

Abstract
κ‐Opioid receptors (KORs) play a predominant role in pain alleviation, itching skin diseases, depression and neurodegenerative disorders such as multiple sclerosis. Therefore, imaging of KOR by a fluorinated PET tracer was envisaged. Two strategies were followed to introduce a F atom into the very potent class of cis,trans‐configured perhydroquinoxalines. Whereas the synthesis of fluoroethyltriazole 2 has already been reported, fluoropyrrolidines 14 (1‐[2‐(3,4‐dichlorophenyl)acetyl]‐8‐[(R)‐3‐fluoropyrrolidin‐1‐yl]‐perhydroquinoxalines) were prepared by SN2 substitution of a cyclic sulfuric acid derivative with hydroxypyrrolidine and subsequent transformation of the OH moiety into a F substituent. Fluoropyrrolidines 14 showed similar low‐nanomolar KOR affinity and selectivity to the corresponding pyrrolidines, but the corresponding alcohols were slightly less active. In the cAMP and β‐arrestin assay, 14b (proton at the 4‐position) exhibited similar KOR agonistic activity as U‐50,488. The fluoro derivatives 14b and 14c (CO2CH3 at the 4‐position) revealed KOR‐mediated anti‐inflammatory activity as CD11c and the IFN‐γ production were reduced significantly in mouse and human dendritic cells. Compounds 14b and 14‐c also displayed anti‐inflammatory and immunomodulatory activity in mouse and human T cells. The PET tracer [18F]‐2 was prepared by 1,3‐dipolar cycloaddition. In vivo, [18F]‐2 did not label KOR due to very fast elimination kinetics. Nucleophilic substitution of a mesylate precursor provided [18F]‐14c. Unfortunately, defluorination of [18F]‐14c occurred in vivo, which was analyzed in detail by in vitro studies.
Keywords: anti-inflammatory activity, effector cells, fluorine, opioid receptor agonists, perhydroquinoxaline, PET tracers
κ‐Opioid receptors (KORs) play a prominent role in pain alleviation, skin diseases, depression and neurodegenerative disorders. Two strategies were pursued to prepare fluorinated tracers for positron emission tomography based on the perhydroquinoxaline class of KOR agonists. The agonists showed high KOR affinity, selectivity, agonistic activity and KOR‐mediated anti‐inflammatory and immunomodulatory activity. One tracer was quickly eliminated, another was defluorinated.
1. Introduction
The analgesic activity of opium had already been reported by Theophrastus in the third century BC, but it took more than 2000 years until the alkaloids of opium responsible for the pharmacological effects as well as the opioid system consisting of opioid receptors and their endogenous ligands were identified. Today, three opioid receptors that alleviate pain upon activation are known: the μ‐opioid receptor (MOR), the δ‐opioid receptor (DOR) and the κ‐opioid receptor (KOR). According to sequence homology, the nociceptive opioid receptor (NOR) also belongs to the class of opioid receptors, but it behaves differently as it induces pain upon activation.1, 2, 3
The clinically used strong analgesics activate predominantly MOR, which is associated with the development of physical and psychical dependence, obstipation and dangerous respiratory depression. In this project, we focus on KOR, as activation of KOR also leads to strong analgesia, but to a modified side‐effect profile without respiratory depression and reduced addiction potential. However, sedation, strong diuresis and dysphoria have been observed after application of KOR agonists.4, 5
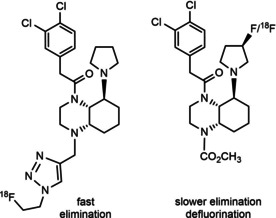
Whereas centrally and peripherally acting KOR agonists can be used for the treatment of severe pain, visceral pain and itching skin diseases, KOR antagonists have antidepressant, anxiolytic, antiaddictive, anticonvulsant and anti‐seizure potential.5, 6, 7, 8, 9, 10 Recent studies showed the central role of KOR in the pathogenesis of multiple sclerosis (MS), an inflammatory neurodegenerative disease.11, 12, 13 A decreased level of KOR mRNA was found in Theiler's encephalomyelitis mouse model mimicking parts of the MS pathology.11 In the myelin oligodendrocyte glycoprotein (MOG) induced experimental autoimmune encephalomyelitis (EAE) mouse model of MS activation of KOR was able to prevent neuronal damage and promote remyelination.12 Very recently, we showed that KOR agonists 1b (R=H) and 2 exhibit potent anti‐inflammatory and immunomodulatory activity in primary mouse and human immune cells. Moreover, both KOR agonists led to almost complete protection of mice from developing MS‐like symptoms in the MOG induced EAE13 (Figure 1).
Figure 1.
Potent KOR agonists 1b (R=H) and 2 ameliorating MS‐like symptoms in MOG induced EAE. The effect of higher polarity of compounds 3 (R2=OH) with an additional OH moiety at the pyrrolidine ring will be studied. Compounds 2 and 3 (R2=F) will be developed into fluorinated PET tracers for imaging of KOR.
Due to their prominent role in pain alleviation, itching skin diseases, depression, anxiety, epilepsy and neurodegenerative disorders such as MS, the localization, expression level, activity and density of KOR is of high interest. Positron emission tomography (PET) represents a non‐invasive modality, which allows imaging of biological targets in vivo upon using appropriate radioligands. In 2005, the first PET tracer addressing KOR, the carbon‐11‐labeled KOR agonist [11C]‐4 ([11C]GR103545) was reported (Figure 2). Although 4 showed high KOR affinity, activity and selectivity and [11C]‐4 led to promising results in various animal models (mice, primates), human studies were not satisfying.14, 15, 16, 17, 18, 19, 20, 21 The 11C‐labeled KOR antagonist [11C]‐5 ([11C]LY2795050) was used successfully in several human studies.22, 23, 24, 25, 26 Due to the short half‐life of carbon‐11 (t 1/2=20 min), fluorine‐18 (t 1/2=110 min) labeled PET tracers were developed as well. In 2017 the synthesis of KOR antagonist [18F]‐6 ([18F]LY2459989) via a iodonium ylide was reported. In first in‐man studies (4 healthy male subjects), [18F]‐6 displayed favorable kinetics and binding characeteristics.27, 28, 29
Figure 2.
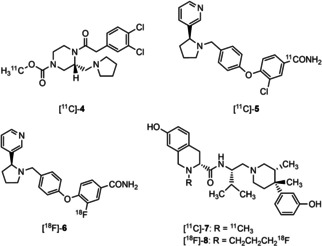
PET tracers for imaging of KORs. Whereas 4 represents a KOR agonist, compounds 5–8 are KOR antagonists. [11C]‐4, [11C]‐5 and [11C]‐7 are labeled with short‐lived carbon‐11 and [18F]‐6 and [18F]‐8, are labeled with fluorine‐18 with a half‐life of 110 min.
In addition to the diphenyl ether based KOR antagonists 5 and 6, JDTic derived PET tracers [11C]‐7 and [18F]‐8 were synthesized and biologically evaluated. In mice, carbon‐11‐labeled KOR antagonist [11C]‐7 was able to cross the blood‐brain‐barrier and accumulate in regions with high KOR density. Binding of [11C]‐7 was reduced by potent KOR ligands, such as U‐50,488 and naltrindole.30 Although the distribution of fluoropropyl JDTic [18F]‐8 correlates nicely with known KOR localization, a significant specific binding of [18F]‐8 at KOR was not observed. Moreover, [18F]‐8 was rapidly metabolized31, 32 (Figure 2).
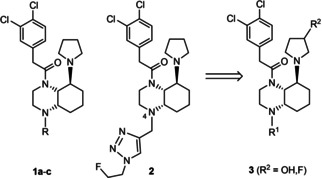
In a recent project,13 we demonstrated the promising activity of 1b and the corresponding fluoroethyltriazole derivative 2, even in vivo in EAE mouse model. Therefore, the biological properties of the compound class of cis,trans‐configured perhydroquinoxalines should be further exploited. Since it was postulated that immune cells were activated predominantly in the periphery, KOR agonists with higher polarity, e. g. bearing an additional hydroxy moiety in the pyrrolidine ring (3, R2=OH) should be synthesized and pharmacologically evaluated. Additionally, fluorinated PET tracers will be developed. It was planned to introduce fluorine‐18 either in the side chain at 4‐position as in compound 2 or at the pyrrolidine ring as shown in compound 3 (R2=F; Figure 1).
2. Synthesis
For the synthesis of hydroxy and fluoro substituted KOR agonists of type 3, the synthetic route previously reported by us to obtain cis,trans‐configured perhydroquinoxalines was pursued.13, 33, 34 At first, the cyclic sulfuric acid ester amide 10 was prepared in seven steps starting from tetrahydroquinoxaline 9.13 Nucleophilic ring opening of 10 by (S)‐3‐hydroxypyrrolidine led to secondary amine 11, which was acylated with 2‐(3,4‐dichlorophenyl)acetyl chloride to afford the amide 12a. Removal of the Boc‐protective group with trifluoroacetic acid (TFA) provided the secondary amine 12b (Scheme 1).
Scheme 1.
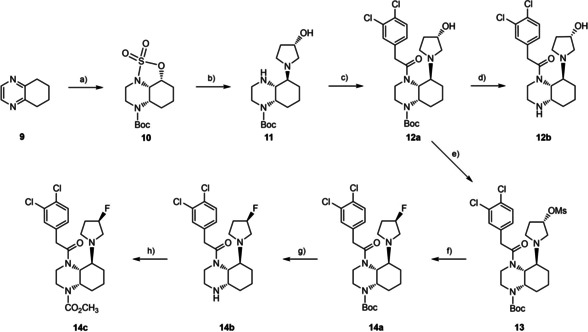
Synthesis of hydroxylated and fluorinated pyrrolidine derivatives 12a,b and 14a–c. a) 7 steps;13 b) (S)‐3‐hydroxpyrrolidine, K2CO3, DMF, 80 °C, 16 h, 81 %; c) 2‐(3,4‐dichlorophenyl)acetyl chloride, iPr2NEt, CH2Cl2, 0 °C→RT, 16 h, 85 %; d) F3CCO2H, CH2Cl2, RT, 16 h, 87 %; e) H3CSO2Cl (MsCl), NEt3, DMAP, CH2Cl2, 0 °C→RT, 17 h, 81 %; f) Bu4NF, H2O, 45 °C, 16 h, 50 %; g) F3CCO2H, CH2Cl2, RT, 17 h, 77 %; h) ClCO2CH3, pyridine, CH2Cl2, 0 °C→RT, 1 h, 18 %; Only one enantiomer of the racemic mixture 10 and only one diastereomer of diastereomeric mixtures 11 ‐ 14 are shown. The configuration of C‐3 of the pyrrolidine ring is defined: S configuration of hydroxy and mesyloxy derivatives 11–13 and R configuration of fluoro derivatives 14a–c.
In order to obtain fluorinated derivatives, the alcohol 12a was transformed into the mesylate 13. Nucleophilic substitution of mesylate 13 with fluoride (Bu4NF) provided the fluoropyrrolidine derivative 14a. Cleavage of the Boc‐protective group with TFA led to secondary amine 14b, which was acylated with methyl chloroformate to obtain the carbamate 14c (Scheme 1).
For the synthesis of hydroxypyrrolidine derivative 12c with a methoxycarbonyl moiety the OH moiety of 12a was protected as TBDPS ether 15. Removal of the Boc‐protective group with TFA and subsequent acylation of 16 with methyl chloroformate afforded the methoxycarbonyl derivative 17, which was reacted with Bu4NF to remove the silyl protective group. The desired alcohol 12c, which should also serve as precursor in the radiosynthesis, was obtained in 33 % yield starting from 12a (Scheme 2).
Scheme 2.
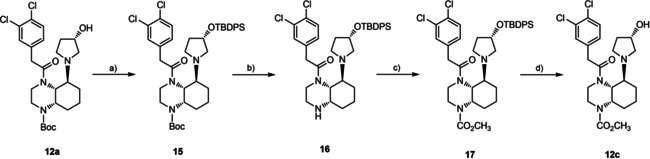
Synthesis hydroxypyrrolidine derivative 12c. a) TBDPSCl (tert‐butyl‐diphenyl‐silyl chloride), imidazole, DMAP, CH2Cl2, RT, 16 h, 74 %; b) F3CCO2H, CH2Cl2, RT, 16 h, 89 %; c) ClCO2CH3, pyridine, CH2Cl2, 0 °C→RT, 1 h, 78 %; d) Bu4NF, THF, RT, overnight, 64 %. Only one diastereomer of diastereomeric mixtures 12 and 15–17 is shown. The S configuration of C‐3 of the pyrrolidine ring is defined by the reagent (S)‐3‐hydroxypyrrolidine.
3. Receptor Affinity, Selectivity and Activity
3.1. KOR affinity
The KOR affinity of the hydroxylated and fluorinated perhydroquinoxalines 12 and 14 was studied in competitive receptor binding assays. In brief, the test compounds compete with the potent and KOR‐selective radioligand [3H]U‐69,593 for a limited number of KORs in a membrane preparation from guinea pig brains. The radioactivity bound by the receptor correlates with the KOR affinity of the test compounds.35, 36, 37, 38 The KOR affinity of the hydroxylated and fluorinated ligands 12 and 14 as well as the unsubstituted ligands 1 and some reference compounds is summarized in Table 1.
Table 1.
Affinities of hydroxylated and fluorinated perhydroquinoxalines and reference compounds towards KOR and related receptors.
|
| |||||||
|---|---|---|---|---|---|---|---|
|
Compd. |
R1 |
R2 |
Ki±SEM [nM]/% inhibition at 1 μM[a,b] |
||||
|
KOR |
MOR |
DOR |
σ1 |
σ2 |
|||
|
[3H]U‐69,593 |
[3H]DAMGO |
[3H]DPDPE |
(+)‐[3H]pentazocine |
[3H]DTG |
|||
|
1a13 |
CO2C(CH3)3 |
H |
3.8±1.2 |
260 |
438 |
12 % |
16 % |
|
1b13 |
H |
H |
0.54±0.15 |
28 % |
205 |
0 % |
5 % |
|
1c13 |
CO2CH3 |
H |
1.3±0.4 |
0 % |
250 |
0 % |
8 % |
|
12 a |
CO2C(CH3)3 |
(S)−OH |
16±2 |
9 % |
5 % |
0 % |
0 % |
|
12 b |
H |
(S)−OH |
2.0±0.5 |
29 % |
246 |
2 % |
0 % |
|
12 c |
CO2CH3 |
(S)−OH |
9.2±6.0 |
5 % |
693 |
0 % |
5 % |
|
14 b |
H |
(R)−F |
1.5±0.4 |
2 % |
251 |
0 % |
0 % |
|
14 c |
CO2CH3 |
(R)−F |
0.82±0.69 |
0 % |
12 % |
8 % |
962 |
|
U‐50,488 |
|
|
0.34±0.07 |
– |
– |
– |
– |
|
Naloxone |
|
|
7.3±0.40 |
2.3±1.1 |
103 |
– |
– |
|
Morphine |
|
|
– |
5.2±1.6 |
– |
– |
– |
|
SNC80 |
|
|
– |
– |
1.2±0.5 |
– |
– |
|
(+)‐pentazocine |
|
|
– |
– |
– |
5.4±0.5 |
– |
|
Haloperidol |
|
|
– |
– |
– |
6.6±0.9 |
78±2.3 |
[a] A value in % reflects the inhibition of the radioligand binding at a test compound concentration of 1 μM. K i values without SEM values represent the mean of two experiments (n=2) and K i values with SEM values represent the mean of three experiments (n=3). [b] Guinea pig brain membrane preparations were used in the KOR, MOR and σ1 assay. In the DOR assay rat brain and in the σ 2 assay rat liver membrane preparations were used.
The KOR affinity of the hydroxylated and fluorinated pyrrolidines 12 and 14 is in the low‐nanomolar range. However, a Boc group at the quinoxaline ring system (12 a: K i=16 nM) seems to be less tolerated by KOR than a methoxycarbonyl moiety or a proton. Although the K i values are below 10 nM, the alcohols 12b and 12c are less active than the fluoro derivatives 14b and 14c and the unsubstituted analogs 1b and 1c. A F atom at the pyrrolidine ring is well tolerated as indicated by the comparable KOR affinities of H/F pairs (e. g., K i ( 14c)=0.82 nM, K i ( 1c)=1.3 nM). Altogether, KOR agonists 1 without a substituent at the pyrrolidine ring show the same KOR affinity as the corresponding F derivatives 14, but the OH derivatives 12 are slightly less active. With exception of 14 b/c, the secondary amines (1b,12b) show higher KOR affinity than the methoxycarbonyl derivatives 1c and 12c.
3.2. Selectivity over MOR, DOR and σ receptors
The affinity towards related opioid receptors MOR and DOR was analyzed in receptor binding studies. In order to broaden the selectivity profile, the affinity to σ receptors was also investigated. In particular, in the class of ethylenediamine KOR agonists variation of the stereochemistry or reduction of the phenylacetamide to a phenylethylamine structural element can change the receptor profile towards σ affinity.39, 40 Membrane preparations from guinea pig brain were used in the MOR and σ1 assay, whereas preparations from rat brain were employed in the DOR assay. For investigation of σ2 affinity rat liver homogenates were used. The following radioligands were employed for selective labeling of the respective receptor: [3H]DAMGO (MOR), [3H]DPDPE (DOR), (+)‐[3H]pentazocine (σ1 receptor) and [3H]di‐o‐tolylguanidine+(+)‐pentazocine (σ2 receptor).35, 36, 37, 38, 41, 42, 43
In Table 1 the interaction of the hydroxy and fluoro derivatives 12 and 14 with related receptors is summarized. The complete competition curves were only recorded, when the specific binding of the radioligand at a test compound concentration of 1 μM was reduced by more than 50 %. In case the specific binding of the radioligand was not reduced by more than 50 %, the inhibition of radioligand binding [%] at a test compound concentration of 1 μM is given in Table 1.
The hydroxy‐ and fluoropyrrolidines 12 and 14 did not show remarkable affinity towards MOR, σ1 and σ2 receptors indicating high selectivity for KOR over MOR, σ1 and σ2 receptors. A slight DOR affinity was recorded for the secondary amines 12b (K i=246 nM) and 14b (K i=251 nM). However, both compounds exhibit a KOR:DOR selectivity of more than 100‐fold. The fluoro derivative 14c, a non‐radioactive counterpart of a potential fluorine‐18‐labeled PET tracer, displays a more than 1000‐fold selectivity over the related MOR, DOR and σ receptors.
3.3. Functional activity
In order to further explore the pharmacological potential of quinoxaline‐based KOR agonists 12 and 14, the agonistic activity at KOR was investigated. The inhibition of cAMP production by the agonists was investigated in a cAMP assay (human HEK293T cells).44 A Tango assay (human HTLA cells)45 was used to study β‐arrestin‐2 recruitment. Both assays are complementary, since they address two different pathways activated by KOR agonists. A potential bias for one of these pathways could be detected. The results are summarized in Table 2.
Table 2.
Activity of selected KOR agonists correlated with KOR affinity.
|
| |||||||
|---|---|---|---|---|---|---|---|
|
compd. |
R1 |
R2 |
KOR[a] |
cAMP[b] |
β‐Arrestin‐2[c] |
||
|
|
|
|
[3H]U‐69,593 |
|
|
|
|
|
|
|
|
Ki±SEM [nM] |
EC50 [nM] |
E max [%][d] |
EC50 [nM] |
E max [%][d] |
|
213 |
CH2‐triazole‐CH2CH2F |
H |
5.6±0.6 |
2.8 |
103 |
43 |
86 |
|
12 b |
H |
(S)−OH |
2.0±0.5 |
0.20 |
102 |
16 |
97 |
|
12 c |
CO2CH3 |
(S)−OH |
9.2±6.0 |
0.19 |
100 |
8.8 |
82 |
|
14 b |
H |
(R)−F |
1.5±0.4 |
0.033 |
100 |
6.9 |
91 |
|
U‐50,488 |
|
|
0.97±0.40 |
0.013 |
100 |
8.0 |
100 |
[a] Guinea pig brain membrane preparations. [b] Human HEK 293T cells. [c] Human HTLA cells. [d] The E max values refer to U‐50,488 (100 % intrinsic activity).
In the cAMP assay, the perhydroquinoxalines 12b and 12c with OH moiety were approximately ten times less active than U‐50,488, whereas the fluoro derivative 14b shows almost the same EC50 value as U‐50,488. All three test compounds represent full agonists with intrinsic activity of 100 % (referred to U‐50,488).
In the β‐arrestin‐2 recruitment assay, the test compounds 12b, 12c and 14b revealed almost the same EC50 values as U‐50,488. However, 12c and 14b reached only 82 and 91 % of the full agonistic activity of U‐50,488.
Referring the activity of the test compounds 12b, 12c and 14b to the activity of the full agonist U‐50,488 in both assays does not indicate any preference for one of the two pathways, that is, no bias for the G protein or β‐arrestin recruitment pathway could be detected.
4. Anti‐inflammatory Activity of the Quinoxaline‐Based KOR Agonists in Vitro
Anti‐inflammatory effects of KOR agonists are mainly based on the inhibition of immune cell activation or the downregulation of cell proliferation. Accordingly, it is well known that KOR agonists prevented the maturation of antigen‐presenting cells resulting in an impaired effector T cell priming.46 We were able to demonstrate that the newly developed KOR agonist 2 significantly decreased the expression of classical activation markers and the secretion of the pro‐inflammatory cytokines interferon‐gamma (IFN‐γ), tumor‐necrosis‐factor‐alpha (TNF‐α), and interleukin 17 A (IL‐17A) in mouse as well as human immune cell subsets. In addition, fluoroethyltriazole 2 had strong immunomodulatory capacities as besides down‐regulating pro‐inflammatory cytokines it was able to increase the production of IL‐10, suggesting that 2 contributed to the switch of immune cells from an effector towards a suppressor phenotype. In line with this, 2 significantly decreased diseases severity and delayed disease onset in a mouse model of experimental autoimmune encephalomyelitis, which was associated with downregulated numbers of effector T cells and upregulated levels of regulatory T cells in 2 treated mice compared to controls.13
Having confirmed the anti‐inflammatory and immunomodulatory properties of cis,trans‐perhydroquinoxaline‐based KOR agonists in general, we now intended to characterize more polar substances belonging to this class of compounds. For this purpose, we treated primary mouse or human T cells and dendritic cells (DCs) with 12b, 12c, 14b and 14c after the cells had been activated with phorbol 12‐myristate 13‐acetate (PMA) in combination with ionomycin (PMA/Iono) to assess the anti‐inflammatory properties of the compounds. The onset and perpetuation of many systemic inflammatory diseases usually involve the activation of antigen‐presenting cells like DCs, which get into contact with exogenous or endogenous triggers and upon maturation, migrate to secondary lymphoid organs, where they induce the priming of pathogenic effector cells, mainly T cells.47, 48 DC activation and maturation is characterized by the upregulation of CD11c, the induction of costimulatory receptors of the TNF‐ and B7 families including CD80, CD86 or CD40 and the secretion of pro‐inflammatory cytokines such as IFN‐γ, IL‐12, IL‐23 or IL‐6.49, 50
Accordingly, we observed markedly increased levels of CD11c and IFN‐γ in PMA/ionomycin‐stimulated mouse DCs compared to non‐activated controls (Figure 3A). However, when the activated DCs were treated with 12c, 14b or 14c, the expression of the maturation marker CD11c and the IFN‐γ production were significantly reduced, indicating an anti‐inflammatory effect of these three compounds, whereas 12b did non modulate the CD11c or IFN‐γ expression. Worth mentioning that 14b seemed to be more potent than 12c or 14c (Figure 3A and B) and that the effect of all three compounds was concentration‐dependent because a more pronounced reduction in DC activation and maturation was observed when the cells were treated with 5 μg/mL instead of 1 μg/mL of the KOR agonists. Interestingly, the anti‐inflammatory capacity of all three compounds was clearly mediated by binding to KOR since the effect was completely abrogated when DCs from KOR‐deficient mice were stimulated with 12c, 14b or 14c (Figure 3B). Next, we assessed whether the KOR agonists were also able to modulate the activation and maturation of human DCs and therefore, purified HLA‐DR+ cells from peripheral blood of healthy human donors. As shown in Figure 3C, 12c, 14b and 14c reduced the expression of CD11c and the production of IFN‐γ in human DCs as well, whereas compound 12b seemed to have only a minor but not significant anti‐inflammatory effect on human antigen‐presenting cells (Figure 3C).
Figure 3.
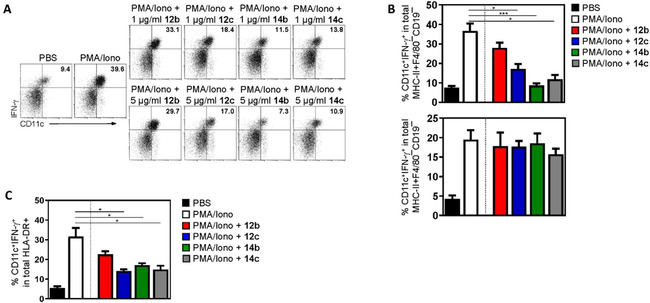
Compounds 12c, 14b and 14c prevented DC maturation and activation. A), B) Mouse DCs were generated from bone marrow precursors in the presence of GM‐CSF and IL‐4, activated with PMA and ionomycin (PMA/Iono) for 12 h, and stimulated with 12b, 12c, 14b or 14c in a concentration of 1 or 5 μg/mL for an additional 48 h. Control cells received an equal amount of PBS. Representative dot‐plots (A) and percentages of cells expressing the maturation marker CD11c as well as the pro‐inflammatory cytokine IFN‐γ after stimulation with 5 μg/mL of the KOR agonists (B) are shown. Data from n=4 wild‐type (top) or n=4 KOR‐deficient mice (bottom) are depicted. Cells are gated on MHC−II+F4/80−CD19− DCs, and IFN‐γ staining was performed after cell permeabilization. Data are presented as mean ±SD; * p<0.05 and *** p<0.01 vs. PMA/Iono treatment. C) Human DCs were purified from the peripheral blood of healthy donors and stimulated with PMA/Iono for 12 h. Subsequently, cells were incubated with compounds 12b, 12c, 14b, and 14c (5 μM/mL each) for an additional 48 h or received an equal amount of PBS. The percentages of cells expressing CD11c and IFN‐γ from n=4 healthy human donors are shown. Cells are gated on HLA‐DR, and IFN‐γ staining was performed after cell permeabilization. Data are presented as mean ±SD; * p<0.05 vs. PMA/Iono stimulation.
After entering secondary lymphoid organs, activated DC get into contact to naive T cells, resulting in the priming of effector cells (T helper or cytotoxic T cells). Effector T cells including Th1 or Th17 cells migrate to the site of inflammation and in case of infection, contribute to the elimination of bacteria or virus‐containing cells. Under sterile inflammatory conditions (e. g., in individuals with multiple sclerosis, psoriasis or rheumatoid arthritis), Th1 and Th17 are thought to mediate tissue destruction.51, 52, 53 Besides the upregulation of surface markers like CD25, CD44 or CD69, these cells are characterized by the expression of pro‐inflammatory cytokines including IFN‐γ, TNF‐α or IL‐17. To quantify the anti‐inflammatory effect of 12b, 12c, 14b and 14c in T cells, total T cells were purified from mouse peripheral lymph nodes, activated with PMA plus ionomycin, and stimulated with the KOR agonists in a concentration of 5 μg/mL. In contrast to our observations in activated DC, only 14b and 14c exhibited a potent anti‐inflammatory capacity by significantly reducing the expression of the surface activation markers CD25, CD44 and CD69 as well as the IFN‐γ production (Figure 4A and B), whereas 12b and 12c did not modulate the activation status of mouse T cells. Of note, also in T cells the effect of 14b and 14c was mediated by binding to KOR because treatment of T cells from KOR‐deficient mice did not result in an altered expression of activation markers or cytokine release (Figure 4B and C). Interestingly, 14b and 14c might not only have anti‐inflammatory properties but also immunomodulatory capacities since besides the downregulated IFN‐γ and IL‐17 levels in cell cultures stimulated with these KOR agonists, we detected a significant upregulation of the anti‐inflammatory cytokine IL‐10 (Figure 3C). This observation could possibly indicate that compounds 14b and 14c might have induced a switch from effector to regulatory T cells, which is in line with published data. Liang et al. described that opioid receptor agonists are able to modulate immune cell functions by inhibiting NF‐κB activation resulting in increased IL‐10 levels.46 Worth mentioning that the immunomodulatory capacity of compounds 14b and 14c was not an off‐target effect but dependent on binding to KOR because we did not observe any impact of these KOR agonists on the cytokine secretion in activated T cells from KOR‐deficient mice (Figure 4C).
Figure 4.
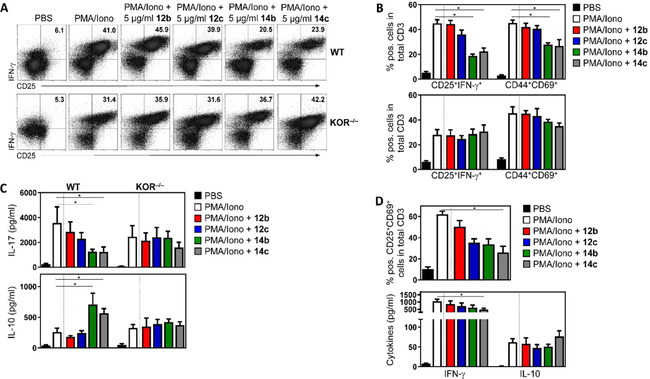
Anti‐inflammatory and immunomodulatory effects of 14b and 14c in T cells. A), B) Mouse T cells were purified from the peripheral lymph nodes of wild‐type (WT) or KOR‐deficient mice (KOR−/−), activated with PMA and ionomycin (PMA/Iono) for 12 h, and stimulated with 12b, 12c, 14b or 14c at a concentration of 5 μg/mL for an additional 48 h. Control cells received an equal amount of PBS. Representative dot‐plots (A) and percentages of cells expressing the typical activation markers CD25, CD69, CD44 as well as the pro‐inflammatory cytokine IFN‐γ (B) are shown. Data from n=4 WT (B, top) or n=4 KOR−/− mice (B, bottom) are depicted. Cells are gated on CD3, and IFN‐γ staining was performed after cell permeabilization. Data are presented as mean ±SD; * p<0.05 vs. PMA/Iono treatment. C) Cytokine quantification in cell culture supernatants from T cells of WT and KOR−/− after activation with PMA/Iono and stimulation with compounds 12b, 12c, 14b or 14c. Data from n=3 mice per group are shown (* p<0.05 vs. PMA/Iono). D) Percentages of CD25+CD69+ cells (top) and cytokine expression (bottom) in total CD3+ T cells from n=4 healthy human donors. IFN‐γ staining (top) was performed after cell permeabilization, and cytokine quantification in cell culture supernatants (bottom) was done using the Legend‐Plex bead‐based immunoassay. Data are presented as means ±SD; * p<0.05 vs. PMA/Iono.
Next, we assessed the anti‐inflammatory and immunomodulatory capacity of 14b and 14c in human T cells and therefore, purified total T cells from peripheral blood of four healthy donors. As shown in Figure 4D, only compound 14c was able to significantly reduce the expression of typical surface activation markers such as CD25 and CD69 and moreover, to decrease the IFN‐γ secretion. However, the stimulation of human T cells with 14c only slightly but not significantly induced IL‐10 (Figure 4D), indicating that at least the anti‐inflammatory effect of the KOR agonist 14c was not limited to mouse cells but also detectable in human primary T cells.
5. Radiosynthesis and Biological Evaluation of KOR‐PET tracers [18F]‐2 and [18F]‐14c
With the long‐term goal of better understanding the role of KOR in MS, a KOR selective PET tracer should be developed. Some of the most important characteristics for a promising KOR PET tracer are high KOR affinity and high selectivity over related receptors. Moreover, the introduction of the radioactive atom in an easy and fast way compatible with the half‐life of the radionuclide at the end of the synthesis should be possible. According to these requirements the fluorinated KOR agonists 2 (K i=5.6 nM) and 14c (K i=0.82 nM) represent suitable candidates to be developed as PET tracers.
5.1. Radiosynthesis, pharmacokinetic properties and in vivo biodistribution of [18F]‐2
The fluorinated PET tracer [18F]‐2 was prepared in a two‐step radiosynthesis. (Scheme 3) In the first step, [18F]‐1‐azido‐2‐fluoroethane was prepared by nucleophilic substitution (SN2) of 2‐azidoethyl 4‐methylbenzenesulfonate with K[18F]F. Subsequent copper(I)‐catalyzed 1,3‐dipolar cycloaddition of the fluorine‐18‐labeled intermediate with the alkyne precursor 18 13 provided the desired product [18F]‐2. [18F]‐2 was purified by preparative HPLC and finally formulated as water/EtOH (9 : 1, v/v) solution. The triazole [18F]‐2 was obtained in radiochemical yields (rcy) of 49.9±8.6 % (n=5, decay‐corrected, based on cyclotron‐derived [18F]fluoride ions (d.c.)). The total synthesis time was 136±12 min from the end of radionuclide production up to final formulation. [18F]‐2 was isolated with radiochemical purities (rcp) of 99.3±0.4 % and a molar activities (A m) of 4.6–14.6 GBq/μmol. (Scheme 3)
Scheme 3.
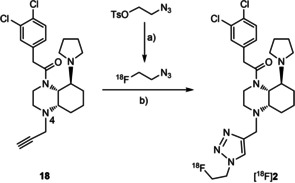
Two‐step radiosynthesis of fluorinated PET tracer [18F]‐2. a) K[18F]F, K222, K2CO3, CH3CN, 110 °C, 3 min; b) CuSO4, Na ascorbate, DMF, H2O, 40 °C, 30 min. [18F]‐2: rcy: 49.9±8.6 % (n=5, d,c.); total synthesis time: 136±12 min; rcp: 99.3±0.4 %; A m: 4.6–14.6 GBq/μmol.
The distribution coefficient at pH 7.4 (logD7.4) was determined by a shake flask method using the radioligand [18F]‐2. After distribution of [18F]‐2 between n‐octanol and phosphate buffered saline (pH 7.4) layers, the radioactivity in both layers was determined and set in ratio. According to this procedure, a logD7.4 value of 1.37±0.06 was determined indicating a moderate lipophilic compound, which should be able to penetrate into the central nervous system.
Enzymes present in blood serum could be able to transform the radioactive PET tracer [18F]‐2. Therefore, the blood serum stability of [18F]‐2 was investigated before testing the tracer in vivo. [18F]‐2 was incubated with mouse and human serum for 90 min at 37 °C. Even after 90 min, radio‐HPLC did not show any additional radioactive compound. Only the parent tracer [18F]‐2 could be detected indicating high stability in mouse and human blood serum (Figure S1 in Supporting Information).
Next, the distribution of the PET tracer [18F]‐2 in mice was investigated. For this purpose, [18F]‐2 was injected into 15‐week‐old C57BL/6 mice. Figure 5 displays representative maximum intensity projections at indicated time points after injection of [18F]‐2. A very fast and efficient clearance of [18F]‐2 from the body was observed following both hepatic and renal pathways. Already after 2 min the radioactivity was almost completely found in liver, kidneys and bladder. In the liver, the radioactivity remained almost constant over the period of 90 min (Figure 6). Besides a transient local increase in radioactivity in the submandibular and harderian glands, no significant accumulation of [18F]‐2 in non‐excreting organs such as heart, lung and muscles as indicator for unspecific binding could be detected. Furthermore, increased radioactivity in the bones was not observed excluding a fast defluorination of [18F]‐2.
Figure 5.
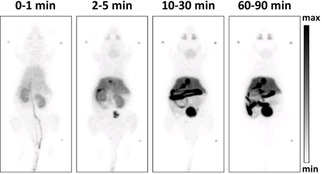
In vivo biodistribution analysis of radioactivity in an adult C57BL/6 mouse after intravenous injection of [18F]‐2. Maximum intensity projections of the biodistribution at indicated time points post injection.
Figure 6.
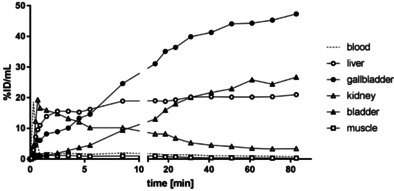
In vivo biodistribution of radioactivity in an adult C57BL/6 mouse after intravenous injection of [18F]‐2. Time–activity concentration curves illustrate tracer dynamics in selected regions of interest (ROI). Activity is displayed as percentage of injected dose per volume during the whole observation period of 90 min.
The PET images do not show any enrichment of radioactivity in the brain or spinal cord at any time point. Obviously, [18F]‐2 was not able to target KOR in the central nervous system. This behavior can be attributed to low penetration of the blood‐brain barrier, fast elimination kinetics or a very fast in vivo biotransformation of [18F]‐2.
In order to investigate the in vivo biotransformation of [18F]‐2, the blood of three mice 90 min after injection of [18F]‐2 was pooled and analyzed. The radio‐HPLC showed 90 % of the intact PET tracer [18F]‐2 and 10 % of a more polar radiometabolite (Figure S2).
It can be concluded that a very fast biotransformation is not the reason for missing accumulation of [18F]‐2 in regions with high KOR density, for example, spinal cord. The logD7.4 value of 1.37 is in a promising range for penetration the blood brain barrier. Therefore, it is assumed that the very fast elimination via liver and kidneys is responsible for the unfavorable imaging properties of [18F]‐2.
5.2. Radiosynthesis, pharmacokinetic properties and in vivo biodistribution of [18F]‐14c
In order to obtain a KOR‐PET tracer with better pharmacokinetics the fluoropyrrolidine derivative 14c showing even higher KOR affinity (K i=0.82 nM) was considered to be prepared in a radioactive form. For this purpose, the synthesis was changed in order to introduce [18F]fluoride at the very end of the synthesis. (Scheme 4). At first, the alcohol 12c was converted into the mesylate 19 upon reaction with mesyl chloride. Subsequent nucleophilic substitution of 19 with K[18F]F in the presence of the cryptand K222 provided the fluorinated PET tracer [18F]‐14c in a radiochemical yield of 20.7±3.4 % (n=5, decay corrected) within a total synthesis time of 128±14 min up to formulation. The radiochemical purity was 98.9±0.7 %. (Scheme 4)
Scheme 4.
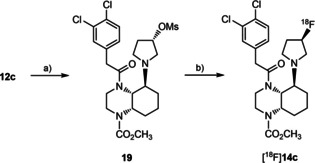
Radiosynthesis of fluorinated PET tracer [18F]‐14c by direct nucleophilic substitution. a) CH3SO2Cl, NEt3, DMAP, CH2Cl2, 0 °C, 30 min, then RT, 16 h, 55 %; b) K[18F]F, K222, K2CO3, CH3CN, 110 °C, 20 min. [18F]‐14c: rcy: 20.7±3.4 % (n=5, d.c.); total synthesis time: 128±14 min; rcp: 98.9±0.7 %; A m: 4.0–111.3 GBq/μmol.
The logD7.4 value of [18F]‐14c was recorded as described above for [18F]‐2. The fluoropyrrolidine [18F]‐14c displayed a higher lipophilicity than [18F]‐2 with a 15‐fold higher concentration of [18F]‐14c in the n‐octanol layer than [18F]‐2 resulting in a logD7.4 value of 2.54±0.22. However, this value is still in a promising range for reaching the central nervous system.
In order to analyze the serum stability [18F]‐14c was incubated with human and mouse serum. After an incubation period of 90 min, radio‐HPLC did not show any new radioactive product indicating high stability in both human and mouse serum.
The in vivo biodistribution of [18F]‐14c was investigated in 10‐week‐old C57BL/6 mice. As reported for [18F]‐2, the clearance of [18F]‐14c from the body was fast following both renal and hepatic pathways. Unfortunately, 10 min after injection, radioactivity in the bones (Ca[18F]F2) was observed indicating fast defluorination of the compound. The high radioactivity detected 60–90 min after injection of [18F]‐14c in the joints, the spine and the skull confirmed the extensive defluorination of [18F]‐14c (Figure 7).
Figure 7.
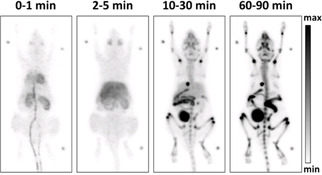
In vivo biodistribution analysis of radioactivity in an adult C57BL/6 mouse after intravenous injection of [18F]‐14c. Maximum intensity projections of the biodistribution at indicated time points post injection.
In Figure 8 the distribution of the radioactivity ([18F]‐14c) in different organs over the time is shown. Two minutes after injection, radioactivity was found only in liver, kidneys and gallbladder. Interestingly, the radioactivity found in liver decreased after 10 min; whereas the radioactivity for the bladder increased (Figure 8).
Figure 8.
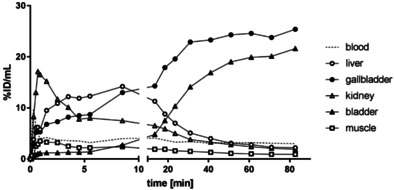
In vivo biodistribution of radioactivity in an adult C57BL/6 mouse after intravenous injection of [18F]‐14c. Time‐activity concentration curves illustrate tracer dynamics in selected regions of interest (ROI). Activity is displayed as percentage of injected dose per volume during the whole observation period of 90 min.
90 min after injection of [18F]‐14c, the animals were sacrificed, and the organs were collected and analyzed ex vivo at the γ‐counter to determine the residual radioactivity in the different organs. It was shown that the radioactivity was mainly present in the excretion organs and in the urine (Figure S3).
5.3. In vitro biotransformation of 14c in the presence of mouse liver microsomes
Due to its defluorination [18F]‐14c does not represent a good candidate to become a PET tracer for selective imaging KORs in the central nervous system. Therefore, [18F]‐14c was not further evaluated in vivo. However, in order to confirm the defluorination by in vitro experiments and to get an idea about possible metabolically labile positons, the non‐radioactive compound 14c was incubated with mouse liver microsomes and the cofactor NADPH. The stability of 14c and the formed metabolites were identified by LC‐MS/MS experiments.
After an incubation period of 90 min, five phase‐I metabolites were identified (Figure 9). Hydroxylation took place at the pyrrolidine ring affording metabolite 14c‐1. Loss of HF gave dihydropyrrole 14c‐2, which was most likely further oxidized (aromatization) to provide pyrrole metabolite 14c‐3. Defluorination by hydroxylation and subsequent elimination of HF to form the respective ketone was already described as defluorination mechanism in literature.54 Therefore, it is assumed that parent compound 14c was hydroxylated at the pyrrolidine ring (e. g., metabolite 14c‐1) leading to a ketone after loss of HF. The ketone was then reduced to an alcohol, which induced elimination of H2O to form dihydropyrrole 14c‐2. Metabolite 14c‐2 was most likely hydroxylated in α‐position of the dihydropyrrole N atom resulting in aromatization by H2O elimination (metabolite 14c‐3). Whereas the metabolite 14c‐1 initiates elimination of fluoride, the metabolites 14c‐2 and 14c‐3 resulted from direct loss of HF. The high amounts of metabolites 14c‐2 and 14c‐3 (Figure S4) correlate nicely with the defluorination observed in vivo during the biodistribution experiments with [18F]‐14c.
Figure 9.
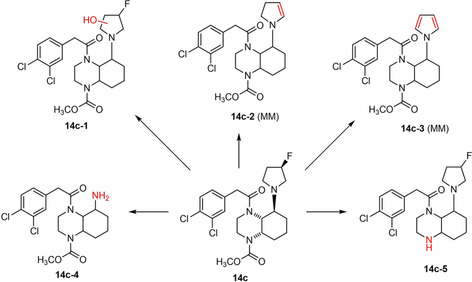
Postulated structures of phase‐I metabolites 14c‐1–5 of KOR agonist 14c produced in vitro using mouse liver microsomes and NADPH. The parent compound 14c represents a mixture two enantiomerically pure diastereomers, the stereochemistry of the metabolites 14c‐1–5 was not determined. MM: main metabolite.
In addition to the metabolites 14c‐1‐3, the primary amine 14c‐4 and the secondary amine 14c‐5 were produced in minor amounts (Figure S4A and B) The primary amine 14c‐4 was formed by double oxidative N‐dealkylation and the secondary amine 14c‐5 by hydrolytic cleavage of the methyl carbamate, whereas hydrolysis of the phenylacetamide was not observed.
6. Conclusion
KORs play a crucial role in the pathology of various central nervous system disorders, including pain, depression, anxiety, Multiple Sclerosis, and, furthermore, itching skin diseases. Anti‐inflammatory effects of KOR agonists are mainly based on inhibition of immune cell activation, which predominantly occurs in the periphery.
In this project, we focused on two issues of KOR agonists: on the one hand side polar KOR agonists should be developed to activate KOR in the periphery with the aim to inhibit immune cell activation and reduce centrally mediated side effects. On the other side, a fluorinated PET tracer should be developed, which allows imaging of KOR non‐invasively in healthy and diseased conditions.
In order to reach these aims an SN2 substitution of cyclic sulfuric acid derivative 10 with hydroxypyrrolidine was performed. A series of three polar hydroxypyrrolidines 12a–c bearing a Boc group, a proton or a methoxycarbonyl moiety at the second quinoxaline N atom was prepared. Conversion of the OH moiety into a F atom led to fluoropyrrolidines 14a–c with the same substitution pattern at the second quinoxaline N‐atom. The KOR affinity of the hydroxypyrrolidines 12b,c was in the low‐nanomolar range, and those of the fluoropyrrolidines 14b,c in the low‐ to sub‐nanomolar range. Hydroxypyrrolidines 12 and fluoropyrrolidines 14 exhibit high selectivity for KOR over related MOR, DOR, σ1 and σ2 receptors. In the cAMP and β‐arrestin recruitment assay, the fluoropyrrolidine 14b was as active as the prototypical KOR agonist U‐50,488, but the hydroxypyrrolidine derivatives 12b and 12c were slightly less potent.
The production of inflammation markers CD11c and INF‐γ was increased upon stimulation of mouse dendritic cells with PMA/ionomycin. Treatment of activated dendritic cells with hydroxypyrrolidine 12c and fluoropyrrolidines 14b and 14c led to reduced formation of CD11c and INF‐γ indicating anti‐inflammatory activity. The most polar KOR agonist 12b did not show this effect, which was attributed to its very high polarity. Worth mentioning that this effect was completely abrogated when stimulated DC from KOR‐deficient mice were treated with 12c, 14b and 14c. This observation proves that the anti‐inflammatory effect was mediated by KOR. The same result was obtained with human DC.
Activated DC are able to activate effector T cells including Th1 and Th17 cells, which reinforce the inflammatory process. Therefore, the effect of KOR agonists 12b, 12c, 14b and 14c on mouse and human T cells activated with PMA/ionomycin was investigated. Whereas the fluoropyrrolidines 14b and 14c led to significantly reduced expression of activation markers CD25, CD44 and CD69 and release of IFN‐γ, the more polar hydroxypyrrolidines 12b and 12c were not active. It was shown that these effects were mediated by KOR, since T cells isolated from KOR‐deficient mice could not be modulated by KOR agonists 14b and 14c.
The anti‐inflammatory effects of fluoropyrrolidines 14b and 14c are mediated by KOR as DC and T cells from KOR‐deficient mice were not influenced by the KOR agonists 14b and 14c. The effects of 14b and 14c on mouse DC and T cells were also observed for human DC and T cells, indicating a translational potential of the anti‐inflammatory effects of KOR agonists to humans. The hydroxypyrrolidines were either not active (12b) or less active (12c) in these experiments. This can be attributed to the high polarity of the hydroxypyrrolidines 12b and 12c or to the lower KOR affinity and KOR activity compared to the fluoropyrrolidines 14b and 14c.
The fluorinated PET tracer [18F]2 was prepared by a 1,3‐dipolar cycloaddition of propargylamine 18 with [18F]fluoroethyl azide (Click reaction). Application of tracer [18F]‐2 to mice led to a very fast elimination via kidney and liver. After 2 min, almost the complete radioactivity was found in liver, kidneys and bladder. Although the polarity of [18F]‐2 (logD7.4=1.37) was in a promising range, KOR rich regions were not labeled by [18F]‐2. Fast metabolism could be excluded as reason of the fast elimination.
As second option, the fluorinated PET tracer [18F]‐14c was prepared by nucleophilic substitution of the mesylate 19 with [18F]fluoride. The second PET tracer [18F]‐14c displayed slightly higher lipophilicity (logD7.4=2.54) than [18F]‐2. In biodistribution studies, [18F]‐14c exhibited slower elimination than [18F]‐2, but labeling of bones was observed, which was attributed to defluorination of the PET tracer [18F]‐2. This defluorination was analyzed in vitro using mouse liver microsomes and NADPH/H+. Altogether, five metabolites were identified and the main metabolites result from defluorination.
Experimental Section
Chemistry
General methods
Oxygen and moisture sensitive reactions were carried out under nitrogen, dried with silica gel with moisture indicator (orange gel, Merck) and in dry glassware (Schlenk flask or Schlenk tube). All solvents were of analytical grade quality. Flash chromatography (FC): Silica gel 60, 40–63 μm (Machery Nagel); parentheses include: diameter of the column (Ø), length of the stationary phase (l), fraction size (v) and eluent. NMR: NMR spectra were recorded on Agilent DD2 400 MHz and 600 MHz spectrometers; chemical shifts (δ) are reported in parts per million (ppm) against the reference substance tetramethylsilane and calculated using the solvent residual peak of the undeuterated solvent. HPLC method to determine the purity of compounds (Method Pu): Equipment 1: Pump: L‐7100, degasser: L‐7614, autosampler: L‐7200, UV detector: L‐7400, interface: D‐7000, data transfer: D‐line, data acquisition: HSM‐Software (all from LaChrom, Merck Hitachi); Equipment 2: Pump: LPG‐3400SD, degasser: DG‐1210, autosampler: ACC‐3000T, UV‐detector: VWD‐3400RS, interface: DIONEX UltiMate 3000, data acquisition: Chromeleon 7 (Thermo Fisher Scientific); column: LiChropher® 60 RP‐select B (5 μm), LiChroCART® 250–4 mm cartridge; flow rate: 1.0 mL/min; injection volume: 5.0 μL; detection at λ=210 nm; solvents: A: demineralized water with 0.05 % (v/v) trifluoroacetic acid, B: acetonitrile with 0.05 % (v/v) trifluoroacetic acid; gradient elution (% A): 0–4 min: 90 %; 4–29 min: gradient from 90 to 0 %; 29–31 min: 0 %; 31–31.5 min: gradient from 0 to 90 %; 31.5–40 min: 90 %. The purity of all test compounds is greater than 95 %.
Synthetic procedures
tert‐Butyl (4aR,5S,8aS)‐ and (4aS,5R,8aR)‐5‐[(S)‐3‐hydroxypyrrolidin‐1‐yl]‐3,4,4 a,5,6,7,8,8 a‐octahydroquinoxaline‐1(2H)‐ carboxylate (11)
Under a N2 atmosphere, (S)‐3‐hydroxypyrrolidine (0.25 mL, 3.1 mmol, 5 equiv) was added to a solution of 10 (200 mg, 0.63 mmol, 1 equiv) and K2CO3 (17 mg, 0.13 mmol, 0.2 equiv) in dry DMF (2 mL). The mixture was stirred at 80 °C for 16 h. Afterwards, HCl (1 M, 5 mL) was added, and the mixture was extracted with CH2Cl2 (2×10 mL). Then, NaOH (1 M, 5 mL) was added (pH 10) and the aqueous layer was extracted with CH2Cl2 (2×20 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (Ø=2 cm, h=15 cm, CH2Cl2/CH3OH/NH3 (25 %) solution 94 : 5 : 1, V=12 mL, R f=0.36 (CH2Cl2/CH3OH/NH3 (25 %) 94 : 5 : 1)). Pale yellow oil, yield 165 mg (81 %). C17H31N3O3 (325.5 g/mol). 1H NMR (600 MHz, CDCl3): δ (ppm)=1.35–1.50 (m, 2H, 7‐CH 2, 8‐CH 2), 1.43 (s, 9H, CO2C(CH 3)3), 1.57–1.64 (m, 2H, 6‐CH 2), 1.66–1.80 (m, 2H, 7‐CH 2, NCH2CH 2CHOH), 1.86–2.01 (m, 1H, 8‐CH 2), 2.07–2.17 (m, 1H, NCH2CH 2CHOH), 2.20–2.38 (m, 3H, 2‐CH 2, 5‐CH, NCH 2CH2CHOH), 2.46–2.57 (m, 1H, NCH 2CHOH), 2.70–2.78 (m, 1H, 3‐CH 2), 2.78–2.84 (m, 1H, NCH 2CHOH), 2.86–2.94 (m, 1H, NCH 2CH2CHOH), 2.95–3.07 (m, 3H, 2‐CH 2, 3‐CH 2, 4a‐CH), 3.71–3.81 (m, 1H, −NH), 4.15–4.24 (m, 1H, 8a‐CH), 4.24–4.32 (m, 1H, NCH2CHOH). A signal for the OH proton is not seen in the spectrum. 13C NMR (151 MHz, CDCl3): δ (ppm)=19.4 (1 C, C‐7), 22.8 (1 C, C‐8), 24.4 (1 C, C‐6), 28.6 (3 C, CO2C(CH3)3, 34.5 (1 C, NCH2 CH2CHOH), 46.5 (1 C, C‐3), 49.2 (1 C, C‐8a), 50.0 (1 C, NCH2CH2CHOH), 50.2 (1 C, C‐2), 56.7 (1 C, C‐4a), 60.5 (1 C, NCH2CHOH), 65.5 (1 C, C‐5), 71.1 (1 C, NCH2 CHOH), 79.7 (1 C, CO2 C(CH3)3), 155.0 (1 C, CO2C(CH3)3). IR (neat): ν (cm−1)=3437 (OH), 3298 (R2NH), 2943 (C−Haliph), 1670 (N−C=O), 1165 (N−C). HRMS (APCI): m/z 326.2430 (calcd. 326.2438 for C17H32N3O3 [M+H]+).
tert‐Butyl (4aR,5S,8aS)‐ and (4aS,5R,8aR)‐4‐[2‐(3,4‐dichlorophenyl)acetyl]‐5‐[(S)‐3‐hydroxypyrrolidin‐1‐yl]‐3,4,4 a,5,6, 7,8,8 a‐octahydroquinoxaline‐1(2H)‐carboxylate (12a)
Diisopropylethylamine (476 mg, 3.7 mmol, 4 equiv) was added to a solution of secondary amine 11 (300 mg, 0.9 mmol, 1 equiv) in CH2Cl2 (8 mL). At 0 °C a solution of 2‐(3,4‐dichlorophenyl)acetyl chloride (515 mg, 2.3 mmol, 2.5 equiv) in CH2Cl2 (3 mL) was added dropwise. The mixture was stirred at room temperature for 16 h. Then, NaOH (0.5 M, 10 mL) was added, and the mixture was extracted with CH2Cl2 (3×15 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (Ø=3 cm, h=10 cm, CH2Cl2/CH3OH/NH3 (25 %) solution 94 : 5 : 1, V=30 mL, R f=0.50 (CH2Cl2/CH3OH/NH3 (25 %) 89 : 10 : 1)). Yellow oil, yield 398 mg (85 %). C25H35Cl2N3O4 (512.5 g/mol). 1H NMR (600 MHz, CDCl3): δ (ppm)=1.20–1.32 (m, 1H, 8‐CH 2), 1.35–1.56 (m, 2H, 6‐CH 2, 7‐CH 2), 1.45 (s, 9H, CO2C(CH 3)3), 1.64–1.80 (m, 1H, 7‐CH 2), 1.82–1.98 (m, 2H, 6‐CH 2, NCH2CH 2CHOH), 2.02–2.19 (m, 1H, NCH2CH 2CHOH), 2.22–2.40 (m, 1H, 8‐CH 2), 3.09–3.30 (m, 2H, 5‐CH, 8a‐CH), 3.38–3.95 (m, 6H, C(=O)CH 2‐aryl, NCH 2CHOH, NCH2CH2CHOH), 4.00–4.17 (m, 1H, 4a‐CH), 4.25–4.42 (m, 1H, NCH2CHOH), 4.50–4.78 (m, 1H, OH), 7.09–7.18 (m, 1H, 6‐CH arom), 7.33–7.41 (m, 2H, 2‐CH arom, 5‐CH arom). Signals for 2‐CH 2 and 3‐CH 2 protons are not seen in the spectrum. 13C NMR (151 MHz, CDCl3): δ (ppm)=19.3 (1 C, C‐7), 28.5 (1 C, C‐6), 28.5 (3 C, CO2C(CH3)3), 30.4 (1 C, C‐8), 34.4 (1 C, NCH2 CH2CHOH), 40.0 (1 C, NCH2CHOH), 40.8 (1 C, C(=O)CH2‐aryl), 41.1 (1 C, NCH2CH2CHOH), 53.6 (1 C, C‐8a), 60.9 (2 C, C‐4a, C‐8), 70.0 (1 C, NCH2 CHOH), 80.9 (1 C, CO2 C(CH3)3), 128.9 (1 C, Carom), 130.6 (2 C, Carom), 131.2 (1 C, Carom), 131.3 (1 C, Carom), 132.6 (1 C, Carom), 155.1 (1 C, CO2C(CH3)3), 169.2 (1 C, NC(=O)benzyl). Signals for C‐2 and C‐3 carbons atoms are not seen in the spectrum. IR (neat): ν (cm−1)=3402 (OH), 2932 (C−Haliph), 1686 (N−C=O), 1639 (N−C=O), 1165 (N−C), 734 and 698 (1,2‐disubst. arom.). Exact Mass (APCI): m/z 512.2070 (calcd. 512.2077 for C25H36 35Cl2N3O4 [M+H]+). HPLC (Method Pu): purity 96 % (t R=19.20 min).
2‐(3,4‐Dichlorophenyl)‐1‐{(4aR,8R,8aR)‐ and (4aS,8S,8aS)‐ 8‐[(S)‐3‐hydroxypyrrolidin‐1‐yl]‐3,4,4 a,5,6,7,8,8 a‐octahydroquinoxalin‐1(2H)‐yl}ethan‐1‐one (12b)
TFA (232 μL, 3.1 mmol, 32 equiv) was added dropwise to a solution of alcohol 12a (50 mg, 0.1 mmol, 1 equiv) in CH2Cl2 (1 mL). The mixture was stirred at room temperature overnight. After addition of toluene, the solvents were removed in vacuo (azeotropic distillation). Then, NaOH (0.5 M, 10 mL) was added and the mixture was extracted with CH2Cl2 (3×15 mL). After removal of the solvent in vacuo the residue was purified by flash column chromatography (Ø=2 cm, h=10 cm, CH2Cl2/CH3OH/NH3 (25 %) 89 : 10 : 1, V=12 mL, R f=0.28 (CH2Cl2/CH3OH/NH3 (25 %) 94 : 5 : 1)). Pale yellow oil, yield 35 mg (87 %). C20H27Cl2N3O2 (412.4 g/mol). 1H NMR (400 MHz, CDCl3): δ (ppm)=1.22–1.32 (m, 1H, 5‐CH 2), 1.52–1.80 (m, 6H, 6‐CH 2, 7‐CH 2, NCH2CH 2CHOH, −NH), 1.85–1.96 (m, 1H, 5‐CH 2), 1.98–2.09 (m, 1H, NCH2CH 2CHOH), 2.50–2.86 (m, 4H, 3‐CH 2, NCH 2CHOH, NCH 2CH2CHOH), 2.90–3.07 (m, 3H, 8‐CH, 3‐CH 2, NCH 2CHOH), 3.20, 3.24 (dt, each, J=12.8/3.6 Hz, 1H, 2‐CH 2), 3.34–3.55 (m, 2H, 2‐CH 2, 4a‐CH), 3.59–3.83 (m, 2H, C(=O)CH 2‐aryl), 4.17–4.27 (m, 1H, NCH2CHOH), 4.51, 4.56 (dd, each, J=11.2/3.4 Hz, 1H, 8a‐CH), 7.08–7.17 (m, 1H, 6‐CH arom), 7.34–7.40 (m, 2H, 2‐CH arom, 5‐CH arom). A signal for the OH proton is not seen in the spectrum. 13C NMR (101 MHz, CDCl3): δ (ppm)=20.0 (1 C, C‐6), 24.2 (1 C, C‐5), 32.1 (1 C, C‐7), 34.6 (1 C, NCH2 CH2CHOH), 40.4 (1 C, C(=O)CH2‐aryl), 42.6 (1 C, NCH2CH2CHOH), 46.5 (1 C, C‐2), 48.2 (1 C, NCH2CHOH), 52.7 (1 C, C‐4a), 53.9 (1 C, C‐8a), 55.6 (1 C, C‐8), 57.4 (1 C, C‐3), 71.1 (m, 1H, NCH2 CHOH), 128.5 (1 C, Carom), 130.6 (2 C, Carom), 130.8 (1 C, Carom), 132.6 (1 C, Carom), 135.7 (1 C, Carom), 169.8 (1 C, C(=O)‐benzyl). IR (neat): ν (cm−1)=3399 (OH), 3310 (R2NH), 2932 (C−Haliph), 1620 C(=O), 1134 (N−C), 733 and 689 (1,2‐disubst. arom.). Exact Mass (APCI): m/z 412.1515 (calcd. 412.1553 for C20H28 35Cl2N3O2 [M+H]+). HPLC (Method Pu): purity 96 % (t R=12.85 min).
tert‐Butyl (4aR,5S,8aS)‐ and (4aS,5R,8aR)‐4‐[2‐(3,4‐dichlorophenyl)acetyl]‐5‐{(S)‐3‐[(methylsulfonyl)oxy]pyrrolidin‐1‐ yl}‐3,4,4 a,5,6,7,8,8 a‐octahydroquinoxaline‐1(2H)‐carboxylate (13)
Triethylamine (30.6 μL, 0.2 mmol, 2.3 equiv) and 4‐(dimethylamino)pyridine (12 mg, 0.1 mmol, 1 equiv) were added to a solution of alcohol 12a (50 mg, 0.1 mmol, 1 equiv) in CH2Cl2 (5 mL). At 0 °C methanesulfonyl chloride (15.1 μL, 0.2 mmol, 2 equiv) was added dropwise. The mixture was stirred at 0 °C for 30 min and then at room temperature for 16 h. Afterwards, H2O (20 mL) was added, and the mixture was extracted with CH2Cl2 (3×15 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (Ø=2 cm, h=15 cm, CH2Cl2/CH3OH/NH3 (25 %) solution 98 : 1 : 1, V=12 mL, R f=0.72 (CH2Cl2/CH3OH/NH3 (25 %) 94 : 5 : 1)). Yellow oil, yield 47 mg (81 %). C26H37Cl2N3O6S (590.6 g/mol). 1H NMR (600 MHz, CDCl3): δ (ppm)=1.35–1.42 (m, 2H, 6‐CH 2, 8‐CH 2), 1.45 (s, 9H, CO2C(CH 3)3), 1.58–1.78 (m, 5H, 6‐CH 2, 7‐CH 2, NCH2CH 2CHOMs), 1.91–2.27 (m, 3H, 8‐CH 2, NCH 2CH2CHOMs), 2.48–2.65 (m, 1H, 2‐CH 2), 2.80–3.19 (m, 4H, 2‐CH 2, 5‐CH, NCH 2CHOMs), 2.98 (s, 3H, CH3 mesyl), 3.45–3.55 (m, 1H, 3‐CH 2), 3.61–3.76 (m, 3H, C(=O)CH 2‐aryl, 8a‐CH), 3.77–3.86 (m, 1H, 3‐CH 2), 4.03–4.16 (m, 1H, 4a‐CH), 5.00–5.21 (m, 1H, NCH2CHOMs), 7.04–7.18 (m, 1H, 6‐CH arom), 7.31–7.46 (m, 2H, 2‐CH arom, 5‐CH arom). 13C NMR (151 MHz, CDCl3): δ (ppm)=19.5 (1 C, C‐7), 23.9 (1 C, C‐6), 28.5 (3 C, CO2C(CH3)3), 30.6 (1 C, C‐8), 32.2 (1 C, NCH2CH2CHOMs), 38.6 (1 C, −CH3 mesyl), 40.2 (1 C, NCH2 CH2CHOMs), 40.3 (1 C, C‐3), 41.1 (1 C, C(=O)CH2‐aryl), 42.4 (1 C, C‐8a), 47.4 (1 C, C‐2), 52.1 (1 C, C‐4a), 55.6 (1 C, NCH2CHOMs), 59.1 (1 C, C‐5), 68.3 (1 C, NCH2 CHOMs), 80.6 (1 C, CO2 C(CH3)3), 128.6 (1 C, Carom), 130.7 (2 C, Carom), 131.1 (1 C, Carom), 132.7 (1 C, Carom), 135.3 (1 C, Carom), 167.9 (1 C, CO2C(CH3)3), 170.0 (1 C, C(=O)‐benzyl). IR (neat): ν (cm−1)=2974 (C−Haliph), 1686 (N−C=O), 1639 (N−C=O), 1165 (N−C), 1254 and 1034 (O=S=O), 729 and 682 (1,2‐disubst. arom.). Exact Mass (APCI): m/z 590.1842 (calcd. 590.1853 for C26H38 35Cl2N3O6S [M+H]+). HPLC (Method Pu): purity 90 % (t R=20.76 min).
tert‐Butyl (4aR,5S,8aS)‐ and (4aS,5R,8aR)‐4‐[2‐(3,4‐dichlorophenyl)acetyl]‐5‐[(R)‐3‐fluoropyrrolidin‐1‐yl]‐3,4,4 a,5,6,7,8,8 a‐ octahydroquinoxaline‐1(2H)‐carboxylate (14a)
TBAF ⋅ 3H2O (63 mg, 0.2 mmol, 2.5 equiv) was added to a suspension of mesylate 13 (47 mg, 0.1 mmol, 1 equiv) in H2O (5 μL). The mixture was stirred at 45 °C for 16 h. Afterwards, the mixture was diluted with EtOAc (10 mL) and the mixture was washed with H2O and brine (3×10 mL). The organic layer was dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (Ø=2 cm, h=15 cm, CH2Cl2/CH3OH/NH3 (25 %) 98 : 1 : 1→94 : 5 : 1, V=12 mL, R f=0.58 (CH2Cl2/CH3OH/NH3 (25 %) 94 : 5 : 1)). Yellowish oil, yield 20 mg (50 %). C25H34Cl2FN3O3 (514.5 g/mol). 1H NMR (400 MHz, CDCl3): δ (ppm)=1.33–1.49 (m, 5H, 6‐CH 2, 7‐CH 2, NCH2CH 2CHF), 1.45 (s, 9H, CO2C(CH 3)3), 1.51–1.60 (m, 1H, 8‐CH 2), 1.66–1.73 (m, 1H, 6‐CH 2), 1.89–2.07 (m, 2H, NCH 2CH2CHF), 2.07–2.19 (m, 1H, 8‐CH 2), 2.71–3.10 (m, 4H, 2‐CH 2, 5‐CH, NCH 2CHF), 3.30–3.44 (m, 1H, NCH 2CHF), 3.44–3.56 (m, 1H, 3‐CH 2), 3.59–3.77 (m, 3H, C(=O)CH 2‐aryl, 8a‐CH), 3.79–3.92 (m, 1H, 3‐CH 2), 4.05–4.17 (m, 1H, 4a‐CH), 4.95–5.22 (m, 1H, NCH2CHF), 7.10 (td, J=8.0, 2.2 Hz, 1H, 6‐CH arom), 7.28–7.44 (m, 2H, 2‐CH arom, 5‐CH arom). 13C NMR (101 MHz, CDCl3): δ (ppm)=19.7 (1 C, C‐7), 28.5 (3 C, CO2C(CH3)3), 29.8 (1 C, C‐6), 30.7 (1 C, C‐8), 32.7 (d, J=17.9 Hz 1 C, NCH2CH2CHF), 33.0 (d, J=18.7 Hz, 1 C, NCH2 CH2CHF), 40.9 (1 C, C‐3), 41.8 (1 C, C(=O)CH2‐aryl), 47.6 (1 C, C‐2), 52.5 (1 C, C‐4a), 56.5 (d, J=23.5 Hz, 1 C, NCH2CHF), 59.3 (1 C, C‐8a), 59.9 (1 C, C‐5), 80.5 (1 C, CO2 C(CH3)3), 93.4 (d, J=174.8 Hz, 1 C, NCH2 CHF), 128.5 (1 C, Carom), 130.6 (2 C, Carom), 131.0 (1 C, Carom), 132.6 (1 C, Carom), 135.4 (1 C, Carom), 155.2 (1 C, CO2C(CH3)3), 170.0 (1 C, NC(=O)‐benzyl). IR (neat): ν (cm−1)=2932 (C−Haliph), 1686 (N−C=O), 1639 (N−C=O), 1157 (N−C), 729 and 683 (1,2‐disubst. arom.). Exact Mass (APCI): m/z 514.2061 (calcd. 514.2034 for C25H35 35Cl2FN3O3 [M+H]+). HPLC (Method Pu): purity 74 % (t R=20.70 min).
2‐(3,4‐Dichlorophenyl)‐1‐{(4aR,8R,8aR)‐ and (4aS,8S,8aS)‐8‐ [(R)‐3‐fluoropyrrolidin‐1‐yl]‐3,4,4 a,5,6,7,8,8 a‐octahydroquinoxalin‐1(2H)‐yl}ethan‐1‐one (14b)
TFA (739 μL, 10 mmol, 32 equiv) was added dropwise to a solution of Boc‐derivative 14a (160 mg, 0.3 mmol, 1 equiv) in CH2Cl2 (3 mL). The mixture was stirred at room temperature overnight. After addition of toluene, the solvents were removed in vacuo (azeotropic distillation). Then, NaOH (0.5 M, 10 mL) was added and the mixture was extracted with CH2Cl2 (3×15 mL). After removal of the solvent in vacuo, the residue was purified by flash column chromatography (Ø=3 cm, h=10 cm, CH2Cl2/CH3OH/NH3 (25 %) 94 : 5 : 1, V=30 mL, R f=0.46 (CH2Cl2/CH3OH/NH3 (25 %) 94 : 5 : 1)). Colorless oil, yield 96 mg (77 %). C20H26Cl2FN3O (414.3 g/mol). 1H NMR (400 MHz, CDCl3): δ (ppm)=1.22–1.37 (m, 1H, 5‐CH 2), 1.43–1.69 (m, 6H, 6‐CH 2, 7‐CH 2, NCH2CH 2CHF), 1.81–2.09 (m, 3H, NCH 2CH2CHF, 5‐CH 2), 2.64–2.86 (m, 2H, NCH 2CHF), 2.94–3.07 (m, 3H, 3‐CH 2, 8‐CH), 3.10–3.22 (m, 1H, C(=O)CH 2‐aryl), 3.30–3.53 (m, 2H, 4a‐CH, C(=O)CH 2‐aryl), 3.60–3.84 (m, 2H, 2‐CH 2), 4.36–4.64 (m, 1H, 8a‐CH), 4.93–5.21 (m, 1H, NCH2CHF), 7.01–7.19 (m, 1H, 6‐CH arom), 7.28–7.44 (m, 2H, 2‐CH arom, 5‐CH arom). A signal for the NH proton is not seen in the spectrum. 13C NMR (101 MHz, CDCl3): δ (ppm)=20.2 (1 C, C‐6), 23.4 (1 C, C‐5), 32.0 (1 C, C‐7), 32.8 (d, J=22.5 Hz,1 C, NCH2 CH2CHF), 33.2 (d, J=21.6 Hz, 1 C, NCH2CH2CHF), 40.6 (1 C, C‐2), 42.4 (1 C, C(=O)CH2‐aryl), 47.1 (1 C, C‐3), 52.3 (1 C, C‐4a), 54.0 (1 C, C‐8a), 54.8 (d, J=22.7 Hz, 1 C, NCH2CHF), 55.6 (1 C, C‐8), 93.7 (d, J=176.1 Hz, 1 C, NCH2 CHF), 128.3 (1 C, Carom), 130.5 (2 C, Carom), 131.4 (1 C, Carom), 132.5 (1 C, Carom), 135.9 (1 C, Carom), 170.1 (1 C, C(=O)benzyl). IR (neat): ν (cm−1)=3313 (R2NH), 2932 (C−Haliph), 1628 (N−C=O), 1134 (N−C), 733 and 679 (1,2‐disubst. arom.). Exact Mass (APCI): m/z 414.1536 (calcd. 414.1510 for C20H27 35Cl2FN3O [M+H]+). HPLC (Method Pu): purity 95 % (t R=13.89 min).
Methyl (4aR,5S,8aS)‐ and (4aS,5R,8aR)‐4‐[2‐(3,4‐dichlorophenyl)acetyl]‐5‐[(R)‐3‐fluoropyrrolidin‐1‐yl]‐3,4,4 a,5,6,7,8,8 a‐ octahydroquinoxaline‐1(2H)‐carboxylate (14c)
Under a N2 atmosphere, pyridine (49 μL, 0.60 mmol, 5 equiv) was added to a solution of secondary amine 14b (50 mg, 0.12 mmol, 1 equiv) in dry CH2Cl2 (0.5 mL). At 0 °C, a solution of methyl chloroformate (56 μL, 0.72 mmol, 6 equiv) in CH2Cl2 (0.5 mL) was added dropwise. The ice bath was removed and the mixture was stirred at room temperature for 1 h. H2O (10 mL) was added and the mixture was extracted with CH2Cl2 (3×10 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (Ø=2 cm, h=14 cm, CH2Cl2/CH3OH/NH3 (25 %) 97 : 2 : 1, V=12 mL, R f=0.8 (CH2Cl2/CH3OH/NH3 (25 %) 94 : 5 : 1). Pale yellow oil, yield 10 mg (18 %). C22H28Cl2FN3O3 (472.4 g/mol). 1H NMR (400 MHz, CDCl3): δ (ppm)=1.32–1.50 (m, 2H, 7‐CH 2), 1.52–1.63 (m, 1H, 8‐CH 2), 1.65–1.81 (m, 2H, 6‐CH 2), 1.84–2.00 (m, 2H, NCH2CH 2CHF), 2.06–2.18 (m, 1H, 8‐CH 2), 2.27–2.58 (m, 1H, NCH 2CH2CHF), 2.69–3.09 (m, 4H, 8‐CH, NCH 2CH2CHF, NCH 2CHF), 3.32–3.49 (m, 1H, 3‐CH 2), 3.49–3.63 (m, 2H, 2‐CH 2, 8a‐CH), 3.64–3.84 (m, 4H, C(=O)CH 2‐aryl, 2‐CH 2, 3‐CH 2), 3.69 (s, 3H, CO2CH 3), 4.13–4.23 (m, 1H, 4a‐CH), 4.97–5.22 (m, 1H, NCH2CHF), 7.09 (td, J=8.0, 2.1 Hz, 1H, 6‐CH arom), 7.28–7.43 (m, 2H, 2‐CH arom, 5‐CH arom). 13C NMR (101 MHz, CDCl3): δ (ppm)=19.4 (1 C, C‐7), 19.5 (1 C, C‐6), 30.5 (1 C, C‐8), 32.6 (d, J=16.5 Hz, 1 C, NCH2 CH2CHF), 32.8 (d, J=17.0 Hz, 1 C, NCH2CH2CHF), 40.2 (1 C, C‐3), 41.6 (1 C, C(=O)CH2‐aryl), 41.9 (1 C, C‐2), 47.7 (1 C, NCH2CH2CHF), 52.5 (2 C, C‐4a, C‐8a), 52.9 (1 C, CO2 CH3), 56.7 (d, J=21.9 Hz, 1 C, NCH2CHOF), 59.5 (1 C, C‐5), 93.4 (d, J=175.1 Hz, 1 C, NCH2 CHF), 128.6 (1 C, Carom), 130.6 (2 C, Carom), 131.0 (1 C, Carom), 132.6 (1 C, Carom), 135.2 (1 C, Carom), 156.4 (1 C, CO2CH3)., 171.2 (1 C, C(=O)‐benzyl). IR (neat): ν (cm−1)=2947 (C−Haliph), 1694 (N−C=O), 1639 (N−C=O), 1126 (N−C), 733 and 683 (1,2‐disubst. arom.). Exact Mass (APCI): m/z 472.1548 (calcd. 472.1565 for C22H29 35Cl2FN3O3 [M+H]+). HPLC (Method Pu): purity 96 % (t R=17.87 min).
tert‐Butyl (4aR,5S,8aS)‐ and (4aS,5R,8aR)‐5‐{(S)‐3‐[(tert‐butyldiphenylsilyl)oxy]pyrrolidin‐1‐yl}‐4‐[2‐(3,4‐dichlorophenyl)‐ acetyl]‐3,4,4 a,5,6,7,8,8 a‐octahydroquinoxaline‐1(2H)‐carboxylate (15)
Imidazole (167 mg, 2.45 mmol, 2.5 equiv), DMAP (6 mg, 0.05 mmol, 0.05 equiv) and TBDPSCl (280 μL, 1.08 mmol, 1.1 equiv) were added successively to a solution of 12a (500 mg, 0.98 mmol, 1 equiv) in CH2Cl2 (8 mL). The mixture was stirred at room temperature overnight. After addition of CH2Cl2 (10 mL), the mixture was washed with brine (3×10 mL). The organic layer was dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (Ø=3 cm, h=14 cm, cHex/EtOAc 7 : 3, V=30 mL, R f=0.48 (EtOAc/cHex 5 : 5)). Pale yellow oil, yield 534 mg (74 %). C41H53Cl2N3O4Si (750.9 g/mol). 1H NMR (600 MHz, CDCl3): δ (ppm)=0.99–1.10 (s, 9H, SiC(CH 3)3), 1.30–1.39 (m, 2H, 6‐CH 2, 7‐CH 2), 1.41–1.56 (m, 2H, 7‐CH 2, 8‐CH 2), 1.44 (s, 9H, CO2C(CH 3)3), 1.68–1.87 (m, 3H, 6‐CH2, NCH2CH 2CHOSi), 2.06–2.15 (m, 1H, 8‐CH 2), 2.52–2.62 (m, 1H, NCH2CH2CHOSi), 2.66–2.72 (m, 1H, NCH 2CHOSi), 2.73–2.83 (m, 1H, NCH 2CHOSi), 2.84–2.93 (m, 2H, 5‐CH, NCH2CH2CHOSi), 3.23–3.39 (m, 1H, 3‐CH 2), 3.41–3.52 (m, 1H, 2‐CH 2), 3.54–3.76 (m, 3H, C(=O)CH 2‐aryl, 2‐CH 2), 3.80–3.90 (m, 1H, 3‐CH 2), 3.98–4.08 (m, 1H, 8a‐CH), 4.24–4.37 (m, 1H, NCH2CHOSi), 6.98–7.07 (m, 1H, 6‐CH arom), 7.22–7.30 (m, 2H, 2‐CH arom, 5‐CH arom), 7.32–7.46 (m, 6H, CH arom), 7.60–7.67 (m, 4H, CH arom). A signal for 4a‐CH proton is not seen in the spectrum. 13C NMR (151 MHz, CDCl3): δ (ppm)=19.2 (1 C, C‐7), 19.7 (1 C, C‐6), 21.2 (1 C, SiC(CH3)3), 27.0 (3 C, SiC(CH3)3), 28.5 (3 C, CO2C(CH3)3), 30.7 (1 C, C‐8), 35.2 (1 C, NCH2 CH2CHOSi), 38.9 (1 C, C(=O)CH2‐aryl), 40.7 (1 C, C‐3), 42.1 (1 C, C‐2), 52.6 (1 C, C‐8a), 56.9 (1 C, NCH2CH2CHOSi), 59.8 (1 C, C‐5), 60.5 (1 C, NCH2CHOSi), 72.5 (1 C, NCH2 CHOSi), 80.4 (1 C, CO2 C(CH3)3), 127.7 (4 C, Carom), 128.5 (2 C, Carom), 128.7 (1 C, Carom), 129.7 (2 C, Carom), 130.5 (2 C, Carom), 130.7 (1 C, Carom), 131.0 (1 C, Carom), 132.6 (1 C, Carom), 135.8 (4 C, Carom), 155.3 (1 C, CO2C(CH3)3), 169.8 (1 C, NC(=O)‐benzyl). A signal for C‐4a carbon atom is not seen in the spectrum. IR (neat): ν (cm−1)=2932 (C−Haliph), 1690 (N−C=O), 1643 (N−C=O), 1107 (N−C), 729 (1,2‐disubst. arom.), 702 (SiC6H5). Exact Mass (APCI): m/z 394.1437 (calcd. 394.1447 for C20H26 35Cl2N3O [M−C21H28O3Si+H]+). HPLC (Method Pu): purity 92 % (t R=28.82 min).
1‐((4aR,8R,8aR)‐ and (4aS,8S,8aS)‐8‐{(S)‐3‐[(tert‐Butyldiphenylsilyl)oxy]pyrrolidin‐1‐yl}‐3,4,4 a,5,6,7,8,8 a‐octahydroquinoxalin‐1(2H)‐yl)‐2‐(3,4‐dichlorophenyl)ethan‐1‐one (16)
TFA (1.68 mL, 22.6 mmol, 32 equiv) was added dropwise to a solution of 15 (520 mg, 0.7 mmol, 1 equiv) in CH2Cl2 (10 mL). The mixture was stirred at room temperature overnight. After addition of toluene, the solvents were removed in vacuo (azeotropic distillation). Then, NaOH (0.5 M, 20 mL) was added and the mixture was extracted with CH2Cl2 (3×20 mL). After removal of the solvent in vacuo, the residue was purified by flash column chromatography (Ø=3 cm, h=10 cm, CH2Cl2/CH3OH/NH3 (25 %) 94 : 5 : 1, V=30 mL, R f=0.60 (CH2Cl2/CH3OH/NH3 (25 %) 94 : 5 : 1)). Yellow oil, yield 410 mg (89 %). C36H45Cl2N3O2Si (650.8 g/mol). 1H NMR (600 MHz, CDCl3): δ (ppm)=0.98–1.11 (m, 9H, SiC(CH 3)3), 1.21–1.40 (m, 2H, 5‐CH 2, 7‐CH 2), 1.54–1.76 (m, 5H, 6‐CH 2, 7‐CH 2, NCH2CH 2CHOSi, NH), 1.84–2.09 (m, 2H, 5‐CH 2, NCH2CH 2CHOSi), 2.42–2.56 (m, 1H, NCH2CH2CHOSi), 2.59–2.88 (m, 4H, 3‐CH 2, NCH 2CHOSi), 2.90–3.08 (m, 2H, NCH2CH2CHOSi, 8‐CH), 3.20–3.38 (m, 2H, 2‐CH 2, 4a‐CH), 3.41–3.49 (m, 1H, 2‐CH 2), 3.57–3.79 (m, 2H, C(=O)CH 2‐aryl), 4.20–4.41 (m, 1H, NCH2CHOSi), 4.46–4.58 (m, 1H, 8a‐CH), 7.01–7.25 (m, 2H, 2‐CH arom, 6‐CH arom), 7.28–7.32 (m, 1H, 5‐CH arom), 7.33–7.45 (m, 6H, CH arom), 7.59–7.69 (m, 4H, CH arom). 13C NMR (151 MHz, CDCl3): δ (ppm)=19.2 (1 C, C‐6), 20.4 (1 C, SiC(CH3)3), 23.2 (1 C, C‐5), 26.7 (1 C, C‐7), 27.0 (3 C, SiC(CH3)3), 32.1 (1 C, NCH2 CH2CHOSi), 40.5 (1 C, C(=O)CH2‐aryl), 42.4 (1 C, C‐2), 44.3 (1 C, NCH2CHOSi), 47.2 (1 C, NCH2CH2CHOSi), 52.5 (1 C, C‐4a), 53.6 (1 C, C‐8a), 56.0 (1 C, C‐8), 59.4 (1 C, C‐3), 72.6 (1 C, NCH2 CHOSi), 127.7 (4 C, Carom), 128.1 (2 C, Carom), 128.7 (1 C, Carom), 129.6 (2 C, Carom), 130.4 (2 C, Carom), 130.6 (1 C, Carom), 131.2 (1 C, Carom), 132.4 (1 C, Carom), 135.8 (4 C, Carom), 169.7 (1 C, NC(=O)‐benzyl). IR (neat): ν (cm−1)=2932 (C−Haliph), 1632 C(=O), 1103 (N−C), 737 (1,2‐disubst. arom.), 702 (SiC6H5). Exact Mass (APCI): m/z 650.2755 (calcd. 650.2731 for C36H46 35Cl2N3O2Si [M+H]+). HPLC (Method Pu): purity 86 % (t R=22.55 min).
Methyl (4aR,5S,8aS)‐ and (4aS,5R,8aR)‐5‐{(S)‐3‐[(tert‐butyldiphenylsilyl)oxy] pyrrolidin‐1‐yl}‐4‐[2‐(3,4‐dichlorophenyl)acetyl]‐3,4,4 a,5,6,7,8,8 a‐octahydroquinoxaline‐1(2H)‐carboxylate (17)
Under a N2 atmosphere, pyridine (32 μL, 0.40 mmol, 5 equiv) was added to a solution of 16 (50 mg, 0.08 mmol, 1 equiv) in dry CH2Cl2 (0.2 mL). At 0 °C, a solution of methyl chloroformate (37 μL, 0.48 mmol, 6 equiv) in CH2Cl2 (0.2 mL) was added dropwise. The ice bath was removed and the mixture was stirred at room temperature for 1 h. H2O (10 mL) was added and the mixture was extracted with CH2Cl2 (3×10 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (Ø=2 cm, h=10 cm, CH2Cl2/CH3OH/NH3 (25 %) 97 : 2 : 1, V=12 mL, R f=0.84 (CH2Cl2/CH3OH/NH3 (25 %) 94 : 5 : 1)). Pale yellow oil, yield 44 mg (78 %). C38H47Cl2N3O4Si (708.8 g/mol). 1H NMR (400 MHz, CDCl3): δ (ppm)=1.01–1.10 (m, 9H, SiC(CH 3)3), 1.18–1.47 (m, 3H, 6‐CH 2, 7‐CH 2, 8‐CH 2), 1.65–1.88 (m, 4H, NCH2CH 2CHOSi, 6‐CH 2, 7‐CH 2), 2.05–2.17 (m, 1H, 8‐CH 2), 2.52–2.63 (m, 1H, NCH2CH2CHOSi), 2.66–2.73 (m, 1H, NCH 2CHOSi), 2.75–2.82 (m, 1H, NCH 2CHOSi), 2.84–2.99 (m, 2H, 5‐CH, NCH2CH2CHOSi), 3.25–3.43 (m, 1H, 3‐CH 2), 3.45–3.89 (m, 4H, 3‐CH 2, 8a‐CH, C(=O)CH 2‐aryl), 3.69 (s, 3H, CO2CH 3), 4.03–4.15 (m, 1H, 4a‐CH), 4.25–4.39 (m, 1H, NCH2CHOSi), 7.02 (d, J=8.0 Hz, 1H, 6‐CH arom), 7.27–7.31 (m, 2H, 2‐CH arom, 5‐CH arom), 7.32–7.47 (m, 6H, CH arom), 7.58–7.73 (m, 4H, CH arom). Signals for 2‐CH 2 protons are not seen in the spectrum. 13C NMR (101 MHz, CDCl3): δ (ppm)=19.2 (1 C, C‐7), 19.6 (1 C, SiC(CH3)3), 26.7 (1 C, C‐6), 27.1 (3 C, SiC(CH3)3), 30.5 (1 C, C‐8), 35.2 (1 C, NCH2 CH2CHOSi), 40.0 (1 C, C(=O)CH2‐aryl), 42.2 (1 C, 3‐CH 2), 49.0 (1 C, NCH2CH2CHOSi), 52.6 (1 C, C‐4a), 52.9 (1 C, CO2 CH3), 59.4 (1 C, C‐5), 59.8 (1 C, NCH2CHOSi), 72.5 (1 C, NCH2 CHOSi), 127.7 (4 C, Carom), 127.9 (2 C, Carom), 128.4 (1 C, Carom), 129.7 (2 C, Carom), 130.6 (2 C, Carom), 131.0 (1 C, Carom), 132.3 (1 C, Carom), 134.9 (1 C, Carom), 135.8 (4 C, Carom), 156.4 (1 C, CO2CH3), 169.8 (1 C, C(=O)‐benzyl). Signals for C‐2 and C‐8a carbon atoms are not seen in the spectrum. IR (neat): ν (cm−1)=2932 (C−Haliph), 1697 (N−C=O), 1643 (N−C=O), 1107 (N−C), 737 (1,2‐disubst. arom.), 702 (SiC6H5). Exact Mass (APCI): m/z 650.2751 (calcd. 650.2731 for C36H46 35Cl2N3O2Si [M−C2H2O2+H]+). HPLC (Method Pu): purity 85 % (t R=26.57 min).
Methyl (4aR,5S,8aS)‐ and (4aS,5R,8aR)‐4‐[2‐(3,4‐dichlorophenyl)acetyl]‐5‐[(S)‐3‐hydroxypyrrolidin‐1‐yl]‐3,4,4 a,5,6,7, 8,8 a‐octahydroquinoxaline‐1(2H)‐carboxylate (12c)
TBAF ⋅ 3H2O (178 mg, 0.56 mmol, 1.1 equiv) was added to a solution of 17 (364 mg, 0.51 mmol, 1 equiv) in THF (5 mL). The mixture was stirred at room temperature overnight. H2O (10 mL) was added and the mixture was extracted with CH2Cl2 (3×10 mL). The combined organic layers were dried (Na2SO4), filtered and the solvent was removed in vacuo. The residue was purified by flash column chromatography (Ø=2 cm, h=13 cm, CH2Cl2/CH3OH 9 : 1, V=12 cm, R f=0.42 (CH2Cl2/CH3OH/NH3 (25 %) 94 : 5 : 1) Colorless oil, yield 154 mg (64 %). C22H29Cl2N3O4 (470.4 g/mol). 1H NMR (600 MHz, CDCl3): δ (ppm)=1.31–1.44 (m, 2H, 6‐CH 2, 7‐CH 2), 1.47–1.59 (m, 1H, 8‐CH 2), 1.65–1.75 (m, 2H, 7‐CH 2, NCH2CH 2CHOH), 1.76–1.86 (m, 1H, 6‐CH 2), 1.98–2.08 (m, 1H, NCH2CH 2CHOH), 2.11–2.24 (m, 1H, 8‐CH 2), 2.39–2.60 (m, 1H, NCH 2CHOH), 2.61–2.75 (m, 1H, NCH2CH2CHOH), 2.76–2.98 (m, 2H, NCH2CH2CHOH, NCH 2CHOH), 3.01–3.16 (m, 1H, 5‐CH), 3.51–3.61 (m, 1H, 3‐CH 2), 3.63–3.80 (m, 3H, 8a‐CH, C(=O)CH 2‐aryl), 3.69 (s, 3H, CO2CH 3), 3.88–4.01 (m, 1H, 3‐CH 2), 4.12–4.34 (m, 2H, NCH2CHOH, 4a‐CH), 4.56 (brs, 1H, OH), 7.07–7.17 (m, 1H, 6‐CH arom), 7.32–7.41 (m, 2H, 2‐CH arom, 5‐CH arom). Signals for 2‐CH 2 protons are not seen in the spectrum. 13C NMR (151 MHz, CDCl3): δ (ppm)=19.4 (1 C, C‐7), 24.2 (1 C, C‐6), 30.5 (1 C, C‐8), 34.7 (1 C, NCH2 CH2CHOH), 40.4 (1 C, C(=O)CH2‐aryl), 41.1 (1 C, C‐3), 42.8 (1 C, C‐8a), 49.0 (1 C, NCH2CH2CHOH), 52.6 (1 C, C‐4a), 53.0 (1 C, CO2 CH3), 56.3 (1 C, NCH2CHOH), 60.0 (1 C, C‐5), 71.0 (1 C, NCH2 CHOH), 128.7 (1 C, Carom), 130.7 (2 C, Carom), 131.0 (1 C, Carom), 132.7 (1 C, Carom), 135.0 (1 C, Carom), 156.4 (1 C, CO2CH3), 170.9 (1 C, C(=O)‐benzyl). A signal for C‐2 carbon atom is not seen in the spectrum. IR (neat): ν (cm−1)=3402 (OH), 2940 (C−Haliph), 1690 (N−C=O), 1632 (N−C=O), 1107 (N−C), 772 and 683 (1,2‐disubst. arom.). Exact Mass (APCI): m/z 470.1565 (calcd. 470.1608 for C22H30 35Cl2N3O4 [M+H]+). HPLC (Method Pu): purity 99 % (t R=16.55 min).
Methyl (4aR,5S,8aS)‐ and (4aS,5R,8aR)‐4‐[2‐(3,4‐dichlorophenyl)acetyl]‐5‐[(S)‐3‐(methylsulfonyloxy)pyrrolidin‐1‐yl]‐ 3,4,4 a,5,6,7,8,8 a‐octahydroquinoxaline‐1(2H)‐carboxylate (19)
Triethylamine (66 μL, 0.5 mmol, 2.3 equiv) and 4‐(dimethylamino)pyridine (26 mg, 0.2 mmol, 1 equiv) were added to a solution of 12c (100 mg, 0.2 mmol, 1 equiv) in CH2Cl2 (10 mL). At 0 °C, methanesulfonyl chloride (33 μL, 0.4 mmol, 2 equiv) was added dropwise. The mixture was stirred at 0 °C for 30 min and then at room temperature for 16 h. After addition of H2O (20 mL), the mixture was extracted with CH2Cl2 (3×20 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (Ø=2 cm, h=15 cm, CH2Cl2/CH3OH 98 : 2, V=12 mL, R f=0.64 (CH2Cl2/CH3OH/NH3 (25 %) 89 : 10 : 1)). Yellow oil, yield 63 mg (55 %). C23H31Cl2N3O4S (548.5 g/mol). 1H NMR (600 MHz, CDCl3): δ (ppm)=1.35–1.50 (m, 2H, 6‐CH 2, 7‐CH 2), 1.65–1.78 (m, 2H, 6‐CH 2, 7‐CH 2), 1.94–2.01 (m, 1H, NCH2CH 2CHOMs), 2.02–2.24 (m, 3H, 8‐CH 2, NCH2CH 2CHOMs), 2.49–2.60 (m, 1H, NCH 2CH2CHOMs), 2.88–2.92 (m, 1H, NCH 2CHOMs), 2.94–3.03 (m, 5H, −CH 3 mesyl, 4a‐CH, NCH 2CH2CHOMs), 3.07–3.16 (m, 1H, NCH 2CHOMs), 3.38–3.50 (m, 1H, 2‐CH 2), 3.52–3.60 (m, 1H, 3‐CH 2), 3.61–3.82 (m, 5H, C(=O)CH 2‐aryl, 2‐CH 2, 3‐CH 2, 8a‐CH), 3.69 (s, 3H, CO2CH 3), 4.09–4.22 (m, 1H, 4a‐CH), 4.98–5.18 (m, 1H, NCH2CHOMs), 7.03–7.15 (m, 1H, 6‐CH arom), 7.31–7.36 (m, 1H, 2‐CH arom), 7.39 (m, 1H, 5‐CH arom). 13C NMR (151 MHz, CDCl3): δ (ppm)=19.4 (1 C, C‐7), 24.1 (1 C, C‐6), 29.8 (1 C, C‐8), 32.2 (1 C, NCH2 CH2CHOMs), 38.6 (1 C, CH3 mesyl), 40.2 (1 C, C(=O)CH2‐aryl), 40.4 (1 C, C‐8a), 41.7 (1 C, C‐3), 42.1 (1 C, C‐2), 47.6 (1 C, NCH2CH2CHOMs), 52.1 (1 C, C‐4a), 52.9 (1 C, CO2 CH3), 55.7 (1 C, NCH2CHOMs), 59.2 (1 C, C‐5), 80.7 (1 C, NCH2 CHOMs), 128.5 (1 C, Carom), 130.7 (2 C, Carom), 131.0 (1 C, Carom), 132.7 (1 C, Carom), 135.2 (1 C, Carom), 156.4 (1 C, CO2CH3), 170.0 (1 C, C(=O)‐benzyl). IR (neat): ν (cm−1)=2936 (C−Haliph), 1694 (N−C=O), 1639 (N−C=O), 1130 (N−C), 1261 and 1030 (O=S=O), 772 and 683 (1,2‐disubst. arom.). Exact Mass (APCI): m/z 548.1421 (calcd. 548.1383 for C23H32 35Cl2N3O4S [M+H]+). HPLC (Method Pu): purity 99 % (t R=18.02 min).
Receptor binding studies
Preparation of membrane homogenates from guinea pig brain41, 42, 43
Five guinea pig brains were homogenized with the potter (500‐800 rpm, 10 up‐and‐down strokes) in 6 volumes of cold 0.32 M sucrose. The suspension was centrifuged at 1200×g for 10 min at 4 °C. The supernatant was separated and centrifuged at 23 500×g for 20 min at 4 °C. The pellet was resuspended in 5–6 volumes of buffer (50 mM Tris, pH 7.4) and centrifuged again at 23500×g (20 min, 4 °C). This procedure was repeated twice. The final pellet was resuspended in 5–6 volumes of buffer and frozen (−80 °C) in 1.5 mL portions containing about 1.5 mg protein/mL.
Determination of KOR affinity (guinea pig brain)35, 36, 37, 38
The assay was performed with the radioligand [3H]U‐69,593 (55 Ci/mmol, Amersham, Little Chalfont, UK). The thawed guinea pig brain membrane preparation (about 100 μg of the protein) was incubated with various concentrations of test compounds, 1 nM [3H]U‐69,593, and TRIS‐MgCl2‐Puffer (50 mM, 8 mM MgCl2, pH 7.4) at 37 °C. The non‐specific binding was determined with 10 μM unlabeled U‐69,593. The Kd‐value of U‐69,593 is 0.69 nM.
Affinity towards MOR, DOR,35‐38 σ1 and σ2 recetors41, 42, 43
Materials, preparation of membrane homogenates from various tissues, protein determination, and the general procedure for binding assays are detailed in the Supporting Information.
In vitro functional assays
cAMP inhibition assay
To measure KOR Gαi‐mediated cAMP inhibition, HEK 293T (ATCC CRL‐11268) cells were co‐transfected with human KOR along with a luciferase‐based cAMP biosensor (GloSensor; Promega) and assays were performed similar to previously described.55 After 16 h, transfected cells were plated into Poly‐lysine coated 384‐well white clear bottom cell culture plates with DMEM+1 % dialyzed FBS at a density of 15 000–20 000 cells per 40 μL per well and incubated at 37 °C with 5 % CO2 overnight. The next day, drug solutions were prepared in fresh drug buffer (20 mM HEPES, 1x HBSS, 0.3 % bovine serum albumine (BSA), pH 7.4) at 3× drug concentration. Plates were decanted and received 20 μL per well of drug buffer (20 mM HEPES, 1x HBSS) followed by addition of 10 μL of drug solution (3 wells per condition) for 15 min in the dark at room temperature. To stimulate endogenous cAMP via β adrenergic‐Gs activation, 10 μL luciferin (4 mM final concentration) supplemented with isoproterenol (400 nM final concentration) were added per well. Cells were again incubated in the dark at room temperature for 15 min, and luminescence intensity was quantified using a Wallac TriLux microbeta (Perkin Elmer) luminescence counter. Results (relative luminescence units) were plotted as a function of drug concentration, normalized to % SalA stimulation, and analyzed using “log(agonist) vs. response” in GraphPad Prism 5.0.
Tango β‐arrestin‐2 recruitment assay
The KOR Tango constructs were designed and assays were performed as previously described.45, 56 HTLA cells expressing TEV fused‐β‐arrestin2 (kindly provided by Dr. Richard Axel, Columbia University) were transfected with the KOR Tango construct. The next day, cells were plated in DMEM supplemented with 1 % dialyzed FBS in poly‐L‐lysine coated 384‐well white clear bottom cell culture plates at a density of 10 000–15 000 cells/well in a total of 40 μL. The cells were incubated for at least 6 h before receiving drug stimulation. Drug solutions were prepared in drug buffer (20 mM HEPES, 1x HBSS, 0.3 % BSA, pH 7.4) at 3× and added to cells (20 μL per well) for overnight incubation. Drug solutions used for the Tango assay were exactly the same as used for the cAMP assay. The next day, medium and drug solutions were removed and 20 μL per well of BrightGlo reagent (purchased from Promega, after 1 : 20 dilution) was added. The plate was incubated for 20 min at room temperature in the dark before being counted using a luminescence counter. Results (relative luminescence units) were plotted as a function of drug concentration, normalized to % U‐50488 stimulation, and analyzed using “log(agonist) vs. response” in GraphPad Prism 5.0.
In vitro studies to assess the anti‐inflammatory activity in immune cells
Immune cell isolation and stimulation
C57BL/6 mice (wild‐type; purchased from Janvier Labs, Le Genest‐Saint‐Isle, France) and kappa‐opioid receptor deficient mouse mutants (KOR−/−, purchased from The Jackson Laboratory, Bar Harbor, ME) were used at the age of 8 to 12 weeks and housed under specific pathogen‐free conditions in microisolator cages. Mice were given chow and water ad libitum and animal experiments were performed with the approval of the State Review Board of North Rhine‐Westphalia according to the German law for animal welfare (reference numbers 81‐02.05.50.17.015). After sacrifice of mice peripheral lymph nodes, femurs and tibias were removed to isolate T cells or bone marrow cells (see below). Single cell suspensions of mouse lymph nodes were prepared according to standard methods. Subsequently, total T cells were isolated from cell suspensions using the Pan T Cell Isolation Kit II (Miltenyi Biotech, Bergisch Gladbach, Germany), activated for 12 h with phorbol 12‐myristate 13‐acetate (PMA, 5 ng/mL) and ionomycin (500 ng/mL) and cultured for 48 h in the presence of compounds 12 b, 12 c, 14 b and 14 c (1 μg/mL or 5 μg/mL each) or PBS as a control. Finally, cells were subjected to flow cytometry or cytokine quantification assays (see below).
For the generation of DC, bone marrow (bm) was collected from tibias and femurs of wild‐type and KOR−/− mice, single cell suspensions were prepared and cultured in the presence of 150 U/mL GM‐CSF and 75 U/mL IL‐4 (Biotechne, Minneapolis, MN) for 9 days. From day 7 to day 9 of culture, cells were stimulated with PMA (5 ng/mL) and ionomycin (500 ng/mL) in the presence or absence of compounds 12b, 12c, 14b or 14c (1 μg/mL or 5 g/mL each). On day 9, bmDC were harvested and used for flow cytometry analyses (see below).
Human DC and T cells were isolated from peripheral blood mononuclear cells (PBMC). Therefore, PBMC were purified from fully anonymized leukapheresis reduction chambers, obtained from the blood bank with the informed consent of healthy donors by Ficoll gradient centrifugation according to standard methods (Ficoll reagent was purchased from Merck, Darmstadt, Germany). Total DC or T cells were negatively enriched using the Pan‐DC Enrichment Kit or the Pan T Cell Isolation Kit (Miltenyi Biotech). All experiments were carried out according to the declaration of Helsinki and were approved by the ethical committee of the University of Münster Medical School (2008‐180‐f‐S). After isolation, human DC and T cells were activated for 12 h with PMA (5 ng/mL) and ionomycin (500 ng/mL) and cultured for additional 48 h in the presence of compounds 12b, 12c, 14b and 14c (1 or 5 g/mL each) or PBS (as a control) and subjected to flow cytometry analyses or cytokine quantification (see below).
Multicolor flow cytometry
The expression of cell surface and intracellular markers was analyzed by multicolor flow cytometry on a GalliosTM flow cytometer (Beckman Coulter, Krefeld, Germany) using the Kaluza software. For flow cytometry, mouse cells were stained in PBS using antibodies against CD3ϵ (clone 145‐2C11), CD25 (clone PC61), MHC‐II (major histocompatibility complex II; clone M5/114), CD69 (clone H1.2F3), F4/80 (clone BM8), and CD19 (clone 6D5), CD44 (clone IM7) and CD11c (clone N418); all purchased from Biolegend. Intracellular staining of IFN‐γ (clone XMG1.2) was performed after cell permeabilization using the Fix/Perm Buffer Set (Biolegend) according to the manufacturer's instructions.
Human cells were stained in PBS using antibodies against CD3 (clone OKT3), CD25 (clone BC96), HLA‐DR (clone L243), CD69 (clone FN50) and CD11c (clone 3.9); all purchased from Biolegend. Intracellular staining of IFN‐γ (clone 4S.B3) was performed after cell permeabilization using the Fix/Perm Buffer Set (Biolegend) according to the manufacturer's instructions. Isotype‐matched controls were included in each staining and apoptotic cells were identified using an annexin V apoptosis assay kit (Abcam, Cambridge, UK).
Cytokine quantification
Cytokine concentrations in cell culture supernatants of T cells or DC treated with 12b, 12c, 14b or 14c were quantified using LEGENDplex™ kits (Biolegend, San Diego, CA) according to the manufacturer's instructions. All samples were analyzed on a Gallios™ flow cytometer using the LEGENDplex™ Data Analysis Software (Biolegend) and cytokine concentrations were determined according to standard curves.
Statistics
All values are expressed as means ±SD. Statistically significant differences were assessed by one‐way analysis of variance (ANOVA) test, comparing more than two groups. The alpha‐level was set at a value of <0.05 in most and at a value of <0.01 in exceptional cases (e. g., Figure 3B) SigmaPlot 14 or GraphPad Prism 8 was used to analyze, plot, and illustrate data.
Radiosynthesis
General
The first step of the radiosynthesis was carried out on a modified PET tracer radiosynthesiser (TRACERLab FxFDG, GE Healthcare). The recorded data were processed by the TRACERLab Fx software (GE Healthcare). Separation and purification of the radiosynthesized compound [18F]‐2 were performed on the following semipreparative radio‐HPLC system A1: K‐500 and K‐501 pump, K‐2000 UV detector (Herbert Knauer GmbH), NaI(TI) Scintibloc 51 SP51 γ‐detector (Crismatec) and an ACE 5 AQ column (250 mm×10 mm). Method A1 started with a linear gradient from 0 to 100 % CH3CN in water (0.1 % TFA) over 45 min, holding for 5 min and followed by a linear gradient from 100 to 0 % CH3CN in water (0.1 % TFA) over 5 min, with λ=220 nm and a flow rate of 5.5 mL ⋅ min−1. Separation and purification of the radiosynthesized compound [18F]‐14c were performed on the following semipreparative radio‐HPLC system A2: two Variopumps (Scintomics), Smartline UV detector 2500 (Herbert Knauer GmbH), a GabiStar γ‐detector (Raytest Isotopenmessgeräte GmbH) and an ACE 5 AQ column (250 mm×10 mm). Method A2 started with a linear gradient from 20 % to 35 % CH3CN in water (0.1 % TFA) over 45 min and then from 35 % to 100 % in 5 min, followed by a linear gradient from 100 to 20 % CH3CN in water (0.1 % TFA) over 5 min, with λ=220 nm and a flow rate of 5.5 mL ⋅ min−1. Radiochemical purities and specific activities were determined using the analytical radio‐HPLC system B: Two Smartline 1000 pumps and a Smartline UV detector 2500 (Herbert Knauer GmbH), a GabiStar γ‐detector (Raytest Isotopenmessgeräte GmbH) and a Nucleosil 100‐5C‐18 column (250 mm×4 mm). Method B started with a linear gradient from 10 to 100 % CH3CN in water (0.1 % TFA) over 15 min, holding for 3 min followed by a linear gradient from 100 % to 10 % CH3CN in water (0.1 % TFA) over 2 min, with λ=210 nm and a flow rate of 1.0 mL ⋅ min−1. The recorded data of both HPLC‐systems were processed by the GINA Star software (Raytest Isotopenmessgeräte GmbH). No‐carrier‐added aqueous [18F]fluoride was produced on a RDS 111e cyclotron (CTI‐Siemens) by irradiation of a 2.8 mL water target using 10 MeV proton beams on 97.0 % enriched [18O]H2O by the 18O(p,n)18F nuclear reaction. After unloading the target, it was rinsed with ultrapure water (2.0 mL). This rinsed water containing [18F]fluoride ions was used for the radiosyntheses.
Radiosynthesis of 18F‐labeled tracers [18F]‐2 and [18F]‐14c
2‐(3,4‐Dichlorophenyl)‐1‐((4aR,8R,8aS)‐ and ((4aS,8S,8aR)‐ 4‐{[1‐(2‐[18F]fluoroethyl)‐1H‐1,2,3‐triazol‐4‐yl]methyl}‐8‐ (pyrrolidin‐1‐yl)‐3,4,4 a,5,6,7,8,8 a‐octahydroquinoxalin‐ 1(2H)‐yl)ethan‐1‐one ([18F]‐2)
In a computer controlled TRACERLab FxFDG Synthesizer the batch of aqueous [18F]fluoride ions (2280–4771 MBq) from the cyclotron target was passed through an anion exchange resin (pre‐conditioned Sep‐Pak® Light QMA cartridge with CO3 2− as counter ion). [18F]Fluoride ions were eluted from the resin to the reactor with a mixture of 1 M K2CO3 (40 μL), water for injection (200 μL), and DNA‐grade CH3CN (800 μL) containing Kryptofix®2.2.2 (K222) (20 mg, 53 μmol). Subsequently, the aqueous K(K222)[18F]F solution was carefully evaporated to dryness in vacuo.
The precursor 2‐azidoethyl 4‐methylbenzenesulfonate (20 mg, 83 μmol) in DNA‐grade CH3CN (500 μL) was added and the mixture was heated at 110 °C for 3 min. The radiolabeled synthon 1‐azido‐2‐[18F]fluoroethane was distilled from the reactor into a 5 mL flask that contained 0.4 M CuSO4 ⋅ 5H2O aqueous solution (120 μL), sodium ascorbate (16 mg, 81 μmol) in water for injection (100 μL) and alkyne 18 (5.0 mg, 11.5 μmol) in DMF (400 μL) and the mixture was cooled to −10 °C. After 30 min stirring at 40 °C, the mixture was passed through a Waters Sep‐Pak Light cartridge filled with quartz wool. The cartridge was rinsed with DMF (200 μL) and the resulting clear solution was purified by gradient‐radio‐HPLC system A1, method A1. The fraction containing compound [18F]‐2 (t R([18F]‐2)=25.5 min) was evaporated to dryness in vacuo and formulated in water for injection/EtOH (9 : 1 v/v; 1 mL). [18F]‐2 was obtained in an overall rcy of 49.9±8.6 % (d.c., based on cyclotron‐derived [18F]fluoride ions, n=5) in 136±12 min from the end of radionuclide production. [18F]‐2 was isolated in rcp of 99.3±0.4 % with A m in the range of 4.6–14.6 GBq/μmol at the end of the synthesis. Rcp and A m of [18F]‐2 (t R([18F]‐2)=8.98–9.15 min) were determined by analytical radio‐HPLC B, method B. Radiochemical identity of [18F]‐2 was determined by co‐elution of this compound with the non‐radioactive counterpart 2, that was added to a preparation of [18F]‐2, at the analytical radio‐HPLC B (method B) detected by the γ detector ([18F]‐2) and the UV detector (2), respectively.
Methyl (4aR,5S,8aS)‐ and (4aS,5R,8aR)‐4‐[2‐(3,4‐dichlorophenyl)acetyl]‐5‐[(R)‐3‐[18F]fluoropyrrolidin‐1‐yl]‐3,4,4 a,5,6,7, 8,8 a‐octahydroquinoxaline‐1(2H)‐carboxylate ([18F]‐14c)
Separation of [18F]fluoride, formation and evaporation of K(K222)[18F]F in the TRACERLab FxFDG Synthesiser were performed as described in the procedure for the preparation of [18F]‐2 (see above). A solution of the precursor mesylate 19 (5.0 mg, 9.1 μmol) in DNA‐grade CH3CN (1.0 mL) was added and the mixture was heated at 110 °C for 20 min. Then, the solution was cooled to 55 °C, water for injection (10 mL) was added and the mixture was passed through a Waters Sep‐Pak C18 Light cartridge (preconditioned with 10 mL ethanol and 10 mL water). The cartridge was washed with water for injection (10 mL) and eluted with a DMF (0.5 mL) that was preheated to 100 °C. The eluate was diluted with water for injection (0.5 mL) and this product solution was purified by gradient‐radio‐HPLC system A2 (method A2). The fraction containing compound [18F]‐14c (t R([18F]‐14c)=35.0 min) was evaporated to dryness in vacuo and formulated in water for injection/EtOH (9 : 1, v/v; 1 mL). [18F]‐14c was obtained in rcy of 20.7±3.4 % (n=5; d.c. based on cyclotron‐derived [18F]fluoride ions) in 128±14 min from the end of radionuclide production. The target compound was isolated rcp of 98.9±0.7 % with A m of 4.0–111.3 GBq/μmol at the end of the synthesis. Rcp and A m of [18F]‐14c (t R([18F]‐14c)=11.33–11.58 min) were determined by analytical radio‐HPLC B, method B. Radiochemical identity of [18F]‐14c was determined analogously to [18F]‐2.
Determination of the distribution coefficient logD7.4
The lipophilicity of radioligands [18F]‐2 and [18F]‐14c was assessed by determination of the water n‐octan‐1‐ol distribution coefficient (logD) following a published procedure.57 In brief, approximately 3.2 MBq of radioligand [18F]‐2 and approximately 0.3 MBq of radiotracer [18F]‐14c, respectively, were mixed with equal amounts (0.6 mL) of phosphate buffered saline (PBS, pH 7.4) and n‐octan‐1‐ol. The resulting biphasic system was mixed vigorously for 3 min at room temperature and subsequently, the tubes were centrifuged (≥5 min). Then 400 μL of the n‐octan‐1‐ol layer was separated and 400 μL of PBS were added. The mixture was mixed vigorously for 10 min at room temperature followed by centrifugation of the tubes for≥5 min. Two samples of 100 μL of each layer were counted in a γ‐counter (2480 Automatic Gamma Counter Wizard2, Perkin‐Elmer). The distribution coefficient was determined by calculating the ratio cpm(n‐octan‐1‐ol)/cpm(PBS) and expressed as logD 7.4 (log(cpmn‐octanol/cpmPBS)). Three independent experiments were performed in duplicate and data were provided as mean values ±standard deviation.
Animal experiments
All animal experiments were performed in accordance with the legal requirements of the European Community (Directive 2010/63/EU) and the corresponding German Animal Welfare Law (TierSchG, TierSchVersV) and were approved by the local authorizing agency (State Office for Nature, Environment and Consumer Protection North Rhine‐Westphalia; in vivo biodistribution studies: permit no. 84‐02. 04. 2013.A046 and 84‐02.05.30.12.A084.
Stability of [18F]‐2 and [18F]‐14c in mouse and human serum (in vitro)
The serum stability of radioligands [18F]‐2 and [18F]‐14c was evaluated by incubation in mouse or human serum at 37 °C for up to 90 min. An aliquot of formulated [18F]‐2 or [18F]‐14c (20 μL, ca. 6.6 MBq and ca.10 MBq, respectively) was added to a sample of mouse serum (200 μL) or human serum (200 μL), and the mixtures were incubated at 37 °C. Samples of 20 μL each were taken after periods of 10, 30, 60 and 90 min and ice‐cold DNA‐grade CH3CN (100 μL) were added. After centrifugation for ≥5 min, the supernatant was analyzed by analytical radio‐HPLC B, method B.
In vivo Biodistribution studies of [18F]‐2and [18F]‐14c
Adult C57BL/6 mice (n=2 male, 15 weeks, 28 g for [18F]‐2; n=2, male, 10 weeks, 25 g for [18F]‐14c) were anesthetized by isoflurane/O2 and one lateral tail vein was cannulated BY using a 27 G needle connected to 15 cm polyethylene catheter tubing. Radiotracers (∼500 kBq/g bodyweight, 15 MBq) were injected as a bolus (50 μL compound flushed with 100 μL saline) via the tail vein and subsequent PET scanning was performed.
Small Animal PET Scanning. PET experiments were carried out using a submillimeter high resolution (0.7 mm full width at half‐maximum) small animal scanner (32 module quadHIDAC, Oxford Positron Systems Ltd., Oxford, UK) with uniform spatial resolution (<1 mm) over a large cylindrical field (165 mm diameter, 280 mm axial length). List‐mode data were acquired for 90 min and reconstructed into dynamic time frames using an iterative reconstruction algorithm. Subsequently, the scanning bed was transferred to the computed tomography (CT) scanner (Inveon, Siemens Medical Solutions, U.S.), and a CT acquisition with a spatial resolution of 80 μm was performed for each mouse. Reconstructed image data sets were coregistered based on extrinsic markers attached to the multimodal scanning bed and the in‐house developed image analysis software MEDgical. Three‐dimensional volumes of interest (VOIs) were defined over the respective organs in CT data sets, transferred to the coregistered PET data, and analyzed quantitatively. Regional uptake was calculated as percentage of injected dose by dividing counts per milliliter in the VOI by total counts in the mouse multiplied by 100 (%ID/mL).
Biostability and biotransformation of [18F]‐2 in mice (in vivo)
Approximately 72 MBq of [18F]‐2 (in a volume of 100 μL) were injected into the tail vein of each of three 11‐week‐old C57/BL6 mice. The animals were sacrificed 90 min after injection. Whole blood of the three mice was pooled (∼2 mL) and centrifuged (3 min) to isolate plasma. Plasma (∼0.7 mL) was separated and mixed with ice‐cold CH3CN (1.4 mL). After further centrifugation for 5 min, the supernatant was separated and evaporated to dryness in vacuo at moderate temperatures (≤60 °C). The residue was dissolved in water for injection (20 μL) and the solution was analyzed by analytical radio‐HPLC B, method B.
Biotransformation of 14c using mouse liver microsomes (in vitro)
LC‐MS setup (Method LC‐MS)
For the determination of exact masses and for conducting MS/MS experiments, an LC system was coupled with a quadrupole time‐of‐flight (qToF) mass spectrometer.
HPLC‐DAD (Thermo Fisher Scientific): Solvent rack (SRD 3600); pump (DGP‐3600RS); autosampler (WPS‐3000RS); column oven (TCC‐3000RS); precolumn: SecurityGuardTM Cartridge AQ C18 (4.0×2.0 mm, 4.0 μm particle size); column: SynergiTM Hydro‐RP (50×2.1 mm, 2.5 μm particle size, Phenomenex®, Aschaffenburg, Germany); temperature: 30 °C and DAD‐detector (DAD‐3000RS). The LC system was coupled with a micrOTOF‐Q II (Bruker Daltonics, Bremen, Germany). The ESI‐qToF was operated in positive ion polarity in the full scan mode (m/z 70–700, 200–1000 or 500–1600) with the following settings: capillary voltage 4500 V; end plate offset −500 V; collision cell RF 300.0 Vpp; nebulizer 2.0 bar; dry heater 200 °C; dry gas 9.0 L/min. To protect the MS from salts or other components of the matrices, a six‐port valve was used to elute the first 2.0 min of each run into the waste (cut‐off). In case of MS/MS experiments the isolation window of the first quadrupole was set to 10 m/z units (for m/z<600) or 20 m/z units (for m/z>600). The collision energy of the second quadrupole was set in the range of 10–35 eV and is given for each experiment. For data handling and control of the system the software Data Analysis and Hystar from Bruker Daltonics (Bremen, Germany) was used. The calibration of the ToF spectra was achieved by injection of LiHCO2 (m/z<700, i‐propanol/H2O 1 : 1, 10 mM) via a 20 μL sample loop within each LC run at 2.0–2.2 min.
LC parameters: mobile phase A: H2O/CH3CN 90 : 10+0.1 % FA; mobile phase B: CH3CN/H2O 90 : 10+0.1 % FA; mobile phase C: H2O+0.1 % FA; pump 1: flow rate: 0–3 min: 0.1 mL/min, 3–3.1 min: from 0.1 mL/min to 0.4 mL/min, 3.1–17.9 min: 0.4 mL/min, 17.9–18 min: from 0.4 mL/min to 0.1 mL/min; gradient elution: (A%): 0–3.1 min: 100 %, 3.1–12 min: gradient from 100 % to 0 %, 12–14.5 min: 0 %, 14.5–15 min: gradient from 0 % to 100 %, 15–18 min: 100 %; pump 2: flow rate: 0–3 min: 0.3 mL/min, 3–3.1 min: from 0.3 mL/min to 0.0 mL/min, 3.1–17.9 min: 0.0 mL/min, 17.9–18 min: from 0.0 mL/min to 0.3 mL/min; isocratic: (C %): 0–18 min: 100 %.
Experimental procedure
Mouse liver microsomes (7.8 mg protein/mL) were added to an Eppendorf cap filled with sodium phosphate buffer pH 7.4 (PBS, 0.1 M), MgCl2 solution (0.05 M), NADPH (2 mg/mL in PBS) and DMSO stock solution, giving a total volume of 200 μL. Final concentrations for the incubations were 75 mM PBS, 0.6 mM NADPH, 1 mg/mL microsomal protein, 50 μM of the respective compound, 12.5 mM MgCl2 and 0.5 % DMSO. For murine serum stability, the respective compound (1 μL, 10 mM DMSO stock solution) was incubated in murine serum (black six mice, 199 μL). The suspension was mixed vigorously and incubated (37 °C, 90 min, 900 rpm). Subsequently, the incubation was stopped by addition of CH3CN/CH3OH (1 : 1, 400 μL), the caps were cooled down (0 °C, 10 min) and the precipitated proteins were separated by centrifugation (4 °C, 15 min, 16000 rpm). Afterward, the supernatant was analyzed by the LC‐MS method described above. With the same procedure, the empty value (without stock solution), blank value (without NADPH), buffer sample (parent incubated in PBS solution) and negative control (solvent (599 μL) and DMSO stock solution (1 μL)) were prepared.
Supporting Information
Supporting Information contains details on the receptor binding studies, quality control of [18F]‐2 and its stability in blood serum, investigation of in vivo radiometabolites of [18F]‐2, distribution of the PET tracer [18F]‐14c in various organs, in vitro biotransformation of 14c, identification of metabolites of 14c formed in vitro and purity data of all test compounds. Moreover, the 1H and 13C NMR spectra of all synthesized compounds are included. Molecular formula strings are added.
Abbreviations
- Am
molar activity
- APCI
atmospheric pressure chemical ionization
- BBB
blood brain barrier
- bm
bone marrow
- bmDC
bone marrow‐derived dendritic cells
- cpm
counts per minute
- CSF
cerebrospinal fluid
- CT
computer tomography
- d.c.
decay corrected
- DC
dendritic cells
- DOR
δ‐opioid receptor
- EAE
experimental autoimmune encephalomyelitis
- H&E
hematoxylin and eosin
- INF‐β
interferone β
- INF‐γ
interferone γ
- IL‐10
interleukin‐10
- IL‐17
interleukin‐17
- Iono
ionomycin
- KOR
κ‐opioid receptor
- l
length of the stationary phase
- MOG
myelin oligodendrocyte glycoprotein
- MOR
μ‐opioid receptor
- MS
multiple sclerosis
- OPC
oligodendrocyte progenitor cells
- PBMC
peripheral blood mononuclear cells
- PBS
phosphate buffered saline
- PET
positron emission tomography
- PMA
phorbol 12‐myristate 13‐acetate
- PML
progressive multifocal leukoencephalopathy
- rcp
radiochemical purity
- rcy
radiochemical yield
- TBAF
tetrabutylammonium fluoride
- TBDPS
tert‐butyldiphenylsilyl
- TH1
T helper cells 1
- TH17
T helper cells 17
- Treg
regulatory T cells
- V
fraction size
- VOI
volume of interest
- WT
wild‐type
Conflict of interest
The authors declare no conflict of interests.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support of this project by the Deutsche Forschungsgemeinschaft (DFG, SFB‐TR 128 project A10 to K.L., SFB 1009 project B07 to K.L., project 817/5‐1 to K.L., Cluster of excellence Cells in Motion, project FF‐2016‐11 to K.L. and B.W.) as well as the Interdisciplinary Center for Clinical Research (IZKF, project Lo2/004/16 to K.L. and IZKF core unit PIX) is gratefully acknowledged. Support was also given by NIDA (Grant PO1 DA035764 to T.C.) and the NIMH Psychoactive Drug Screening Program for binding assays. We would like to thank Meike Steinert, Kerstin Vischedyk, Christine Bätza, Steffi Bouma, Sarah Köster, and Roman Priebe for excellent technical assistance. Open access funding enabled and organized by Projekt DEAL.
G. Tangherlini, F. Börgel, D. Schepmann, S. Slocum, T. Che, S. Wagner, K. Schwegmann, S. Hermann, N. Mykicki, K. Loser, B. Wünsch, ChemMedChem 2020, 15, 1834.
References
- 1. Pasternak G. W., Pan Y.-X., Pharmacol. Rev. 2013, 65, 1257–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pasternak G., Pan Y. X., Acta Anaesthesiol. Taiwanica 2011, 49, 21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dhawan B. N., Cesselin F., Raghubir R., Reisine T., Bradley P. B., Portoghese P. S., Hamon M., Pharmacol. Rev. 1996, 48, 567–592. [PubMed] [Google Scholar]
- 4. Vanderah T. W., Clin. J. Pain 2010, 26 (suppl.10), 10–15. [DOI] [PubMed] [Google Scholar]
- 5. Chavkin C., Neuropsychopharmacology 2011, 36, 369–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rios M., Neuropsychopharmacology 2011, 36, 368–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mague S. D., Pliakas A. M., Todtenkopf M. S., Tomasiewicz H. C., Zhang Y., Jr Stevens W. C., Jones R. M., Portoghese P. S., Jr Carlezon W. A., Pharmacol. Ther. 2003, 305, 323–330. [DOI] [PubMed] [Google Scholar]
- 8. Metcalf M. D., Coop A., AAPS J. 2005, 7, Article 71, E704–E271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Agostimho A. S., Mietzsch M., Zangrandi L., Kmec I., Mutti A., Kraus L., Fidzinski P., Schneider U. C., Holtkamp M., Heilbronn R., Schwarzer C., EMBO Mol. Med. 2019, 11, e9963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lalanne L., Ayranci G., Kieffer B. L., Lutz P. E., Front. Psychiatry 2014, 5, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lynch J. L., Alley J. F., Wellman L., Beitz A. J., Brain Res. 2008, 1191, 180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Du C., Duan Y., Wei W., Cai Y., Chai H., Lv J., Du X., Zhu J., Xie X., Nat. Commun. 2016, 7, 11120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tangherlini G., Kalinin D. V., Schepmann D., Che T., Mykicki N., Ständer S., Loser K., Wünsch B., J. Med. Chem. 2019, 62, 893–907. [DOI] [PubMed] [Google Scholar]
- 14. Talbot P. S., Narendran R., Butelman E. R., Huang Y., Ngo K., Slifstein M., Martinez D., Laruelle M., Hwang D.-R., J. Nucl. Med. 2005, 46, 484–494. [PubMed] [Google Scholar]
- 15. Hayes A. G., Birch P. J., Hayward N. J., Sheehan M. J., Rogers H., Tyers M. B., Judd D. B., Scopes D. I., Naylor A., Br. J. Pharmacol. 1990, 101, 944–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nabulsi N. B., Zheng M.-Q., Ropchan J., Labaree D., Ding Y.-S., Blumberg L., Huang Y., Nucl. Med. Biol. 2011, 38, 215–221. [DOI] [PubMed] [Google Scholar]
- 17. Schoultz B. W., Hjornevik T., Willoch F., Marton J., Noda A., Murakami Y., Miyoshi S., Nishimura S., Årstad E., Drzezga A., Matsunari I., Henriksen G., Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1174–1180. [DOI] [PubMed] [Google Scholar]
- 18. Naganawa M., Jacobsen L. K., Zheng M.-Q., Lin S.-F., Banerjee A., Byon W., Weinzimmer D., Tomasi G., Nabulsi N., Grimwood S., Badura L. L., Carson R. E., McCarthy T. J., Huang Y., NeuroImage 2014, 99, 69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Placzek M. S., Van de Bittner G. C., Wey H.-Y., Lukas S. E., Hooker J. M., Neuropsychopharmacology, 2015, 40, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miller J. M., Zanderigo F., Purushothaman P. D., DeLorenzo C., Rubin-Falcone H., Ogden R. T., Keilp J., Oquendo M. A., Nabulsi N., Huang Y. H., Parsey R. V., Carson R. E., Mann J. J., Synapse, 2018, 72, e22042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li S., Zheng M.-Q., Naganawa M., Kim S., Gao H., Kapinos M., Labaree D., Huang Y., J. Nucl. Med. 2019, 60, 1023–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zheng M.-Q., Nabulsi N., Kim S. J., Tomasi G., Lin S.-F., Mitch C., Quimby S., Barth V., Rash K., Masters J., Navarro A., Seest E., Morris E. D., Carson R. E., Huang Y., J. Nucl. Med. 2013, 54, 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mitch C. H., Quimby S. J., Diaz N., Pedregal C., De La Torre M. G., Jimenez A., Shi Q., Canada E. J., Kahl S. D., Statnick M. A., McKinzie D. L., Benesh D. R., Rash K. S., Barth C. N., J. Med. Chem. 2011, 54, 8000–8012. [DOI] [PubMed] [Google Scholar]
- 24. Naganawa M., Zheng M.-Q., Henry S., Nabulsi N., Lin S.-F., Ropchan J., Labaree D., Najafzadeh S., Kapinos M., Tauscher J., Neumeister A., Carson R. E., Huang Y., J. Nucl. Med. 2015, 56, 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Naganawa M., Dickinson G. L., Zheng M. Q., Henry S., Vandenhende F., Witcher J., Bell R., Nabulsi N., Lin S. F., Ropchan J., Neumeister A., Ranganathan M., Tauscher J., Huang Y., Carson R. E., J. Pharmacol. Exp. Ther. 2016, 356, 260–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim S. J., Zheng M.-Q., Nabulsi N., Labaree D., Ropchan J., Najafzadeh S., Carson R. E., Huang Y., Morris E. D., J. Nucl. Med. 2013, 54, 1668–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cai Z., Li S., Pracitto R., Navarro A., Shirali A., Ropchan J., Huang Y., ACS Chem. Neurosci. 2017, 8, 12–16. [DOI] [PubMed] [Google Scholar]
- 28. Li S., Cai Z., Zheng M.-Q., Holden D., Naganawa M., Lin S.-F., Ropchan J., Labaree D., Kapinos M., Lara-Jaime T., Navarro A., Huang Y., J. Nucl. Med. 2018, 59, 140–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Naganawa M., Li S., Nabulsi N., Najafzadeh S., Gao H., Cai Z., Kapinos M., Ropchan J., Carson R., Huang Y., J. Nucl. Med. 2019, 60, supplement 1, 577.30602593 [Google Scholar]
- 30. Poinsel G., Oueslati F., Dhilly M., Delamare J., Perrio C., Debruyne D., Barré L., Nucl. Med. Biol. 2008, 35, 561–569. [DOI] [PubMed] [Google Scholar]
- 31. Schmitt S., Colloc'h N., Perrio C., Eur. J. Med. Chem. 2015, 90, 742–750. [DOI] [PubMed] [Google Scholar]
- 32. Schmitt S., Delamare J., Tirel O., Fillesoye F., Dhilly M., Perrio C., Nucl. Med. Biol. 2016, 44, 50–61. [DOI] [PubMed] [Google Scholar]
- 33. Molenveld P., Bouzanne Des Mazery R., Sterk G. J., Storcken R. P. M., Autar R., Van Oss B., Van Der Haas R. N. S., Fröhlich R., Schepmann D., Wünsch B., Soeberdt M., Bioorg. Med. Chem. Lett. 2015, 25, 5326–5330. [DOI] [PubMed] [Google Scholar]
- 34. Soeberdt M., Molenveld P., Storcken R. P. M., Bouzanne Des Mazery R., Sterk G. J., Autar R., Bolster M. G., Wagner C., Aerts S. N. H., Van Holst F. R., Wegert A., Tangherlini G., Frehland B., Schepmann D., Metze D., Lotts T., Knie U., Lin K. Y., Huang T. Y., Lai C. C., Ständer S., Wünsch B., Abels C., J. Med. Chem. 2017, 60, 2526–2551. [DOI] [PubMed] [Google Scholar]
- 35. Bourgeois C., Werfel E., Galla F., Lehmkuhl K., Torres-Gómez H., Schepmann D., Kögel B., Christoph T., Straßburger W., Englberger W., Soeberdt M., Hüwel S., Galla H. J., Wünsch B., J. Med. Chem. 2014, 57, 6845–6860. [DOI] [PubMed] [Google Scholar]
- 36. Wittig C., Schepmann D., Soeberdt M., Daniliuc C. G., Wünsch B., Org. Biomol. Chem. 2017, 15, 6520–6540. [DOI] [PubMed] [Google Scholar]
- 37. Geiger C., Zelenka C., Lehmkuhl K., Schepmann D., Englberger W., Wünsch B., J. Med. Chem. 2010, 53, 4212–4222. [DOI] [PubMed] [Google Scholar]
- 38. Kracht D., Rack E., Schepmann D., Fröhlich R., Wünsch B., Org. Biomol. Chem. 2010, 8, 212–225. [DOI] [PubMed] [Google Scholar]
- 39. Costa B. R., De Bowen W. D., Hellewell S. B., George C., Rothman R. B., Reid A. A., Walker J. M., Jacobson A. E., Rice K. C., J. Med. Chem. 1989, 32, 1996–2002. [DOI] [PubMed] [Google Scholar]
- 40. Radesca L., Bowen W. D., Di Paolo L., de Costa B. R., J. Med. Chem. 1991, 34, 3058–3065. [DOI] [PubMed] [Google Scholar]
- 41. Hasebein P., Frehland B., Lehmkuhl K., Fröhlich R., Schepmann D., Wünsch B., Org. Biomol. Chem. 2014, 12, 5407–5426. [DOI] [PubMed] [Google Scholar]
- 42. Meyer C., Neue B., Schepmann D., Yanagisawa S., Yamaguchi J., Würthwein E.-U., Itami K., Wünsch B., Bioorg. Med. Chem. 2013, 21, 1844–1856. [DOI] [PubMed] [Google Scholar]
- 43. Miyata K., Schepmann D., Wünsch B., Eur. J. Med. Chem. 2014, 83, 709–716. [DOI] [PubMed] [Google Scholar]
- 44. Zheng Z., Huang X.-P., Mangano T. J., Zou R., Chen X., Zaidi S. A., Roth B. L., Stevens R. C., Katritch V., J. Med. Chem. 2017, 60, 3070–3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kroeze W. K., Sassano M. F., Huang X.-P., Lansu K., McCorvy J. D., Giguère P. M., Sciaky N., Roth B. L., Nat. Struct. Mol. Biol. 2015, 22, 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liang X., Liu R., Chen C., Ji F., Li T., Transl. Perioper. pain Med. 2016, 1, 5–13. [PMC free article] [PubMed] [Google Scholar]
- 47. Tai Y., Wang Q., Korner H., Zhang L., Wei W., Front. Pharmacol. 2018, 9, 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sozzani S., Del Prete A., Bosisio D., J. Autoimmune Dis. 2017, 85, 126–140. [DOI] [PubMed] [Google Scholar]
- 49. Reis e Sousa C., Curr. Opin. Immunol. 2004, 16, 21–25. [DOI] [PubMed] [Google Scholar]
- 50. Joffre O., Nolte M. A., Spörri R., Reis e Sousa C., Immunol. Rev. 2009, 227, 234–247. [DOI] [PubMed] [Google Scholar]
- 51. Kunkl M., Frascolla S., Amormino C., Volpe E., Tuosto L., Cells 2020, 9, E482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rendon A., Schäkel K., Int. J. Mol. Sci. 2019, 20, E1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yang P., Qian F. Y., Zhang M. F., Xu A. L., Wang X., Jiang B. P., Zhou L. L., J. Leukocyte Biol. 2019, 106, 1233–1240. [DOI] [PubMed] [Google Scholar]
- 54. Park K. B., Kitteringham N. R., O'Neill P. M., Annu. Rev. Pharmacol. Toxicol. 2001, 41, 443–4470. [DOI] [PubMed] [Google Scholar]
- 55. Che T., Majumdar S., Zaidi S. A., Ondachi P., McCorvy J. D., Wang S., Mosier P. D., Uprety R., Vardy E., Krumm B. E., Han G. W., Lee M. Y., Pardon E., Steyaert J., Huang X. P., Strachan R. T., Tribo A. R., Pasternak G. W., Carroll F. I., Stevens R. C., Cherezov V., Katritch V., Wacker D., Roth B. L., Cell 2018, 172, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu W., Wacker D., Gati C., Han G. W., James D., Wang D., Nelson G., Weierstall U., Katritch V., Barty A., Zatsepin N. A., Li D., Messerschmidt M., Boutet S., Williams G. J., Koglin J. E., Seibert M. M., Wang C., Shah S. T. A., Basu S., Fromme R., Kupitz C., Rendek K. N., Grotjohann I., Fromme P., Kirian R. A., Beyerlein K. R., White T. A., Chapmann H. N., Caffrey M., Spence J. C. H., Stevens R. C., Cherezov V., Science 2013, 342, 1521–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Prante O., Hocke C., Löber S., Hübner H., Gmeiner P., Kuwert T., Nuklearmedizin 2006, 45, 41–48. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary