

Abstract
Ambient temperature ruthenium‐catalyzed C−H arylations were accomplished by visible light without additional photocatalysts. The robustness of the ruthenium‐catalyzed C−H functionalization protocol was reflected by a broad range of sensitive functional groups and synthetically useful pyrazoles, triazoles and sensitive nucleosides and nucleotides, as well as multifold C−H functionalizations. Biscyclometalated ruthenium complexes were identified as the key intermediates in the photoredox ruthenium catalysis by detailed computational and experimental mechanistic analysis. Calculations suggested that the in situ formed photoactive ruthenium species preferably underwent an inner‐sphere electron transfer.
Keywords: arylation, C−H activation, photocatalysis, photochemistry, ruthenium
Light‐enabled ruthenium‐catalyzed C−H activation enables direct arylations at room temperature. Biscyclometalated ruthenium intermediates were identified as photoredox catalysts through a metal‐to‐ligand charge transfer (MLCT), intersystem crossing (ISC) and inner‐sphere electron transfer (ISET) manifold.

Introduction
During the recent decade, transition metal‐catalyzed C−H functionalizations have emerged as a powerful tool in molecular syntheses1 with notable advances by ruthenium catalysis.2 Particularly, C−H arylations3 have played an important role in material sciences, crop protection, and drug discovery.4 Ruthenium‐catalyzed C−H arylations have hence been exploited for the synthesis of biologically active compounds, such as Anacetrapib, Valsartan, and Candesartan by Ouellet at Merck,5 Ackermann,6 and Seki,7 respectively (Figure 1 a). In addition, ruthenium‐catalyzed C−H arylations have enabled late‐stage peptide8 and nucleoside functionalizations.9
Figure 1.

(a) Bioactive biaryls and ruthenium‐catalyzed C−H arylations: (b) under thermal reaction condition and (c) under photoredox condition.
Despite of major advances, ruthenium‐catalyzed C−H arylations continue to be restricted to high reaction temperatures of typically 100–140 °C (Figure 1 b).10 The mechanism for ruthenium‐catalyzed direct arylations with haloarenes was thus far generally accepted to occur through a ruthenium(II/IV) pathway by oxidative addition and reductive elimination.10k
Visible‐light photoredox catalysis11 allowed for direct transformations at ambient temperature, however, additional iridium12 or ruthenium13 photocatalysts were required in these transformations among others as reported by MacMillan, Molander and Doyle. In contrast, Ackermann14 and Greaney15 recently disclosed photo‐induced ruthenium‐catalyzed remote C−H alkylations. After submission of the present manuscript, Greaney has reported on a ruthenium‐catalyzed arylation of phenylpyridines using visible light, which was proposed to occur by an oxidative addition/reductive elimination process.16 Within our program on sustainable C−H activation,17 we have now devised the first light‐driven ruthenium‐catalyzed C−H arylations at ambient temperature (Figure 1 c). Notable features of our strategy include (i) a versatile dual‐functional ruthenium(II) biscarboxylate catalyst for selective direct arylations, (ii) visible‐light‐induced C−H activation, (iii) photocatalysts‐free ambient temperature transformations, (iv) versatile C−H arylations on transformable arenes and biorelevant purines, and (v) mechanistic insights by experiment and computation, being supportive of a ruthenium(II/III/IV) mechanism.
Results and Discussion
We commenced our studies by probing various reaction conditions for the envisioned C−H arylations of arene 1 a with 4‐iodoanisole (2 a) (Table 1).18 Carboxylate assistance19 was found to be key to achieve the ortho‐C−H arylation (entries 1–4). Specifically, we found that [Ru(OAc)2(p‐cymene)] (4) in 1,4‐dioxane was the most effective catalyst (entry 1). Cationic ruthenium(II) complexes in DMA as the solvent also provided the ortho‐arylated product 3 aa, albeit only in moderate yield (entries 5–6). Other ruthenium sources, such as Ru3(CO)12 and RuCl3⋅n H2O, failed to give the desired product 3 aa (entry 7). Control experiments verified the essential role of the ruthenium catalyst (entry 8), the base (entry 9), and the blue light (entry 10). In the dark, the reaction was sluggish even at a reaction temperature of 100 °C, reflecting the great influence of visible light on the C−H arylation. In addition, aryl bromides, chlorides, and triflates also furnished the corresponding product 3 aa in good yields (entry 11).18 While thermal C−H arylations had been realized with different halides by a ruthenium(II/IV) pathway,10l sustainable light energy allowed first ambient temperature arylations, being superior to thermal reactions even at 100 °C.
Table 1.
Optimization of photo‐induced ruthenium(II)‐catalyzed C−H arylation

|
Entry |
Deviation from the standard conditions |
Yield [%][a] |
|---|---|---|
|
1 |
none |
94 |
|
2 |
[RuCl2(p‐cymene)]2 instead of 4 |
(6) |
|
3 |
[RuCl2(p‐cymene)]2 instead of 4 and KOAc instead of K2CO3 |
90 |
|
4 |
n‐Bu4NOAc instead of K2CO3 |
78 |
|
5 |
[Ru(NCt‐Bu)6][PF6]2 instead of 4 and MesCO2H (30 mol %) as the additive |
(4) |
|
6 |
[Ru(NCt‐Bu)6][PF6]2 instead of 4 and MesCO2H (30 mol %) as the additive, DMA instead of 1,4‐dioxane |
(52) |
|
7 |
Ru3(CO)12, RuCl3⋅n H2O instead of 4 and MesCO2H (30 mol %) as the additive |
(0), (0) |
|
8 |
no [Ru], at 30–35 °C |
(0) |
|
9 |
no K2CO3, at 30–35 °C |
(14) |
|
10 |
no light, at 30–35, 40, 60, 80, 100 °C |
(12), (9), (15), (32), (49) |
|
11 |
ArBr, ArCl, ArOTf instead of ArI |
68, (43), 39 |
[a] Reaction conditions: 1 a (0.50 mmol), ArI 2 a (0.75 mmol), [Ru(OAc)2(p‐cymene)] (10 mol %), K2CO3 (1.00 mmol), 1,4‐dioxane (2.0 mL), 30–35 °C, 24 h, under N2, blue LEDs; yield of isolated products. The yield in the parentheses were determined by 1H‐NMR spectroscopy using 1,3,5‐trimethoxybenzene as the internal standard.
With the optimized reaction conditions in hand, we probed its versatility in the direct C−H arylation of 2‐arylpyridine 1 a and a variety of aryl iodides 2 (Scheme 1). Electron‐donating and electron‐withdrawing groups of para‐ and meta‐substituted aryl halides were well tolerated, affording the corresponding arylated product 3 with moderate to high efficacy. Sterically hindered 2‐iodoanisole (2 s) also delivered the desired product 3 as. It is noteworthy that the ruthenium‐catalyzed C−H arylation proved widely applicable, tolerating sensitive functional groups, including hydroxyl (3 af), chloro, bromo (3 ah–3 ai), ketone (3 ak and 3 aq), ester (3 al) and nitrile (3 ar). In addition, the ruthenium(II) catalysis was effective for (NH)‐free indole (3 aw) and carbazole (3 az).20 Besides monohaloarenes, the room temperature ruthenium catalysis regime proved applicable to the twofold (5 aa′–5 ac′)20 or threefold C−H functionalization (5 ad′).
Scheme 1.
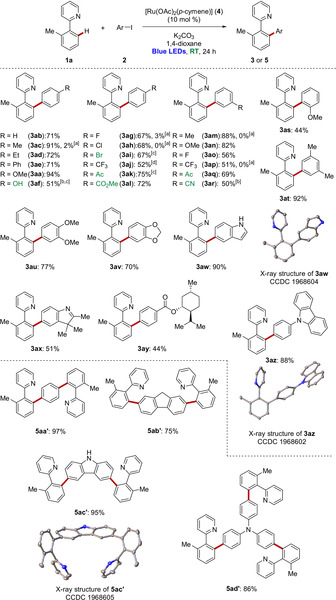
Photo‐induced ruthenium(II)‐catalyzed C−H arylation at ambient temperature. [a] No light at 30–35 °C. [b] DMA as solvent. [c] 48 h. [d] 4‐Bromobenzotrifluoride.
The C−H arylation was not restricted to the assistance by pyridines. Indeed, arenes bearing pyrimidines 1 e, transformable imidates 1 f–1 g, and removable pyrazoles 1 h–1 j were efficiently converted into the desired products 3 (Scheme 2 a).20 In addition, ketimine 1 k underwent the photo‐induced arylation followed by acidic hydrolysis to afford the ortho‐arylated acetophenone 3 ka (Scheme 2 b).
Scheme 2.
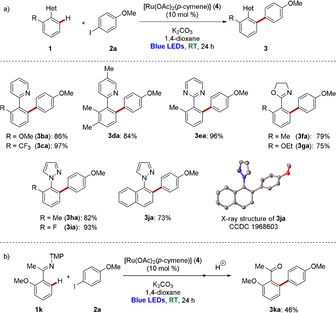
Room‐temperature visible‐light‐enabled ruthenium‐catalyzed C−H arylation with a) heteroarenes 1 and b) ketimine 1 k. TMP=3,4,5‐trimethoxyphenyl.
Notably, the application of the room temperature direct arylation with substituted click‐triazole21 1 l selectively provided the corresponding products 3 la, 3 lg, 3 lv (Scheme 3).
Scheme 3.

Ruthenium‐catalyzed C−H arylation with substituted click‐triazole 1 l.
The unique potential of the direct photoredox arylation was mirrored by the improved chemo‐selectivities in the reactions with 4‐iodophenyl triflate (2 e′) or 1‐bromo‐3‐iodobenzene (2 f′), predominantly affording the monoarylation products, while leaving the valuable electrophilic (pseudo)halides intact (Scheme 4 a,b). In sharp contrast, the thermal reactions delivered the diarylated products 5, lacking a discrimination of the complementary aryl halides. Furthermore, the light‐induced C−H arylation of naphthyl‐pyrazole 1 j with 2‐bromo‐5‐iodotoluene (2 g′) provided the desired monoarylated product 3 jg′ (Scheme 4 c). In stark contrast, undesired protodebromination was observed by conventional thermal arylations.
Scheme 4.

Improved chemo‐selectivity by visible‐light‐induced C−H arylation.
Given the synthetic utility of the mild photo‐induced ruthenium(II)‐catalyzed direct C−H arylations, we became attracted to delineating its mode of action. To this end, intermolecular competition experiments of ortho‐substituted arenes 1 showed a minor electronic bias,18 while experiments of meta‐substituted arenes 1 highlighted electron‐poor arenes to be more efficiently converted (Scheme 5 a). Moreover, a significant amount of free p‐cymene (6) was observed by decoordination from the ruthenium precatalyst (Scheme 5 b). An arene‐ligand‐free ruthenacycle 7, as previously exploited by Larrosa and our group,3c, 3h, 22 was only effective in the presence of KOAc (Scheme 5 c), which suggested that a carboxylate‐modified ruthenacycle was involved in the photo‐induced C−H arylation.
Scheme 5.

Key mechanistic studies.
To elucidate the role of the blue light, we conducted an on/off light experiment (Figure 2).18 The conversion of the direct ortho‐arylation was completely suppressed in the absence of light, being indicative of the photo‐induced ruthenium(II)‐catalyzed arylation not involving a radical chain process. In addition, we obtained the quantum yield of Φ=0.087 for the photoredox ruthenium‐catalyzed arylation.18
Figure 2.

On/off light experiment.
In order to gain detailed insights into the reaction mechanism, DFT calculations were performed for biscyclometalated ruthenacycle enabled ortho‐C−H arylations at the PBE0‐D3(BJ)/6‐311++G(d,p),def2‐TZVP(Ru,I),SDD(Ru,I)+SMD(1,4‐dioxane)//B3LYP‐D3(BJ)/6‐31G(d),def2‐SVP(Ru,I), SDD(Ru,I) level of theory.18 Initially we studied the absorption properties23 of the monocyclometalated ruthenacycles 8 and 9 as well as the biscyclometalated complexes 10 and 11 (Figure 3 a). The TD‐DFT calculations of complexes 10 and 11 confirmed the existence of a strong metal‐to‐ligand charge‐transfer (MLCT) with a maximum absorption at λ=461.44 (oscillator strength of 0.1810) and 461.41 nm (oscillator strength of 0.1601), respectively. These findings are in good agreement with an orbital analysis of complex 11 (Figure 3 b). In sharp contrast, the monocyclometalated complexes 8 and 9 featured a maximum absorption band outside the visible‐light range.
Figure 3.

Computational analysis: a) absorption wavelength of mono‐ and biscyclometalated ruthenacycles, b) HOMO and LUMO of biscyclometalated ruthenium complex 11.
On the basis of our findings, the complexes 10 and 11 were further studied for the photoredox arylations (Figure 4). The excitation of iodoarene‐coordinated ruthenacycle complex 11 followed by intersystem crossing (ISC) leads to a long‐lived triplet complex 11** with an energy of 30.7 kcal mol−1 as compared to the complex 10 (Figure 4 a). Afterwards, inner‐sphere electron transfer (ISET) to iodoarene occurs through a low barrier transition state TS1 to form phenyl radical and ruthenium(III) intermediate 12. The radical recombination of complex 12 affords a stable ruthenium(IV) intermediate 14. Apart from the ISET process, we considered potentially viable outer‐sphere electron transfer (OSET) from the triplet state 10** to iodoarene molecule (Figure 4 b). The OSET transition state TS3 has a barrier of 63.1 kcal mol−1, as computed using Marcus theory24 and Savéant‘s model,25 and thereby generates cationic ruthenium complex 16 with an energy of 57.6 kcal mol−1 as compared to the complex 10. Therefore, the ISET mechanism is considered to be favorable over the OSET pathway.
Figure 4.

Relative Gibbs free energy profile in kcal mol−1 with respect to 10 at thePBE0‐D3(BJ)/6‐311++G(d,p),def2‐TZVP(Ru,I),SDD(Ru,I)+SMD(1,4dioxane)//B3LYP‐D3(BJ)/6‐31G(d),def2‐SVP(Ru,I),SDD(Ru,I) level of theory for photo‐induced ruthenium‐catalyzed C−H arylation via a) inner‐sphere electron transfer (ISET) and b) outer‐sphere electron transfer (OSET) pathways. L=AcOH.
Based on our findings, a plausible catalytic cycle commences by carboxylate‐assisted C−H ruthenation and dissociation of p‐cymene,22c generating the corresponding biscyclometalated complex 10 (Scheme 6). The coordination of iodoarene to complex 10 leads to ruthenacycle 11, which is excited by blue‐light‐absorption to form singlet excited species 11*. Relaxation through intersystem crossing (ISC) affords a long‐lived triplet complex 11**. Subsequently, an inner‐sphere electron transfer (ISET) to iodoarene generates a phenyl radical (13) and ruthenium(III) intermediate 12. They readily recombine to form stable ruthenium(IV) intermediate 14. Reductive elimination and ligand exchange deliver the arylated product 3 and ruthenium complex 15, which finally undergoes C−H ruthenation to regenerate the photocatalytically active ruthenium(II) complex 10.
Scheme 6.

Proposed catalytic cycle.
Finally, the power of the photo‐induced ruthenium‐catalyzed arylation at ambient temperature was highlighted by the late‐stage diversification of biorelevant purines 1 o–1 p, sensitive nucleoside 1 q and nucleotide 1 r (Scheme 7).
Scheme 7.

Late‐stage diversification: photo‐induced ruthenium‐catalyzed C−H arylation with biorelevant purines at room temperature.
Conclusion
In summary, we have reported on ambient temperature ruthenium‐catalyzed C−H arylations enabled by visible light. The versatile photoredox ruthenium catalysis proved viable with a broad range of functional groups and applicable to synthetically useful pyrazoles, triazoles and sensitive nucleosides and nucleotides. Mechanistic studies by experiment and computation were suggestive of an inner‐sphere electron transfer from photocatalytically active biscyclometalated ruthenacycles for the photoredox catalysis.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Generous support by the DAAD (fellowship to K.K.) and the DFG (Gottfried‐Wilhelm‐Leibniz prize) is gratefully acknowledged. We thank Dr. Christopher Golz (University Göttingen) for the X‐ray diffraction analysis. Open access funding enabled and organized by Projekt DEAL.
K. Korvorapun, J. Struwe, R. Kuniyil, A. Zangarelli, A. Casnati, M. Waeterschoot, L. Ackermann, Angew. Chem. Int. Ed. 2020, 59, 18103.
In memory of Rolf Huisgen
Contributor Information
Korkit Korvorapun, http://www.ackermann.chemie.uni‐goettingen.de/.
Prof. Dr. Lutz Ackermann, Email: Lutz.Ackermann@chemie.uni-goettingen.de.
References
- 1.
- 1a. Rej S., Ano Y., Chatani N., Chem. Rev. 2020, 120, 1788–1887; [DOI] [PubMed] [Google Scholar]
- 1b. Gandeepan P., Müller T., Zell D., Cera G., Warratz S., Ackermann L., Chem. Rev. 2019, 119, 2192–2452; [DOI] [PubMed] [Google Scholar]
- 1c. Hu Y., Zhou B., Wang C., Acc. Chem. Res. 2018, 51, 816–827; [DOI] [PubMed] [Google Scholar]
- 1d. Sambiagio C., Schönbauer D., Blieck R., Dao-Huy T., Pototschnig G., Schaaf P., Wiesinger T., Zia M. F., Wencel-Delord J., Besset T., Maes B. U. W., Schnürch M., Chem. Soc. Rev. 2018, 47, 6603–6743; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1e. Leitch J. A., Frost C. G., Synthesis 2018, 50, 2693–2706; [Google Scholar]
- 1f. Park Y., Kim Y., Chang S., Chem. Rev. 2017, 117, 9247–9301; [DOI] [PubMed] [Google Scholar]
- 1g. Huang Z., Lim H. N., Mo F., Young M. C., Dong G., Chem. Soc. Rev. 2015, 44, 7764–7786; [DOI] [PubMed] [Google Scholar]
- 1h. Gao K., Yoshikai N., Acc. Chem. Res. 2014, 47, 1208–1219; [DOI] [PubMed] [Google Scholar]
- 1i. Wencel-Delord J., Glorius F., Nat. Chem. 2013, 5, 369–375; [DOI] [PubMed] [Google Scholar]
- 1j. Engle K. M., Mei T.-S., Wasa M., Yu J.-Q., Acc. Chem. Res. 2012, 45, 788–802; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1k. Arockiam P. B., Bruneau C., Dixneuf P. H., Chem. Rev. 2012, 112, 5879–5918; [DOI] [PubMed] [Google Scholar]
- 1l. Ackermann L., Vicente R., Kapdi A. R., Angew. Chem. Int. Ed. 2009, 48, 9792–9826; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9976–10011. [Google Scholar]
- 2.
- 2a. Korvorapun K., Kuniyil R., Ackermann L., ACS Catal. 2020, 10, 435–440; [Google Scholar]
- 2b. Li J., Korvorapun K., De Sarkar S., Rogge T., Burns D. J., Warratz S., Ackermann L., Nat. Commun. 2017, 8, 15430; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Teskey C. J., Lui A. Y. W., Greaney M. F., Angew. Chem. Int. Ed. 2015, 54, 11677–11680; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11843–11846; [Google Scholar]
- 2d. Paterson A. J., St John-Campbell S., Mahon M. F., Press N. J., Frost C. G., Chem. Commun. 2015, 51, 12807–12810; [DOI] [PubMed] [Google Scholar]
- 2e. Wang L., Ackermann L., Chem. Commun. 2014, 50, 1083–1085; [DOI] [PubMed] [Google Scholar]
- 2f. Rouquet G., Chatani N., Chem. Sci. 2013, 4, 2201–2208; [Google Scholar]
- 2g. Schinkel M., Marek I., Ackermann L., Angew. Chem. Int. Ed. 2013, 52, 3977–3980; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4069–4072; [Google Scholar]
- 2h. Ackermann L., Novák P., Vicente R., Hofmann N., Angew. Chem. Int. Ed. 2009, 48, 6045–6048; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6161–6164; [Google Scholar]
- 2i. Murai S., Kakiuchi F., Sekine S., Tanaka Y., Kamatani A., Sonoda M., Chatani N., Nature 1993, 366, 529–531; [Google Scholar]
- 2j. Lewis L. N., Smith J. F., J. Am. Chem. Soc. 1986, 108, 2728–2735. [Google Scholar]
- 3.
- 3a. Rogge T., Ackermann L., Angew. Chem. Int. Ed. 2019, 58, 15640–15645; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 15787–15792; [Google Scholar]
- 3b. Yetra S. R., Rogge T., Warratz S., Struwe J., Peng W., Vana P., Ackermann L., Angew. Chem. Int. Ed. 2019, 58, 7490–7494; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 7569–7573; [Google Scholar]
- 3c. Simonetti M., Cannas D. M., Just-Baringo X., Vitorica-Yrezabal I. J., Larrosa I., Nat. Chem. 2018, 10, 724–731; [DOI] [PubMed] [Google Scholar]
- 3d. Moselage M., Li J., Kramm F., Ackermann L., Angew. Chem. Int. Ed. 2017, 56, 5341–5344; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5425–5428; [Google Scholar]
- 3e. Nareddy P., Jordan F., Szostak M., ACS Catal. 2017, 7, 5721–5745; [Google Scholar]
- 3f. Simonetti M., Cannas D. M., Panigrahi A., Kujawa S., Kryjewski M., Xie P., Larrosa I., Chem. Eur. J. 2017, 23, 549–553; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3g. Mei R., Zhu C., Ackermann L., Chem. Commun. 2016, 52, 13171–13174; [DOI] [PubMed] [Google Scholar]
- 3h. Simonetti M., Perry G. J. P., Cambeiro X. C., Juliá-Hernández F., Arokianathar J. N., Larrosa I., J. Am. Chem. Soc. 2016, 138, 3596–3606; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3i. Kumar N. Y. P., Jeyachandran R., Ackermann L., J. Org. Chem. 2013, 78, 4145–4152. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Ackermann L., Org. Process Res. Dev. 2015, 19, 260–269; [Google Scholar]
- 4b. Horton D. A., Bourne G. T., Smythe M. L., Chem. Rev. 2003, 103, 893–930; [DOI] [PubMed] [Google Scholar]
- 4c. Hassan J., Sévignon M., Gozzi C., Schulz E., Lemaire M., Chem. Rev. 2002, 102, 1359–1470. [DOI] [PubMed] [Google Scholar]
- 5. Ouellet S. G., Roy A., Molinaro C., Angelaud R., Marcoux J.-F., O'Shea P. D., Davies I. W., J. Org. Chem. 2011, 76, 1436–1439. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Hubrich J., Ackermann L., Eur. J. Org. Chem. 2016, 3700–3704; [Google Scholar]
- 6b. Diers E., Phani Kumar N. Y., Mejuch T., Marek I., Ackermann L., Tetrahedron 2013, 69, 4445–4453. [Google Scholar]
- 7. Seki M., ACS Catal. 2014, 4, 4047–4050. [Google Scholar]
- 8. Schischko A., Ren H., Kaplaneris N., Ackermann L., Angew. Chem. Int. Ed. 2017, 56, 1576–1580; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1598–1602. [Google Scholar]
- 9. Gayakhe V., Sanghvi Y. S., Fairlamb I. J. S., Kapdi A. R., Chem. Commun. 2015, 51, 11944–11960. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Spiewak A. M., Weix D. J., J. Org. Chem. 2019, 84, 15642–15647; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Li J.-W., Wang L.-N., Li M., Tang P.-T., Luo X.-P., Kurmoo M., Liu Y.-J., Zeng M.-H., Org. Lett. 2019, 21, 2885–2889; [DOI] [PubMed] [Google Scholar]
- 10c. Boyaala R., Touzani R., Roisnel T., Dorcet V., Caytan E., Jacquemin D., Boixel J., Guerchais V., Doucet H., Soulé J.-F., ACS Catal. 2019, 9, 1320–1328; [Google Scholar]
- 10d. Drev M., Grošelj U., Ledinek B., Perdih F., Svete J., Štefane B., Požgan F., Org. Lett. 2018, 20, 5268–5273; [DOI] [PubMed] [Google Scholar]
- 10e. Hu F., Szostak M., Org. Lett. 2016, 18, 4186–4189; [DOI] [PubMed] [Google Scholar]
- 10f. Hubrich J., Himmler T., Rodefeld L., Ackermann L., ACS Catal. 2015, 5, 4089–4093; [Google Scholar]
- 10g. Li B., Darcel C., Dixneuf P. H., ChemCatChem 2014, 6, 127–130; [Google Scholar]
- 10h. Ackermann L., Diers E., Manvar A., Org. Lett. 2012, 14, 1154–1157; [DOI] [PubMed] [Google Scholar]
- 10i. Li W., Arockiam P. B., Fischmeister C., Bruneau C., Dixneuf P. H., Green Chem. 2011, 13, 2315–2319; [Google Scholar]
- 10j. Ackermann L., Lygin A. V., Org. Lett. 2011, 13, 3332–3335; [DOI] [PubMed] [Google Scholar]
- 10k. Ackermann L., Vicente R., Potukuchi H. K., Pirovano V., Org. Lett. 2010, 12, 5032–5035; [DOI] [PubMed] [Google Scholar]
- 10l. Ackermann L., Vicente R., Althammer A., Org. Lett. 2008, 10, 2299–2302. [DOI] [PubMed] [Google Scholar]
- 11.For representative reviews, see:
- 11a. Wang C.-S., Dixneuf P. H., Soulé J.-F., Chem. Rev. 2018, 118, 7532–7585; [DOI] [PubMed] [Google Scholar]
- 11b. Twilton J., Le C., Zhang P., Shaw M. H., Evans R. W., MacMillan D. W. C., Nat. Rev. Chem. 2017, 1, 0052; [Google Scholar]
- 11c. Fabry D. C., Rueping M., Acc. Chem. Res. 2016, 49, 1969–1979; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11d. Narayanam J. M. R., Stephenson C. R. J., Chem. Soc. Rev. 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Zhang X., MacMillan D. W. C., J. Am. Chem. Soc. 2017, 139, 11353–11356; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Shaw M. H., Shurtleff V. W., Terrett J. A., Cuthbertson J. D., MacMillan D. W. C., Science 2016, 352, 1304–1308; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12c. Heitz D. R., Tellis J. C., Molander G. A., J. Am. Chem. Soc. 2016, 138, 12715–12718; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12d. Cuthbertson J. D., MacMillan D. W. C., Nature 2015, 519, 74–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Natarajan P., Kumar N., Sharma M., Org. Chem. Front. 2016, 3, 1265–1270; [Google Scholar]
- 13b. Kalyani D., McMurtrey K. B., Neufeldt S. R., Sanford M. S., J. Am. Chem. Soc. 2011, 133, 18566–18569; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c. Osawa M., Nagai H., Akita M., Dalton Trans. 2007, 827–829. [DOI] [PubMed] [Google Scholar]
- 14. Gandeepan P., Koeller J., Korvorapun K., Mohr J., Ackermann L., Angew. Chem. Int. Ed. 2019, 58, 9820–9825; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9925–9930. [Google Scholar]
- 15. Sagadevan A., Greaney M. F., Angew. Chem. Int. Ed. 2019, 58, 9826–9830; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9931–9935. [Google Scholar]
- 16. Sagadevan A., Charitou A., Wang F., Ivanova M., Vuagnat M., Greaney M. F., Chem. Sci. 2020, 11, 4439–4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Ackermann L., Acc. Chem. Res. 2020, 53, 84–104; [DOI] [PubMed] [Google Scholar]
- 17b. Ackermann L., Acc. Chem. Res. 2014, 47, 281–295. [DOI] [PubMed] [Google Scholar]
- 18.For detailed information, see the Supporting Information.
- 19. Ackermann L., Chem. Rev. 2011, 111, 1315–1345. [DOI] [PubMed] [Google Scholar]
- 20.Deposition numbers 1968604 (3aw), 1968602 (3az), 1968605 (5ac′), and 1968603 (3ja) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 21.
- 21a. Ackermann L., Potukuchi H. K., Org. Biomol. Chem. 2010, 8, 4503–4513; [DOI] [PubMed] [Google Scholar]
- 21b. Meldal M., Tornøe C. W., Chem. Rev. 2008, 108, 2952–3015; [DOI] [PubMed] [Google Scholar]
- 21c. Kolb H. C., Finn M. G., Sharpless K. B., Angew. Chem. Int. Ed. 2001, 40, 2004–2021; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 2056–2075; [Google Scholar]
- 21d. Huisgen R., Angew. Chem. Int. Ed. Engl. 1963, 2, 565–598; [Google Scholar]; Angew. Chem. 1963, 75, 604–637. [Google Scholar]
- 22.
- 22a. Simonetti M., Kuniyil R., Macgregor S. A., Larrosa I., J. Am. Chem. Soc. 2018, 140, 11836–11847; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22b. Ackermann L., Althammer A., Born R., Tetrahedron 2008, 64, 6115–6124; [Google Scholar]
- 22c. Ackermann L., Althammer A., Born R., Synlett 2007, 2833–2836; see also: [Google Scholar]
- 22d. Fernandez S., Pfeffer M., Ritleng V., Sirlin C., Organometallics 1999, 18, 2390–2394. [Google Scholar]
- 23. Ryabov A. D., Estevez H., Alexandrova L., Pfeffer M., Lagadec R. L., Inorg. Chim. Acta 2006, 359, 883–887. [Google Scholar]
- 24.
- 24a. Marcus R. A., Angew. Chem. Int. Ed. Engl. 1993, 32, 1111–1121; [Google Scholar]; Angew. Chem. 1993, 105, 1161–1172; [Google Scholar]
- 24b. Marcus R. A., J. Chem. Phys. 1956, 24, 966–978. [Google Scholar]
- 25.
- 25a. Saveant J. M., Acc. Chem. Res. 1993, 26, 455–461; [Google Scholar]
- 25b. Saveant J. M., J. Am. Chem. Soc. 1987, 109, 6788–6795. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary