sphere enforced by the sulfur‐based scorpionate. Cation formation also occurs in the parent halide complexes [WE(C2R2)(TmMe)Br] (E=CO; R=H 1, Me 2; E=O; R=Me 5; E=S, R=H 6, Me 7) upon dissolving in acetonitrile. The bromido ligand is partially replaced by the coordinating solvent molecule forming the respective monomeric cation with a bromide counter ion (Figure S43–S46). This is evidenced by the 1H NMR spectra of [WO(C2Me2)(TmMe)Br] (5) and [WO(C2Me2)(MeCN)(TmMe)](OTf) (13) in CD3CN (Figure 7) which reveals the presence of the cation in the spectrum of the bromide complex 5. The latter show no additional resonances when dissolved in the non‐coordinating solvent CH2Cl2. This is surprising as partial substitution of one of the methimazole arms and a switch to a κ2‐S,S‐ or κ3‐S,S,H‐binding mode was expected as such flexibility in the coordination mode is known for this scorpionate ligand.30 Comparison of the acetylene shifts in the 1H NMR spectrum of the complexes [WE(C2H2)(TmMe)Br] (E=CO 1, O 4, S 6) and the corresponding cations revealed a minor shift of maximum 0.33 ppm which is within the range observed for the complex [WO(C2H2)(H2O)(Tp′)](OTf).14 Similarly, the 13C NMR shifts of the alkynes are not significantly influenced by the introduced positive charge. Surprisingly, a general shift to more electron‐rich alkyne carbons can be observed. This is possibly caused by the CO ligand which may adjust its π‐acidity upon cation formation so that the overall electronic situation on the alkyne ligand remains similar. The CO stretching frequency in IR spectroscopy is in accordance with this assumption. Such an adjustment of electron donation may also be considered in the oxido and sulfido complexes in which also a slight increase of the W≡E stretching frequencies is observed.14 In sulfido complex 6, with no donating methyl substituents on the alkyne, this behavior is most pronounced leading to an significant increase in the W≡S stretching by 35 cm−1.
Figure 7.
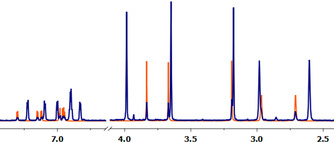
Superimposed 1H NMR spectra of [WO(C2Me2)(TmMe)Br] (5, blue) and [WO(C2Me2)(MeCN)(TmMe)](OTf) (13, orange) in CD3CN.
All acetylene complexes [WE(C2H2)(TmMe)Br] (E=CO 1, O 4, S 6) and [WE(C2H2)(MeCN)(TmMe)](OTf) (E=CO 10, O 12, S 14) were treated with the wet NMR solvents CD3CN or CD2Cl2 to investigate their reactivity toward the second AH substrate, water. 1H NMR spectroscopy represents a suitable tool for this evaluation as the ultimate goal is the formation of volatile acetaldehyde which can easily be detected by a characteristic quadruplet at around 9.5 ppm and a doublet at around 2.1 ppm. However, in none of these experiments, acetaldehyde was detected.
Also the addition of NEt3, in order to increase the nucleophilic character of water, did not lead to the desired nucleophilic attack on acetylene. Nevertheless, it is interesting to note that the NMR data did not even reveal the formation of H2O coordinated species, neither within the bromido series nor in the cationic complexes which holds also true for the 2‐butyne complexes. Also, addition of water (approx. 10 equiv) prior to addition of TlOTf did not lead to aqua complexes. This is in contrast to the nitrogen‐based scorpionate system for which the aqua complexes [WO(C2R2)(H2O)(Tp′)](OTf) (R=H, Me) were isolated, but also no acetaldehyde was observed.14 However, in wet acetonitrile, we observe quite high stability of all complexes, except the sulfido compounds which are converted into their oxido analogues. For example, the complex [WO(C2H2)(MeCN)(TmMe)](OTf) (12) turned out to be stable in wet solution for at least 10 days. Surprisingly, also the dinuclear complex [W2(μ‐S)2(C2Me2)(TmMe)2](OTf)2 (9) is stable as all other sulfido complexes turned out to be very sensitive.
In wet CD2Cl2 the complex [WO(C2Me2)(MeCN)(TmMe)](OTf) form the dinuclear species [W2O(μ‐O)(C2Me2)2(TmMe)2](OTf)2 (8) whereas the corresponding sulfido complex decomposes revealing that water is not able to coordinate to tungsten in this sulfur‐rich system. For this reason, it is not surprising that the cationic acetylene complex deteriorates under these conditions as observed in the direct synthesis (see above).
The well‐considered design of our model complexes allows in principle the coordination of both substrates of AH, which is in contrast to most other models, except for the hard scorpionate complex [WO(C2H2)(Tp′)I] which does not provide the enzyme's sulfur‐rich coordination sphere. The delivered data clearly demonstrates that in the sulfur‐based coordination surrounding acetylene is significantly better coordinated than a hard water molecule. While this might point toward a first‐shell mechanism in AH, future models capable of nucleophilic attack of water will have to provide support. The knowledge gained here as well as in our previous work on intramolecular nucleophilic attack on acetylene20 is nevertheless interesting as it represents the basis for the development of such future models.
Molecular structures
WII alkyne complexes
The molecular structures of the WII complexes [W(CO)(C2R2)(TmMe)Br] (R=H 1, Me 2, Ph 3) and the precursors [W(CO)(C2R2)2(MeCN)Br2] (R=Me, Ph) were determined by single crystal X‐ray diffraction analysis and are depicted in Figure S68–S71 and Figure S80–S83. Selected bond lengths are given in Table S8–S11 and Table S20–S23. The obtained bis‐alkyne complexes constitute different space groups or cell parameters, respectively, relative to the published ones.31 For complex 1 two crystal structures from acetonitrile and CH2Cl2/heptane, respectively were obtained, with the later one showing a 10 % disorder of the Br and CO ligands. All compounds display distorted octahedral geometry with the center of the alkyne occupying one position and with the carbonyl ligand orientated parallel to the alkyne which is common for this type of alkyne complex.21, 32 In the three complexes the three methimazole rings of the facial coordinated scorpionate ligand are arranged mutually approximately orthogonal. Moreover, the structures share a significant shorter W−S bond trans to the bromide (2.3907(15)–2.4065(5) Å vs. 2.6330(13)–2.5808(16) Å). The C atoms of the bound alkynes in the three complexes are sp2‐rehybridized resulting in a C≡C‐R angle different from 180° and are therefore activated (C−C‐R=133.1(6)°–147.1(7)°). Relative to free C2R2 (R=H: Ø 1.175(6) Å,33 Me: 1.211 Å,34 Ph: 1.198(4) Å35) the C≡C bonds are significantly elongated. In regard to similar systems like [W(CO)(C2H2)(S‐Phoz)2]13 (1.327(3) Å) and [W(CO)(C2H2)(S2CNEt2)]36 (1.29(1) Å) the unsubstituted acetylene complex displays a C≡C bond length in between (1.252(7) and 1.306(3) Å). The butyne complex 2 shows the same activation of the alkyne as [W(CO)(C2Me2)(S‐Phoz)2]13 (1.314 Å) but less than the hard scorpionate complex [W(CO)(C2Me2)(Tp′)Br] with 1.277(6) Å.37 With a C≡C bond length of 1.332(8) Å the phenyl substituted complex 3 has a higher cyclopropane character than the diphenylacetylene in [W(CO)(C2Ph2)(S‐Phoz)2]13 (Ø 1.307(6) Å).
WIV alkyne complexes
Solid‐state structures of the complexes [WE(C2R2)(TmMe)Br] (E=O; R=H 4, Me 5; E=S, R=H 6, Me 7) were obtained by single crystal X‐ray diffraction analysis. Molecular views are presented in Figure 4 and 5 and selected bond lengths are given in Table S12–S15. Single crystals of 4, 5 and 7 were obtained from CH2Cl2/heptane at −35 °C and of complex 6 from CH2Cl2/THF/heptane at −35 °C. The acetylene complex 4 contains two molecules in the asymmetric unit and in complex 6 the WBrSC2H2 fragment is disordered (15 %). As for the carbonyl compounds the W−S bond trans to the halide is significantly elongated in all complexes. The W≡S bond lengths of 2.1497(13) Å (6) and 2.1730(18) Å (7) are similar to that of [WS(C2Ph2)(S2CNEt2)2] with 2.147(2) Å,27b one of the two other W≡S alkyne structure. In [WS(C2Ph2)(S2CNEt2)(SCNEt2)]27a the value of 2.138(5) Å is slightly shorter but similar to that of [WS(CO)(S2PPh2)(Tp′)] (2.135(4) Å)28 which is coordinated by a carbonyl instead of diphenylacetylene both being π‐accepting ligands.
The W≡O bond distance in complex 4 with 1.725(6) Å is within those of [WO(C2H2)(S2CNEt2)2]12 and the S‐Phoz complexes [WO(C2R2)(S‐Phoz)2] (R=H, Me, Ph) and [WO(S‐Phoz)2].38 The 2‐butyne complex [WO(C2Me2)(TmMe)Br] (5) displays a significant longer bond of 1.791(4) Å which can be attributed to the +I‐effect of the methyl substituents and thus decreased π‐donation of the oxygen lone pair.
In general, in the structures of the oxidized complexes the alkynes is rotated by 90° relative to the CO complexes in order to optimize orbital overlap in the three‐center four‐electron bond between the alkyne, the W center and the oxido/sulfido ligand.13, 14, 26 The higher oxidation states and the thereby decreased electron density on W results in an elongation of the W‐C2R2 bonds due to lower π‐back‐bonding capability into the alkynes π* which is accompanied by a shortening of the C≡C bond. In contrast to the corresponding S‐Phoz complexes oxidation had a bigger influence on the 2‐butyne complexes.38 Despite the significant shift of the acetylene carbon atoms in the 13C NMR spectra, the structural differences of the alkynes between the sulfido and the oxido complex is within three σ and can thus be regarded as similar. Although the S‐Phoz and the TmMe ligand both provide a sulfur‐rich first coordination sphere in the W≡E complexes, the influence on the alkynes vary significantly. Therefore, it is important to collect more data on model complexes to fully understand how to fine‐tune the properties of the bound alkyne in order to develop truly functional model for acetylene hydratase.
Dinuclear WIV alkyne complexes
Molecular structures of the dinuclear compounds [W2O(μ‐O)(C2Me2)2(TmMe)2](OTf)2 (8) and [W2(μ‐S)2(C2Me2)(TmMe)2](OTf)2 (9) are presented in Figure 6 and were determined by single crystal X‐ray diffraction analysis. Single crystals were obtained from a saturated CH2Cl2 solution at room temperature (8) and from CH2Cl2/heptane at −35 °C (9). In both complexes all W atoms have a distorted octahedral geometry. One W is thereby coordinated by the TmMe‐ligand in a κ3‐S,S,H fashion whereas the second W comprises a κ3‐S,S,S ligand and the remaining donor arm of the other ligand. The two W atoms in 8 are linked by an unsymmetric μ‐oxido group (W1‐O2 2.157(6) Å, W2‐O2 1.772(6) Å) where the shorter W−O distance is in the order of that of the oxido group bonded to W1 (W1–O1 1.715(6) Å). This is in accordance with the fact that the dimer can be cleaved by addition of the stronger σ‐donor acetonitrile by substituting the weaker σ‐donating W≡O moiety of the second cation. The two oxygen ligands are in trans positions to each other (O1‐W1‐O2 165.9(3)°). The C≡C bond of the two 2‐butynes are only marginally affected by the different trans ligands (C1−C2 1.265(13) Å, C3−C4 1.256(14) Å). In the sulfido complex [W2(μ‐S)2(C2Me2)(TmMe)2](OTf)2 (9) the strong σ‐donating W≡S moiety substituted the second alkyne ligand forming an almost planar W2S2 ring (torsion angle of 0.99(11)°) with significantly different W‐S distances (W1‐S12 2.326(2) Å, W1‐S21 2.380(2) Å vs. W2‐S21 2.254(3) Å, W2‐S12 2.262(2) Å). The hydrido W‐H−B bond is trans to the 2‐butyne which shows a significantly elongated C≡C bond of 1.313(15) Å resulting from unrivaled π‐back‐donation. Complex 9 represents the second example of a bis‐μ‐S bridged homo‐nuclear WIV complex, besides [W2(μ‐S)2(StBu)4(PMe2Ph)2] which displays also two significantly different W−S bonds of 2.254(6) and 2.345(7) Å.39 The four W‐S bond lengths of sulfur‐rich WV complexes like [W2S4(S2CNEt2)2] on the other hand are very similar and only range between 2.299(9)–2.318(8) Å. In the asymmetric WIV complex [Cp(S2C2(COOMe2)2W(μ‐S)2WOCp]40 the W−S bond lengths ((2.288(5) and 2.282(5) Å vs. 2.377(5) and 2.402(5) Å) differ even more relative to our complex 9. This bigger difference most likely origins from the more diverse substitution on the two W centers. In general, such asymmetric μ‐S dinuclear W complexes are scarce.
Conclusions
The soft scorpionate ligand TmMe was found suitable for model chemistry of the unique tungstoenzyme acetylene hydratase. Via the formation of WII complexes of the type [W(CO)(C2R2)(TmMe)Br] (R=H 1, Me 2, Ph 3) and subsequent oxidation using pyridine‐N‐oxide or methylthiirane, respectively, the biologically relevant WIV alkyne complexes of the type [WE(C2R2)(TmMe)Br] (E=O, R=H 4, Me 5; E=S, R=H 6, Me 7) could be isolated and structurally and spectroscopically characterized. Complex [WS(C2H2)(TmMe)Br] (6) thereby represents the first example employing the WS(C2H2) motif which increases the already sulfur‐rich first coordination sphere even more. The obtained compounds represent sulfur analogues of the complex [WO(C2H2)(Tp′)I] where the Tp′ ligand exhibits a κ3− N,N,N coordination mode which allowed direct comparison of the influence of a hard and soft coordination sphere on coordinated alkynes.14
The structural situation with a remaining halide within these type of complexes facilitates the preparation of cationic compounds. Upon iodine abstraction, the respective aqua complex [WO(C2H2)(H2O)(Tp′)](OTf) could be obtained from [WO(C2H2)(Tp′)I].14 With the sulfur‐based ligand a drastically different reactivity was found to alter, making the aqua‐complexes not accessible. Instead, dimerization of the 2‐butyne model complexes to [W2O(μ‐O)(C2Me2)2(TmMe)2](OTf)2 (8) and [W2(μ‐S)2(C2Me2)(TmMe)2](OTf)2 (9) was observed. In contrast to 9, the μ‐oxido dimer 8 can be cleaved to monomeric [WO(C2Me2)(MeCN)(TmMe)](OTf) (13) when MeCN is added which was found to be fully reversible. In addition, two complete alkyne‐series of solvent stabilized cationic complexes of the type [WE(C2R2)(MeCN)(TmMe)](OTf) (E=CO, H 10, Me 11; E=O, R=H 12, Me 13; E=S, R=H 14, Me 15) were synthesized which revealed that the π‐acidic CO and the π‐basic O or S ligand compensate the altered electronic situation upon halide abstraction, as indicated by their IR stretching frequencies.14
In our complexes, we found that introducing a positive charge has no significant influence on the electrophilic character of the alkyne carbon atoms while the introduction of a sulfido ligand increases their electropositivity. Thus, if a first‐shell mechanism is occurring our results provides support for the need of a sulfur‐rich environment as found in acetylene hydratase (two molybdopterin co‐factors and a cysteine) facilitating the nucleophilic attack on acetylene. Activation of acetylene is additionally supported by the absence of coordination of the second substrate water in our biologically relevant sulfur‐based model system. The observed preference of C2H2 at tungsten are in line with the DFT calculations considering the H2O molecule in the enzymes solid state structure to be a place holder. Although the new WIV complexes, as yet, only qualify as structural models, they display increased stability toward hydrolysis without oxidative decomposition to for example, disulfide as observed in our S‐Phoz complexes.38
Experimental Section
General
All experiments were carried out under inert atmosphere employing standard Schlenk and glovebox techniques unless otherwise stated. All chemicals were purchased from commercial sources and with the exception of acetylene, diphenylacetylene and pyridine‐N‐oxide all were used without further purification. Acetylene 2.6 was washed with water and concentrated H2SO4 and dried with CaCl2 and KOH. Molecular oxygen (3.5) was dried by passing through CaCl2, P2O5 and molecular sieve (3 Å) prior to use. Diphenylacetylene was recrystallized from ethanol. Pyridine‐N‐oxide was purified by sublimation. All solvents were purified by a Pure Solv Solvent Purification System and stored over activated molecular sieve (3 Å). NMR spectra were recorded using a Bruker Avance III spectrometer. 1H NMR spectra were recorded at 300.13 MHz and 13C NMR at 75.48 MHz and were referenced to the respective solvent peak. The chemical shifts δ are given in ppm. The multiplicity of peaks is denoted as broad singlet (bs), singlet (s), doublet (d), triplet (t), quadruplet (q), multiplet (m) and doublet of quadruplet (dq). Coupling constants J are given in Hertz. Mass spectroscopy measurements using electron impact ionization (EI‐MS) have been performed with an Agilent 5973 MSD with push rod for direct sample measurement. High resolution atmospheric pressure electrospray ionization mass spectrometry (HR‐ESI‐MS) measurements were performed on a Q‐Exactive Hybrid Quadrupole‐Orbitrap MS after flow injection on a Dionex Ultimate 3000 series instrument (Thermo Fisher Sci., Erlangen, Germany). For analysis samples were re‐dissolved in MeCN (5–9 mg mL−1). The HR‐MS was furnished with a heated HESI‐II ionization source using nitrogen as nebulizer and drying gas. Measurements were performed in positive and negative ionization modes using ionization energies of +3.5 kV and −3.2 kV, respectively. Drying gas temperature was 300 °C and the resolution was set to 70 000 (FWHM). The LC‐system was operated in flow injection mode (1 μL injection volume) using pure not absolute acetonitrile as mobile phase with a flow of 0.2 mL min−1. Ions were recorded in scan mode within a mass range of m/z 100–1500. IR spectra were recorded in the solid state at a resolution of 2 cm−1 on a Bruker ALPHA‐P Diamant ATR‐FTIR. Signal intensity was assigned as strong (s), medium (m) and weak (w). Elemental analyses (C,H,N,S) were carried out by the Microanalytical Laboratory, Department of Chemistry, University of Vienna and the Department of Inorganic Chemistry at the Graz University of Technology (Heraeus Vario Elementar automatic analyzer).
Bis(1‐methylimidazolium‐2‐yl)disulfide,41 [W2(CO)7Br4],42 [W(CO)3(MeCN)2Br2],22b [W(CO)(C2Me2)2(MeCN)Br2],22 [W(CO)(C2Ph2)2(MeCN)Br2]22b [W(CO)(C2H2)(MeCN)2Br2]/ [W(CO)(C2H2)2(MeCN)Br2]20 and [WO(C2H2)(S2CNEt2)2] were prepared according to literature. NaTmMe and was prepared according to a modified published procedure.24
Compound syntheses
NaTmMe
In a nitrogen flushed Schlenk flask 1.134 g (30.0 mmol, 1.0 equiv) NaBH4 were mixed with 17.314 g (152 mmol, 5.0 equiv) methimazole. The flask was equipped with a bubbler and the neat mixture was heated at 150 °C with an oil bath for about 40 min until H2 formation had ceased. After cooling to RT 100 mL of MgSO4‐dried ethyl acetate/cyclohexane 3+1 were added and the solidified blue‐grey melt was crushed and ground into a paste with a pestle. The solid was filtered off and repeatedly suspended with 60 mL portions of MgSO4‐dried ethyl acetate/cyclohexane 3+1 in the ultrasonic bath and filtered. The product was dried in vacuo to yield 9.535 g (25.5 mmol, 85 %) of NaTmMe as white powder. 1H NMR (300 MHz, CD3CN): δ=3.46 (s, 3 H, N‐CH3), 4.17 (bs, 1 H, BH), 6.31 (d, J=2.2 Hz, 1 H, CH), 6.71 ppm (d, J=2.2 Hz, 1 H, CH). 13C NMR (75 MHz, CD3CN): δ=34.59 (N‐CH3), 118.15 (CH), 120.93 (CH), 164.77 ppm (C=S). IR: = 2481 cm−1 (w, BH). EI‐MS: m/z 374.1 [M +].
[W(CO)(C2H2)(TmMe)Br] (1)
A mixture of [W(CO)(C2H2)2(MeCN)Br2] and [W(CO)(C2H2)(MeCN)2Br2]20 (1.035 g, approx. 2.30 mmol, 1.0 equiv) and 879 mg NaTmMe (2.35 mmol, 1.0 equiv) were suspended in 20 mL CH2Cl2 in a 50 mL Schlenk flask. The mixture was stirred for 3 h at RT before it was evaporated to dryness. The product was dissolved in ∼25 mL CH2Cl2 and filtered. The solution was concentrated and repeatedly recrystallized from CH2Cl2/heptane. The blue‐green product was isolated by filtration and washed 2× with 10 mL pentane. Evaporation to dryness gave 1.101 g (1.65 mmol, approx. 75 %) of crystalline product. Single crystals for X‐ray diffraction analysis were obtained from a saturated MeCN solution at RT. 1H NMR (300 MHz, CD2Cl2): δ=3.05 (s, 3 H, N‐CH3), 3.83 (s, 3 H, N‐CH3), 3.91 (s, 3 H, N‐CH3), 6.82 (d, J=2.1 Hz, 1 H, CH), 6.85 (d, J=2.1 Hz, 1 H, CH), 6.87 (d, J=2.1 Hz, 1 H, CH), 6.91 (d, J=2.1 Hz, 1 H, CH), 6.95 (d, J=2.0 Hz, 1 H, CH), 7.04 (d, J=2.0 Hz, 1 H, CH), 12.13 (d, J W‐H=1.0 Hz, 1 H, ≡‐H), 12.71 ppm (d, J W‐H=1.0 Hz, 1 H, ≡‐H). 13C NMR (75 MHz, CD2Cl2): δ=33.78 (N‐CH3), 35.33 (N‐CH3), 37.08 (N‐CH3), 120.46 (C=C), 120.72 (C=C), 121.71 (C=C), 123.23 (C=C), 123.39 (C=C), 124.79 (C=C), 159.46 (C=S), 168.37 (C=S), 160.27 (C=S) is obscured, 192.95 (C≡C), 197.32 (C≡C), 235.28 ppm (CO). IR: =2428 (w, BH), 2404 (w, BH), 1903 cm−1 (s, CO). Elemental analysis calcd (%) for C15H18BN6OS3BrW⋅1.20 CH2Cl2: C 25.24, H 2.67, N 10.90, S 12.47; found: C 25.20, H 2.58, N 10.99, S 12.34. EI‐MS: m/z no assignable signals.
[W(CO)(C2Me2)(TmMe)Br] (2)
[W(CO)(C2Me2)2(MeCN)Br2]22b (2.373 mg, 4.56 mmol, 1.0 equiv) was dissolved in a 50 mL Schlenk flask in 10 mL CH2Cl2. NaTmMe (1.709 g, 4.57 mmol, 1.0 equiv) was added portion‐wise and the suspension was then stirred at RT overnight. The mixture was filtered and the product was precipitated by addition of heptane. Repeated recrystallization from CH2Cl2/heptane gave 3.110 g (98 %, 4.45 mmol) of blue crystals. Single crystals for X‐ray diffraction analysis were obtained by recrystallization from CH2Cl2/heptane at −35 °C. 1H NMR (300 MHz, CD2Cl2): δ=3.00 (s, 3 H, ≡‐CH3), 3.05 (s, 3 H, ≡‐CH3), 3.09 (s, 3 H, N‐CH3), 3.82 (s, 3 H, N‐CH3), 3.91 (s, 3 H, N‐CH3), 6.79 (d, J=2.0 Hz, 1 H, CH), 6.84 (d, J=2.1 Hz, 1 H, CH), 6.86 (d, J=2.0 Hz, 1 H, CH), 6.91 (d, J=1.8 Hz, 1 H, CH), 6.95 (d, J=2.0 Hz, 1 H, CH), 7.01 ppm (d, J=2.0 Hz, 1 H, CH). 13C NMR (75 MHz, CD2Cl2): δ=17.69 (J W‐H=25.3 Hz, ≡‐CH3), 21.42 (J W‐H=28.7 Hz, ≡‐CH3), 34.23 (N‐CH3), 35.24 (N‐CH3), 36.92 (N‐CH3), 120.32 (C=C), 120.63 (C=C), 121.32 (C=C), 123.11 (C=C), 123.17 (C=C), 124.74 (C=C), 159.67 (C=S), 160,77 (C=S), 167.94 (C=S), 197.30 (C≡C), 203.17 (C≡C), 237.02 ppm (J C‐W=146 Hz, CO). IR: =2430 (w, BH), 2401 (w, BH), 1882 cm−1 (s, CO). Elemental analysis calcd (%) for C17H22BN6OS3BrW⋅0.10 CH2Cl2⋅0.15 C7H16: C 30.25, H 3.44, N 11.66, S 13.35; found: C 30.25, H 3.53, N 11.38, S 13.68. EIMS m/z: 668.1 (M +‐CO), 616.0 (M +‐HBr), 534.0 (M +‐CO‐C2Me2‐Br).
[W(CO)(C2Ph2)(TmMe)Br] (3)
Precursor [W(CO)(C2Ph2)2(MeCN)Br2]22b (1.000 g, 1.39 mmol, 1.0 equiv) was suspended in 50 mL MeCN in a 100 mL Schlenk flask. Then 522 mg NaTmMe (1.39 mmol, 1.0 equiv) were added portion‐wise while stirring. The reaction mixture was stirred at RT for 5 h, evaporated to dryness and the residue was dissolved in 10 mL CH2Cl2 and stirred for another 2 h. The solution was filtered over Celite and then triturated with heptane. The precipitate was filtered and recrystallized from CH2Cl2/heptane. Filtration, washing with 10 mL pentane and drying in vacuo yielded 894 mg (1.09 mmol, 78 %) of green crystalline product. Single crystals for X‐ray diffraction analysis were obtained from CH2Cl2/heptane. 1H NMR (300 MHz, CD2Cl2): δ=3.07 (s, 3 H, N‐CH3), 3.85 (s, 3 H, N‐CH3), 4.00 (s, 3 H, N‐CH3), 6.83 (d, J=1.9 Hz, 1 H, CH), 6.87 (t, J=2.0 Hz, 2 H, CH), 6.91 (d, J=1.9 Hz, 1 H, CH), 6.93 (d, J=2.1 Hz, 1 H, CH), 7.04 (d, J=1.9 Hz, 1 H, CH), 7.29 (m, 4 H, ≡‐Ph), 7.43 (q, 4 H, ≡‐Ph), 7.60 ppm (m, 2 H, ≡‐Ph). 13C NMR (75 MHz, CD2Cl2): δ=34.58 (N‐CH3), 35.23 (d, J=4.0 Hz, N‐CH3), 37.05 (d, J=3.4 Hz, N‐CH3), 120.59 (C=C), 120.80 (C=C), 121.55 (C=C), 123.19 (C=C), 123.46 (C=C), 124.99 (C=C), 127.11 (2C, Ph), 128.37 (Ph), 128.61 (2C, Ph), 128.73 (2C, Ph), 128.84 (Ph), 129.08 (2C, Ph), 139.12 (Cq, Ph), 142.44 (Cq, Ph), 159.09 (C=S), 160,16 (C=S), 168.52 (C=S), 203.33 (C≡C), 204.72 (C≡C), 237.28 ppm (CO). IR: =2432 (w, BH), 2409 (w, BH), 1913 cm−1 (s, CO). Elemental analysis calcd (%) for C27H26BN6OS3BrW⋅0.05 CH2Cl2⋅0.20 C7H16: C 40.41, H 3.49, N 9.94, S 11.37; found: C 40.39, H 3.43, N 9.79, S 11.48. EI‐MS: m/z no assignable signals.
[WO(C2H2)(TmMe)Br] (4)
Complex [W(CO)(C2H2)(TmMe)Br] (1) (1.071 g, 1.60 mmol) and pyridine‐N‐oxide (191 mg, 2.01 mmol) were suspended in 100 mL MeCN. The flask was covered with aluminum foil and equipped with a bubbler before it was heated at 44 °C for 5 h. During the reaction time vacuum was applied 2× for about 1 min. The formed suspension was dried in vacuo, resuspended in CH2Br2 and filtered over Celite. The Celite was washed with another 10 mL CH2Br2 before 25 mL of toluene were added. The mixture was concentrated in vacuo to precipitate two fractions. The second fraction was repeatedly recrystallized from CH2Cl2/heptane and CH2Cl2/toluene to give 250 mg (19 % of complex, 305 μmol) of a bright yellow powder of a 1:1 mixture of [WO(C2H2)(TmMe)Br] (with 5 % of the chloride derivative [WO(C2H2)(TmMe)Cl] and PyHBr. Single crystals for X‐ray diffraction analysis were obtained from CH2Cl2/heptane at −25 °C. 1H NMR (300 MHz, CD2Cl2): δ=[WO(C2H2)(TmMe)Br]: 3.23 (s, 3 H, N‐CH3), 3.74 (s, 3 H, N‐CH3), 4.06 (s, 3 H, N‐CH3), 6.77 (d, J=2.1 Hz, 1 H, CH), 6.80 (d, J=2.0 Hz, 1 H, CH), 6.85 (d, J=2.1 Hz, 1 H, CH), 6.91 (d, J=2.0 Hz, 1 H, CH), 7.07 (d, J=1.9 Hz, 1 H, CH), 7.11 (d, J=2.0 Hz, 1 H, CH), 8.00 (d, J=13.8 Hz, 2 H, Py), 8.50 (d, J=15.6 Hz, 1 H, Py), 8.80 (d, J=5.8 Hz, 2 H, Py), 10.17 (s, JH‐W=11.9 Hz, 1 H, ≡‐H), 10.77 (s, JH‐W=11.6 Hz, 1 H, ≡‐H), 17.43 (s, 1 H, HPyBr). [WO(C2H2)(TmMe)Cl]: 3.23 (obscured, 3 H, N‐CH3), 3.74 (obscured, 3 H, N‐CH3), 4.01 (s, 3 H, N‐CH3), 6.89 (obscured 1 H, CH), 6.96 (obscured 1 H, CH), 7.04 (obscured 1 H, CH), 3 CH obscured, 10.32 (s, 1 H, ≡‐H), 10.60 ppm (s, 1 H, ≡‐H). 13C NMR (75 MHz, CD2Cl2): δ=[WO(C2H2)(TmMe)Br]: 34.54 (N‐CH3), 35.45 (N‐CH3), 36.72 (N‐CH3), 120.21 (C=C), 121.36 (C=C), 122.15 (C=C), 123.04 (C=C), 123.70 (C=C), 124.84 (C=C), 145.46 (J W‐C=26.4 Hz, C≡C), 155.53 (J W‐C=32.5 Hz, C≡C), 156.97 (C=S), 159.20 (C=S), 159.62 ppm (C=S). IR: =2434 (w, BH), 2413 (w, BH), 933 cm−1 (s, W≡O). Elemental analysis calcd (%) for C14H18BN6OS3Br0.95Cl0.05W⋅0.05 C15H18BN6OS3BrW⋅1.0 PyHBr⋅0.10 CH2Cl2⋅0.50 C7H8: C 30.75, H 3.35, N 11.72, S 11.64; found: C 30.85, H 3.18, N 11.30, S 11.37. EI‐MS: m/z no assignable signals.
[WO(C2Me2)(TmMe)Br] (5)
Method A
In a 120 mL Schlenk tube [W(CO)(C2Me2)(TmMe)Br] (2) (2.009 g, 2.88 mmol) and 331 mg pyridine‐N‐oxide (3.48 mmol) were dissolved in 110 mL MeCN. The tube was covered with aluminum foil and equipped with a bubbler before it was heated at 44 °C for 5.5 h. During the reaction time vacuum was applied 2× for about 1 min. The formed suspension was dried in vacuo, resuspended in CH2Cl2 and filtered over Celite. To the filtrate 40 mL of toluene and 12 mL of heptane were added and then concentrated in vacuo. Fractionate precipitation gave two fractions of solid and the dried filtrate. All fractions were combined again in 100 mL of CH2Cl2 and after addition of 50 mL heptane the mixture was concentrated in vacuo. The resulting supernatant was filtered off and dried in vacuo. The residue was again dissolved in 75 mL CH2Cl2 and 40 mL heptane were added. The resulting suspension was concentrated in vacuo and the supernatant filtered off. The filtrates were combined repeatedly recrystallized from CH2Cl2 and heptane to give 482 mg of yellow crystals (24 %, 0.70 mmol).
Method B
In a 100 mL Schlenk flask 204 mg of [W(CO)(C2Me2)(TmMe)Br] (2) (0.293 mmol) were dissolved in 15 mL MeCN and the flask was purged with O2 for 16 min. The reaction was heated at 45 °C and stirred under O2 atmosphere for 19 h before it was dried in vacuo. The residue was taken up in CH2Br2 and filtered over Celite before it was layered with heptane. Next day, the mixture was dried in vacuo and fractionated precipitated from CH2Cl2/toluene/heptane to give 88 mg (44 %) of the halide scrambled complex [WO(C2Me2)(TmMe)Br0.80Cl0.20].1H NMR (300 MHz, CD2Cl2): δ=[WO(C2Me2)(TmMe)Br]: 2.64 (d, J=1.1 Hz, 3 H, ≡‐CH3); 3.06 (d, J=1.2 Hz, 3 H, ≡‐CH3), 3.21 (s, 3 H, N‐CH3), 3.70 (s, 3 H, N‐CH3), 4.04 (s, 3 H, N‐CH3), 6.76 (d, J=2.1 Hz, 1 H, CH), 6.77 (d, J=2.1 Hz, 1 H, CH), 6.86 (d, J=2.4 Hz, 1 H, CH), 6.87 (d, J=2.1 Hz, 1 H, CH), 7.05 (d, J=2.0 Hz, 1 H, CH), 7.09 ppm (d, J=2.0 Hz, 1 H, CH). [WO(C2Me2)(TmMe)Cl]: 2.64 (obscured 3 H, ≡‐CH3); 2.93 (d, 3 H, ≡‐CH3), 3.22 (obscured 3 H, N‐CH3), 3.71 (s, obscured, 3 H, N‐CH3), 3.98 (s, 3 H, N‐CH3), 6.75 (obscured 1 H, CH), 6.77 (obscured 1 H, CH), 6.86 (obscured 1 H, CH), 6.87 (obscured 1 H, CH), 7.06 (obscured 1 H, CH), 7.06 ppm (obscured 1 H, CH). 13C NMR (75 MHz, CD2Cl2): δ=13.43 (≡‐CH3), 18.11 (≡‐CH3), 34.40 (N‐CH3), 35.31 (N‐CH3), 36.58 (N‐CH3), 119.82 (C=C), 121.07 (C=C), 122.08 (C=C), 123.11 (C=C), 123.41 (C=C), 124.81 (C=C), 148.63 (J W‐C=26.8 Hz, C≡C), 157.31 (C=S), 159.30 (J W‐C=34.8 Hz, C≡C), 159.68 (C=S), 159.86 ppm (C=S). IR: =2415 (w, BH), 2339 (w, BH), 919 cm−1 (s, W≡O). Elemental analysis calcd (%) for C16H22BN6OS3Br0.8Cl0.2W⋅0.2 CH2Cl2: C 28.14, H 3.26, N 12.21, S 13.97; found: C 28.37, H 3.22, N 12.19, S 13.70. EI‐MS: m/z no assignable signals.
[WS(C2H2)(TmMe)Br] (6)
Complex [W(CO)(C2H2)(TmMe)Br] (1) (1.196 g, 1.79 mmol) was suspended in 140 mL MeCN and methylthiirane (160 μL, 1.02 mmol) was added. The tubes were equipped with a bubbler and wrapped in aluminum foil before they were heated at 43 °C. After 4 h of stirring at 43 °C the dark brown solutions were combined and concentrated in vacuo to about 30 mL. To the suspension 25 mL of toluene were added and three fractions were precipitated upon concentration of the mixture. All fractions were dissolved in CH2Cl2 and combined again before 25 mL of THF and 15 mL of heptane were added. By concentrating the mixture in vacuo three fractions were precipitated. The third fraction was washed with 1×6 mL and 2×1 mL of MeCN before it was recrystallized from CH2Cl2/THF to give 31 % (368 mg, 0.55 mmol) of red‐brown crystals. 1H NMR (300 MHz, CD2Cl2): δ=3.19 (s, 3 H, N‐CH3), 3.73 (s, 3 H, N‐CH3), 4.08 (s, 3 H, N‐CH3), 6.77 (d, J=2.1 Hz, 1 H, CH), 6.78 (d, J=2.1 Hz, 1 H, CH), 6.88 (dd, J=1.6 Hz, J=2.1 Hz, 2 H, CH), 7.10 (d, J=2.0 Hz, 1 H, CH), 7.11 (d, J=2.0 Hz, 1 H, CH), 11.40 (s, J W‐H=11 Hz, 1 H, ≡‐H), 11.96 ppm (s, J W‐H=10 Hz, 1 H, ≡‐H). 13C NMR (75 MHz, CD2Cl2): δ=34.50 (N‐CH3), 34.62 (N‐CH3), 36.75 (N‐CH3), 120.44 (C=C), 121.07 (C=C), 122.32 (C=C), 123.28 (2 C=C), 124.93 (C=C), 158.08 (C=S), 158.66 (C=S), 159.53 (C=S), 162.14 (J W‐c=23 Hz, C≡C), 173.96 ppm (J W‐c=28 Hz, C≡C). IR: =2460 (w, BH), 2440 (w, BH), 479 cm−1 (s, W≡S). Elemental analysis calcd (%) for C14H18BBrN6S4W . 0.1 C15H18BBrN6OS3W⋅1.0 CH2Cl2⋅1.0 C4H8O: C 27.53, H 3.38, N 10.29, S 15.34; found: C 27.68, H 3.19, N 10.36, S 15.24. EI‐MS: m/z no assignable signals.
[WS(C2Me2)(TmMe)Br] (7)
In a 120 mL Schlenk tube blue [W(CO)(C2Me2)(TmMe)Br] (2) (998 mg, 1.43 mmol) was dissolved in 50 mL MeCN. Methylthiirane (121 μL, 1.54 mmol) was added with a piston‐operated pipette and the tube was equipped with a bubbler and wrapped in aluminum foil before it was heated at 41 °C. After 3.5 h of heavy stirring the amber mixture was dried in vacuo. The residue was suspended in 120 mL CH2Cl2 and stirred for 1 h before it was concentrated to about 15 mL. The orange‐brown precipitate was filter‐cannulated, washed with 4 mL of CH2Cl2 and dried in vacuo to yield 539 mg (54 %) of micro‐crystalline product. 1H NMR (300 MHz, CD2Cl2): δ=2.84 (s, 3 H, ≡‐CH3), 3.20 (s, 3 H, N‐CH3), 3.32 (s, 3 H, ≡‐CH3), 3.68 (s, 3 H, N‐CH3), 4.05 (s, 3 H, N‐CH3), 6.73 (d, J=2.1 Hz, 1 H, CH), 6.76 (d, J=2.1 Hz, 1 H, CH), 6.83 (d, J=2.1 Hz, 1 H, CH), 6.90 (d, J=2.1 Hz, 1 H, CH), 7.08 ppm (s, 2 H, CH). 13C NMR (75 MHz, CD2Cl2): δ=17.09 (≡‐CH3), 21.82 (≡‐CH3), 34.56 (2 C, N‐CH3), 36.69 (N‐CH3), 120.03 (C=C), 120.81 (C=C), 122.25 (C=C), 123.02 (C=C), 123.52 (C=C), 125.01 (C=C), 158.03 (C=S), 158.43 (C=S), 159.59 (C=S), 174.10 (≡‐CH3), 186.16 ppm (≡‐CH3). IR: =2442 (w, BH), 2413 (w, BH), 479 cm−1 (s, W≡S). Elemental analysis calcd (%) for C16H22BN6S4BrW⋅1.2 CH2Cl2: C 25.72, H 3.06, N 10.46, S 15.97; found: C 25.93, H 2.99, N 10.61, S 15.76. EI‐MS: m/z no assignable signals.
[W2O2(C2Me2)2(TmMe)2](OTf)2 (8)
A suspension of 203 mg of [WO(C2Me2)(TmMe)Br0.82Cl0.18] (300 μmol) in 8 mL CH2Cl2 was added to a suspension of TlOTf (110 mg, 311 μmol) in 6 mL of CH2Cl2. The mixture was reacted for 1.25 h before it was filtered over Celite and dried in vacuo. The residue was suspended in 5 mL CH2Cl2 and treated with the ultrasonic bath. After concentrating to about 2 mL the solution was filtered off and the yellow microcrystalline powder was washed 2 mL CH2Cl2 to yield 139 mg (64 %, 95.6 μmol) of product. 1H NMR (300 MHz, CD2Cl2): δ=1.96 (bs, 3 H, ≡‐CH3), 2.96 (bs, 3 H, ≡‐CH3), 3.03 (bs, 3 H, ≡‐CH3), 3.06 (bs, 3 H, ≡‐CH3), 3.14 (s, 3 H, N‐CH3), 3.55 (s, 3 H, N‐CH3), 3.78 (s, 6 H, N‐CH3), 3.85 (s, 3 H, N‐CH3), 3.97 (s, 3 H, N‐CH3), 6.86 (bs, 2 H, CH), 6.90 (d, J=1.9 Hz, 1 H, CH), 7.05 (d, J=2.1 Hz, 1 H, CH), 7.08 (bs, 1 H, CH), 7.20 (bs, 1 H, CH), 7.24 (d, J=2.1 Hz, 1 H, CH), 7.25 (bs, 1 H, CH), 7.38 (bs, 1 H, CH), 7.68 (bs, 1 H, CH), 7.90 (bs, 1 H, CH), 8.26 ppm (bs, 1 H, CH). IR: =2445 (w, BH), 1255 (s, SO3), 1028 (s, SO3), 827 (m, W‐O), 803 cm−1 (m, W‐O). Elemental analysis calcd (%) for C34H44B2F6N12O8S8W2⋅0.30 CH2Cl2⋅0.15 H2O: C 26.81, H 2.94, N 10.94, S 16.69; found: C 26.44, H 2.81, N 10.78, S 17.04. Sample showed a mass increase during sample preparation presumably due to hygroscopic behavior. HR‐MS (m/z): [M/2−MeCN]+ calcd for C16H22BN6OS3W, 605.061; found, 605.058.
[W2S2(C2Me2)(TmMe)2](OTf)2 (9)
In a 50 mL Schlenk flask 102 mg (145 μmol) of [WS(C2Me2)(TmMe)Br] (7) were suspended in 10 mL CH2Cl2 and a solution of 58 mg (164 μmol) of TlOTf in 2 mL CH2Cl2 were added. The mixture was left for 42 h before it was filtered over Celite and 2× recrystallized from CH2Cl2/heptane to yield 25 % (26 mg, 17 μmol) of purple needle‐shaped crystals. 1H NMR (300 MHz, CD2Cl2): δ=−3.01 (bq, J=70.4 Hz, 1 H, BH), 2.25 (s, 3 H, N‐CH3), 3.21 (s, 6 H, ≡‐CH3), 3.69 (s, 3 H, N‐CH3), 3.81 (s, 3 H, N‐CH3), 3.96 (s, 3 H, N‐CH3), 3.97 (s, 3 H, N‐CH3), 4.31 (s, 3 H, N‐CH3), 6.26 (d, J=2.1 Hz, 1 H, CH), 6.49 (d, J=2.2 Hz, 1 H, CH), 6.72 (d, J=2.2 Hz, 1 H, CH), 6.82 (d, J=2.1 Hz, 1 H, CH), 6.89 (d, J=2.1 Hz, 1 H, CH), 6.98 (d, J=2.0 Hz, 1 H, CH), 7.02 (d, J=2.2 Hz, 1 H, CH), 7.04 (d, J=2.0 Hz, 1 H, CH), 7.05 (d, J=2.0 Hz, 1 H, CH), 7.08 (d, J=2.2 Hz, 1 H, CH), 7.34 (d, J=2.0 Hz, 1 H, CH), 7.36 ppm (d, J=2.0 Hz, 1 H, CH). 13C NMR (75 MHz, CD2Cl2): δ=21.67 (br, 2 ≡‐CH3), 32.43 (N‐CH3), 36.03 (N‐CH3), 36.06 (N‐CH3), 36.35 (N‐CH3), 36.55 (N‐CH3), 37.12 (N‐CH3), 119.66 (C=C), 119.97 (C=C), 121.99 (C=C), 122.67 (C=C), 122.94 (C=C), 123.30 (C=C), 123.75 (C=C), 124.45 (2 C=C), 124.59 (C=C), 125.91 (C=C), 126.11 (C=C), 149.04 (br Cq), 149.60 (Cq), 156.71 (Cq), 157.93 (Cq), 159.23 (br Cq), 160.82 ppm (br Cq), 2 Cq and 2 CF3 obscured. Elemental analysis calcd (%) for C30H38B2F6N12O6S10W2⋅0.2 C7H16⋅2.10 H2O: C 24.42, H 2.96, N 10.88, S 20.75; found: C 24.24, H 2.62, N 10.54, S 20.75. Sample showed a mass increase during sample preparation presumably due to hygroscopic behavior. IR: =2448 (w, BH), 1267 (s, SO3), 1028 cm−1 (s, SO3). HR‐MS (m/z): [M]2+ calcd for C28H38B2N12S8W2, 594.015; found, 594.012.
[W(CO)(C2H2)(MeCN)(TmMe)](OTf) (10)
A solution of 106 mg (299 μmol) of TlOTf in 2 mL MeCN was added to a suspension of 207 mg (309 μmol) of [W(CO)(C2H2)(TmMe)Br] (1) in 10 mL of MeCN. After 1 h the resulting suspension was double‐filtered over Celite. Drying in vacuo gave 81 % (184 mg without MeCN, 249 μmol) of turquoise product. 1H NMR (300 MHz, CD3CN): δ=3.01 (s, 3 H, N‐CH3), 3.70 (s, 3 H, N‐CH3), 3.85 (s, 3 H, N‐CH3), 7.00 (d, J=2.2 Hz, 1 H, CH), 7.02 (d, J=2.2 Hz, 1 H, CH), 7.14 (t, J=1.9 Hz, J=1.7 Hz, 3 H, CH), 7.17 (d, J=2.0 Hz, 1 H, CH), 12.19 (bs, 1 H, ≡‐H), 12.97 ppm (bs, 1 H, ≡‐H). 13C NMR (75 MHz, CD3CN): δ=34.03 (N‐CH3), 35.45 (N‐CH3), 35.51 (N‐CH3), 122.08 (C=C), 122.68 (C=C), 123.47 (C=C), 124.65 (C=C), 125.13 (C=C), 125.79 (C=C), 156.75 (C=S), 158.67 (C=S), 164.62 (C=S), 192.66 (C≡C), 196.29 (C≡C), 227.11 ppm (J W‐C=142 Hz, CO). IR: =2441 (w, BH), 1921 (s, CO), 1258 (s, SO3), 1028 cm−1 (s, SO3). Elemental analysis calcd (%) for C17H22BF3N6O3S5W⋅0.55 MeCN⋅1.5 H2O: C 26.07, H 2.90, N 11.64, S 16.28; found: C 25.76, H 2.55, N 11.54, S 15.92. Sample showed a mass increase during sample preparation presumably due to hygroscopic behavior. HR‐MS (m/z): [M−MeCN]+ calcd for C15H18BN6OS3W, 589.029; found, 589.026.
[W(CO)(C2Me2)(MeCN)(TmMe)](OTf) (11)
A solution of 132 mg (373 μmol) of TlOTf in 3 mL MeCN was added to a suspension of 258 mg (370 μmol) of [W(CO)(C2Me2)(TmMe)Br] (2) in 10 mL of MeCN. After 1.5 h the resulting fine suspension was double‐filtered over Celite. Drying in vacuo gave 78 % (221 mg without MeCN, 288 μmol) of turquoise product. 1H NMR (300 MHz, CD3CN): δ=3.06 (s, 3 H, N‐CH3), 3.09 (bs, 6 H, ≡‐CH3), 3.70 (s, 3 H, N‐CH3), 3.83 (s, 3 H, N‐CH3), 6.96 (d, J=2.1 Hz, 1 H, CH), 7.00 (d, J=2.1 Hz, 1 H, CH), 7.12 (m, 3 H, CH), 7.15 ppm (d, J=2.0 Hz, 1 H, CH). 13C NMR (75 MHz, CD3CN): δ=17.24 (≡‐CH3), 21.37 (≡‐CH3), 34.50 (N‐CH3), 35.39 (2 N‐CH3), 122.03 (C=C), 122.53 (C=C), 123.14 (C=C), 124.58 (C=C), 124.90 (C=C), 125.62 (C=C), 156.96 (C=S), 159.02 (C=S), 164.31 (C=S), 198.70 (C≡C), 203.16 (C≡C), 229.09 ppm (CO), CF3 obscured. IR: =2440 (w, BH), 2410 (w, BH), 1907 (s, CO), 1258 (s, SO3), 1029 cm−1 (s, SO3). Elemental analysis calcd (%) for C17H22BF3N6O3S5W⋅0.95 MeCN⋅1.10 H2O: C 28.97, H 3.30, N 11.80, S 15.54; found: C 28.86, H 3.01, N 11.55, S 15.26. Sample showed a mass increase during sample preparation presumably due to hygroscopic behavior. HR‐MS (m/z): [M−MeCN]+ calcd for C17H22BN6OS3W, 617.061; found, 617.053.
[WO(C2H2)(MeCN)(TmMe)](OTf) (12)
A solution of 21 mg (59.4 μmol) of TlOTf in 1 mL MeCN was added to a suspension of 37 mg (56.3 μmol) of [WO(C2H2)(TmMe)Br] (4) in 4 mL of MeCN. After 15 min the resulting colloidal suspension was centrifuged for 20 min at 13 500 rpm (RZB: 12 100×g), decantated and double‐filtered over Celite. The filtrate was dried in vacuo and, resuspended in 2 mL MeCN and the residual TlBr was allowed to settle for 2 h before it was again centrifuged for 10 min at 13 500 rpm (RZB: 12 100×g). The supernatant was decantated of and dried in vacuo to give 95 % (39 mg without MeCN, 53.7 μmol) 1H NMR (300 MHz, CD3CN): δ=3.23 (s, 3 H, N‐CH3), 3.69 (s, 3 H, N‐CH3), 3.86 (s, 3 H, N‐CH3), 6.96 (t, J=2.1 Hz, 2 H, CH), 6.99 (d, J=2.2 Hz, 1 H, CH), 7.14 (d, J=2.1 Hz, J=1.7 Hz, 1 H, CH), 7.17 (d, J=2.0 Hz, 1 H, CH), 7.31 (d, J=2.0 Hz, 1 H, CH), 10.30 (s, J W‐H=11.7 Hz, 1 H, ≡‐H), 11.02 ppm (s, J W‐H=12.4 Hz, 1 H, ≡‐H). 13C NMR (75 MHz, CD3CN): δ=34.71 (N‐CH3), 35.33 (N‐CH3), 35.51 (N‐CH3), 122.50 (C=C), 122.87 (C=C), 123.93 (C=C), 124.80 (C=C), 125.34 (C=C), 125.94 (C=C), 142.73 (C≡C), 147.73 (C≡C), 155.19 (C=S), 156.01 (C=S), 157.00 ppm (C=S), IR: =2442 (w, BH), 1252 (s, SO3), 1028 (s, SO3), 935 cm−1 (m, W≡O). Elemental analysis calcd (%) for C17H22BF3N6O3S5W⋅0.2 TlBr⋅0.3 MeCN: C 23.56, H 2.39, N 11.09, S 16.12; found: C 23.19, H 2.50, N 10.64, S 16.51. Sample showed a mass increase during sample preparation presumably due to hygroscopic behavior. HR‐MS (m/z): [M−MeCN]+ calcd for C14H18BN6OS3W, 577.029; found, 577.027.
[WO(C2Me2)(MeCN)(TmMe)](OTf) (13)
Method A
A solution of 80 mg (226 μmol) of TlOTf in 3 mL MeCN was added to a solution of 153 mg (232 μmol) of [WO(C2Me2)(TmMe)Br0.43Cl57] (8) in 12 mL of MeCN. After 30 min the resulting fine colloidal suspension was centrifuged for 20 min. at 2.000 rpm before the supernatant was filter‐cannulated and dried in vacuo to yield 79 % (137 mg without MeCN, 182 μmol) of yellow–orange complex 16.
Method B
In a 25 mL Schlenk tube 90 mg of [W2O2(C2Me2)2(TmMe)2](OTf)2 were dissolved in 12 mL of MeCN and dried in vacuo to yield 80 mg (89 %, 106 μmol) of yellow–orange complex. 1H NMR (300 MHz, CD3CN): δ=2.71 (d, J=1.1 Hz, 3 H, ≡‐CH3); 2.97 (d, J=1.2 Hz, 3 H, ≡‐CH3), 3.19 (s, 3 H, N‐CH3), 3.67 (s, 3 H, N‐CH3), 3.83 (s, 3 H, N‐CH3), 6.95 (dd, J=2.1 Hz, J=2.5 Hz, 2 H, CH), 6.99 (d, J=2.1 Hz, 1 H, CH), 7.13 (d, J=2.1 Hz, 1 H, CH), 7.15 (d, J=2.0 Hz, 1 H, CH), 7.31 ppm (d, J=2.0 Hz, 1 H, CH). 13C NMR (75 MHz, CD3CN): δ=12.93 (≡‐CH3), 16.80 (≡‐CH3), 34.52 (N‐CH3), 35.21 (N‐CH3), 35.39 (N‐CH3), 122.26 (C=C), 122.63 (C=C), 123.89 (C=C), 124.73 (C=C), 125.13 (C=C), 125.95 (C=C), 145.23 (J W‐C=26.5 Hz, C≡C), 152.64 (J W‐C=28.0 Hz, C≡C), 155.58 (C=S), 156.07 (C=S) 157.46 ppm (C=S). IR: =2446 (w, BH), 1258 (s, SO3), 1028 (s, SO3), 925 cm−1 (s, W≡O). Elemental analysis calcd (%) for C17H22BF3N6O4S4W⋅0.35 MeCN⋅0.65 H2O: C 27.24, H 3.150, N 11.40, S 16.43; found: C 27.10, H 3.00, N 11.28, S 16.48. Sample showed a mass increase during sample preparation presumably due to hygroscopic behavior. HR‐MS (m/z): [M−MeCN]+ calcd for C16H22BN6OS3W, 605.061; found, 605.058.
[WS(C2H2)(MeCN)(TmMe)](OTf) (14)
A solution of 39 mg (110 μmol) of TlOTf in 1 mL MeCN was added to a suspension of 71 mg (56.3 μmol) of [WS(C2H2)(TmMe)Br] (4) in 4 mL of MeCN. After 30 min the resulting colloidal suspension was centrifuged for 10 min at 13 500 rpm (RZB: 12 100×g), decantated and double‐filtered over Celite. Drying in vacuo gave 74 % (58 mg without MeCN, 78.2 μmol) of a 1:9 Mixture of 1 and 14 as a brown powder. 1H NMR (300 MHz, CD3CN): δ=3.19 (s, 3 H, N‐CH3), 3.69 (s, 3 H, N‐CH3), 3.88 (s, 3 H, N‐CH3), 6.92 (d, J=2.1 Hz, 1 H, CH), 7.01 (s, 2 H, CH), 7.11 (d, J=2.1 Hz, 1 H, CH), 7.18 (d, J=2.1 Hz, 1 H, CH), 7.32 (d, J=2.1 Hz, 1 H, CH), 11.35 (s, J W‐C=10.9 Hz 1 H, ≡‐H), 12.10 ppm (s, J W‐C=11.2 Hz 1 H, ≡‐H). 13C NMR (75 MHz, CD3CN): δ=34.65 (2 N‐CH3), 35.49 (N‐CH3), 122.57 (C=C), 122.72 (C=C), 124.06 (C=C), 124.88 (C=C), 125.12 (C=C), 125.90 (C=C), 155.72 (C=S), 156.28 (C=S), 157.79 (C=S), 158.80 (J W‐C=21.8 Hz C≡C), 164.60 ppm (J W‐C=25.0 Hz C≡C). IR: =2440 (w, BH), 1255 (s, SO3), 1027 (s, SO3), 515 cm−1 (m, W≡S). Elemental analysis calcd (%) for C17H22BF3N6O3S5W⋅0.2 10⋅0.35 TlBr: C 21.95, H 2.19, N 10.13, S 19.32; found: C 22.69, H 2.38, N 10.41, S 18.50. Sample showed a mass increase during sample preparation presumably due to hygroscopic behavior. HR‐MS (m/z): [M−MeCN]+ calcd for C14H18BN6S4W, 595.009; found, 595.006.
[WS(C2Me2)(MeCN)(TmMe)](OTf) (15)
A solution of 102 mg (289 μmol) of TlOTf in 5 mL MeCN was added to a suspension of 195 mg (278 μmol) of [WS(C2Me2)(TmMe)Br] (7) in 10 mL of MeCN. After 1 h the resulting colloidal suspension was centrifuged for 20 min. at 13 500 rpm (RZB: 12 100×g) before the supernatant was taken off and filtered over Celite. Drying in vacuo gave 90 % (193 mg without MeCN, 250 μmol) of orange‐red product. 1H NMR (300 MHz, CD3CN): δ=2.92 (d, J=1.1 Hz, 3 H, ≡‐CH3), 3.12 (d, J=1.1 Hz, 3 H, ≡‐CH3), 3.15 (s, 3 H, N‐CH3), 3.66 (s, 3 H, N‐CH3), 3.85 (s, 3 H, N‐CH3), 6.90 (d, J=2.1 Hz, 1 H, CH), 7.00 (q, J=2.1 Hz, 2 H, CH), 7.10 (d, J=2.1 Hz, 1 H, CH), 7.16 (d, J=2.0 Hz, 1 H, CH), 7.31 ppm (d, J=2.0 Hz, 1 H, CH). 13C NMR (CD3CN): δ=16.28 (≡‐CH3), 19.50 (≡‐CH3), 34.54 (2× N‐CH3), 35.38 (N‐CH3), 122.32 (C=C), 122.47(C=C), 124.03 (C=C), 124.62 (C=C), 125.06 (C=C), 125.91 (C=C), 155.87 (C=S), 156.63 (C=S) 158.09 (C=S), 167.54 (J W‐C=22.5 Hz, C≡C), 175.20 ppm (J W‐C=24.5 Hz, C≡C). IR: =2444 (w, BH), 1257 (s, SO3), 1029 (s, SO3), 484 cm−1 (m, W≡S). Elemental analysis calcd (%) for C17H22BF3N6O3S5W⋅0.7 MeCN⋅0.85 H2O: C 27.14, H 3.19, N 11.52, S 19.68; found: C 27.08, H 3.00, N 11.59, S 19.51. Sample showed a mass increase during sample preparation presumably due to hygroscopic behavior. HR‐MS (m/z): [M−MeCN]+ calcd for C16H22BN6S4W, 621.038; found, 621.032.
mt‐S‐S‐mt
This compound was prepared for reference purposes. In a 50 mL round bottom flask 510 mg (4.47 mmol, 2.0 equiv) methimazole were dissolved in 25 mL benchtop CH2Cl2 under ambient conditions and 476 mg (2.1 equiv., 4.70 mmol) trimethylamine were added. To the stirred solution 565 mg iodine (2.23 mmol, 1.0 equiv) were added portion‐wise over a period of 25 min and stirred for another 30 min. The reaction was quenched by addition of 30 mL water. The yellow organic phase was collected, washed 2× with 10 mL water and dried over MgSO4. The solvent was removed in vacuo yielding 373 mg (1.65 mmol, 74 %) of yellow crystals. 1H NMR (300 MHz, CDCl3): δ=3.56 (s, 3 H, N‐CH3), 7.05 (d, J=0.8 Hz, 1 H, CH), 7.13 ppm (d, J=0.8 Hz, 1 H, CH). 13C NMR (75 MHz, CDCl3): δ=34.11 (N‐CH3), 124.90 (C=C), 131.01 (C=C), 139.57 ppm (C−S). EI‐MS: m/z 226.1 [M +].
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors thank the NAWI Graz for financial support, Bernd Werner for NMR measurements, and Alina Hambrusch for contributing.
C. Vidovič, F. Belaj, N. C. Mösch-Zanetti, Chem. Eur. J. 2020, 26, 12431.
References
- 1.
- 1a. Majumdar A., Dalton Trans. 2014, 43, 8990–9003; [DOI] [PubMed] [Google Scholar]
- 1b. Hille R., Trends Biochem. Sci. 2002, 27, 360–367; [DOI] [PubMed] [Google Scholar]
- 1c. Majumdar A., Sarkar S., Coord. Chem. Rev. 2011, 255, 1039–1054. [Google Scholar]
- 2.
- 2a. Johnson M. K., Rees D. C., Adams M. W. W., Chem. Rev. 1996, 96, 2817–2839; [DOI] [PubMed] [Google Scholar]
- 2b. Bevers L. E., Hagedoorn P.-L., Hagen W. R., Coord. Chem. Rev. 2009, 253, 269–290. [Google Scholar]
- 3.
- 3a. Rosner B. M., Schink B., J. Bacteriol. 1995, 177, 5767–5772; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Molybdenum and Tungsten Enzymes: Biochemistry (Eds.: R. Hille, C. Schulzke, M. L. Kirk), Royal Society of Chemistry, Cambridge, 2016. [Google Scholar]
- 4. Seiffert G. B., Ullmann G. M., Messerschmidt A., Schink B., Kroneck P. M. H., Einsle O., Proc. Natl. Acad. Sci. USA 2007, 104, 3073–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kroneck P. M. H., J. Biol. Inorg. Chem. 2016, 21, 29–38. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Boll M., Einsle O., Ermler U., Kroneck P. M. H., Ullmann G. M., J. Mol. Microbiol. Biotechnol. 2016, 26, 119–137; [DOI] [PubMed] [Google Scholar]
- 6b. The Metal-Driven Biogeochemistry of Gaseous Compounds in the Environment (Eds.: P. M. H. Kroneck, M. E. Sosa Torres), Springer, Dordrecht, 2014. [Google Scholar]
- 7.
- 7a. Liao R.-Z., Yu J.-G., Himo F., Proc. Natl. Acad. Sci. USA 2010, 107, 22523–22527; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Liu Y.-F., Liao R.-Z., Ding W.-J., Yu J.-G., Liu R.-Z., J. Biol. Inorg. Chem. 2011, 16, 745–752; [DOI] [PubMed] [Google Scholar]
- 7c. Liao R.-Z., Thiel W., J. Comput. Chem. 2013, 34, 2389–2397. [DOI] [PubMed] [Google Scholar]
- 8. Liao R.-Z., Thiel W., J. Chem. Theory Comput. 2012, 8, 3793–3803. [DOI] [PubMed] [Google Scholar]
- 9. Najafian A., Cundari T. R., Polyhedron 2018, 154, 114–122. [Google Scholar]
- 10. Yadav J., Das S. K., Sarkar S., J. Am. Chem. Soc. 1997, 119, 4315–4316. [Google Scholar]
- 11. Schreyer M. A., Hintermann L., Beilstein J. Org. Chem. 2017, 13, 2332–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Templeton J. L., Ward B. C., Chen G. J. J., McDonald J. W., Newton W. E., Inorg. Chem. 1981, 20, 1248–1253. [Google Scholar]
- 13. Peschel L. M., Belaj F., Mösch-Zanetti N. C., Angew. Chem. Int. Ed. 2015, 54, 13018–13021; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13210–13213. [Google Scholar]
- 14. Crane T. W., White P. S., Templeton J. L., Inorg. Chem. 2000, 39, 1081–1091. [DOI] [PubMed] [Google Scholar]
- 15. Garner M., Reglinski J., Cassidy I., Spicer M. D., Kennedy A. R., Chem. Commun. 1996, 1975–1976. [Google Scholar]
- 16. Wallace D., Gibson L. T., Reglinski J., Spicer M. D., Inorg. Chem. 2007, 46, 3804–3806. [DOI] [PubMed] [Google Scholar]
- 17. Hill A. F., Tshabang N., Willis A. C., Eur. J. Inorg. Chem. 2007, 3781–3785. [Google Scholar]
- 18. Baker P. K., Clark A. I., Coles S. J., Drew M. G. B., Durrant M. C., Hursthouse M. B., Richards R. L., J. Chem. Soc. Dalton Trans. 1998, 1281–1287. [Google Scholar]
- 19.
- 19a. Kersting M., El-Kholi A., Mueller U., Dehnicke K., Chem. Ber. 1989, 122, 279–285; [Google Scholar]
- 19b. Al-jahdali M., Baker P. K., Lavery A. J., Meehan M. M., Muldoon D. J., J. Mol. Catal. A 2000, 159, 51–62. [Google Scholar]
- 20. Vidovič C., Peschel L. M., Buchsteiner M., Belaj F., Mösch-Zanetti N. C., Chem. Eur. J. 2019, 25, 14267–14272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Templeton J. L. in Advances in Organometallic Chemistry V29, Advances in Organometallic Chemistry, Vol. 29 (Eds.: F. G. A. Stone, R. West), Academic Press, San Diego, 1989, pp. 1–100. [Google Scholar]
- 22.
- 22a. Baker P. K., Muldoon D. J., Lavery A. J., Shawcross A., Polyhedron 1994, 13, 2915–2921; [Google Scholar]
- 22b. Braendle A., Vidovič C., Mösch-Zanetti N. C., Niederberger M., Caseri W., Polymers 2018, 10, 881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ehweiner M. A., Vidovič C., Belaj F., Mösch-Zanetti N. C., Inorg. Chem. 2019, 58, 8179–8187. [DOI] [PubMed] [Google Scholar]
- 24. Garner M., Lehmann M.-A., Reglinski J., Spicer M. D., Organometallics 2001, 20, 5233–5236. [Google Scholar]
- 25. Templeton J. L., Ward B. C., Inorg. Chem. 1980, 19, 1753–1759. [Google Scholar]
- 26. Crane T. W., White P. S., Templeton J. L., Organometallics 1999, 18, 1897–1903. [Google Scholar]
- 27.
- 27a. Brower D. C., Tonker T. L., Morrow J. R., Rivers D. S., Templeton J. L., Organometallics 1986, 5, 1093–1097; [Google Scholar]
- 27b. Morrow J. R., Tonker T. L., Templeton J. L., Kenan W. R., Organometallics 1985, 4, 745–750. [Google Scholar]
- 28. Thomas S., Tiekink E. R. T., Young C. G., Inorg. Chem. 2006, 45, 352–361. [DOI] [PubMed] [Google Scholar]
- 29. Abernethy R. J., Hill A. F., Neumann H., Willis A. C., Inorg. Chim. Acta 2005, 358, 1605–1613. [Google Scholar]
- 30. Spicer M. D., Reglinski J., Eur. J. Inorg. Chem. 2009, 1553–1574. [Google Scholar]
- 31.
- 31a. Armstrong E. M., Baker P. K., Drew M. G. B., Organometallics 1988, 7, 319–325; [Google Scholar]
- 31b. Drew M. G. B., Baker P. K., Clark A. I., Richards R. L., Durrant M. C., Gazz. Chim. Ital. 1997, 127, 601–603. [Google Scholar]
- 32. Templeton J. L., Ward B. C., J. Am. Chem. Soc. 1980, 102, 3288–3290. [Google Scholar]
- 33. McMullan R. K., Kvick Å., Popelier P., Acta Crystallogr. Sect. B 1992, 48, 726–731. [Google Scholar]
- 34. Pignataro E., Post B., Acta Crystallogr. 1955, 8, 672–674. [Google Scholar]
- 35. Mavridis A., Moustakali-Mavridis I., Acta Crystallogr. Sect. B 1977, 33, 3612–3615. [Google Scholar]
- 36. Ricard L., Weiss R., Newton W. E., Chen G. J. J., McDonald J. W., J. Am. Chem. Soc. 1978, 100, 1318–1320. [Google Scholar]
- 37. Adams C. J., Bartlett I. M., Carlton S., Connelly N. G., Harding D. J., Hayward O. D., Orpen A. G., Patron E., Ray C. D., Rieger P. H., Dalton Trans. 2007, 62–72. [DOI] [PubMed] [Google Scholar]
- 38. Peschel L. M., Vidovič C., Belaj F., Neshchadin D., Mösch-Zanetti N. C., Chem. Eur. J. 2019, 25, 3893–3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blower P. J., Dilworth J. R., Hutchinson J., Nicholson T., Zubieta J. A., J. Chem. Soc. Dalton Trans. 1985, 2639. [Google Scholar]
- 40. Hossain M. M., Lin H.-M., Shyu S.-G., Organometallics 2007, 26, 685–691. [Google Scholar]
- 41. Figueroa J. S., Yurkerwich K., Melnick J., Buccella D., Parkin G., Inorg. Chem. 2007, 46, 9234–9244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Peschel L. M., Schachner J. A., Sala C. H., Belaj F., Mösch-Zanetti N. C., Z. Anorg. Allg. Chem. 2013, 639, 1559–1567. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary