

Abstract
Mediator‐based extracellular electron transfer (EET) pathways can balance the redox metabolism of microbes. However, such electro‐biosynthesis processes are constrained by the unknown underlying EET mechanisms. In this paper, Pseudomonas putida was studied to systematically investigate its EET pathway to transition metal complexes (i. e., [Fe(CN)6]3−/4− and [Co(bpy)3]3+/2+; bpy=2,2′‐bipyridyl) under anaerobic conditions. Comparative proteomics showed the aerobic respiratory components were upregulated in a bioelectrochemical system without oxygen, suggesting their potential contribution to EET. Further tests found inhibiting cytochrome c oxidase activity by NaN3 and NADH dehydrogenase by rotenone did not significantly change the current output. However, the EET pathway was completely blocked, while cytochrome c reductase activity was inhibited by antimycin A. Although it cannot be excluded that cytochrome c and the periplasmic subunit of cytochrome c oxidase donate electrons to the transition metal complexes, these results strongly demonstrate that cytochrome c reductase is a key complex for the EET pathway.
Keywords: bioelectrochemical system, cytochrome c reductase, electron transfer, proteomics, redox mediator
Liquid wires for biohybrid systems: The electron transfer pathway between redox metal‐complexes and biological respiratory proteins is systematically investigated and revealed. Such a pathway can be readily applied to many non‐electrogenic microorganisms and thus expand the application of microbial electrochemical technology in many fields such as industrial biotechnology and biosensors.
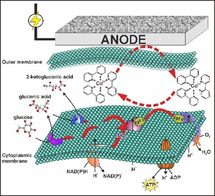
Introduction
Biotechnology plays a key role in reducing our dependence on fossil fuels.1, 2 However, only few bio‐based products are currently competitive in the market, compared to chemicals derived from fossil fuels. The majority of biosynthetic processes suffer from redox imbalance caused by the degree of reduction (DoR) of substrate(s) and products, which typically exhibit low product yields, rates and titres and the formation of by‐products.3, 4, 5, 6 This limitation cannot be solved by genetic elimination of a by‐product forming pathway. The microbial hosts will simply respond by secretion of other side products and even multiple pathway intermediates.7
A new technology, termed microbial electrochemical technology (MET), is emerging as a promising concept to overcome these inherent limitations and enhance the efficiency of bio‐based production.8, 9, 10 MET uses reaction vessels equipped with electrodes that serve as a source (cathode) or a sink (anode) for electrons. Such reactors, termed bioelectrochemical systems (BES),11 can (at least partially) uncouple redox metabolism from carbon core metabolism and consequently elevate product yield.12, 13 This concept has been experimentally validated, where electrodes acting as electron sinks or donors could alter the cellular metabolism and product profile.14, 15, 16, 17, 18, 19, 20 However, the industrialisation of MET for biochemical production is largely limited. On one hand, although the direct extracellular electron transfer (EET) routes have been well‐studied for the model MET strains, that is, Shewanella and Geobacter,21, 22 these model strains unfortunately are not yet applicable in synthetic biology. On the other hand, transferring the known direct EET knowledge into common industrial hosts has also only reached limited success so far, in terms of maintaining the electron transfer rate23, 24 and also translocating the outer membrane complex from model MET strains into the target host.
Compared to the constraints of direct EET, indirect EET via redox mediators can be more pronounced. Artificial redox chemicals can be readily applied for even non‐exoelectrogens which can expand the application of MET technology. For instance, mediator‐based EET has been applied for Pseudomonas fluorescens to perform the bioelectrocatalytic hydroxylation of nicotinic acid and to establish an in situ biochemical oxygen demand sensor.25, 26, 27 Recently, Pseudomonas putida (P. putida), an obligate aerobic host for biochemical production,28, 29 was also demonstrated to use external redox mediators as the final electron acceptor in BES,20, 30 while no oxygen was present. The redox power from the mediator/anode could drive anoxic glucose metabolism and enable “redox‐imbalanced” fermentation of glucose (DoR of 4) into 2‐ketogluconic acid (DoR of 3.33) at high yield (>90 %), with acetate (DoR of 4) as the only minor by‐product. Moreover, this electro‐bioprocess could also be steered by the rational selection of mediators based on their redox potentials. Higher redox potential mediators provide stronger thermodynamic driving force and thus facilitate the anoxic carbon turnover rate of P. putida in BES.20 Furthermore, mediator‐based EET also enables planktonic reactor setups, which will benefit process scale‐up. Considering the cost and potential toxicity effects of such chemicals to the environment, recycling the mediators in the downstream process and/or rational design of environmentally friendly mediators (e. g., biodegradable molecules) should probably be targeted in the future.
However, no clear understanding presently exists on how mediators interact with intracellular electron carriers to withdraw electrons in mediator‐based EET. The poor knowledge of indirect EET prevents further rational optimization to improve electron transfer rates and thus the metabolic turnover rate for targeted products. In this paper, our target was to investigate and understand mediator‐based EET routes at the protein level. Proteomics was firstly applied to screen potential targets and then specific electron transfer inhibitors were introduced to target different sites on the electron transfer chain. All the results demonstrated cytochrome c reductase, the respiratory complex III commonly present in many microorganisms, was the key protein involved in the EET pathway from P. putida cells to external mediators (both [Fe(CN)6]3−/4− and [Co(bpy)3]3+/2+; bpy=2,2′‐bipyridyl).
Results and Discussion
Oxidative phosphorylation pathway of P. putida upregulated in a BES
P. putida could use synthetic redox mediators as electron acceptors to perform anoxic catabolism of glucose. However, the cells could not grow anaerobically in a BES and the electrode‐driven glucose consumption rate was only about 5 % of that measured for aerobic growth culture.31, 32 Thus, the cells could only gain limited energy for cell maintenance (≈17–75 % of the non‐growth associated maintenance).20 Consequently, this posed significant stress on protein synthesis, since this process is very energy‐intensive in bacteria.33, 34 With limited energy supply, the cells would have to restrict protein maintenance to only essential components required for driving the anoxic glucose oxidation, where mediator‐based EET was the sole pathway providing redox driving force. Moreover, it was also observed that the current density was gradually increasing in the first 100 h after inoculation in the BES while the biomass was decreasing.20, 30 This indicated an optimization of the electron transfer in the remaining P. putida cells. Since this could point towards a changed gene expression, analysing the proteome of P. putida cells in the BES could possibly reveal key proteins involved in electron transfer to external mediators.
Comparative proteomics was thus applied to investigate the proteome rearrangement of P. putida cells induced in a BES. In general, the proteome (in total 1000 proteins identified) of P. putida F1 was gradually rearranged until end‐of‐batch after inoculation (Figure 1B), with the majority of proteins being down‐regulated or statistically constant. All ribosome subunits were down‐regulated, indicating a decreased efficiency of protein biosynthesis machinery of P. putida in the BES. However, a number of translocation units, which were related to Sec‐ and Tat‐protein secretion systems across the cytoplasmic membrane,35 were surprisingly up‐regulated (see Figure S1). This might indicate a change in the membrane proteome. Indeed, after analysing the protein localizations according to the Pseudomonas database,36 it was found that 96 out of the 285 upregulated proteins were located on the cell envelope (Figure 1C). This concurred with the phenomenon that anoxic glucose oxidation mainly happens in the periplasmic space. Further analysis of the proteins also showed that almost all identified oxidative phosphorylation pathway components and the corresponding adenosine triphosphate (ATP) synthase were significantly upregulated (Figure 1D). These results indicate that the aerobic respiration system might play an important role in electron transfer towards mediators, even in the absence of oxygen. Further analysis would be required to identify the respective contribution of each component.
Figure 1.
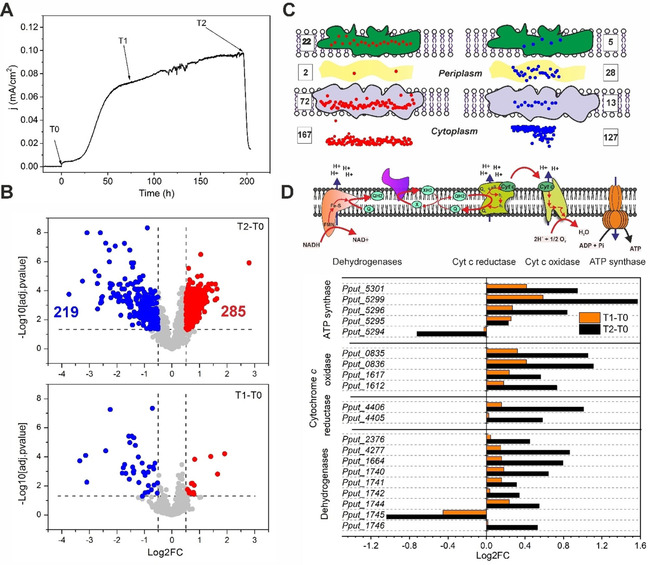
Comparative proteome analysis of P. putida F1 in BES. A) Sampling time points for proteomics analysis. T0 of inoculation as the control, and T1 and T2 of reaching peak current density or end‐of‐batch respectively as experimental group. B) Volcano plot of the proteins identified and quantified by SWATH‐MS after manual curation of the peptide transition chromatographs. Log2FC represents the binary logarithm of the fold changes of protein abundance at different time points. Log10[adj.pvalue] represents the common logarithm of the statistical significance calculated by MSstat (v3.6.0). Cut‐off values for significant difference were adj.pvalue <0.05 and abs(Log2FC) >0.5 (the dashed lines shown in the figure). Six samples (3 independent biological reactors and 2 technical replicates from each reactor) were used for statistical analysis. C) Location distributions of the up‐ (in red) and down‐regulated (in blue) proteins for time T2 against T0 (T2–T0). Protein location information were extracted from Pseudomonas genome database.36 D) Protein abundance changes of the membrane respiration and energization machinery of P. putida F1 in BES. Dehydrogenases include the complex I, complex II and alternative dehydrogenases exiting in P. putida (e. g., PQQ‐glucose dehydrogenase and FAD‐gluconate dehydrogenase).
Cytochrome c oxidase activity inhibited by sodium azide showed no effect in the extracellular electron transfer
Despite the absence of oxygen, the upregulation of cytochrome c oxidases (Pput_1612, Pput_1617, Pput_0835, Pput_0836) was observed. However, P. putida neither has an anaerobic respiration chain nor fermentative pathways and thus, genetically interrupting the cytochrome c oxidases (or any other aerobic respiratory components) would result in cell death. Therefore we used sodium azide (NaN3) to specifically inhibit all cytochrome c oxidases to investigate their function in EET. Previous studies have shown that NaN3 forms a bridge between two transmembrane subunits of the cytochrome c oxidase (i. e., the Fea3 and CuB) and therefore disrupts its activity.37, 38, 39, 40 NaN3 affects the growth of strains such as Pseudomonas aeruginosa and Pseudomonas fluorescens from a concentration as low as 0.02 % (≈3 μm).41 Moreover, glucose uptake (and thus growth) of P. putida CSV86 was completely blocked in the presence of 25 mm NaN3.42 In a similar fashion 3 mm NaN3 completely eliminated the aerobic growth of P. putida F1 in DM9 medium (Figure 2A). No glucose consumption could be measured under such conditions.
Figure 2.
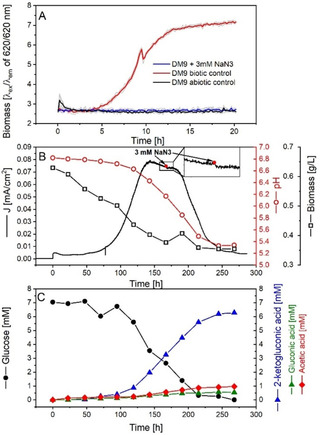
Effects of NaN3 on the growth, extracellular electron transfer and metabolic activity of P. putida F1 in aerobic shaking flasks and the BES. A) growth inhibition effects of 3 mm NaN3 on P. putida F1 in DM9 medium determined using BioLector cultivation system. The coloured lines were averaged from 4 biologic replicates (shown in light grey lines in the background) for each condition. B) interruption test of NaN3 on the performance of BES reactor. P. putida was inoculated at time 0. C) glucose consumption and product profile of the BES reactor corresponding to the conditions described in B). Extracellular concentrations were measured.
However, same dosage of NaN3 did not have any effect on P. putida cells in the BES (Figure 2B). The current density did not change before or after injection, and also the pH trend remained unaffected. The glucose consumption was also not inhibited, and the product profile was the same as observed before (Figure 2C).20, 30 An abiotic control also confirmed no electrochemical interaction between NaN3 and [Fe(CN)6]3− (see Figure S2). These results suggested that inhibiting transmembrane subunits of the cytochrome c oxidases did not interrupt EET from P. putida F1 to [Fe(CN)6]3−. However, it is also not possible to rule out the involvement of CuA or ccoO/ccoP in EET. These are the subunits of the cytochrome c oxidase complex exposed to the periplasmic space and responsible to interact with and take electrons from soluble cytochrome c for aerobic respiration. They can still be active even if the CuB−Fea3 centre is inhibited. Moreover, thermodynamically electron transfer from CuA and cco/ccoP to ferricyanide could also be feasible, considering their redox potential of about 270 mV vs. standard hydrogen electrode (SHE).43 However, such an electron transfer pathway may not be the sole EET pathway for P. putida F1, since our previous study demonstrated compounds with a redox potential as low as 208 mV vs. SHE could still successfully accept electrons from the cells.20 However, the difference in redox potential (i. e., 62 mV) was again too small to obtain a conclusive answer here. A specific inhibitor that can bind to the CuA or ccoO/ccoP subunit should be screened in the future, in order to evaluate the function cytochrome c oxidase in the EET.
Membrane dehydrogenases are unlikely targets for redox mediator
P. putida is rich in membrane dehydrogenases, including nicotinamide adenine dinucleotide (NADH) dehydrogenase (complex I), succinate dehydrogenase (complex II), and dehydrogenases involved in sugar oxidation. Succinate dehydrogenase was not investigated in this study, because there was no accumulation of any other carbon product in the BES apart from 2‐ketogluconic acid, gluconic acid and acetic acid and the carbon and electron balances were both closed with these three products.20, 30 This indicated that succinate dehydrogenase was not functioning under BES conditions. Therefore, the focus in this work was on the complex I NADH dehydrogenase and other dehydrogenases involved in glucose oxidation.
The electron flux via NADH dehydrogenase should be quite small, based on the estimated flux balance conducted before.20 Only less than 10 % of the carbon was catabolized through cytosolic glycolysis towards acetate where NADH would be produced. Nevertheless, rotenone, a specific inhibitor of complex I,44 was applied to show if this complex plays a role in reducing the mediator (i. e., [Fe(CN)6]3− in this case). Rotenone can bind to type I NADH dehydrogenase (NDH‐I) and consequently inhibit electron transfer from the iron‐sulfur centre of complex I to the ubiquinone pool.44 It can function in diverse organisms ranging from bacteria, plants, animals to human cells.45, 46, 47, 48 However, its activity seems to be highly dependent on the species and conditions (e. g., in vivo or in vitro, dosing quantity, etc.). For instance, a dosing of 1.0 μm rotenone can impair over 50 % of cell viability for human B lymphoma cell line PW,49 while only 23 % inhibition was detected for 100 μm rotenone dosing for isolated P. putida membrane fractions.50 An aerobic growth test of P. putida F1 with rotenone of 0–400 μm (dissolved in DMSO) was conducted in this work. Cell growth was not significantly changed by DMSO up to 0.33 % v/v (i. e., 0 μm rotenone) and rotenone up to 200 μm (see Figure S3). The effects for higher concentrations (≥300 μm) could unfortunately not be quantified because the medium became too cloudy for cell density measurement. Thus, as a balance between maximizing rotenone inhibition and minimizing the interference to optical measurement, 250 μm was chosen for subsequent experiments.
Upon injection of rotenone, a sharp decrease in current output was observed (Figure 3A). While rotenone did not show any visible electrochemical interaction with [Fe(CN)6]3− and anode (Figure S4), this current drop indicated a change in the biologic function of P. putida F1 cells induced by rotenone. However, this effect only lasted for about 2.5 h, and then the current density started to recover gradually back to the original value before injection within the next 24 h. Similar changes of the current density profile were observed for acetate production (Figure 3B). The production rate slowed after rotenone injection, compared to the value before, and then recovered after 24 h. One possible reason for these phenomena might be the internal compensation of NADH dehydrogenase activity after rotenone inhibition by alternative dehydrogenase (e. g., the rotenone‐insensitive type II NADH dehydrogenase, NDH‐II). P. putida has both NDH‐I and NDH‐II, while the NDH‐I is typically the major complex.51, 52 This was observed for Arabidopsis,53 where the respiration rate was inhibited by rotenone for the first 4 h and then progressively recovered to initial levels within 32 h. Similar internal compensation of NADH dehydrogenase activity was also confirmed for P. taiwanensis VLB120,54 a strain that shares 98.9 % genome similarity with P. putida.55 Nevertheless, the results confirmed the inhibition of rotenone on NADH dehydrogenase activity and consequently confirmed the active role of NADH dehydrogenase in the EET route.
Figure 3.
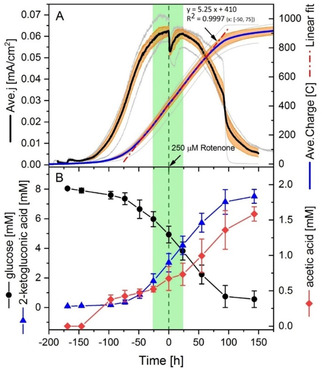
Effects of rotenone injection on the behaviour of P. putida F1 cells in BES. A) the effect of rotenone on electron output. Left vertical axis indicates the current density normalized to project anode surface area and right axis indicates the charge output. Solid black and blue lines were the averaged value of four biologic replicates (in grey), and the orange shading indicated the standard errors of the averaged value. The dash‐dot red line was a linear regression of the charge output between time −50 h and 75 h, with the formula given in the figure. B) Corresponding glucose consumption and product formation secreted by the cells. Data presented were the average of two biologic replicates with standard errors. Time 0 h indicated the time when 250 μm rotenone (in 200 μL DMSO) was injected into the reactor.
In contrast to NADH dehydrogenases, it is quite challenging to address whether the pyrroloquinoline quinone (PQQ) dependent glucose dehydrogenase and flavin adenine dinucleotide (FAD) dependent gluconate dehydrogenase were able to transfer electrons to the mediator directly. Purified enzyme assays have confirmed that both enzymes could use [Fe(CN)6]3− as an electron acceptor under in vitro conditions.56, 57 However, those proteins are embedded in the cytoplasmic membrane for the in vivo case, and the hydrophobic cell membrane would quite likely block diffusion of the highly charged and hydrophilic [Fe(CN)6]3− to reach the enzymes for direct electron transfer. Moreover, testing knockout mutants or screening with specific activity inhibitors of these two dehydrogenases also cannot reveal the in vivo interaction between them and mediators, because these enzymes were both essential for carbon and electron flux. It would not be possible to distinguish whether the current output drop is attributed to the elimination of glucose oxidation or the inhibition of mediator electron transfer within the cell.
In summary, the NADH‐, glucose‐, and gluconate‐ dehydrogenases were confirmed to be essential for the EET to the mediator, but the present results could not answer whether they could directly transfer electrons to the mediator (while the further tests discussed below excluded this possibility).
Inhibiting cytochrome c reductase activity by antimycin A completely blocked the extracellular electron transfer pathway
Respiratory complex III, that is, cytochrome c reductase, was theoretically a possible target for redox mediators, according to the thermodynamic feasibility. An effective mediator for EET should have a minimum formal redox potential of 0.078–0.208 V vs. SHE,20 which overlapped with the redox potential range for subunits of cytochrome c reductase.58 To further investigate this hypothesis, antimycin A, which selectively binds to the Qi site of complex III,59, 60 was applied to interrupt cytochrome c reductase function. The active function of antimycin A to P. putida F1 was confirmed by an aerobic growth test. Treating cells with antimycin A slowed down the aerobic growth rate of P. putida F1 in mineral medium and lowered the biomass yield (Figure 4B, Figure S5). The effects gradually increased with higher dosing quantity, but 20 μg mL−1 (about 36 μm) was chosen as the working condition in this study to minimize the inference of insoluble antimycin A in optical measurements (Figure S5).
Figure 4.
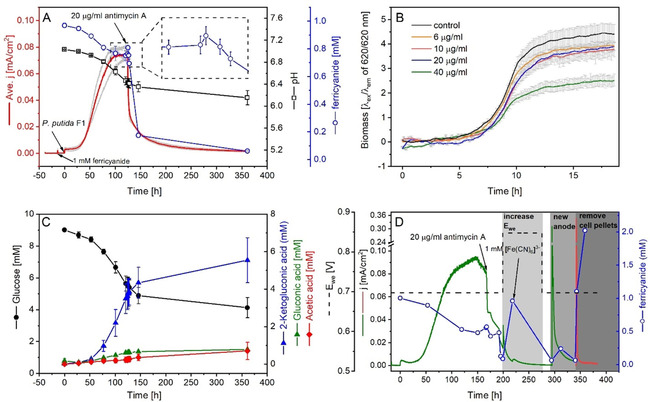
Effects of antimycin A on P. putida F1 in aerobic flask and BES reactor with [Fe(CN)6]3−. A) The current density, pH, and [Fe(CN)6]3− concentration changes upon antimycin A injection in BES and C) the corresponding metabolite profiles. Data presented were averaged from 3 biologic replicates. Grey lines in the background gave the original current profiles of the 3 biologic replicates. B) Growth inhibition of antimycin A on P. putida F1 under aerobic condition determined by Bio‐lector. Data presented were the averaged data of 4 biologic replicates subtracting the respective abiotic controls (see Figure S5). Error bars were given in the grey line. D) [Fe(CN)6]3− concentration at different conditions in the BES after adding antimycin A.
Unlike rotenone or NaN3, antimycin A was reported to react with [Fe(CN)6]3−.61 The chemical reaction between the two substances was confirmed in an abiotic control in this study (Figure S6A). A positive current was observed upon the addition of antimycin A into [Fe(CN)6]3− solution without bacteria cells, and the concentration of [Fe(CN)6]3− decreased slightly. However, this could be by‐passed if the cell suspensions were added prior to antimycin A and absorption of antimycin A on the hydrophobic cell membrane could prevent the compound from being deactivated by [Fe(CN)6]3−.61 Indeed, injecting antimycin A into the BES after inoculation showed a very strong inhibition of the current output (Figure 4A). The current dropped sharply immediately after the injection and could not be recovered afterwards, demonstrating a permanent interruption to the electron transfer route by antimycin A. The pH change terminated and also the consumption of glucose and product formation (Figure 4C). All these results demonstrated antimycin A completely blocked the electron transfer route of P. putida F1 in the BES.
Surprisingly, the [Fe(CN)6]3− concentration was not changed as expected (Figure 4A,D). In principle, blocking electron transfer from P. putida F1 cells to the mediator should result in it staying in its oxidized form in the medium due to the applied anodic potential. However, with only a slight increase within the first half hour, the concentration of [Fe(CN)6]3− continuously decreased towards zero. The total concentration of [Fe(CN)6]3−/4− was constant, meaning [Fe(CN)6]3− was being reduced but not degraded in the reactor. Meanwhile, the current drop further suggested [Fe(CN)6]4− could not be re‐oxidized by the working electrode, since otherwise a current output should be detected. Cyclic voltammetry tests were also conducted for the conditions before and after the addition of mediator, biomass and antimycin A (Figure S7), and the results also showed the ferrocyanide was not able to be oxidised when present with P. putida F1 cells and antimycin A. After excluding antimycin A would interfere with the optical quantification of [Fe(CN)6]3−/4− (Figure S6B), the electrode surface property, passivated changed by the undissolved antimycin A, was firstly thought to be the reason. However, raising the working electrode potential to 0.847 V and even replacing the (putatively passivated) anode with a clean electrode did not improve the situation (Figure 4D). These results excluded the working electrode as the reason for this phenomenon.
Unexpectedly, the oxidation of [Fe(CN)6]4− recovered by removing the cells from the medium (Figure 4D). [Fe(CN)6]4− was quickly oxidized to [Fe(CN)6]3− after removing the cell pellets by centrifugation. This phenomenon was quite confusing, as it suggested the antimycin A‐treated P. putida F1 cells could strongly reduce the [Fe(CN)6]3− and also block re‐oxidation at the electrode. While the increased cellular ROS level induced by antimycin A dosing might explain the driving force for the reduction of [Fe(CN)6]3−,62, 63, 64, 65 the mechanism of P. putida F1 cells to “preserve” the soluble mediator in the periplasmic space (or elsewhere) would be unprecedented to the best of our knowledge. This would also be rapidly disrupted by centrifugation to explain the measurement of [Fe(CN)6]4− along the whole batch and the oxidation of [Fe(CN)6]4− after removing the cells by centrifugation (Figure 4D). Further studies are necessary to understand this phenomenon.
Because of the concerns regarding the [Fe(CN)6]3− behaviour discussed above, we also tested another mediator, [Co(bpy)3]3+/2+ (E f (formal redox potential) of 0.31 V). It exhibited a similar performance to [Fe(CN)6]3− for P. putida F1 in the BES, in terms of current profile and metabolic products from glucose.20 In contrast to [Fe(CN)6]3−, this cobalt complex was unaffected by antimycin A addition, since no interference in the current output could be observed in the abiotic control upon antimycin A addition (Figure 5B). While tested in the BES reactor with P. putida F1 cells, the current output sharply decreased immediately after adding antimycin A (Figure 5A), which was the same as observed for the case of [Fe(CN)6]3−. This further indicated the key role of cytochrome c reductase for the electron transfer route from cells towards external mediators.
Figure 5.
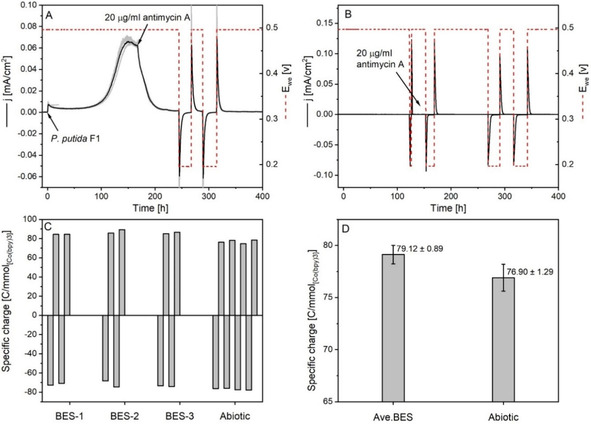
Effects of antimycin A on the extracellular electron transfer of P. putida F1 in BES with [Co(bpy)3]3+/2+. A) Current output and working electrode potential (E we, V) profiles of the BES reactors before and after adding antimycin A. The current output data presented (solid black line) were averaged from three biological replicates (in grey), and the red dashed line indicated the corresponding working electrode potential during the batch. B) The abiotic BES control of antimycin A and [Co(bpy)3]3+/2+. Current output: black solid line; working electrode potential: red dashed line. C) The specific charge output of each [Co(bpy)3]3+/2+ oxidation‐reduction cycle normalized to the absolute quantity. D) The averaged specific charge outputs of the BES group and abiotic control. The Ave.BES data were averaged based on the two oxidation‐reduction cycles of three individual biologic replicates and the abiotic data were the average of the four oxidation‐reduction cycles from one replicate.
However, the redox status of the cobalt mediator had to be determined, to further demonstrate the function of cytochrome c reductase in EET. While the current dropped back to the baseline value after adding antimycin A, the working electrode potential was changed from 0.497 to 0.197 V. If the cobalt mediator was in its oxidized form, this potential would then be sufficient to reduce this mediator to its CoII form. Indeed, a strong cathodic (reducing) current was detected after adjusting the working electrode potential (Figure 5A), confirming the presence of [Co(bpy)3]3+ in the medium after adding antimycin A. The specific charge output/input, which could be used to determine the ratio of [Co(bpy)3]3+ to [Co(bpy)3]2+, was also calculated for each redox cycle (Figure 5C,D). By comparing the data for the BES reactor against the abiotic control, it could be concluded that the cobalt mediator was fully in its oxidized or reduced form under high or low working electrode potentials, respectively. This further demonstrated that inhibition of cytochrome c reductase activity by antimycin A completely blocks electron transfer from P. putida cells to the mediator.
It should also be noted that water‐soluble cytochrome c, the electron carrier between cytochrome c reductase and oxidase,66 might also be involved in the mediator‐based EET chain. The in vitro electron transfer reaction between cytochrome c and ferricyanide has been well studied.67, 68 Even for [Co(bpy)3]3+/2+, which has a redox potential slightly higher than cytochrome c, in vitro studies show the equilibrium favours cytochrome c oxidation (redox equilibrium constant K E≈3).69 These results suggested cytochrome c could potentially bridge the cytochrome c reductase and the redox mediator. However, such a hypothesis, to the best of our knowledge, is unfortunately not able to be validated or excluded. Genetic modification of cytochrome c would interrupt the cellular growth and moreover, there is no targeted inhibition method available so far on for soluble cytochrome c. However, despite the uncertainty of cytochrome c, our results still demonstrate that cytochrome c reductase is a key enzyme which connects all the intracellular electron transfer pathways of P. putida F1 cells to external mediators in the BES.
Conclusions
Synthetic biology processes often suffer from redox imbalances between substrates and products in microbial metabolism. This intrinsic limitation can be solved by providing an external electrode to balance the electron flux of the biosynthesis process with the assistance of artificial redox mediators. However, the mechanism of this mediator‐assisted extracellular electron transfer (EET) has remained unknown, preventing further rational design and targeted optimization. In this paper, we systematically investigated the electron transfer route between Pseudomonas putida and artificial redox mediators. Proteomics suggested the respiratory chain was involved in the EET of P. putida to the mediator (and then anode). Further targeted analysis on individual respiratory components using specific inhibitors demonstrated that inhibiting respiratory complex III, cytochrome c reductase, with antimycin A completely blocked the electron transfer pathway from P. putida cells to the redox mediator. It was still not clear whether the water‐soluble cytochrome c and the periplasmic subunit (CuA or ccoO/ccoP) of cytochrome c oxidase can interact with the mediator. However, the results demonstrated that cytochrome c reductase is a key enzyme involved in such a mediator‐based EET pathway for P. putida, which converges all intracellular electron transfer pathways to the external mediator. This work for the first time highlights a key enzyme for redox mediator‐dependent EET for a microorganism, and will lay the foundation for future optimization for practical applications such as microbial electrosynthesis or biosensors.
Experimental Section
Strain and growth conditions
Pseudomonas putida F1 used in this paper was provided by Dr. Nicholas Coleman from University of Sydney, Australia. Lysogeny‐Broth agar plate was used to activate the cryo stock culture and a defined mineral medium (DM9) was used for liquid culture, as described elsewhere.30 Glucose was used as sole carbon source. The precultures were grown in baffled shaking flasks overnight (≈16 h) using DM9 medium. Growth conditions were 30 °C, and 200 rpm aerobically in an orbital shaking incubator (Multitron, Infors, Switzerland). Cell pellets were then harvested by centrifugation at 7000 g, 30 °C for 10 min, resuspended in DM9 buffer (without glucose) and injected into the BES reactor using a syringe. The cell density was measured from the optical density at 600 nm and then converted to dry weight using the following formula: CDW [g L−1]=0.476×OD600.
Bioelectrochemical system
The setup of the bioelectrochemical system was the same as reported before.20 A detailed description of the system was reported elsewhere.70 Briefly, a fully autoclavable three‐electrode electrochemical system was used. Carbon cloth electrode of 25 cm2 (1071, FuelCellStore, USA) was used as working electrode and poised at 0.697 V vs. SHE while using [Fe(CN)6]3−/4− as mediator and 0.497 V for [Co(bpy)3]2+/3+ using a potentiostat (VMP3, BioLogic, France). Mediators of 1 mm final concentration were used for all batches tested. All potentials reported in this work were against SHE.
Extracellular metabolite quantification
The extracellular metabolites were quantified using HPLC with the methods detailed described elsewhere.71 Samples were prepared by centrifugation at 17 000 g, 4 °C, and 10 min.
Growth inhibition test by respiration inhibitors
The growth inhibition tests of electron transfer inhibitors were conducted in 48‐well microtiter plate (MTP‐48‐BOH, m2p‐labs GmbH, Baesweiler, Germany) using an online monitored microbioreactor system (Biolector Pro, m2p‐labs GmbH, Baesweiler, Germany). A total of 1.2 mL DM9 medium was added to each well, containing respective concentrations of different inhibitors, and then inoculated with pre‐grown liquid culture. Stock solutions of the inhibitors were freshly prepared in different solvents: rotenone in DMSO, antimycin A in ethanol and NaN3 in MilliQ water. Abiotic controls were conducted with the respective pure solvent and water. A sterile gas permeable sealing foil (F‐GP‐10, m2p‐labs GmbH, Baesweiler, Germany) was used to cover the plate for keeping sterile conditions as well as allowing oxygen supply. Growth conditions were set up at 30 °C, 1200 rpm of 3 mm orbit, cycling time of 3 min and biomass gain of 1. O2, CO2, and humidity controls were not applied. Replicates of 3 or 4 wells were conducted for each condition, and growth curves were averaged and then smoothed if necessary (10 Points of Window, Savitzky‐Golay method) using Origin Pro 9.
Quantification of mediators
Ferricyanide ([Fe(CN)6]3−)and ferrocyanide ([Fe(CN)6]4−) were quantified colorimetrically at 420 nm and 320 nm as described before.20 Supernatants were tested after removing of cells (17000 g, 4 °C, 10 min).
For the quantification of the different redox status of [Co(bpy)3]3+/2+, an electrochemical method was developed and applied. Although a color change of the solution could be visualized (Figure S8), the reduced and oxidized forms could not be quantified colorimetrically as their UV/Vis spectra are very similar (Figure S9). The redox status of this mediator was determined coulometrically by calculating the specific charge required to oxidize or reduce the complex under a specific condition. To achieve this in the BES reactor, the working electrode was firstly biased at 0.497 V to oxidize or 0.197 V vs. SHE to reduce this compound. Afterwards, the total charge output or input was calculated and normalized to the absolute quantity of the mediator present in the system at a specific time, here termed “specific charge”. The redox status of this compound could thus be determined by comparing the specific charge at a specific time of bio‐batch to the abiotic control. The equation to calculate the specific charge can be expressed as below:
where Q represents the charge output/input at a specific time between t0 (the time the working electrode potential changed) and t1 (the time when the reducing/oxidizing current became essentially zero), and m refers to the absolute quantity [mmol] of the mediator calculated based on the total amount added into the reactor subtracting the loss due to sampling. An estimated water loss of 0.021 mL h−1 for the BES reactor was also included to compensate the calculations.
SWATH‐MS proteomics analysis
Time‐related proteome changes were analysed using a data‐independent acquisition proteomics method, called sequential windowed acquisition of all theoretical fragment ion mass spectra (SWATH‐MS),72 and the BES reactors were sampled at T0 (time of inoculation), T1 (time of the current reaching peak value) and T2 (time of the end of BES batch) (see Figure 1A). Cell pellets were harvested by centrifugation at 14000 g, 4 °C, for 5 min. The pellets were washed once with fresh DM9 medium, centrifuged again and then resuspended in 5 mL lysis buffer. Recipe of the lysis buffer (pH 7.5) was 7 m urea, 2 m thiourea, 4 % CHAPS (3‐[(3‐cholamidopropyl)dimetehylammonio]‐1‐propanesulfate), 25 mm HEPES (4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid), 30 mm dithiothreitol, 1 mm ethylenediaminetetraacetic acid (EDTA) and 1X protease inhibitor (5892791001, cOmplete Ultra Tablet, EDTA‐free, Roche). Cells in suspension were lysed using a pre‐cooled French press at 1500 psi (high position) for three times (Aminco FA‐073, Thermo Schientific), and then centrifuged at 14000 g, 4 °C for 10 min to remove cell debris. Supernatants were collected and stored in −80 °C until analysis. All solutions used were pre‐cooled on ice, and the samples were kept on ice throughout the protocol.
Protein concentrations in the supernatant were quantified using a detergent compatible commercial assay kit (2‐D Quant kit, 80‐6483‐56, GE Healthcare Life Science), following the manufacturer's instructions. Protein was digested by trypsin using filter‐aided sample preparation (FASP) protocol,73 followed by Ziptip cleaning (ZTC18S096, Millipore). The final peptides were suspended in aqueous formic acid (0.1 %) before being injected into nanoLC (Prominence, Shimadzu, Kyoto, Japan) and mass spectrometry (TripleTof 5600, AB Sciex, USA) for analysis. The data were processed using Skyline software and MSstat package in Rstudio.74, 75
Author contributions
B.L. and J.O.K developed the concept, designed the experiment and drafted the manuscript. P.V.B. synthesized the cobalt mediator and contributed to scientific discussions. B.L. performed the experiment and data analysis. All authors contributed to the editing of the paper, read and approved the final manuscript.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
B.L. thanks Dr. Amanda Nouwens, from School of Chemistry and Molecular Biosciences, The University of Queensland, Australia for the SWATH‐MS analysis and Dr. Yang Lu from Advanced Water Management Centre, The University of Queensland, Australia for help with statistical analysis of proteomics data using R. B. L. also appreciates fruitful and useful discussions about protein structure and function with Prof. Kunrong Mei from the School of Pharmaceutical Science and Technology, Tianjin University, P.R. China. The authors appreciate the use of the facilities of the Centre for Biocatalysis (MiKat) at the Helmholtz Centre for Environmental Research, which is supported by European Regional Development Funds (EFRE, Europe funds Saxony) and the Helmholtz Association. Open access funding enabled and organized by Projekt DEAL.
B. Lai, P. V. Bernhardt, J. O. Krömer, ChemSusChem 2020, 13, 5308.
Contributor Information
Dr. Bin Lai, Email: bin.lai@ufz.de.
Dr. Jens O. Krömer, Email: jens.kroemer@ufz.de.
References
- 1. McGlade C., Ekins P., Nature 2015, 517, 187–190. [DOI] [PubMed] [Google Scholar]
- 2. von Braun J., Global Food Security 2018, 19, 81–83. [Google Scholar]
- 3. Chen X., Li S., Liu L., Trends Biotechnol. 2014, 32, 337–343. [DOI] [PubMed] [Google Scholar]
- 4. Wang Y., San K.-Y., Bennett G. N., Curr. Opin. Biotechnol. 2013, 24, 994–999. [DOI] [PubMed] [Google Scholar]
- 5. van Hoek M. J., Merks R. M., BMC Syst. Biol. 2012, 6, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu G., Yan Q., Jones J. A., Tang Y. J., Fong S. S., Koffas M. A., Trends Biotechnol. 2016, 34, 652–664. [DOI] [PubMed] [Google Scholar]
- 7. Becker J., Reinefeld J., Stellmacher R., Schäfer R., Lange A., Meyer H., Lalk M., Zelder O., von Abendroth G., Schröder H., Haefner S., Wittmann C., Biotechnol. Bioeng. 2013, 110, 3013–3023. [DOI] [PubMed] [Google Scholar]
- 8. Rabaey K., Rozendal R. A., Nat. Rev. Microbiol. 2010, 8, 706–716. [DOI] [PubMed] [Google Scholar]
- 9. Kracke F., Lai B., Yu S., Krömer J. O., Metab. Eng. 2018, 45, 109–120. [DOI] [PubMed] [Google Scholar]
- 10. Su L., Ajo-Franklin C. M., Curr. Opin. Biotechnol. 2019, 57, 66–72. [DOI] [PubMed] [Google Scholar]
- 11. Schröder U., Harnisch F., Angenent L. T., Energy Environ. Sci. 2015, 8, 513–519. [Google Scholar]
- 12. Rosenbaum M. A., Henrich A. W., Curr. Opin. Biotechnol. 2014, 29, 93–98. [DOI] [PubMed] [Google Scholar]
- 13. Kracke F., Vassilev I., Krömer J. O., Front. Microbiol. 2015, 6, 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim T. S., Kim B. H., Biotechnol. Lett. 1988, 10, 123–128. [Google Scholar]
- 15. Schuppert B., Schink B., Trösch W., Appl. Microbiol. Biotechnol. 1992, 37, 549–553. [Google Scholar]
- 16. Vassilev I., Giesselmann G., Schwechheimer S. K., Wittmann C., Virdis B., Krömer J. O., Biotechnol. Bioeng. 2018, 115, 1499–1508. [DOI] [PubMed] [Google Scholar]
- 17. Xafenias N., Kmezik C., Mapelli V., Bioelectrochemistry 2017, 117, 40–47. [DOI] [PubMed] [Google Scholar]
- 18. Nevin K. P., Woodard T. L., Franks A. E., Summers Z. M., Lovley D. R., mBio 2010, 1(2), e00103-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kracke F., Virdis B., Bernhardt P. V., Rabaey K., Krömer J. O., Biotechnol. Biofuels 2016, 9, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lai B., Yu S., Bernhardt P. V., Rabaey K., Virdis B., Krömer J. O., Biotechnol. Biofuels 2016, 9, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pirbadian S., Barchinger S. E., Leung K. M., Byun H. S., Jangir Y., Bouhenni R. A., Reed S. B., Romine M. F., Saffarini D. A., Shi L., Gorby Y. A., Golbeck J. H., El-Naggar M. Y., PNAS 2014, 111, 12883–12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang F., Gu Y., O'Brien J. P., Yi S. M., Yalcin S. E., Srikanth V., Shen C., Vu D., Ing N. L., Hochbaum A. I., Egelman E. H., Malvankar N. S., Cell 2019, 177, 361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jensen H. M., Albers A. E., Malley K. R., Londer Y. Y., Cohen B. E., Helms B. A., Weigele P., Groves J. T., Ajo-Franklin C. M., PNAS 2010, 107, 19213–19218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Su L., Fukushima T., Prior A., Baruch M., Zajdel T. J., Ajo-Franklin C. M., ACS Synth. Biol. 2020, 9, 115–124. [DOI] [PubMed] [Google Scholar]
- 25. Takayama K., Kurosaki T., Ikeda T., Nagasawa T., J. Electroanal. Chem. 1995, 381, 47–53. [Google Scholar]
- 26. Yoshida N., Hoashi J., Morita T., McNiven S. J., Nakamura H., Karube I., J. Biotechnol. 2001, 88, 269–275. [DOI] [PubMed] [Google Scholar]
- 27. Yoshida N., Yano K., Morita T., McNiven S. J., Nakamura H., Karube I., Analyst 2000, 125, 2280–2284. [DOI] [PubMed] [Google Scholar]
- 28. Nikel P. I., de Lorenzo V., Metab. Eng. 2018, 50, 142–155. [DOI] [PubMed] [Google Scholar]
- 29. Nikel P. I., Martinez-Garcia E., de Lorenzo V., Nat. Rev. Microbiol. 2014, 12, 368–379. [DOI] [PubMed] [Google Scholar]
- 30. Yu S., Lai B., Plan M. R., Hodson M. P., Lestari E. A., Song H., Krömer J. O., Biotechnol. Bioeng. 2018, 115, 145–155. [DOI] [PubMed] [Google Scholar]
- 31. Nikel P. I., Chavarria M., Fuhrer T., Sauer U., de Lorenzo V., J. Biol. Chem. 2015, 290, 25920–25932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kohlstedt M., Wittmann C., Metab. Eng. 2019, 54, 35–53. [DOI] [PubMed] [Google Scholar]
- 33. Neijssel O., Teixeira De Mattos M., Tempest D., Escherichia coli and Salmonella: Cellular and Molecular Biology, 2nd ed. (Eds.: F. Neidhart, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, H. E. Umbarger), American Society for Microbiology, Washington DC: 1996, pp. 1683–1692. [Google Scholar]
- 34. Klumpp S., Scott M., Pedersen S., Hwa T., PNAS 2013, 110, 16754–16759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Natale P., Brüser T., Driessen A. J. M., Biochim. Biophys. Acta Biomembr. 2008, 1778, 1735–1756. [DOI] [PubMed] [Google Scholar]
- 36. Winsor G. L., Griffiths E. J., Lo R., Dhillon B. K., Shay J. A., Brinkman F. S., Nucleic Acids Res. 2016, 44, D646–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bennett M. C., Mlady G. W., Kwon Y. H., Rose G. M., J. Neurochem. 1996, 66, 2606–2611. [DOI] [PubMed] [Google Scholar]
- 38. Tsubaki M., Yoshikawa S., Biochemistry 1993, 32, 164–173. [DOI] [PubMed] [Google Scholar]
- 39. Blumenthal D. C., Kassner R. J., J. Biol. Chem. 1979, 254, 9617–9620. [PubMed] [Google Scholar]
- 40. Yoshikawa S., Shinzawa-Itoh K., Nakashima R., Yaono R., Yamashita E., Inoue N., Yao M., Fei M. J., Libeu C. P., Mizushima T., Yamaguchi H., Tomizaki T., Tsukihara T., Science 1998, 280, 1723–1729. [DOI] [PubMed] [Google Scholar]
- 41. Lichstein H. C., Soule M. H., J. Bacteriol. 1944, 47, 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Basu A., Phale P. S., FEMS Microbiol. Lett. 2006, 259, 311–316. [DOI] [PubMed] [Google Scholar]
- 43. Melin F., Xie H., Meyer T., Ahn Y. O., Gennis R. B., Michel H., Hellwig P., Biochim. Biophys. Acta Bioenerg. 2016, 1857, 1892–1899. [DOI] [PubMed] [Google Scholar]
- 44. Fendel U., Tocilescu M. A., Kerscher S., Brandt U., Biochim. Biophys. Acta Bioenerg. 2008, 1777, 660–665. [DOI] [PubMed] [Google Scholar]
- 45. McEwan A. G., Ferguson S. J., Jackson J. B., Arch. Microbiol. 1983, 136, 300–305. [DOI] [PubMed] [Google Scholar]
- 46. Ravanel P., Tissut M., Douce R., Plant Physiol. 1984, 75, 414–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Heinz S., Freyberger A., Lawrenz B., Schladt L., Schmuck G., Ellinger-Ziegelbauer H., Sci. Rep. 2017, 7, 45465–45465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Newhouse K., Hsuan S. L., Chang S. H., Cai B., Wang Y., Xia Z., Toxicol. Sci. 2004, 79, 137–146. [DOI] [PubMed] [Google Scholar]
- 49. Armstrong J. S., Hornung B., Lecane P., Jones D. P., Knox S. J., Biochem. Biophys. Res. Commun. 2001, 289, 973–978. [DOI] [PubMed] [Google Scholar]
- 50. Sweet W. J., Peterson J. A., J. Bacteriol. 1978, 133, 217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ebert B. E., Kurth F., Grund M., Blank L. M., Schmid A., Appl. Environ. Microbiol. 2011, 77, 6597–6605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Melo A. M. P., Bandeiras T. M., Teixeira M., Microbiol. Mol. Biol. Rev. 2004, 68, 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Garmier M., Carroll A. J., Delannoy E., Vallet C., Day D. A., Small I. D., Millar A. H., Plant Physiol. 2008, 148, 1324–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nies S. C., Dinger R., Chen Y., Wordofa G. G., Kristensen M., Schneider K., Büchs J., Petzold C. J., Keasling J. D., Blank L. M., Ebert B. E., Appl. Environ. Microbiol. 2020, 86(11), e03038-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang L. T., Tai C. J., Wu Y. C., Chen Y. B., Lee F. L., Wang S. L., Int. J. Syst. Evol. Microbiol. 2010, 60, 2094–2098. [DOI] [PubMed] [Google Scholar]
- 56. Matsushita K., Ohno Y., Shinagawa E., Adachi O., Ameyama M., Agric. Biol. Chem. 1980, 44, 1505–1512. [Google Scholar]
- 57. Matsushita K., Shinagawa E., Adachi O., Ameyama M., J. Biochem. 1979, 86, 249–256. [PubMed] [Google Scholar]
- 58. Biochemical Pathways: An Atlas of Biochemistry and Molecular Biology, 2nd ed. (Eds.: G. Michal, D. Schomburg), Wiley, Hoboken: 2013. [Google Scholar]
- 59. Huang L.-S., Cobessi D., Tung E. Y., Berry E. A., J. Mol. Biol. 2005, 351, 573–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Potter V. R., Reif A. E., J. Biol. Chem. 1952, 194, 287–297. [PubMed] [Google Scholar]
- 61. Walter P., Lardy H. A., Biochemistry 1964, 3, 812–816. [DOI] [PubMed] [Google Scholar]
- 62. Han Y. H., Kim S. H., Kim S. Z., Park W. H., Oncol. Rep. 2008, 20, 689–693. [PubMed] [Google Scholar]
- 63. Lanciano P., Khalfaoui-Hassani B., Selamoglu N., Ghelli A., Rugolo M., Daldal F., Biochim. Biophys. Acta Bioenerg. 2013, 1827, 1332–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bleier L., Dröse S., Biochim. Biophys. Acta Bioenerg. 2013, 1827, 1320–1331. [DOI] [PubMed] [Google Scholar]
- 65. Katafias A., Impert O., Kita P., Transition Met. Chem. 2008, 33, 1041–1046. [Google Scholar]
- 66. Kokhan O., Wraight C. A., Tajkhorshid E., Biophys. J. 2010, 99, 2647–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. McCray J. A., Kihara T., Biochim. Biophys. Acta Bioenerg. 1979, 548, 417–426. [DOI] [PubMed] [Google Scholar]
- 68. Eley C. G., Ragg E., Moore G. R., J. Inorg. Biochem. 1984, 21, 295–310. [DOI] [PubMed] [Google Scholar]
- 69. Meier M., van Eldik R., Chem. Eur. J. 1997, 3, 39–46. [Google Scholar]
- 70. Lai B., Nguyen A. V., Krömer J. O., Methods Protoc. 2019, 2, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lai B., Plan M., Hodson M., Krömer J., Fermentation 2016, 2, 6. [Google Scholar]
- 72. Gillet L. C., Navarro P., Tate S., Rost H., Selevsek N., Reiter L., Bonner R., Aebersold R., Mol. Cell. Proteomics 2012, 11, O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wisniewski J. R., Zougman A., Nagaraj N., Mann M., Nat. Methods 2009, 6, 359–362. [DOI] [PubMed] [Google Scholar]
- 74. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J., Bioinformatics 2010, 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Choi M., Chang C.-Y., Clough T., Broudy D., Killeen T., MacLean B., Vitek O., Bioinformatics 2014, 30, 2524–2526. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary