

Abstract
Ruthenium(II)biscarboxylate complexes enabled the selective alkylation of C−H and C−C bonds at the ortho‐ or meta‐position. ortho‐C−H Alkylations were achieved with 4‐, 5‐ as well as 6‐membered halocycloalkanes. Furthermore, the judicious choice of the directing group allowed for a full control of ortho‐/meta‐selectivities. Detailed mechanistic studies by experiment and computation were performed and provided strong support for an oxidative addition/reductive elimination process for ortho‐alkylations, while a homolytic C−X cleavage was operative for the meta‐selective transformations.
Keywords: alkylation, C−C activation, C−H activation, decarboxylation, ruthenium
Ortho vs. meta: C−H alkylations of arylpyrazoles are achieved through ruthenium‐catalyzed C−H or C−C activation. The site‐selectivity is controlled by the pyrazole substituents and the steric hindrance of the alkyl moiety.

Introduction
Methods for the direct modification of otherwise inert C−H bonds gained enormous attention throughout the last decade.1 For the development of synthetically useful molecular transformations, the full control of positional selectivity is of prime importance for C−H functionalization reactions.2 One important strategy for site‐selective C−H activations is the use of chelation‐assistance through the introduction of directing groups, thus allowing for proximity‐induced ortho‐C−H metalation.3 During the past years, ruthenium catalysis was particularly recognized as an efficient tool for C−H functionalizations and a plethora of ruthenium‐catalyzed C−H transformations was developed.4 Especially, site‐selective ortho‐,5 meta‐6 as well as para‐alkylations7 of arenes were devised by ruthenium catalysis, with major contributions by the groups of Frost,8 and Ackermann,9 among others.10 Typically, secondary and tertiary alkyl halides result in C−H alkylations at the meta‐ or para‐position with excellent levels of selectivity. In contrast, ortho‐alkylated arenes were thus far predominantly obtained with primary alkyl halides (Scheme 1 a).
Scheme 1.
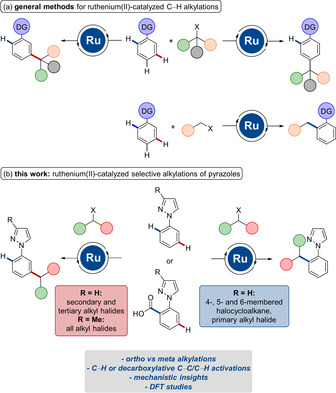
Ruthenium‐catalyzed site‐selective alkylations.
Likewise, ruthenium catalysis proved to be powerful for C−C bond transformations, with notable progress by inter alia Dong and Hartwig.11 Inspired by the versatility and robustness of the ruthenium catalyst, we became intrigued whether this C−C bond functionalization could be exploited for alkylation with unactivated alkyl halides.
Within our program on sustainable C−H activations,12 we have now unraveled ruthenium‐catalyzed ortho‐ or meta‐alkylations through C−H or decarboxylative C−C/C−H activations (Scheme 1 b). Notable feature of our strategy include (i) versatile ruthenium‐catalyzed meta‐ as well as ortho‐alkylations with secondary alkyl bromides, (ii) functionalization of synthetically useful pyrazoles through C−H or decarboxylative C−C/C−H activations, (iii) detailed mechanistic insights by experiment, and (iv) DFT studies for ruthenium‐catalyzed ortho‐C−H alkylations.
Results and Discussion
In orienting experiments, we first examined the C−H alkylation of 2‐phenylpyridine (1) with bromocyclohexane (2 a), which provided the corresponding meta‐alkylated product 4 in moderate yield (Scheme 2 a). However, ortho‐C−H alkylated product 6 aa was obtained when pyrazole 5 a was reacted with secondary alkyl bromide 2 a (Scheme 2 b).
Scheme 2.

Site‐selective C−H alkylations.
Intrigued by these unexpected results, we became interested in investigating the C−H alkylation of arylpyrazole 5 a. To this end, different reaction conditions were probed for the ruthenium‐catalyzed C−H alkylation with bromocyclohexane (2 a) (Table 1).13 PhCMe3 9f proved to be the optimal solvent (entries 1–2). Furthermore, carboxylic acids14 were found to be critical for achieving high conversions (entry 3). Previously, we and Larrosa had employed p‐cymene‐ligand‐free ruthenium complexes for C−H activation.5a, 15 Cationic ruthenium(II) complexes could also here be employed as catalysts (entries 4–8). In addition, cyclohexyl chloride or iodide also afforded products 6 aa and 7 aa with positional selectivity, albeit in somewhat reduced yield (entries 9–10).
Table 1.
Ruthenium‐catalyzed C−H alkylation of pyrazole 5 a

|
Entry |
Deviation from the standard conditions |
6 aa [%] |
7 aa [%] |
|---|---|---|---|
|
1 |
none |
60 |
12 |
|
2 |
o‐xylene instead of PhCMe3 |
50 |
– |
|
3 |
without MesCO2H |
28 |
– |
|
4 |
[Ru(NCt‐Bu)6][BF4]2 instead of 3 |
60 |
8 |
|
5 |
[Ru(NCt‐Bu)6][PF6]2 instead of 3 |
62 |
10 |
|
6 |
[Ru(NCt‐Bu)6][SbF6]2 instead of 3 |
63 |
9 |
|
7 |
[Ru(NCMe)6][PF6]2 instead of 3 |
65 |
12 |
|
8 |
[Ru(NCMe)6][PF6]2 instead of 3 and without MesCO2H |
– |
– |
|
9 |
Cy‐Cl instead of 2 a |
38 |
5 |
|
10 |
Cy‐I instead of 2 a |
53 |
7 |
[a] Reaction conditions: 5 a (0.5 mmol), 2 a (1.5 mmol), [Ru] (5.0 mol %), MesCO2H (30 mol %), K2CO3 (1.0 mmol), PhCMe3 (1.0 mL), 120 °C, 16 h, yields of isolated products.
We next examined the effect of the halocycloalkane 2 ring size on the site‐selectivity of the C−H alkylation reaction (Scheme 3). The reaction of unsubstituted phenylpyrazole 5 a with bromocyclobutane (2 b) and bromocyclohexane (2 a) afforded the ortho‐alkylated products 6 aa and 6 ab as the major product, whereas bromocycloheptane (2 d) and bromocyclooctane (2 e) preferentially furnished the meta‐alkylated products 7 (Scheme 3 a). In contrast, bromocyclopentane (2 c) yielded a mixture of the ortho‐ and meta‐alkylated products 6 ac and 7 ac. Then, we probed the alkylation of arylpyrazoles 5 with primary as well as secondary alkyl bromides 2 (Scheme 3 b). The alkylation reaction of arylpyrazoles 5 with exo‐2‐bromonorbornane (2 f) or neopentyl bromide (2 i) afforded the ortho‐alkylated products 6 af, 6 ai, and 6 bi exclusively. Acyclic secondary alkyl bromides 2 g and 2 h were smoothly converted into meta‐alkylated products 7 ag and 7 ah with excellent levels of positional selectivity.
Scheme 3.

(a) Site‐selectivity of ruthenium‐catalyzed C−H alkylations of pyrazole 5 a with various bromocycloalkanes 2, (b) scope for C−H alkylation of pyrazoles 5. [a] The yield of meta‐alkylated product 7 is given in parentheses. [b] o‐Xylene was used as solvent.
Next, the electronic effect on the site‐selectivity was studied with differently substituted arylpyrazoles 5 with cyclohexyl bromide (2 a) (Scheme 4). Electron‐donating groups at the para‐position led to a mixture of ortho‐ and meta‐alkylated products 6 and 7, whereas electron‐withdrawing groups exclusively afforded the ortho‐alkylated products (6 da–6 fa). The connectivity of product 6 fa was unambiguously assigned by X‐ray diffraction analysis.16
Scheme 4.

Electronic effect on the site‐selectivity of ruthenium‐catalyzed C−H alkylations of arylpyrazoles 5 with bromocyclohexane (2 a). [a] The yield of meta‐alkylated product 7 is given in parentheses. [b] Dialkylated product was obtained in 29 % yield.
In contrast to arylpyrazoles 5 a–5 f, the direct alkylation of 3,5‐dimethyl‐1‐phenyl‐1H‐pyrazole (5 g) with cyclic and acyclic secondary alkyl bromides 2 exclusively provided the meta‐alkylated products 7 (Scheme 5). In addition, the alkylation with neopentyl bromide (2 i) selectively furnished the meta‐alkylated adduct 7 gi, albeit in lower yield.
Scheme 5.

Ruthenium‐catalyzed C−H alkylation of phenylpyrazole 5 g. [a] The yield of di‐meta‐alkylated product is given in parentheses.
To understand the nature of the reaction mechanism, the alkylation reaction was conducted in the presence of typical radical scavengers (Scheme 6 a). While 2,2,6,6‐tetramethylpiperidin‐1‐oxyl (TEMPO) fully inhibited the catalytic reaction, the use of 1,1‐diphenylethylene significantly reduced the yield of the corresponding product 6 aa. The isolation of adduct 8 was supportive of a homolytic C−X bond cleavage. The reaction mechanism was further elucidated by the use of diastereomerically pure electrophiles 2 j and 2 k (Scheme 6 b). The reaction with endo‐2‐bromobornane (endo‐2 j) provided ortho‐alkylated product endo‐6 fj as well as a diastereomeric mixture of meta‐alkylated products 7 fj. Similarly, the stereochemistry of tert‐butylcyclohexyl bromide cis‐2 k and trans‐2 k 9f, 17 translated directly into the corresponding ortho‐alkylated products cis‐6 fk and trans‐6 fk, respectively. These findings thus provide strong support for a concerted oxidative addition/reductive elimination mechanism to be operative for the ortho‐alkylation. In contrast, the meta‐functionalized product 7 fk was obtained as cis‐ and trans‐isomers from the reaction with the single isomer cis‐2 k, which is indicative of the formation of an alkyl radical via a single‐electron transfer (SET) process. The stereochemistry and site‐selectivity of products 6 and 7 were confirmed by X‐ray analysis.16
Scheme 6.

Key mechanistic studies: (a) reaction in the presence of radical scavengers, (b) C−H alkylations with diastereomerically pure alkyl bromides 2.
Furthermore, we prepared the well‐defined cationic cyclometalated ruthenium complexes Ru I and Ru II,13 which showed high catalytic activity in the presence of MesCO2H (Scheme 7 a). In contrast to the standard condition, the reaction of phenylpyrazole 5 g in the absence of an acid additive resulted in a mixture of ortho‐ and meta‐alkylated products 6 ga and 7 ga. In addition, a substantial amount of decoordinated p‐cymene was observed in the initial period of the alkylation reaction (Scheme 7 b).
Scheme 7.

(a) Reactions with cyclometalated complex as catalysts, (b) detection of free p‐cymene. [a] The yield in parentheses was determined by 1H‐NMR using 1,3,5‐trimethoxybenzene as the internal standard.
Mechanistic studies by means of density functional theory (DFT) calculations were next conducted at the PW6B95‐D3(BJ)/def2‐TZVP+COSMO(o‐xylene)//TPSS‐D3(BJ)/def2‐TZVP level of theory.18 These findings reveal a facile oxidative addition of cyclohexyl bromide/reductive elimination process to occur on biscyclometalated ruthenium(II) intermediates with an energy barrier of only 17.6 kcal mol−1 (Figure 1). Calculations with various substituted arylpyrazoles indicated a rather minor influence of the substrate's electronic properties on the energy barriers for the oxidative addition/reductive elimination elementary steps.
Figure 1.

Relative Gibbs free energy profile for the oxidative addition/reductive elimination elementary step at the PW6B95‐D3(BJ)/def2‐TZVP+COSMO(o‐xylene)//TPSS‐D3(BJ)/def2‐TZVP level of theory.
A distortion energy analysis of TS2 with different directing groups revealed a substantially increased distortion energy, when the 3,5‐dimethylpyrazole was employed (Figure 2).13
Figure 2.

Distortion energy (a) for reductive elimination with different heterocycles, (b) for radical addition with N‐heterocycles.
Furthermore, the ruthenium(II)carboxylate catalysis was also found to facilitate decarboxylative alkylation reactions. Here various reaction conditions for the envisioned decarboxylative alkylation reaction of acid 10 a with bromocycloheptane (2 d) were tested first (Table 2).13 Carboxylate assistance significantly improved the catalytic efficacy, with MesCO2H being the optimal acid additive (entries 1–4).14b The reaction without an acid additive gave a reduced yield (entry 2), presumably because the substrate 10 a can itself act as carboxylate ligand. Control experiments verified the essential role of the ruthenium catalyst (entry 5). Furthermore, the well‐defined complex [Ru(O2CMes)2(p‐cymene)]19 turned out to be a competent catalyst (entry 6). To our delight, the reaction also proceeded under arene‐ligand‐free conditions using ruthenium‐nitrile complexes (entries 7–8). Other ruthenium sources such as Ru3(CO)12 and RuCl3 ⋅(H2O)n failed to facilitate any conversion (entries 9–10). Moreover, no product formation was observed when the reaction was attempted with palladium, rhodium, cobalt, or nickel complexes.13
Table 2.
Optimization of ruthenium‐catalyzed decarboxylative C−C alkylation of 10 a

|
Entry |
Deviation from the standard conditions |
Yield (%) |
|---|---|---|
|
1 |
none |
73 |
|
2 |
without MesCO2H |
39 |
|
3 |
1‐AdCO2H instead of MesCO2H |
49 |
|
4 |
PivOH instead of MesCO2H |
56 |
|
5 |
without 3 |
– |
|
6 |
[Ru(O2CMes)2(p‐cymene)] instead of 3 and without MesCO2H |
70 |
|
7 |
[Ru(NCt‐Bu)6][BF4]2 instead of 3 |
60 |
|
8 |
[Ru(NCt‐Bu)6][SbF6]2 instead of 3 |
49 |
|
9 |
RuCl3⋅(H2O)n instead of 3 |
– |
|
10 |
Ru3(CO)12 instead of 3 |
– |
|
11 |
Pd(OAc)2, [Cp*RhCl2]2, [Cp*Co(CO)I2] or [Ni(cod)2] as catalysts |
– |
[a] Reaction conditions: 10 a (0.5 mmol), 2 d (1.5 mmol), [Ru] (5.0 mol %), additive (30 mol %), K2CO3 (1.0 mmol), o‐xylene (1.0 mL), 120 °C, 16 h, yields of isolated products.
Having identified the optimal reaction conditions, we tested the versatility towards different alkyl bromides 2 (Scheme 8). With primary alkyl bromides 2 i–2 m, the C−H alkylation took place at the ortho‐position with excellent levels of regioselectivity. It is noteworthy that the reaction of acid 10 i and neopentyl bromide (2 i) afforded 40 % of the desired product 6 ii and 37 % of the ortho‐xylylated product 6 ii′ as a side‐product,16 which presumably forms via H‐atom abstraction from the o‐xylene solvent followed by benzylation. Similar to the C−H alkylation reaction (vide supra), the decarboxylative alkylation of bromocyclohexane (2 a) and exo‐2‐bromonorbornane (2 f) furnished the ortho‐alkylated products 6 aa, 6 af, and 6 if with excellent levels of site‐selectivity. In contrast, reactions with a broad range of acyclic alkyl bromides as well as cyclic alkyl bromides resulted in a preferred meta‐alkylation.16 Inspired by a recent meta‐selective alkylation with α‐bromoesters from our group,9c we probed whether this reaction can be combined with a C−C cleavage step. Indeed, slightly modified reaction conditions allowed for the formation of the products 7 ap–7 jp via C−C/C−H activation in high yields. Moreover, tertiary alkyl bromides reacted in the decarboxylative alkylation regime solely with meta‐selectivity.
Scheme 8.

Ruthenium‐catalyzed decarboxylative C−C alkylation. [a] [RuCl2(p‐cymene)]2 (5.0 mol %). [b] HCl adduct. [c] n‐Octane instead of o‐xylene as solvent. [d] PPh3 (5.0 mol %), PhCMe3 instead of o‐xylene.
Finally, ozonolysis20 of the alkylated arenes 6 or 7 provided access to synthetically useful meta‐alkylated acetanilides 11 in remarkably good yields, highlighting the versatility of the ruthenium‐catalyzed direct C−H alkylation (Scheme 9).16
Scheme 9.
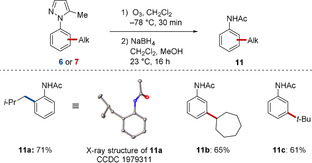
Product diversification by ozonolysis.
Given the broad applicability of this decarboxylative alkylation reaction, we became interested in unraveling its mode of action. To this end, detailed mechanistic studies were performed (Scheme 10). Reactions with radical scavengers led to a complete or partial inhibition of the catalytic activity (Scheme 10 a). In the presence of TEMPO, the alkyl‐TEMPO adduct 12 could be detected and isolated, which is in line with a radical C−X bond cleavage. Reactions in the presence of deuterated co‐solvents clearly indicated the organometallic character of the C−C cleavage (Scheme 10 b). In the absence of an alkyl bromide, almost complete decarboxylation took place and significant deuterium incorporation was observed at the ortho‐position and partly at the C3 and C5 position of the pyrazole, presumably due to electrophilic activation. In the presence of alkyl bromide 2 d, a deuterium incorporation of 46 % and 47 % was observed at the ortho‐position of alkylated product [D]n‐7 ad. A considerable decoordination of p‐cymene was detected during the initial period of the decarboxylative alkylation (Scheme 10 c).
Scheme 10.
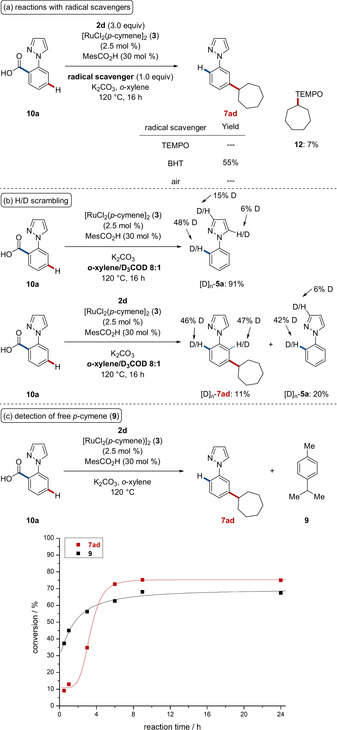
Key mechanistic findings: (a) reaction in the presence of radical scavengers, (b) H/D scrambling experiments, (c) detection of free p‐cymene.
On the basis of our findings, a plausible catalytic cycle for the ortho‐selective alkylation commences by a carboxylate‐assisted C−H ruthenation and dissociation of p‐cymene, thereby forming the cyclometalated complex 14 (Scheme 11, left). A second molecule of phenylpyrazole 5 coordinates to ruthenium complex 14 and undergoes C−H activation to form biscyclometalated complex 15. The oxidative addition of alkyl bromide 2 to complex 15 generates the stable ruthenium(IV) intermediate 16/B (Figure 1). Finally, reductive elimination and ligand exchange deliver the ortho‐alkylated product 6 and ruthenacycle 14. In contrast, meta‐C−H alkylation occurs through a SET process from ruthenium(II) complex 14 to alkyl bromide 2, forming ruthenium(III) intermediate 18 and a stabilized alkyl radical 19 (Scheme 11, right). Subsequently, 19 preferentially attacks the position para to ruthenium, thus leading to the formation of triplet ruthenium intermediate 20.9a, 9c Ligand‐to‐metal electron transfer and rearomatization furnishes ruthenacycle 21, which undergoes protodemetalation and C−H activation to furnish the desired meta‐alkylated product 7 and regenerates the active ruthenium species 14.
Scheme 11.

Proposed catalytic cycle for ruthenium‐catalyzed ortho‐ or meta‐alkylation.
Conclusion
In summary, we have reported on a ruthenium‐catalyzed C−H and C−C activation allowing for ortho‐ and meta‐alkylations of synthetically useful pyrazoles. The steric properties of the employed alkyl bromides and pyrazoles had a significant influence on the position‐selectivity of the alkylation reaction. Mechanistic studies were suggestive of two distinct mechanisms, an oxidative addition/reductive elimination event for the ortho‐C−H alkylation, while a SET pathway is proposed for meta‐functionalization. Moreover, an arene‐ligand‐free ruthenacycles was identified as the key intermediate in this transformation. Furthermore, computational studies and experiments with diastereomerically pure alkyl bromides unraveled an energetically favorable novel mechanism for ortho‐C−H secondary alkylations.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Generous support by the DAAD (fellowship to K.K.) and the DFG (SPP1807 and Gottfried‐Wilhelm‐Leibniz prize) is gratefully acknowledged. We thank Dr. Christopher Golz (University Göttingen) for the X‐ray diffraction analysis. Open access funding enabled and organized by Projekt DEAL.
K. Korvorapun, M. Moselage, J. Struwe, T. Rogge, A. M. Messinis, L. Ackermann, Angew. Chem. Int. Ed. 2020, 59, 18795.
Contributor Information
Korkit Korvorapun, http://www.ackermann.chemie.uni‐goettingen.de/.
Prof. Dr. Lutz Ackermann, Email: Lutz.Ackermann@chemie.uni-goettingen.de.
References
- 1.For selected reviews, see:
- 1a. Rej S., Ano Y., Chatani N., Chem. Rev. 2020, 120, 1788–1887; [DOI] [PubMed] [Google Scholar]
- 1b. Gandeepan P., Müller T., Zell D., Cera G., Warratz S., Ackermann L., Chem. Rev. 2019, 119, 2192–2452; [DOI] [PubMed] [Google Scholar]
- 1c. Wang C.-S., Dixneuf P. H., Soulé J.-F., Chem. Rev. 2018, 118, 7532–7585; [DOI] [PubMed] [Google Scholar]
- 1d. Newton C. G., Wang S.-G., Oliveira C. C., Cramer N., Chem. Rev. 2017, 117, 8908–8976; [DOI] [PubMed] [Google Scholar]
- 1e. Park Y., Kim Y., Chang S., Chem. Rev. 2017, 117, 9247–9301; [DOI] [PubMed] [Google Scholar]
- 1f. Gensch T., Hopkinson M. N., Glorius F., Wencel-Delord J., Chem. Soc. Rev. 2016, 45, 2900–2936; [DOI] [PubMed] [Google Scholar]
- 1g. Ritleng V., Sirlin C., Pfeffer M., Chem. Rev. 2002, 102, 1731–1770. [DOI] [PubMed] [Google Scholar]
- 2.For selected reviews, see:
- 2a. Khake S. M., Chatani N., Trends Chem. 2019, 1, 524–539; [Google Scholar]
- 2b. Kommagalla Y., Chatani N., Coord. Chem. Rev. 2017, 350, 117–135; [Google Scholar]
- 2c. Colby D. A., Tsai A. S., Bergman R. G., Ellman J. A., Acc. Chem. Res. 2012, 45, 814–825; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d. Neufeldt S. R., Sanford M. S., Acc. Chem. Res. 2012, 45, 936–946; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2e. Colby D. A., Bergman R. G., Ellman J. A., Chem. Rev. 2010, 110, 624–655; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2f. Lyons T. W., Sanford M. S., Chem. Rev. 2010, 110, 1147–1169; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2g. Ackermann L., Vicente R., Kapdi A. R., Angew. Chem. Int. Ed. 2009, 48, 9792–9826; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9976–10011. [Google Scholar]
- 3.For selected reviews on chelation-assisted C−H functionalization, see:
- 3a. Sambiagio C., Schönbauer D., Blieck R., Dao-Huy T., Pototschnig G., Schaaf P., Wiesinger T., Zia M. F., Wencel-Delord J., Besset T., Maes B. U. W., Schnürch M., Chem. Soc. Rev. 2018, 47, 6603–6743; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Chen Z., Wang B., Zhang J., Yu W., Liu Z., Zhang Y., Org. Chem. Front. 2015, 2, 1107–1295; [Google Scholar]
- 3c. Huang Z., Lim H. N., Mo F., Young M. C., Dong G., Chem. Soc. Rev. 2015, 44, 7764–7786; [DOI] [PubMed] [Google Scholar]
- 3d. Zhang F., Spring D. R., Chem. Soc. Rev. 2014, 43, 6906–6919. [DOI] [PubMed] [Google Scholar]
- 4.For selected reviews on ruthenium-catalyzed C−H functionalization, see:
- 4a. Shan C., Zhu L., Qu L.-B., Bai R., Lan Y., Chem. Soc. Rev. 2018, 47, 7552–7576; [DOI] [PubMed] [Google Scholar]
- 4b. Nareddy P., Jordan F., Szostak M., ACS Catal. 2017, 7, 5721–5745; [Google Scholar]
- 4c. Ackermann L., Acc. Chem. Res. 2014, 47, 281–295; [DOI] [PubMed] [Google Scholar]
- 4d. De Sarkar S., Liu W., Kozhushkov S. I., Ackermann L., Adv. Synth. Catal. 2014, 356, 1461–1479; [Google Scholar]
- 4e. Arockiam P. B., Bruneau C., Dixneuf P. H., Chem. Rev. 2012, 112, 5879–5918; [DOI] [PubMed] [Google Scholar]
- 4f. Kakiuchi F., Chatani N., Adv. Synth. Catal. 2003, 345, 1077–1101. [Google Scholar]
- 5.
- 5a. Wang G.-W., Wheatley M., Simonetti M., Cannas D. M., Larrosa I., Chem 2020, 6, 1459–1468; [Google Scholar]
- 5b. Ackermann L., Hofmann N., Vicente R., Org. Lett. 2011, 13, 1875–1877; [DOI] [PubMed] [Google Scholar]
- 5c. Ackermann L., Novák P., Vicente R., Hofmann N., Angew. Chem. Int. Ed. 2009, 48, 6045–6048; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6161–6164; [Google Scholar]
- 5d. Ackermann L., Novák P., Org. Lett. 2009, 11, 4966–4969. [DOI] [PubMed] [Google Scholar]
- 6.For selected reviews on ruthenium-catalyzed meta-C−H functionalization, see:
- 6a. Mihai M. T., Genov G. R., Phipps R. J., Chem. Soc. Rev. 2018, 47, 149–171; [DOI] [PubMed] [Google Scholar]
- 6b. Leitch J. A., Frost C. G., Chem. Soc. Rev. 2017, 46, 7145–7153; [DOI] [PubMed] [Google Scholar]
- 6c. Li J., De Sarkar S., Ackermann L., Top. Organomet. Chem. 2016, 55, 217–257. [Google Scholar]
- 7.
- 7a. Yuan C., Zhu L., Chen C., Chen X., Yang Y., Lan Y., Zhao Y., Nat. Commun. 2018, 9, 1189; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Yuan C., Zhu L., Zeng R., Lan Y., Zhao Y., Angew. Chem. Int. Ed. 2018, 57, 1277–1281; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1291–1295; [Google Scholar]
- 7c. Wang X.-G., Li Y., Zhang L.-L., Zhang B.-S., Wang Q., Ma J.-W., Liang Y.-M., Chem. Commun. 2018, 54, 9541–9544; [DOI] [PubMed] [Google Scholar]
- 7d. Leitch J. A., McMullin C. L., Paterson A. J., Mahon M. F., Bhonoah Y., Frost C. G., Angew. Chem. Int. Ed. 2017, 56, 15131–15135; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15327–15331. [Google Scholar]
- 8.
- 8a. Paterson A. J., Heron C. J., McMullin C. L., Mahon M. F., Press N. J., Frost C. G., Org. Biomol. Chem. 2017, 15, 5993–6000; [DOI] [PubMed] [Google Scholar]
- 8b. Paterson A. J., St John-Campbell S., Mahon M. F., Press N. J., Frost C. G., Chem. Commun. 2015, 51, 12807–12810. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Korvorapun K., Kuniyil R., Ackermann L., ACS Catal. 2020, 10, 435–440; [Google Scholar]
- 9b. Gandeepan P., Koeller J., Korvorapun K., Mohr J., Ackermann L., Angew. Chem. Int. Ed. 2019, 58, 9820–9825; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9925–9930; [Google Scholar]
- 9c. Korvorapun K., Kaplaneris N., Rogge T., Warratz S., Stückl A. C., Ackermann L., ACS Catal. 2018, 8, 886–892; [Google Scholar]
- 9d. Fumagalli F., Warratz S., Zhang S.-K., Rogge T., Zhu C., Stückl A. C., Ackermann L., Chem. Eur. J. 2018, 24, 3984–3988; [DOI] [PubMed] [Google Scholar]
- 9e. Ruan Z., Zhang S.-K., Zhu C., Ruth P. N., Stalke D., Ackermann L., Angew. Chem. Int. Ed. 2017, 56, 2045–2049; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 2077–2081; [Google Scholar]
- 9f. Li J., Korvorapun K., De Sarkar S., Rogge T., Burns D. J., Warratz S., Ackermann L., Nat. Commun. 2017, 8, 15430; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9g. Li J., Warratz S., Zell D., De Sarkar S., Ishikawa E. E., Ackermann L., J. Am. Chem. Soc. 2015, 137, 13894–13901; [DOI] [PubMed] [Google Scholar]
- 9h. Hofmann N., Ackermann L., J. Am. Chem. Soc. 2013, 135, 5877–5884. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Li G., Jia C., Cai X., Zhong L., Zou L., Cui X., Chem. Commun. 2020, 56, 293–296; [DOI] [PubMed] [Google Scholar]
- 10b. Jia C., Wang S., Lv X., Li G., Zhong L., Zou L., Cui X., Eur. J. Org. Chem. 2020, 1992–1995; [Google Scholar]
- 10c. Sagadevan A., Greaney M. F., Angew. Chem. Int. Ed. 2019, 58, 9826–9830; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9931–9935; [Google Scholar]
- 10d. Li B., Fang S.-L., Huang D.-Y., Shi B.-F., Org. Lett. 2017, 19, 3950–3953; [DOI] [PubMed] [Google Scholar]
- 10e. Li G., Gao P., Lv X., Qu C., Yan Q., Wang Y., Yang S., Wang J., Org. Lett. 2017, 19, 2682–2685; [DOI] [PubMed] [Google Scholar]
- 10f. Li G., Li D., Zhang J., Shi D.-Q., Zhao Y., ACS Catal. 2017, 7, 4138–4143; [Google Scholar]
- 10g. Li G., Ma X., Jia C., Han Q., Wang Y., Wang J., Yu L., Yang S., Chem. Commun. 2017, 53, 1261–1264; [DOI] [PubMed] [Google Scholar]
- 10h. Li Z.-Y., Li L., Li Q.-L., Jing K., Xu H., Wang G.-W., Chem. Eur. J. 2017, 23, 3285–3290. [DOI] [PubMed] [Google Scholar]
- 11.For selected examples of transition metal-catalyzed C−C activations, see:
- 11a. Hou S.-H., Prichina A. Y., Zhang M., Dong G., Angew. Chem. Int. Ed. 2020, 59, 7848–7856; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 7922–7930; [Google Scholar]
- 11b. Hu Z., Hu X.-Q., Zhang G., Gooßen L. J., Org. Lett. 2019, 21, 6770–6773; [DOI] [PubMed] [Google Scholar]
- 11c. Zhu J., Chen P.-h., Lu G., Liu P., Dong G., J. Am. Chem. Soc. 2019, 141, 18630–18640; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11d. Zhu J., Wang J., Dong G., Nat. Chem. 2019, 11, 45–51; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11e. Deng L., Fu Y., Lee S. Y., Wang C., Liu P., Dong G., J. Am. Chem. Soc. 2019, 141, 16260–16265; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11f. Sun T., Zhang Y., Qiu B., Wang Y., Qin Y., Dong G., Xu T., Angew. Chem. Int. Ed. 2018, 57, 2859–2863; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 2909–2913; [Google Scholar]
- 11g. Zhu Z., Li X., Chen S., Chen P.-h., Billett B. A., Huang Z., Dong G., ACS Catal. 2018, 8, 845–849; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11h. Wang H., Choi I., Rogge T., Kaplaneris N., Ackermann L., Nat. Catal. 2018, 1, 993–1001; [Google Scholar]
- 11i. Moselage M., Li J., Kramm F., Ackermann L., Angew. Chem. Int. Ed. 2017, 56, 5341–5344; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5425–5428; [Google Scholar]
- 11j. Kumar N. Y. P., Bechtoldt A., Raghuvanshi K., Ackermann L., Angew. Chem. Int. Ed. 2016, 55, 6929–6932; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7043–7046; [Google Scholar]
- 11k. Zhang J., Shrestha R., Hartwig J. F., Zhao P., Nat. Chem. 2016, 8, 1144–1151; [DOI] [PubMed] [Google Scholar]
- 11l. Huang L., Biafora A., Zhang G., Bragoni V., Gooßen L. J., Angew. Chem. Int. Ed. 2016, 55, 6933–6937; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7047–7051; [Google Scholar]
- 11m. Ozkal E., Cacherat B., Morandi B., ACS Catal. 2015, 5, 6458–6462; [Google Scholar]
- 11n. Ishida N., Ikemoto W., Murakami M., J. Am. Chem. Soc. 2014, 136, 5912–5915; [DOI] [PubMed] [Google Scholar]
- 11o. Souillart L., Cramer N., Angew. Chem. Int. Ed. 2014, 53, 9640–9644; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9794–9798; [Google Scholar]
- 11p. Seiser T., Cramer N., J. Am. Chem. Soc. 2010, 132, 5340–5341; [DOI] [PubMed] [Google Scholar]
- 11q. Seiser T., Roth O. A., Cramer N., Angew. Chem. Int. Ed. 2009, 48, 6320–6323; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6438–6441; [Google Scholar]
- 11r. Chatani N., Ie Y., Kakiuchi F., Murai S., J. Am. Chem. Soc. 1999, 121, 8645–8646. [Google Scholar]
- 12. Ackermann L., Acc. Chem. Res. 2020, 53, 84–104. [DOI] [PubMed] [Google Scholar]
- 13.For detailed information, see the Supporting Information.
- 14.
- 14a. Davies D. L., Macgregor S. A., McMullin C. L., Chem. Rev. 2017, 117, 8649–8709; [DOI] [PubMed] [Google Scholar]
- 14b. Ackermann L., Chem. Rev. 2011, 111, 1315–1345. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Rogge T., Ackermann L., Angew. Chem. Int. Ed. 2019, 58, 15640–15645; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 15787–15792; [Google Scholar]
- 15b. Simonetti M., Cannas D. M., Just-Baringo X., Vitorica-Yrezabal I. J., Larrosa I., Nat. Chem. 2018, 10, 724–731; [DOI] [PubMed] [Google Scholar]
- 15c. Simonetti M., Cannas D. M., Panigrahi A., Kujawa S., Kryjewski M., Xie P., Larrosa I., Chem. Eur. J. 2017, 23, 549–553; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15d. Ackermann L., Althammer A., Born R., Tetrahedron 2008, 64, 6115–6124; [Google Scholar]
- 15e. Ackermann L., Althammer A., Born R., Synlett 2007, 2833–2836; [Google Scholar]
- 15f. Ackermann L., Born R., Álvarez-Bercedo P., Angew. Chem. Int. Ed. 2007, 46, 6364–6367; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 6482–6485. [Google Scholar]
- 16.Deposition numbers 1979314 (6fa), 2016734 (endo-6fj), 2016645 (exo-7fj), 2016646 (endo-7fj), 2016647 (cis-6fk), 2016735 (trans-6fk), 1979319 (6ii′), 1979310 (7 cd), and 1979311 (11 a) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 17. Cera G., Haven T., Ackermann L., Angew. Chem. Int. Ed. 2016, 55, 1484–1488; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1506–1510. [Google Scholar]
- 18.
- 18a. Grimme S., Ehrlich S., Goerigk L., J. Comput. Chem. 2011, 32, 1456–1465; [DOI] [PubMed] [Google Scholar]
- 18b. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104; [DOI] [PubMed] [Google Scholar]
- 18c. Sinnecker S., Rajendran A., Klamt A., Diedenhofen M., Neese F., J. Phys. Chem. A 2006, 110, 2235–2245; [DOI] [PubMed] [Google Scholar]
- 18d. Zhao Y., Truhlar D. G., J. Phys. Chem. A 2005, 109, 5656–5667; [DOI] [PubMed] [Google Scholar]
- 18e. Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297–3305; [DOI] [PubMed] [Google Scholar]
- 18f. Tao J., Perdew J. P., Staroverov V. N., Scuseria G. E., Phys. Rev. Lett. 2003, 91, 146401; [DOI] [PubMed] [Google Scholar]
- 18g. Klamt A., Jonas V., Bürger T., Lohrenz J. C. W., J. Phys. Chem. A 1998, 102, 5074–5085; [Google Scholar]
- 18h. Klamt A., J. Phys. Chem. 1995, 99, 2224–2235. [Google Scholar]
- 19. Ackermann L., Vicente R., Potukuchi H. K., Pirovano V., Org. Lett. 2010, 12, 5032–5035. [DOI] [PubMed] [Google Scholar]
- 20. Kashima C., Hibi S., Maruyama T., Harada K., Omote Y., J. Heterocycl. Chem. 1987, 24, 637–639. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary