

Abstract
The homoleptic group 5 carbonylates [M(CO)6]− (M=Nb, Ta) serve as ligands in carbonyl‐terminated heterobimetallic AgmMn clusters containing 3 to 11 metal atoms. Based on our serendipitous [Ag6{Nb(CO)6}4]2+ (4 a 2+) precedent, we established access to such AgmMn clusters of the composition [Agm{M(CO)6}n]x (M=Nb, Ta; m=1, 2, 6; n=2, 3, 4, 5; x=1−, 1+, 2+). Salts of those molecular cluster ions were synthesized by the reaction of [NEt4][M(CO)6] and Ag[Al(ORF)4] (RF=C(CF3)3) in the correct stoichiometry in 1,2,3,4‐tetrafluorobenzene at −35 °C. The solid‐state structures were determined by single‐crystal X‐ray diffraction methods and, owing to the thermal instability of the clusters, a limited scope of spectroscopic methods. In addition, DFT‐based AIM calculations were performed to provide an understanding of the bonding within these clusters. Apparently, the clusters 3 + (m=6, n=5) and 4 2+ (m=6, n=4) are superatom complexes with trigonal‐prismatic or octahedral Ag6 superatom cores. The [M(CO)6]− ions then bind through three CO units as tridentate chelate ligands to the superatom core, giving overall structures related to tetrahedral AX4 (4 2+) or trigonal bipyramidal AX5 molecules (3 +).
Keywords: carbonyl clusters, charge, DFT, heterobimetallic compounds, silver
Anything is possible! [Agm{M(CO)6}n]x clusters (M=Nb, Ta; m=1, 2, 6; n=2, 3, 4, 5; x=1−, 1+, 2+) were synthesized by the reaction of [NEt4][M(CO)6] and Ag[Al(ORF)4] (RF=C(CF3)3) and the structure determined by single‐crystal X‐ray diffraction methods. The [M(CO)6] fragments serve as ligands in the obtained clusters. DFT‐based AIM calculations were performed to provide an understanding of the bonding situation and the cores of the cations were interpreted as superatoms.
Introduction
With the discovery of Ni(CO)4,1 the first homoleptic transition‐metal carbonyl complex, a whole family of diversified complexes was born. This still growing family does not only include mononuclear,2 but also bi‐3 and polynuclear4, 5 complexes with terminal, bridging, and even capping CO ligands. Owing to the synergistic bonding between the CO ligand and the metal atom, the carbonylates2, 6 and the neutral complexes7 are the most stable and the most studied compounds of this group, whereas cationic8, 9 homoleptic carbonyl complexes are much more reactive. One reason for the continuing intensive interest in these complexes is certainly their wide range of applications in catalysis,8 biochemical processes,10 and medicine.11
In coordination chemistry, carbonyl complexes can also act as ligands for main‐group or transition metals, thus forming heterometallic clusters. Carbonylates are used frequently to donate electrons to a main‐group12, 13, 14, 15, 16 or transition‐metal17 acceptor, whereas the capacity of neutral carbonyl complexes to serve as electron donors is less pronounced. However, there are a few examples that contain neutral carbonyl complexes as donor ligands to main‐group12, 18 or transition metals,19, 20, 21 such as [(OC)5Fe→GaCl3]22 as well as [Ag{Fe(CO)5}2]+.19, 23 Overall, the heterometallic transition‐metal carbonyl complexes are dominated by anionic clusters, with just a few neutral examples such as [Ag4{Co(CO)4}4].24 To the best of our knowledge, [Ag{Fe(CO)5}2]+[19, 23] is the only, albeit rather small, cationic and structurally characterized heterobimetallic homoleptic transition‐metal carbonyl cluster so far.
In many of the larger transition‐metal carbonyl cluster anions, a M6 octahedral core is the central structural motif. Figure 1 shows a selection of small carbonyl cluster anions containing an octahedral core, in which every second triangular surface is capped, thus creating a super‐tetrahedron. The highly symmetric monometallic [Os20(CO)40]2−[5, 25] (Figure 1 a) even shows one more layer forming a “super‐super‐tetrahedron”. The structural motif shown in Figure 1 b mainly appears in heterobimetallic cluster salts. Note that the ratio and positions of the transition metals vary in the published complexes.17, 26
Figure 1.
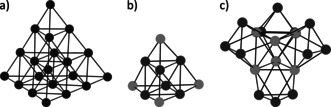
Metallic core of transition‐metal clusters containing an octahedron, which is capped on every second triangular surface. The shown examples are the simplified crystal structures of (a) [Os20(CO)40]2−,5 (b) [Cu6{Fe(CO)4}4]2−,17 (c) [Au6{Ni3(CO)6}4]2−.27, 28 The counterions and CO ligands were omitted for clarity.
The highly symmetric [Au6{Ni3(CO)6}4]2−[27, 28] also belongs to this group (Figure 1 c). Three Ni atoms cap every other triangular face of the octahedral Au6 core, building a super‐tetrahedron with T d symmetry. Thus, the [Ni3(CO)6] unit can be seen as a tridentate pseudo‐ligand towards an Au6 superatom. Yet, it is challenging to fully understand the structural and bonding properties of such clusters. The superatom model29 is one concept used to address these questions for molecular clusters. Thus, the calculated molecular orbitals of the cluster [Au6{Ni3(CO)6}4]2− are isolobal with those of the simple molecule CH4. Therefore, the concept of hybridization was extended and the Au6 cluster orbitals were classified as sp3 hybrid orbitals.28 By removing one, two, or three (pseudo‐)ligands, analogies to NH3, H2O, and HCl are obtained. However, the question remains as to what extent the concept can be transferred from metal‐based pseudo‐ligands to other systems. Sticking to this bonding concept, in principle, hitherto unknown coordination numbers of the M6 core exceeding four could be achieved, that is, five corresponding to a trigonal bipyramidal AX5 molecule.
Turning to known heterobimetallic carbonyl clusters with a silver core, the subject of this contribution, the large majority of structurally characterized examples contain the water‐stable [Fe(CO)4]2− ligand as a building block. The metallic core of all the known (anionic) structures with 3 to 13 silver atoms and with exterior [Fe(CO)4] units are collected in Figure 2.
Figure 2.
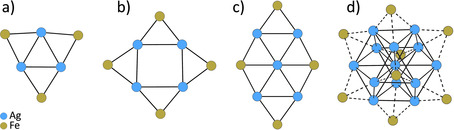
Metallic core arrangement of typical structurally characterized anionic homoleptic silver‐ironcarbonylate clusters. (a) [Ag3{μ2‐Fe(CO)4}3]3−[21]; (b) [Ag4{μ2‐Fe(CO)4}4]4−[21, 30]; (c) [Ag5{μ2‐Fe(CO)4}2{μ3‐Fe(CO)4}2]3−[30]; (d) [Ag13{Fe(CO)4}8]4−[31], [Ag12(μ12‐Ag){μ3‐Fe(CO)4}8]5−.32
Further examples of silver clusters with transition‐metal carbonylates other than [Fe(CO)4]2− are [Ag9Os13(CO)48]−,33 [Ag16Ni24(CO)40]4−,34 and [Ag4{Co(CO)4}4].24 In addition to the discussed heterobimetallic homoleptic carbonyl clusters, several related heteroleptic carbonyl complexes are known. For example, the first heterobimetallic carbonyl complexes, published by Nyholm and co‐workers,35 contained phosphine or cyclopentadienyl ligands. Yet, here, we focus on homoleptic clusters only including CO ligands.
In this publication, we present crystal structures and spectroscopic as well as bonding analyses of several heterobimetallic AgmMn clusters (M=Nb, Ta) containing 3 to 11 metal atoms. They obey the general formula [Agm{M(CO)6}n]x (M=Nb, Ta, m=1, 2, 6; n=2, 3, 4, 5; x=1−, 1+, 2+). Of those, only the structure of the super‐tetrahedral cluster cation [Ag6{Nb(CO)6}4]2+ was communicated recently.36 In these complexes, niobium or tantalum hexacarbonylates serve as ligands for the silver atoms. For group 5 elements, structurally characterized heterometallic clusters are yet limited to only a few heteroleptic complexes. The neutral trimer [AgM(CO)4(dmpe)]3 (dmpe=1,2‐bis(dimethylphosphino)ethane, M=Nb, Ta)37 appears to be the closest literature reference to the compounds in this work.
Results and Discussion
During our continuing efforts to prepare novel transition‐metal carbonyl cations19, 38 as salts of suitable and very weakly coordinating anions (WCAs),39 we determined the crystal structure of [Ag6{M(CO)6}4]2+([Al(ORF)4]−)2 (4 a; RF=C(CF3)3) by serendipity. The structure of the cluster cation adheres to Figure 1 b. It is an isolable intermediate towards the preparation of the heptacarbonyl salt [Nb(CO)7]+[Al(ORF)4]−.36 The efforts towards the synthesis of further and related cluster salts are presented here.
Syntheses and molecular structures of 1–4
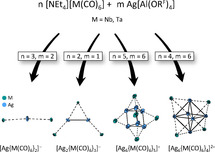
With the intention to prepare homoleptic carbonyl cations, cluster salt 4 a was initially obtained from a reaction of [NEt4][Nb(CO)6] with Ag[Al(ORF)4] under 3 bar CO pressure.36 Using this work as a starting point, we found that all syntheses to the heterobimetallic cluster ions described herein could be carried out in the absence of CO gas. This prevents the further reaction to homoleptic carbonyl cations as far as possible. With this route and using the polar, but non‐coordinating solvent 4FB (1,2,3,4‐F4C6H2), a series of heterobimetallic cluster ions [Agm{M(CO)6}n]x (M=Nb, Ta, m=1, 2, 6; n=2, 3, 4, 5; x=1−, 1+, 2+) was synthesized. Their composition only represents the stoichiometric ratio used [Eq. (1)–(4); Scheme 1].
Scheme 1.
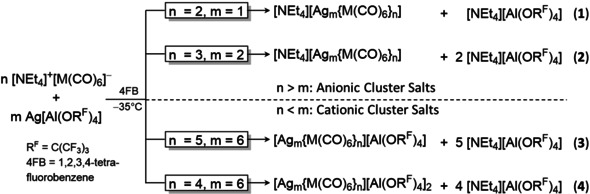
Synthesis routes to [NEt4]+[Ag{Nb(CO)6}2]− (1 a M=Nb, 1 b M=Ta), [NEt4]+[Ag2{M(CO)6}3]− (2 a M=Nb; 2 b M=Ta), [Ag6{Nb(CO)6}5]+[Al(ORF)4]− (3 a), and [Ag6{M(CO)6}4]2+([Al(ORF)4]−)2 (4 a M=Nb; 4 b M=Ta).
Neutral clusters like the hypothetical [Ag3{M(CO)6}3]—related to the trimer [AgM(CO)4(dmpe)]3 37—were inaccessible. Rather decomposition was observed when using an equimolar ratio of the starting materials. All syntheses were carried out at −35 °C, close to the melting point of the solvent 4FB (m.p. −42 °C). The intensely red salts were crystallized by storage at −30 °C after careful layering the reaction mixture with cold pentane at −30 °C. Powders were obtained at the same temperature by precipitation upon addition of cold pentane. Starting at about −15 °C, the compounds decomposed quickly with evolution of CO, leaving a brown solid, shown exemplary in Figure 3 for crystals of 4 b.
Figure 3.
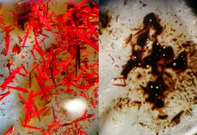
Photographs of crystals of 4 b at −30 °C (left) and their quick decomposition at −15 °C with concomitant CO evolution (right).
Molecular structures
Crystals suitable for single‐crystal structure determinations of all compounds were obtained (Figure 5). For a better understanding, first the metallic cores of the cluster ions 1 −, 2 −, 3 +, and 4 2+ including the schematic structural formulae of 1 to 4 are shown in Figure 4. Figure 5 then contains full ellipsoid plots of the carbonyl‐terminated cluster ions. In all structures, both the [NEt4]+ or the [Al(ORF)4]− counterions are well separated and clearly non‐coordinating, exhibiting their typical structural parameters. For brevity, they are not discussed.
Figure 5.
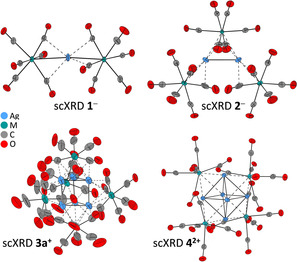
Molecular structures of the cluster ions within the single‐crystal structures of 1–4 (anionic: 1 a − (M=Nb), 1 b − (M=Ta), 2 a − (M=Nb), and 2 b − (M=Ta); cationic 3 a + (M=Nb); dicationic 4 a 2+ (M=Nb) and 4 b 2+ (M=Ta). As the structures are almost identical for M=Nb and Ta, only one representation with M=Nb is shown. The counterions [NEt4]+ or [Al(ORF)4]− were omitted for clarity. All thermal ellipsoids are drawn at the 50 % probability level.
Figure 4.
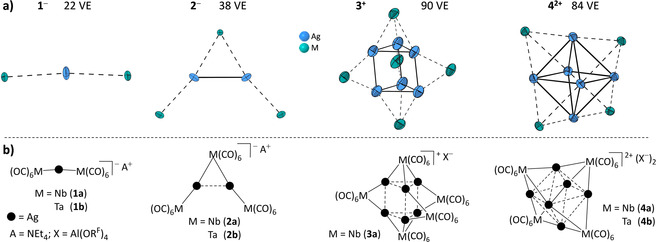
(a) Sum of the formally available valence electrons (VE) of the metal cores (Ag+: 10 VE and M−: 6 VE). Structures of the metallic cores of the anionic clusters 1 a − (M=Nb) and 1 b − (M=Ta), 2 a − (M=Nb) and 2 b − (M=Ta), as well as structures of the cationic cluster cores of 3 a + (M=Nb), 4 a 2+ (M=Nb), and 4 b 2+ (M=Ta). The counterions [NEt4]+ or [Al(ORF)4]− and the CO ligands are omitted for clarity in all structures. Ellipsoids were drawn at the 50 % probability level. (b) Schematic structural formulae for 1, 2, 3, and 4.
Single‐crystal X‐ray structure determinations (scXRD) of mono‐clinic 1 a, space group P21/c, revealed the formation of a C 2‐symmetric anion 1 a −. In the anion 1 a −, two [Nb(CO)6] moieties coordinate one Ag atom almost linearly (“μ1”; Nb‐Ag‐Nb 174.03(1)°). Two almost equidistant Ag−Nb bonds at 2.8359(5) and 2.8467(5) Å are the key structural features (Figure 5). The closely related crystal structure of 1 b includes eight different pairs of ions in the asymmetric unit (Figure S16, Figure S17 in the Supporting Information), probably owing to the low Ag–Ta rotation barrier as previously described for the related cationic [Ag{Fe(CO)5}2]+.19 Because of disorder and the large unit cell size, only X‐ray data of inferior quality was acquired. Therefore, no structural parameters of this structure are discussed in the following. Nevertheless, the data clearly showed that the desired compound [NEt4][Ag{Ta(CO)6}2] was obtained.
Isomorphous 2 a and 2 b crystallized in the monoclinic space group C 2/c with the isostructural anions having almost C 2 symmetry. Both structures contain two Ag atoms, each of which is linearly coordinated by two Nb or Ta atoms from [M(CO)6] units. One apical [M(CO)6] unit is bound to two silver atoms (“μ2”), whereas the others interact only with one silver atom (“μ1”). The overall Ag2M3 core is reminiscent of an “A‐frame” structure (Figure 4).40 The central Ag–Ag distance is equal to the metal–metal distances in metallic silver (d Ag–Ag=2.889(1) Å (2 a −), 2.882(1) Å (2 b −), d Ag–metal=2.88 Å).41 In addition, the Ag–M distances (2.8081(8) to 2.8710(7) Å) are in a similar range to the Ag–Ag separations. This suggests that 2 a − and 2 b − are delocalized formally 38 VE metal cluster anions.
Compound 3 a crystallized in the triclinic space group P . The structural core of 3 a + includes six Ag atoms arranged as a trigonal prism, which is capped by five [Nb(CO)6] fragments. Two are located on the triangular faces (“μ3”) whereas the other three are coordinating to the edges (“μ2”, Figure 4; Figure 5). Unfortunately, we could not get crystallographic data of sufficiently high quality for in depth discussion of light atom bond lengths and angles (R1=9.45 %). Apparently, the CO ligands and also the heavy atoms are in motion, as visible from the size of the ellipsoids in the crystal structure (Figure 5). Yet, the metallic Nb5Ag6 heavy atom core structure with formally 90 VE is well determined, exhibits almost D 3h symmetry and includes similar Ag–Ag (av. 2.85 Å) and Ag–Nb (av. 2.96 Å) distances. Both are close to the values in silver or niobium metal (d Nb–metal=2.94 Å). All attempts to obtain a similar structure with tantalum were futile.
Cluster salts 4 a and 4 b (Scheme 1) crystallized isomorphously in the orthorhombic space group Pbca. In the cluster dications 4 a 2+ and 4 b 2+, four [M(CO)6] fragments cap a regular Ag6 octahedron on every other triangular face (“μ3”; Figure 4; Figure 5). The structure cores of the dications are highly symmetric, adopt almost T d symmetry, and form a hetero‐bimetallic Ag6M4 super‐tetrahedron. Like in the other compounds, the very similar Ag–Ag (av. 2.90 Å) and Ag–Nb (av. 2.99 Å) or Ag–Ta (av. 2.98 Å) distances are close to the values found in the pure metals (Ag: 2.88 Å; Nb and Ta: 2.94 Å). This observation suggests 4 a 2+ and 4 b 2+ to be delocalized formal 84 VE metal cluster dications. All bond lengths and angles given are summarized in Table S13 (in the Supporting Information).
Compared with the isolated octahedral anions [M(CO)6]−, the averaged M−C bond lengths of compounds 1 to 4 are elongated, and in accordance the respective C−O bond lengths are shortened with a higher Ag/M ratio leading to shorter CO bonds culminating in the dicationic structures 4 2+ (Table 1, Table S14 in the Supporting Information). IR spectroscopic analysis in the next section supports this observation.
Table 1.
Experimental range of relevant bond lengths [Å] within the clustered ions of 1–4.[a]
|
Bond |
|
1 a − |
2 a − |
2 b − |
3 a +[b] |
4 a 2+ |
4 b 2+ |
|---|---|---|---|---|---|---|---|
|
d Ag–M |
μ1 |
2.836–2.847 |
2.808 |
2.817 |
– |
– |
– |
|
|
μ2 |
‐ |
2.867 |
2.871 |
2.891–2.952 |
– |
– |
|
|
μ3 |
– |
– |
– |
2.941–3.067 |
2.966–3.015 |
2.966–3.016 |
|
|
|
|
|
|
|
|
|
|
d Ag–C |
μ1 |
2.544–2.669 |
2.505–2.629 |
2.524–2.597 |
– |
– |
– |
|
|
μ2 |
– |
2.551 |
2.568 |
2.57–2.70 |
– |
– |
|
|
μ3 |
– |
– |
– |
2.57–2.69 |
2.592–2.701 |
2.582–2.706 |
|
|
|
|
|
|
|
|
|
|
d M–C |
μ1 |
2.111–2.150 |
2.107–2.149 |
2.091–2.128 |
– |
– |
– |
|
|
μ2 |
– |
2.124–2.164 |
2.110–2.141 |
2.07–2.21 |
– |
– |
|
|
μ3 |
– |
– |
– |
2.07–2.23 |
2.110–2.219 |
2.102–2.173 |
|
|
|
|
|
|
|
|
|
|
d CO,Ag |
μ1 |
1.146–1.157 |
1.157–1.160 |
1.139–1.157 |
– |
– |
– |
|
|
μ2 |
– |
1.152 |
1.152 |
1.15–1.20 |
– |
– |
|
|
μ3 |
– |
– |
– |
1.13–1.21 |
1.096–1.150 |
1.119–1.161 |
|
|
|
|
|
|
|
|
|
|
d CO,free |
μ1 |
1.137–1.140 |
1.133–1.164 |
1.124–1.150 |
– |
– |
– |
|
|
μ2 |
– |
1.126–1.149 |
1.131–1.151 |
1.10–1.24 |
– |
– |
|
|
μ3 |
– |
– |
– |
1.10–1.17 |
1.098–1.166 |
1.113–1.153 |
[a] For comparison, the M−C bonds in [M(CO)6]− are found on average at 2.089 [Nb42]/2.103 Å [Ta43] and the C−O bonds at 1.163 [Nb42]/1.149 Å [Ta44]. All structural parameters of the cluster ions are in very good agreement with the DFT calculated structures optimized at the BP86‐D3(BJ)/def2‐TZVPP level of theory. See the Supporting Information and Table S15 to Table S18 for a comparison. [b] Owing to poor quality of the crystallographic data, the values written in italics are only guidelines for reference purposes.
Characterization by IR spectroscopy
Owing to the temperature sensitivity of the products, the IR spectroscopic analyses of the products 1, 2, 3, and 4 were performed on the precipitated powders. Thus, the starting materials were dissolved in the correct ratio in cold 4FB in the glovebox and stirred at −35 °C for 30 min. Then, the products were precipitated with cold pentane and were allowed to settle for another 30 min at −35 °C. Afterwards, the solutions were decanted from the solid products and the remaining solvent was evaporated prior to the measurements on the precooled attenuated total reflection (ATR) unit of a FTIR spectrometer situated inside the glovebox. Figure 6 a shows the ν(CO) range from experimental and calculated (BP86‐D3(BJ)/def2‐TZVPP) IR spectra of the starting material [NEt4][Nb(CO)6] and the cluster ions 1 a −, 2 a −, 3 a +, 4 a 2+. In the adjacent graphic on the right (Figure 6 b), the corresponding data with tantalum is shown. It must be noted that the calculated structures of 1 − include imaginary frequencies (1 a −: −3.12 cm−1, 1 b −: −15.07 cm−1) which despite several attempts could not be relaxed. It appears that the potential energy surface is very flat and, despite tight conversion criteria, the converged structures are still slightly off the true minimum. However, this will not affect the vibrational and bonding analysis as indicated by the good agreement between optimized and experimental structures (cf. Table S3 to Table S6 in the Supporting Information). Furthermore, 1 b − converged only in the experimentally unrelated symmetry D 3d. This observation indicates a flat hypersurface and is consistent with the flexibility in the experimental crystal data of 1 b − discussed above (eight ion pairs in the asymmetric unit).
Figure 6.
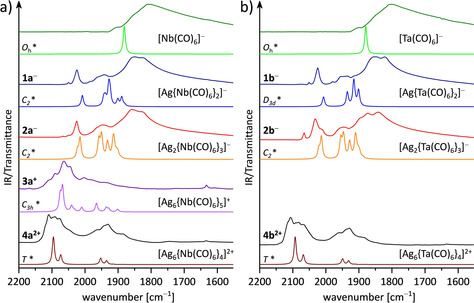
Section of the ν CO range from experimental and calculated IR spectra (* BP86/D3(BJ)/def2‐TZVPP). IR spectra of the starting materials [NEt4][M(CO)6] (top traces) and the precipitated powders with stoichiometries targeting at 1 a, 2 a, 3 a, and 4 a (a) or 1 b, 2 b, and 4 b (b).
A precise assignment of the experimental bands was not possible, as the bands of the different clusters are quite broad and too similar to differentiate clearly. It is very likely that unreacted starting material was present in the samples of 1 and 2, where the niobium/tantalum to silver ratio was quite high, resulting in very broad bands ranging between 1920–1530 cm−1. In addition, it may be possible that different clusters exist in parallel and some of the precipitated samples could include mixtures. However, in agreement with single‐crystal analysis, the patterns of the measured IR spectra confirm that stoichiometry influences at least the ratio of the resulting molecules. Moreover, from the rather good qualitative agreement between experimental and calculated IR spectra, it appears very likely that at least for the cluster cations 3 a +, 4 a 2+, and 4 b 2+, the majority of the material corresponds to the assigned clusters. Additionally, the experimental and calculated IR spectra demonstrate that the CO bands of the cationic clusters 3 a +, 4 a 2+, and 4 b 2+ are blueshifted compared with the CO bands in the anionic compounds 1 − and 2 − (Figure 6, Figure S1, Figure S2, Table S2 in the Supporting Information). This observation is in agreement with a reduced M–CO π‐back bonding character similar to the well examined bonding situation in homoleptic transition‐metal carbonyl complexes.45
Atoms in molecules (AIM) analyses
A first formal rationalization of the cluster ions [Agm{M(CO)6}n]x yields their charges based on the interaction of the charged constituents: m Ag+ silver ions and n [M(CO)6]− carbonylates yield the x=(m−n) cluster charge. To investigate how the charge is distributed within the [M(CO)6] moieties and the silver cluster core and how the charges change upon complexation to silver, an atoms in molecules (AIM) analysis of the cluster ions 1 − to 4 2+ was applied (Figure 7). For comparison, isolated [M(CO)6]− was also assessed.
Figure 7.
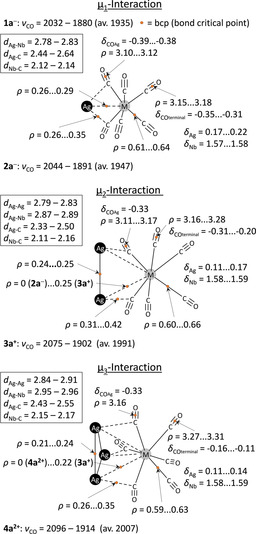
Schematic representations of the three μ1‐, μ2‐, and μ3‐bonding situations encountered within the cluster ions 1 a–4 a. For clarity, only the niobium compounds were considered. AIM‐calculated bond critical points (bcps), electron densities residing on bcps ρ [e Å−3], partial charges on the metal atoms δ M or CO moieties δ CO [e], DFT‐calculated bond lengths d [Å], and CO stretching frequencies ν CO [cm−1] are displayed.
In addition, we performed natural bond orbital (NBO) analyses, but—as found previously46—this type of analysis trying to describe the bonding in the moiety in question as a set of electron precise natural Lewis structures, is not suitable to account for the heavily delocalized nature of the bonding in the clusters in question. Therefore, we concentrated here on the analysis of the topology of the electron density with AIM.
The charges of the integrated AIM metal atom basins within free [M(CO)6]− (Nb +1.53; Ta +1.69) increase upon coordination to Ag+ only very slightly by +0.02 to +0.06. This holds regardless of the structure and total charge of the clusters (Table S7 in the Supporting Information). On the other hand, the positive charge residing on the silver atoms in each cluster is rather low and is found to be in the range +0.11 (3 a +) to +0.22 (1 a −; Table S8 in the Supporting Information). This finding indicates that the entire [M(CO)6] fragment needs to be considered to deduce the bonding entities and their associated charge transfer relevant for the interaction with the silver core (Table S9 in the Supporting Information). Upon coordination, the charge residing on the [M(CO)6] fragments is reduced from −1 in the free carbonylate down to at most +0.28/+0.31 in 4 2+. This removal of electron density follows the M/Ag ratio: the lower this ratio, the higher is the positive charge on silver. Another trend is noticeable when the hapticity of the [M(CO)6] fragments is analyzed (Figure 7). The higher the hapticity, the higher is the net amount of electron transfer from the [M(CO)6] fragments to the silver atoms. As almost no change of the charges at Nb/Ta atoms was found upon complexation, the CO ligands have to contribute considerably (Table S10 in the Supporting Information). Within the free [M(CO)6]− complexes, the π‐back bonding is dominant and thereby delocalizes the anionic charge to a large extent towards the CO ligands, that is, the negative charge on those reaches −0.42 (Nb) or −0.45 (Ta). This transfer results in rather redshifted CO stretching frequencies (ν CO) well below 1900 cm−1. Upon coordination to silver, these negative charges gradually decrease depending on the M/Ag ratio and go down to −0.11 (Nb) or −0.12 (Ta) in 4 2+. Further, we note that the charges residing on the CO units differ for terminal (COterminal) or additionally silver‐bound ligands (COAg). The COAg ligands deliver less electron density than the terminal ones and remain considerably more negatively charged at −0.33 to −0.39. This observation suggests that the [M(CO)6] units coordinate via the COAg moieties as chelating tridentate ligands to the silver atom(s). Apparently, cluster formation is strongly supported by the ligands. According to the AIM analysis, the negative charge transferred to the silver cores is delivered by the COterminal ligands (Figure 7).
This picture, based on the integrated charges of the AIM basins and included with Figure 7, is in full agreement with the calculated electron densities residing on the bond‐critical points (bcps) of the clusters 1 −, 2 −, 3 +, and 4 2+ and the parent ion [M(CO)6]− (Table S12 in the Supporting Information). The isolated hexacarbonylate complexes [M(CO)6]− show an electron density of 0.62/0.65 e Å−3 (M=Nb/Ta) residing at the M–C critical points and 3.09/3.08 e Å−3 (M=Nb/Ta) on the C−O bond paths (Figure S3 in the Supporting Information). In comparison, the charge density residing on these C–O bcps in the clusters increases with increasing positive charge and to a greater extent within the COterminal than within the COAg ligands. This corresponds well to the experimental IR data, which indicate stronger CO bonds with increasing positive charge as seen by the blueshifted CO frequencies (ν CO) described earlier and included with Figure 7. The electron densities on the M−C bond paths remain almost constant in all complexes. Turning to the cluster core, the electron density between the silver atoms decreases slightly with increasing number of clustered atoms. The electron density at the Ag−M and Ag−C bcps are comparable but typically higher for the Ag−C bond paths. The [M(CO)6] fragments thus appear to bind more strongly to silver via the CO ligands than via the metal atom M. Interestingly, the standard parameters of the AIM analysis did not find Ag–M bcps for cluster 4 2+ (Figure S7 in the Supporting Information). In 2 − (Figure S5 in the Supporting Information), only the M−Ag bond paths with the hapticity of one showed a clear bcp (Table S12 in the Supporting Information, marked with *). This missing Ag−M interaction is apparently overcompensated by the coordination of the COAg ligands, which seems to allow the formation of the cluster 4 2+ (Figure 8).
Figure 8.
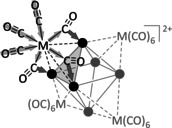
Schematic representation of the bonding situation in the cluster ion 4 2+. The arrows indicate the shift of the electron density from the COterminal ligands through bridging COAg ligands to the Ag6 cluster core. Note that in the AIM picture, a direct M−Ag interaction is absent as evidenced by the missing Ag−M bcp. Thus, the description as a super‐tetrahedron as in Figure 1 b is only topological, but without bonding contribution.
Overall, it may be stated that the clusters form owing to electron transfer from the [M(CO)6] moieties to the silver core. The CO ligands play two major roles in the formation of these ions: 1) they serve as chelating anchors tethering the [M(CO)6]− ligands to the silver core; 2) the COterminal ligands provide the electron density for the bonding within the larger trigonal prismatic or octahedral delocalized Ag6 cores. Roughly, five electrons are altogether transferred from the [M(CO)6] ligands to the Ag6 cores according to the AIM picture (Table S11 in the Supporting Information).
The interaction described in 2) apparently leads to the rather strong blueshift of the experimental CO stretching frequencies. In addition, the Ag−M interactions, if present at all, are rather weak. This observation is supported by the fact that—to our knowledge—binary silver–niobium and silver–tantalum alloys are unknown. Thus, the Ag−M interaction appears to be rather weak. Given the calculated charges at M (≈+1.7) and Ag (≈+0.2), such a bond would actually be expected to be much shorter than the observed distances of 2.81 to 3.07 Å (Table 1). Adding up the radii for MIII (0.72 Å; radius for MII is unknown) and Ag0 (1.44 Å) gives approximately 2.16 Å as an approximation for a binding interaction. Altogether, this leads to the unexpected notion that the description of 4 2+ as a super‐tetrahedron as shown in Figure 1 b is only topological, but without Ag−M bonding contribution. Thus, the central cluster unit in the large 3 + and 4 2+ systems is the Ag6 core. This may be understood in terms of the superatom model, as presented in the next section.
Structures 3+ and 42+ in the context of the superatom model
Cluster ion 4 2+ may be compared with literature clusters, which were examined in more detail with regard to the superatom model. The octahedral Ag6 core of 4 2+, which is capped by Nb/Ta atoms on every second triangular face forming the highly symmetric super‐tetrahedron, has a structure related to the cluster [Au6X4]2+ (X=F, Cl, Br, I) previously investigated with theoretical methods.47 This cluster was considered as an analog of a tetrahedral molecule CX4 and sp3‐hybridization of the Au6 core was deduced. However, the earlier discussed AIM analysis of cluster 4 a 2+ showed that in our case the CO ligands have the decisive role in the bonding rather than the Nb/Ta atoms. This reasoning allows for a comparison of compound 4 2+ with the cluster [Au6{Ni3(CO)6}4]2−[27, 28] in Figure 9. The latter was also shown to be an analog of tetrahedral molecule CH4.28
Figure 9.
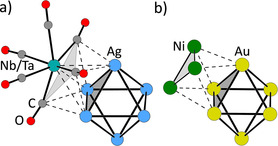
Schematic representations of a part of the structures of (a) [Ag6{M(CO)6}4]2+ (4 2+) and (b) [Au6{Ni3(CO)6}4]2−.27, 28
Every second face of the gold octahedron in Figure 9 b is capped by a [Ni3(CO)6] unit. Using the published electron count of an [Au6]2+ cluster core, this would include four [Ni3(CO)6].− radical anions as chelating pseudo‐ligands. In 4 2+, the chelating pseudo‐ligand [Ni3(CO)6].− is exchanged for the tridentate M(CO)6 . radical—isoelectronic to the known 17 valence electron metalloradical V(CO)6 .—as a ligand to the sp3 [Ag6]2+ superatom.
By contrast, in structure 3 a +, the prismatic Ag6 core is capped by five [Nb(CO)6] fragments, overall giving a D 3h trigonal bipyramid (Figure 10 b). The comparison with other clusters is quite difficult as only very few molecular clusters with this structural motif are known.48 Nevertheless, according to the lines rationalizing the super‐tetrahedron in 4 2+ as sp3, and being related to CH4 or generalized as AX4,27, 28 it is reasonable to argue that 3 a + would correspond to trigonal bipyramidal SiH5 −,49 NH5,50 PH5,51 or generalized to an AX5 molecule as shown in Figure 10.
Figure 10.
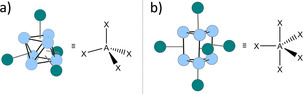
Schematic representations of the metal core of 4 2+ (a) and 3 + rationalized by the superatom model as simple molecules generalized as AX4 (a) or AX5 (b).
However, to enable a firm classification of the cluster cores of compounds 3 + and 4 2+ as superatoms, further intensive theoretical investigations are necessary and we would be very pleased if another research group with more experience in this field would take up this topic.
Conclusion
Several heterobimetallic formal AgmMn clusters (M=Nb, Ta) with the general formula [Agm{M(CO)6}n]x (M=Nb, Ta; m=1, 2, 6; n=2, 3, 4, 5; x=1−, 1+, 2+) were prepared, structurally and spectroscopically analyzed, and their bonding was investigated. As an addition to known anionic and neutral heterobimetallic Ag−M carbonyl clusters, we present the first examples including the [M(CO)6] moiety as a component of anionic clusters. Furthermore, the first larger cationic clusters of this kind were obtained, stabilized by a weakly coordinating anion. They include a D 3h‐symmetric Ag6Nb5 or T d‐symmetric Ag6M4 core (M=Nb, Ta). The discussion of the crystal structures, IR data, and quantum calculations showed indications for a peculiar bonding situation in these clusters: The [M(CO)6] groups can be seen as chelate ligands to the silver core. The CO ligands of the M carbonylates play a significant role, as they provide the electron density for the bonding and act as chelating anchors to the silver core. Thus, apparently the [M(CO)6] units in 3 + or 4 2+ serve as ligands to the Ag6 superatom. In 4 2+, this follows the lines as described for [Au6X4]2+ (X=F, Cl, Br, I),47 which was considered as an AX4 analogue with sp3‐hybridization of the [Au6]2+ core. The structure of 3 + possibly is the first example of an extension to an AX5 analogue. The hybridization of the underlying trigonal prismatic Ag6 superatom would be interesting to be analyzed by groups with a particular focus on theoretical chemistry. Thus, we encourage other research groups to further investigate the clusters quantum chemically in terms of the superatom model or with other suitable approaches.
Experimental Section
Crystallographic data: Deposition numbers 1998854, 1998851, 1998852, 1998853, 1909279, and 1998922 (1 a, 2 a, 2 b, 3 a, 4 a, and 4 b) contain(s) the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service..
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Open access funding was enabled and organized by Projekt DEAL.
W. Unkrig, K. Kloiber, B. Butschke, D. Kratzert, I. Krossing, Chem. Eur. J. 2020, 26, 12373.
References
- 1. Mond L., Langer C., Quincke F., J. Chem. Soc. Trans. 1890, 57, 749. [Google Scholar]
- 2. Maher J. M., Beatty R. P., Cooper N. J., Organometallics 1985, 4, 1354. [Google Scholar]
- 3. Handy L. B., Ruff J. K., Dahl L. F., J. Am. Chem. Soc. 1970, 92, 7312. [Google Scholar]
- 4.
- 4a. Jackson P. F., Johnson B. F. G., Lewis J., McPartlin M., Nelson W. J. H., J. Chem. Soc. Chem. Commun. 1979, 735; [Google Scholar]
- 4b. Johnson B. F. G., Coord. Chem. Rev. 1999, 190–192, 1269. [Google Scholar]
- 5. Gade L. H., Johnson B. F. G., Lewis J., McPartlin M., Powell H. R., Raithby P. R., Wong W.-T., J. Chem. Soc. Dalton Trans. 1994, 521. [Google Scholar]
- 6.
- 6a. Beck W., Angew. Chem. Int. Ed. Engl. 1991, 30, 168; [Google Scholar]; Angew. Chem. 1991, 103, 173; [Google Scholar]
- 6b. Ellis J. E., Organometallics 2003, 22, 3322. [Google Scholar]
- 7.
- 7a. Sumner G. G., Klug H. P., Alexander L. E., Acta Crystallogr. 1964, 17, 732; [Google Scholar]
- 7b. Hanson A. W., Acta Crystallogr. 1962, 15, 930. [Google Scholar]
- 8. Xu Q., Coord. Chem. Rev. 2002, 231, 83. [Google Scholar]
- 9.
- 9a. Willner H., Aubke F., Organometallics 2003, 22, 3612; [Google Scholar]
- 9b. Willner H., Aubke F., Chem. Eur. J. 2003, 9, 1668; [DOI] [PubMed] [Google Scholar]
- 9c. Geier J., Willner H., Lehmann C. W., Aubke F., Inorg. Chem. 2007, 46, 7210; [DOI] [PubMed] [Google Scholar]
- 9d. Finze M., Bernhardt E., Willner H., Lehmann C. W., Aubke F., Inorg. Chem. 2005, 44, 4206. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Jaouen G., Vessieres A., Top S., Ismail A. A., Butler I. S., J. Am. Chem. Soc. 1985, 107, 4778; [Google Scholar]
- 10b. Monney A., Albrecht M., Coord. Chem. Rev. 2013, 257, 2420. [Google Scholar]
- 11.
- 11a. Johnson T. R., Mann B. E., Clark J. E., Foresti R., Green C. J., Motterlini R., Angew. Chem. Int. Ed. 2003, 42, 3722; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 3850; [Google Scholar]
- 11b. Mann B. E., Organometallics 2012, 31, 5728. [Google Scholar]
- 12. Bauer J., Braunschweig H., Dewhurst R. D., Chem. Rev. 2012, 112, 4329. [DOI] [PubMed] [Google Scholar]
- 13. Fischer R. A., Miehr A., Hoffmann H., Rogge W., Boehme C., Frenking G., Herdtweck E., Z. Anorg. Allg. Chem. 1999, 625, 1466. [Google Scholar]
- 14. Clarkson L. M., McCrudden K., Norman N. C., Farrugia L. J., Polyhedron 1990, 9, 2533. [Google Scholar]
- 15. Leiner E., Hampe O., Scheer M., Eur. J. Inorg. Chem. 2002, 584. [Google Scholar]
- 16. Rutsch P., Renner G., Huttner G., Sandhöfner S., Z. Naturforsch. B 2002, 57, 757. [Google Scholar]
- 17. Doyle G., Eriksen K. A., van Engen D., J. Am. Chem. Soc. 1985, 107, 7914. [DOI] [PubMed] [Google Scholar]
- 18. Bissert R., Braunschweig H., Dewhurst R. D., Schneider C., Organometallics 2016, 35, 2567. [Google Scholar]
- 19. Malinowski P. J., Krossing I., Angew. Chem. Int. Ed. 2014, 53, 13460; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13678. [Google Scholar]
- 20. Albano V. G., Azzaroni F., Iapalucci M. C., Longoni G., Monari M., Mulley S., MRS Proc. 1992, 272, 115. [Google Scholar]
- 21. Berti B., Bortoluzzi M., Cesari C., Femoni C., Iapalucci M. C., Mazzoni R., Vacca F., Zacchini S., Inorg. Chem. 2019, 58, 2911. [DOI] [PubMed] [Google Scholar]
- 22. Braunschweig H., Dewhurst R. D., Hupp F., Kaufmann C., Phukan A. K., Schneider C., Ye Q., Chem. Sci. 2014, 5, 4099. [Google Scholar]
- 23. Wang G., Ceylan Y. S., Cundari T. R., Dias H. V. R., J. Am. Chem. Soc. 2017, 139, 14292. [DOI] [PubMed] [Google Scholar]
- 24. Klüfers P., Z. Kristallogr. Cryst. Mater. 1984, 166, 143. [Google Scholar]
- 25. Dyson P. J., McIndoe J. S., Transition-Metal Carbonyl Cluster Chemistry, CRC Press, Amsterdam, 2019. [Google Scholar]
- 26. Chihara T., Sato M., Konomoto H., Kamiguchi S., Ogawa H., Wakatsuki Y., J. Chem. Soc. Dalton Trans. 2000, 2295. [Google Scholar]
- 27. Whoolery A. J., Dahl L. F., J. Am. Chem. Soc. 1991, 113, 6683. [Google Scholar]
- 28. Muñoz-Castro A., Chem. Sci. 2014, 5, 4749. [Google Scholar]
- 29.
- 29a. Khanna S. N., Jena P., Phys. Rev. B Condens. Matter 1995, 51, 13705; [DOI] [PubMed] [Google Scholar]
- 29b. Khanna S. N., Jena P., Phys. Rev. Lett. 1992, 69, 1664; [DOI] [PubMed] [Google Scholar]
- 29c. Reveles J. U., Khanna S. N., Roach P. J., Castleman A. W., Proc. Natl. Acad. Sci. USA 2006, 103, 18405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Albano V. G., Azzaroni F., Iapalucci M. C., Longoni G., Monari M., Mulley S., Proserpio D. M., Sironi A., Inorg. Chem. 1994, 33, 5320. [Google Scholar]
- 31. Albano V. G., Grossi L., Longoni G., Monari M., Mulley S., Sironi A., J. Am. Chem. Soc. 1992, 114, 5708. [Google Scholar]
- 32. Collini D., Femoni C., Iapalucci M. C., Longoni G., C. R. Chim. 2005, 8, 1645. [Google Scholar]
- 33. Lee Y.-B., Wong W.-T., Chem. Commun. 2007, 3924. [DOI] [PubMed] [Google Scholar]
- 34. Zhang J., Dahl L. F., J. Chem. Soc. Dalton Trans. 2002, 1269. [Google Scholar]
- 35. Coffey C. E., Lewis J., Nyholm R. S., J. Chem. Soc. 1964, 1741. [Google Scholar]
- 36. Unkrig W., Schmitt M., Kratzert D., Himmel D., Krossing I., Nat. Chem. 2020, 12, 647. [DOI] [PubMed] [Google Scholar]
- 37. Calderazzo F., Pampaloni G., Englert U., Strähle J., Angew. Chem. Int. Ed. Engl. 1989, 28, 471; [Google Scholar]; Angew. Chem. 1989, 101, 469. [Google Scholar]
- 38.
- 38a. Schaefer J., Kraft A., Reininger S., Santiso-Quinones G., Himmel D., Trapp N., Gellrich U., Breit B., Krossing I., Chem. Eur. J. 2013, 19, 12468; [DOI] [PubMed] [Google Scholar]
- 38b. Meier S. C., Himmel D., Krossing I., Chem. Eur. J. 2018, 24, 19348; [DOI] [PubMed] [Google Scholar]
- 38c. Bohnenberger J., Derstine B., Daub M., Krossing I., Angew. Chem. Int. Ed. 2019, 58, 9586; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9687; [Google Scholar]
- 38d. Bohnenberger J., Krossing I., Angew. Chem. Int. Ed. 2020, 59, 5581; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 5629; [Google Scholar]
- 38e. Bohnenberger J., Schmitt M., Feuerstein W., Krummenacher I., Butschke B., Czajka J., Malinowski P. J., Breher F., Krossing I., Chem. Sci. 2020, 11, 3592; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38f. Meier S. C., Holz A., Schmidt A., Kratzert D., Himmel D., Krossing I., Chem. Eur. J. 2017, 23, 14658; [DOI] [PubMed] [Google Scholar]
- 38g. Bohnenberger J., Feuerstein W., Himmel D., Daub M., Breher F., Krossing I., Nat. Commun. 2019, 10, 624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.
- 39a. Krossing I., Chem. Eur. J. 2001, 7, 490; [DOI] [PubMed] [Google Scholar]
- 39b. Riddlestone I. M., Kraft A., Schaefer J., Krossing I., Angew. Chem. Int. Ed. 2018, 57, 13982; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14178. [Google Scholar]
- 40.
- 40a. Puddephatt R. J., Chem. Soc. Rev. 1983, 12, 99; [Google Scholar]
- 40b. Kubiak C. P., Eisenberg R., J. Am. Chem. Soc. 1977, 99, 6129; [Google Scholar]
- 40c. Ferguson G. S., Wolczanski P. T., Parkanyi L., Zonnevylle M. C., Organometallics 1988, 7, 1967; [Google Scholar]
- 40d. Kubiak C. P., Eisenberg R., Inorg. Chem. 1980, 19, 2726. [Google Scholar]
- 41. Bondi A., J. Phys. Chem. 1964, 68, 441. [Google Scholar]
- 42. Calderazzo F., Pampaloni G., Pelizzi G., J. Organomet. Chem. 1982, 233, C41. [Google Scholar]
- 43. Cotton F. A., Murillo C. A., Wang X., Inorganica Chim. Acta 2000, 300–302, 1. [Google Scholar]
- 44. Calderazzo F., Englert U., Pampaloni G., Pelizzi G., Zamboni R., Inorg. Chem. 1983, 22, 1865. [Google Scholar]
- 45. Willner H., Aubke F., Angew. Chem. Int. Ed. Engl. 1997, 36, 2402; [Google Scholar]; Angew. Chem. 1997, 109, 2506. [Google Scholar]
- 46. Riddlestone I. M., Weis P., Martens A., Schorpp M., Scherer H., Krossing I., Chem. Eur. J. 2019, 25, 10546. [DOI] [PubMed] [Google Scholar]
- 47. Muñoz-Castro A., King R. B., Int. J. Quantum Chem. 2017, 117, e25331. [Google Scholar]
- 48.
- 48a. Calabrese J. C., Dahl L. F., Chini P., Longoni G., Martinengo S., J. Am. Chem. Soc. 1974, 96, 2614; [Google Scholar]
- 48b. Chen Y.-T., Krytchankou I. S., Karttunen A. J., Grachova E. V., Tunik S. P., Chou P.-T., Koshevoy I. O., Organometallics 2017, 36, 480. [Google Scholar]
- 49. Hajdasz D. J., Ho Y., Squires R. R., J. Am. Chem. Soc. 1994, 116, 10751. [Google Scholar]
- 50. Olah G. A., Donovan D. J., Shen J., Klopman G., J. Am. Chem. Soc. 1975, 97, 3559. [Google Scholar]
- 51. Shih S.-K., Peyerimhoff S. D., Buenker R. J., J. Chem. Soc. Faraday Trans. 2 1979, 75, 379. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary