

Abstract
RNA‐cleaving deoxyribozymes can serve as selective sensors and catalysts to examine the modification state of RNA. However, site‐specific endonuclease deoxyribozymes that selectively cleave post‐transcriptionally modified RNA are extremely rare and their specificity over unmodified RNA is low. We report that the native tRNA modification N6‐isopentenyladenosine (i6A) strongly enhances the specificity and has the power to reconfigure the active site of an RNA‐cleaving deoxyribozyme. Using in vitro selection, we identified a DNA enzyme that cleaves i6A‐modified RNA at least 2500‐fold faster than unmodified RNA. Another deoxyribozyme shows unique and unprecedented behaviour by shifting its cleavage site in the presence of the i6A RNA modification. Together with deoxyribozymes that are strongly inhibited by i6A, these results highlight that post‐transcriptional RNA modifications modulate the catalytic activity of DNA in various intricate ways.
Keywords: deoxyribozymes, epitranscriptomics, in vitro selection, RNA modification, site-specific RNA cleavage
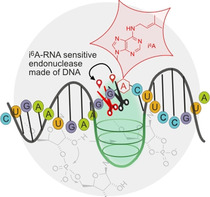
The natural tRNA modification i6A carries an isopentenyl (prenyl) group at the N 6 position of adenine. This modified nucleotide finetunes the active site of RNA‐cleaving deoxyribozymes, resulting in a distinct shift of the cleavage site. DNA catalysts are reported to exclusively cut post‐transcriptionally modified RNAs.
Introduction
Cellular RNAs contain a variety of chemically diverse post‐transcriptional modifications with important cellular functions.1 Besides sophisticated analytical methods that generate transcriptome‐wide modification maps, simple tools are required to validate predictions and to examine the modification state of distinct sites.2 Focusing on N 6‐methyladenosine (m6A) as one of the most abundant mRNA modifications, we have recently reported m6A‐sensitive RNA‐cleaving (endonuclease) deoxyribozymes for the site‐specific interrogation of m6A modification levels.3 Deoxyribozymes with endonuclease activity bind to the target RNA via Watson–Crick base‐pairing and catalyse site‐specific RNA cleavage by promoting the attack of a specific 2′‐hydroxy group onto the adjacent phosphodiester linkage, resulting in the formation of 2′,3′‐cyclic phosphate and 5′‐hydroxy termini in the fragments of the target RNA.4 Inhibition of this mechanisms by 2′‐O‐methylated nucleotides was used to analyse ribose methylation in rRNA.5 The m6A‐sensitive DNAzymes were generally applicable to analyse the presence of m6A in DGACH sequence motifs, as shown for lncRNAs and a set of C/D box snoRNAs,3 and these analyses were recently extended to other RNAs.6 The detailed mechanism, how DNA enzymes recognize m6A and modulate the cleavage response is currently unknown. Best discrimination was observed by the VMC10 DNA enzyme, which was inhibited by m6A and cleaved unmodified RNA up to 150‐fold faster. In contrast, DNA enzymes that preferred cleavage of m6A‐RNA were less specific and reached only 5–10‐fold faster cleavage rates for modified versus unmodified RNA. From these observations, we concluded that a modified nucleotide like m6A can easily interfere with formation of a catalytically competent DNA structure, but the opposite direction is more difficult: it is challenging to evolve catalytic DNA that is strictly dependent on the presence of a small chemical modification in the RNA target. Along these lines, one may expect that larger RNA modifications could establish energetically favourable interactions, and result in deoxyribozymes that more effectively discriminate modified from unmodified RNA. To explore this hypothesis, we chose to evolve deoxyribozymes that selectively cleave N 6‐isopentenyladenosine (i6A)‐modified RNA and compared their activity to m6A‐sensitive endonucleases.
N 6‐isopentenyladenosine (i6A) is a structural analogue of m6A that contains the bulkier dimethylallyl group as N 6‐substituent. The i6A modification is conserved in certain tRNAs in bacteria and eukaryotes.7 Located at position 37 next to the anticodon, i6A is suggested to enhance translation efficiency and fidelity by stabilizing cognate codon‐anticodon interactions,8 and may facilitate RNA localization by associating with membranes.9 In some cases, i6A is further modified, for example by thiomethylation to ms2i6A in mitochondrial tRNAs7 and potentially in other (nuclear) RNAs,10 or by oxidation/hydroxylation to ms2io6A, which is found for example, in salmonella tRNAs.7, 11 Interestingly, the i6A modification is necessary for the expression of human selenoproteins,12 and it has recently been shown as an essential determinant for the installation of additional tRNA anticodon modifications, including m3C at position 32, and Um at position 34.13 Given the importance of i6A in many natural contexts, i6A‐sensitive deoxyribozymes could become diagnostically useful tools for the analysis of tRNA modification states.
Here we report the discovery and characterization of three classes of RNA‐cleaving deoxyribozymes with distinct responses to the natural tRNA modification i6A (Figure 1). First, we found that the activity of three AA deoxyribozymes is abolished by i6A. Second, the AB08 deoxyribozyme is strongly activated by i6A and shows unprecedented selectivity in cleaving only i6A‐modified RNA next to the site of modification. Third, we found the deoxyribozyme AC17 for which the modified nucleotide i6A causes a distinct shift of the cleavage site by one nucleotide.
Figure 1.
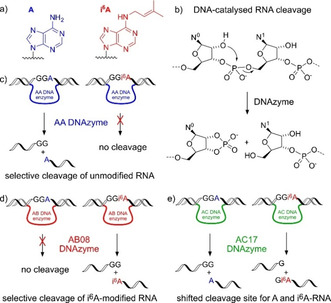
a) N 6‐isopentenyladenosine (red) in comparison to unmodified adenosine (blue, only nucleobases shown). b) RNA‐cleaving deoxyribozymes catalyse the cleavage of the phosphodiester backbone by transesterification. c–e) Depiction of three different classes of DNAzymes identified in this study; AA DNAzymes are inhibited by i6A (c), the AB08 DNAzyme cleaves only i6A‐modified RNA (d). The cleavage site of the AC17 DNAzyme is influenced by the modification state of the RNA; that is, modified and unmodified RNA are cleaved at different positions (e).
Results and Discussion
In vitro selection of RNA‐cleaving deoxyribozymes
A synthetic DNA library with 20 random nucleotides was used for the in vitro selection of RNA‐cleaving deoxyribozymes, following established procedures (Supporting Information, Figure S1).3 The 28‐nt long RNA substrates were prepared by solid‐phase synthesis (Supporting Information, Table S1), and covalently ligated to the DNA library (Table S2). The modified nucleotide i6A was incorporated using a suitable i6A phosphoramidite building block, which was obtained by regioselective N 6‐alkylation of an N 6‐acetylated intermediate (Supporting Information, Scheme S1).14
During 15 rounds of separation and amplification, catalytically active DNA sequences were trained to discriminate modified and unmodified RNA. To achieve the desired selectivity, two separate selection experiments were carried out, in which positive and negative selections were altered to favour unmodified RNA (AA selection) or i6A‐modified RNA (AB selection), respectively.
AA DNAzymes are inhibited by i6A
After cloning of the enriched AA selection library, three DNA enzymes (AA07, AA14 and AA17) were identified that catalysed site‐specific cleavage of unmodified RNA (R1) (Figure 2). RNA cleavage products were only observed with R1 as substrate (Figures 2 b,c), but not with i6A‐containing RNA (R2). The cleavage sites were assigned by comparison to alkaline hydrolysis and RNase T1 digestion ladders and confirmed by high‐resolution electrospray ionisation mass spectrometry (HR‐ESI‐MS) of both cleavage products (Figure 2 d). AA17 cleaved R1 at position G16, while the other two DNA enzymes cleaved one nucleotide further upstream at G15. Interestingly, the i6A at position 17 in the substrate RNA strongly inhibited the catalytic activity of all three DNA enzymes, independent of their cleavage site. Previously, m6A was shown to inhibit the cleavage activity of DNAzymes when located directly next to the cleavage site (e.g. in VMC10),3 but such a strong effect of a natural RNA modification at a more remote location, as is the case of i6A on AA07 and AA14 activities, has not been observed before. The larger size of the isopentenyl group in i6A compared to the methyl group in m6A is likely responsible for this long‐range effect. Indeed, when the AA DNAzymes were tested on m6A‐modified RNA (R3, same sequence as R2, with i6A replaced by m6A), AA17 was strongly inhibited by m6A, while AA07 and AA14 retained substantial activity (Figure S3).
Figure 2.
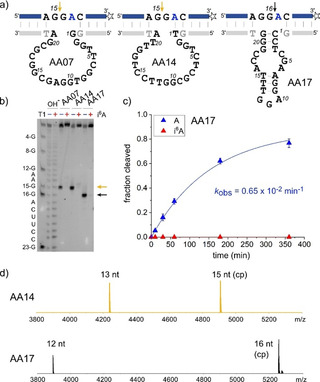
a) Sequences and predicted secondary structures of three AA DNAzymes, AA07, AA14, and AA17. b) PAGE analysis of the cleavage reaction with unmodified RNA (R1, labelled with blue −) and i6A‐modified RNA (R2, labelled with red +) shows selective cleavage of unmodified RNA, and identifies the cleavage site; AA07 and AA14 cleave at G15, and AA17 cleaves at G16. Experiments used 3′‐fluorescein‐labelled RNA. c) Single‐turnover kinetics for AA17. Graphs for others and gel images are shown in Figure S3. d) HR‐ESI‐MS analyses of cleaved products. The longer 15/16‐nt products contain a 2′,3′‐cyclic phosphate, and the shorter 12/13‐nt fragments contain 5′‐OH and 3′‐C6‐NH2‐termini.
AB08 DNAzyme cleaves only i6A RNA
The AB selection revealed the deoxyribozyme AB08, which showed high activity for cleaving i6A‐RNA, but it did not cut unmodified RNA to any significant extent (Figure 3). This notable finding reports the first DNA enzyme that specifically cleaves a post‐transcriptionally modified RNA. The AB08 DNA enzyme strictly required the modified nucleotide in the substrate RNA and yielded 80 % cleaved product after 6 h at pH 7.5 with 20 mm MgCl2. The cleavage site of AB08 was located directly next to the modified nucleotide i6A, as confirmed by high‐resolution mass spectrometry of the 12‐nt 5′‐OH‐terminated i6A‐containing fragment (Figure 3 c). These findings raised the question if AB08 could be activated by m6A rather than i6A in the RNA substrate. Using the analogous m6A‐modified RNA R3, no cleavage product was observed upon incubation with AB08 (Figure S4). Thus, the AB08 DNA enzyme is indeed specific for i6A‐modified RNA.
Figure 3.
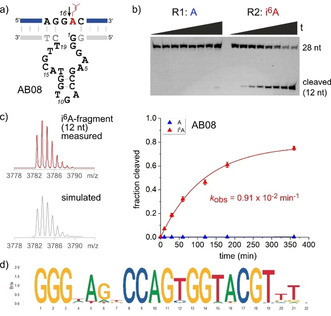
a) Secondary structure prediction of AB08 and indicated cleavage site of i6A RNA. b) Gel image and kinetic graph demonstrating selectivity for i6A‐modified RNA. Time points are: 0 and 10 min; 0.5, 1, 2, 3, 6, and 22 h. c) HR‐ESI‐MS confirming the cleavage site of R2. Top: measured spectrum of 12‐nt i6A‐modified fragment (i6ACUUCCGUAACU). Bottom: simulated spectrum for C117H149N40O83P11 with an exact mass of 3782.58 amu. d) Sequence logo for alignment of 54 sequences (within a Levenshtein distance of 4 from AB08), represented in the active fraction of the enriched DNA library.
To learn more about the abundance of AB08 and potential related sequences, the enriched selection library was analysed by next generation sequencing (NGS). The round 15 library was separated into active and inactive fractions for cleavage of i6A‐RNA; this corresponded to one additional round of selection (round 16). Round 15 and both fractions of round 16 were amplified with the corresponding Illumina NGS primers (Figure S2), and 1.3 million reads were analysed (Table S3).15 AB08 and its relatives with Levenshtein distance ≤4 represented 6.4 % of reads in round 15. This fraction increased to 10.9 % in round 16. Interestingly, the number of variants decreased from 73 to 54.16 Comparison of the most enriched AB08 variants (Figure S5) and the sequence logo17 plotted for the alignment of 54 relatives of AB08 shown in Figure 3 d identified the nucleotides with highest variability at position 4 and at the 3′‐end. The catalytic activities of several synthetic AB08 variants confirmed the conserved motif (Figure S6, Table S4), and may suggest a distant relationship of AB08 to the 8–17 DNA enzyme family, which has been identified in several in vitro selection experiments.4c The AGY triloop (position 10–12 in Figure 3 d) and a conserved CG dinucleotide in the 3′‐bulge (position 17–18 in the sequence logo) may be involved in a pseudoknot motif related to the one observed in the crystal structure of an 8–17 variant.18 The highly invariable guanosine triple in the 5′‐bulge is a distinctive feature of AB08 and is likely responsible for the astonishing selectivity for cleaving only i6A RNA. In the absence of any structural insights into the organization of the catalytic core, the detailed mechanism of i6A recognition remains unknown.
i6A shifts the cleavage site of AC17 DNAzyme
The AB selection surfaced an additional highly interesting DNA enzyme, which was named AC17.19 Upon screening the catalytic activity, it was observed that AC17 yielded cleavage products with both i6A‐modified and unmodified RNAs (R1 and R2), and that these products exhibited different migration on the analysis gel (Figure 4). Comparison of the migration behaviour with the already assigned cleavage products of AA14 and AB08 revealed that AC17 cuts R1 at G16, but R2 at G15 (Figure 4 b). Inspection of the sequence and secondary structure prediction of AC17 revealed that it is highly related to the previously identified VMC10 DNA enzyme, which showed strong inhibition by m6A.3 A single point mutation (C12T) converted AC17 into VMC10. Therefore, a more detailed characterization of the AC17 DNA enzyme in comparison to VMC10 was undertaken. First, AC17 was tested on m6A‐RNA and was found to yield only 10 % cleaved RNA, and this cleavage occurred at G16 (Figure 4 c, Figure S7). Thus, m6A inhibited the ability of AC17 to cut at G16, but at the same time did not activate it to cut at G15 either. This confirmed that the unrivalled shift of the cleavage site is specific to i6A. On the other hand, we also checked the activity of VMC10 to cut i6A RNA and found about 25 % cleavage at G15 (Figure 4 d, Figure S7). Thus, both AC17 and VMC10 cut i6A RNA at G15, but a cytidine at position 12 (C12) of the catalytic core enabled higher rate and yield compared to thymidine (T12).
Figure 4.
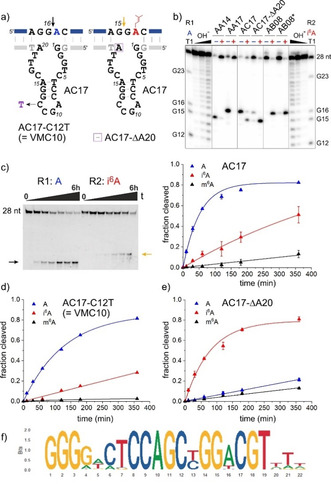
a) Predicted secondary structure of AC17 shown to cut unmodified RNA at G16 and i6A‐RNA at G15. Sites of important mutations are indicated. b) PAGE analysis of cleavage sites of AC17 in comparison to AA14, AA17 and AB08. Note that this analysis was performed with 5′‐32P‐labelled RNA (in contrast to the 3′‐fluorescein‐labelled RNAs used in Figures 2 b and 3 b). c) PAGE analysis with 3′‐fluorescein‐labelled RNAs and kinetic graphs for cleavage of R1 (A), R2 (i6A) and R3 (m6A) RNA). d,e) kinetic graphs for cleavage of R1 (A), R2 (i6A) and R3 (m6A) RNA with VMC10 (d) and AC17‐ΔA20 (e). f) Sequence logo for alignment of 45 AC17 variants with >50 reads and maximal 4 mutations.
Intrigued by the activity of AC17 to cut both RNAs, we analysed the enriched DNA library for AC17 abundance and potential relatives. The round 15 AB pool was separated into active and inactive fractions, now using unmodified RNA. This corresponded to a “negative” selection round 16. Both fractions were amplified with the corresponding NGS primers, and 1.4 million reads were analysed (Table S3). In accordance with the faster cleavage rate of R1, a larger number of AC17 variants (with Levenshtein distance ≤4) were found in the cleaved fraction (1177 variants versus 426 variants in the uncleaved fraction; the numbers were significantly reduced when filtered for more than 50 reads: 45 and 7 variants, respectively). The sequence logo for the alignment of 45 AC17 variants is shown in Figure 4 f. The 3′‐terminal nucleotides of the catalytic core showed highest variability. The importance of this region was investigated using synthetic variants of AC17. Deleting T19 resulted in a DNA enzyme that cleaved both R1 and R2 with comparable rates, and the ratio of the cleavage products can be directly translated into modification levels (Figure S8). Mutation at position 20 resulted in a series of DNA enzymes with very different behaviour: the activity of the A20G mutant was highly comparable to the parent deoxyribozyme, while the A20C variant showed strongly preferred cleavage of unmodified RNA with a 35‐fold faster rate (Table S4, Figure S8). On the other hand, the deletion of A20 completely changed the activity of the deoxyribozyme.
Surprisingly, the AC17‐ΔA20 variant showed highly accelerated cleavage of i6A‐RNA, while unmodified RNA R1 was cleaved only to a minor extent (Figure 4 e, Figure S7). In other words, AC17‐ΔA20 is the second i6A‐RNA‐selective DNA enzyme discovered in this study, with comparable kinetic activity to AB08, but cleaving one nucleotide further upstream. Consistent with this result, the number of reads for AC17‐ΔA20 (and its relatives with up to maximal four mutations) was almost twice as high in the active fraction of the i6A NGS library compared to the uncleaved fraction (Table S2). Interestingly, AC17 contains conserved features reminiscent of the AGY/CG motif and the guanosine triple at the 5′‐bulge, which were also found in AB08 (compare sequence logos in Figures 3 d and 4 f, and alignments in the Supporting Information). The relative location of these motifs seems preserved, but the position of the intervening GG dinucleotide involved in the formation of the base‐paired region is shifted by one nucleotide. In other words, the AGY motif is located in a predicted triloop in AB08 and a tetraloop in AC17. It remains to be seen if these motifs are directly involved in the catalytic mechanism, and how the DNA nucleotides interact with the RNA bases near the cleavage site to distinctly modulate the activation of a specific 2′‐OH group in response to the modification state of the adenosine under investigation. Future crystal structure analyses may also reveal the nucleotides contacting the cleavage site, enabling rational engineering in analogy to the structure‐based design of RNA‐ligating deoxyribozymes.20
Conclusion
In summary, this study reported three new classes of RNA‐cleaving DNA enzymes that respond in distinct ways to the presence of the natural modified nucleotide i6A in the target RNA. The AB08 DNA enzyme absolutely requires the presence of i6A in the target RNA for catalytic activity. The finding that i6A can finetune the active site of the DNA enzyme AC17 for cleavage at distinct positions is exceptional and may be further explored for analytical applications; for example, as a direct readout of chemically modulated adenosine isopentenylation states.21 In the future, it will be interesting to investigate how other tRNA modifications, including hydroxylated or thiomethylated i6A analogues are recognized and how they influence the catalytic activities of AB08, AC17 and future evolved variants for the study of native tRNAs.22
The presented data demonstrate the surprising plasticity of DNA's catalytic ability. The reported modes of action are distinct from previously observed responses of DNA enzymes to m6A in RNA, and no other DNA catalysts sensitive to nucleobase modifications have been reported. The direct comparison of how i6A and m6A affect the catalytic activity supports the conclusion that the larger and more hydrophobic isopentenyl group permits better discrimination due to larger energetic and kinetic differences. This observation correlates with the larger thermodynamically stabilizing effect of i6A compared to m6A in RNA hairpin structures.23 Structural and mechanistic investigations will follow to allow deeper insights into the architecture of these new DNA catalysts and their modes of i6A recognition.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, SPP1784 Chemical Biology of Native Nucleic Acid modifications) and by the European Research Council (ERC No. 682586). A.L. acknowledges funding by a PhD scholarship from the German Academic Exchange Service (DAAD). M.V.S. thanks the Graduate School for Life Sciences at the University of Würzburg for a Postdoc Plus fellowship. We thank Michaela Schraut and Tim Rieseberg for contributions to i6A synthesis, and Sebastian Mayer for mass spectrometry. Illumina sequencing was performed at the Core Unit Systems Medicine at the University of Würzburg. Open access funding enabled and organized by Projekt DEAL.
A. Liaqat, C. Stiller, M. Michel, M. V. Sednev, C. Höbartner, Angew. Chem. Int. Ed. 2020, 59, 18627.
References
- 1.
- 1a. Boccaletto P., Machnicka M. A., Purta E., Piatkowski P., Baginski B., Wirecki T. K., de Crecy-Lagard V., Ross R., Limbach P. A., Kotter A., Helm M., Bujnicki J. M., Nucleic Acids Res. 2018, 46, D303–D307; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. McCown P. J., Ruszkowska A., Kunkler C. N., Breger K., Hulewicz J. P., Wang M. C., Springer N. A., Brown J. A., Wiley Interdiscip. Rev. RNA 2020, 10.1002/wrna.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Helm M., Motorin Y., Nat. Rev. Genet. 2017, 18, 275; [DOI] [PubMed] [Google Scholar]
- 2b. Linder B., Jaffrey S. R., Cold Spring Harbor Perspect. Biol. 2019, 11, a032201; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Hartstock K., Rentmeister A., Chem. Eur. J. 2019, 25, 3455–3464. [DOI] [PubMed] [Google Scholar]
- 3. Sednev M. V., Mykhailiuk V., Choudhury P., Halang J., Sloan K. E., Bohnsack M. T., Höbartner C., Angew. Chem. Int. Ed. 2018, 57, 15117–15121; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 15337–15341. [Google Scholar]
- 4.
- 4a. Breaker R. R., Joyce G. F., Chem. Biol. 1994, 1, 223–229; [DOI] [PubMed] [Google Scholar]
- 4b. Silverman S. K., Nucleic Acids Res. 2005, 33, 6151–6163; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Schlosser K., Li Y., ChemBioChem 2010, 11, 866–879. [DOI] [PubMed] [Google Scholar]
- 5. Buchhaupt M., Peifer C., Entian K.-D., Anal. Biochem. 2007, 361, 102–108. [DOI] [PubMed] [Google Scholar]
- 6. Bujnowska M., Zhang J., Dai Q., Heideman E. M., Fei J., J. Biol. Chem. 2020, 295, 6992–7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schweizer U., Bohleber S., Fradejas-Villar N., RNA Biol. 2017, 14, 1197–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Maraia R. J., Iben J. R., RNA 2014, 20, 977–984; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Lamichhane T. N., Arimbasseri A. G., Rijal K., Iben J. R., Wei F. Y., Tomizawa K., Maraia R. J., RNA 2016, 22, 583–596; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Yarham J. W., Lamichhane T. N., Pyle A., Mattijssen S., Baruffini E., Bruni F., Donnini C., Vassilev A., He L., Blakely E. L., PLoS Genet. 2014, 10, e1004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Janas T., Janas T., Yarus M., RNA 2012, 18, 2260–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Reiter V., Matschkal D. M., Wagner M., Globisch D., Kneuttinger A. C., Müller M., Carell T., Nucleic Acids Res. 2012, 40, 6235–6240; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Fakruddin M., Wei F. Y., Emura S., Matsuda S., Yasukawa T., Kang D., Tomizawa K., Nucleic Acids Res. 2017, 45, 11954–11961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mathevon C., Pierrel F., Oddou J. L., Garcia-Serres R., Blondin G., Latour J. M., Menage S., Gambarelli S., Fontecave M., Atta M., Proc. Natl. Acad. Sci. USA 2007, 104, 13295–13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Warner G. J., Berry M. J., Moustafa M. E., Carlson B. A., Hatfield D. L., Faust J. R., J. Biol. Chem. 2000, 275, 28110–28119. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Zhou M., Long T., Fang Z. P., Zhou X. L., Liu R. J., Wang E. D., RNA Biol. 2015, 12, 900–911; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Arimbasseri A. G., Iben J., Wei F. Y., Rijal K., Tomizawa K., Hafner M., Maraia R. J., RNA 2016, 22, 1400–1410; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c. Kim L. K., Matsufuji T., Matsufuji S., Carlson B. A., Kim S. S., Hatfield D. L., Lee B. J., RNA 2000, 6, 1306–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tararov V. I., Kolyachkina S. V., Alexeev C. S., Mikhailov S. N., Synthesis 2011, 2483–2489. [Google Scholar]
- 15.Analysis of the sequencing data was performed using fastaptamer perl scripts: Alam K. K., Chang J. L., Burke D. H., Mol. Ther. Nucleic Acids 2015, 4, e230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Only variants with more than 50 reads were counted.
- 17. Wagih O., Bioinformatics 2017, 33, 3645–3647. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Liu H., Yu X., Chen Y., Zhang J., Wu B., Zheng L., Haruehanroengra P., Wang R., Li S., Lin J., Li J., Sheng J., Huang Z., Ma J., Gan J., Nat. Commun. 2017, 8, 2006; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Santoro S. W., Joyce G. F., Proc. Natl. Acad. Sci. USA 1997, 94, 4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.This sequence was identified from clone number 17 of the AB selection (AB17), but because of its distinct properties and the sequence similarity to VMC10, it was renamed to AC17.
- 20. Ponce-Salvatierra A., Wawrzyniak-Turek K., Steuerwald U., Höbartner C., Pena V., Nature 2016, 529, 231–234. [DOI] [PubMed] [Google Scholar]
- 21. Cheng H. P., Yang X. H., Lan L., Xie L. J., Chen C., Liu C., Chu J., Li Z. Y., Liu L., Zhang T. Q., Luo D. Q., Cheng L., Angew. Chem. Int. Ed. 2020, 59, 10645–10650; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 10732–10737. [Google Scholar]
- 22.AB08 and AC17 are not directly applicable for the examination of natural tRNAs, because of their preferred i6A sequence context. Natural isopentenyl transferase enzymes preferably prenylate the central adenosine of a triple AAA motif,[24] while the DNA enzymes reported in this study require a guanine next to i6A. The RNA substrate containing the Gi6A motif was chosen to allow for direct comparison with m6A-sensitive DNA enzymes. Analogous experiments with Ai6AA-containing RNAs are expected to yield DNA enzymes in the future that can be used on native tRNA sequences.
- 23.
- 23a. Kierzek E., Kierzek R., Nucleic Acids Res. 2003, 31, 4472–4480; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23b. Denmon A. P., Wang J., Nikonowicz E. P., J. Mol. Biol. 2011, 412, 285–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Soderberg T., Poulter C. D., Biochemistry 2000, 39, 6546–6553. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary