

Abstract
Oncogenic c-Myc plays a critical role in cell proliferation, apoptosis, and tumorigenesis, but the precise mechanisms that drive this activity remain largely unknown. P27Kip1 (CDKN1B) arrests cells in G1, and SAP155 (SF3B1), a subunit of the essential splicing factor 3b (SF3b) subcomplex of the spliceosome, is required for proper P27 pre-mRNA splicing. FUSE-binding protein-interacting repressor (FIR), a splicing variant of PUF60 lacking exon5, is a c-Myc transcriptional target that suppresses the DNA helicase p89 (ERCC3) and is alternatively spliced in colorectal cancer lacking the transcriptional repression domain within exon 2 (FIRΔexon2). FIR and FIRΔexon2 form a homo- or hetero-dimer that complexes with SAP155. Our study indicates that the FIR/FIRΔexon2/SAP155 interaction bridges c-Myc and P27 expression. Knockdown of FIR/FIRΔexon2 or SAP155 reduced p27 expression, inhibited its pre-mRNA splicing, and reduced CDK2/Cyclin E expression. Moreover, spliceostatin A, a natural SF3b inhibitor, markedly inhibited P27 expression by disrupting its pre-mRNA splicing and reduced CDK2/Cyclin E expression. The expression of P89, another FIR target, was increased in excised human colorectal cancer tissues. Knockdown of FIR reduced P89; however, the effects on P27 and P89 expression are not simply or directly related to altered FIR expression levels, indicating that the mechanical or physical interaction of the SAP155/FIR/FIRΔexon2 complex is potentially essential for sustained expression of both P89 and P27. Together, the interaction between SAP155 and FIR/FIRΔexon2 not only integrates cell-cycle progression and c-Myc transcription by modifying P27 and P89 expression but also suggests that the interaction is a potential target for cancer screening and treatment.
Introduction
c-Myc plays a critical role in cell proliferation, tumorigenesis, apoptosis, and cell-cycle regulation, but its precise cellular mechanisms are largely unknown. The far-upstream element (FUSE) is a sequence required for proper transcriptional regulation of the human c-myc gene (1). FUSE is located 1.5 kb upstream of the c-myc promoter P1 and is recognized by the FUSE-binding protein (FBP). FBP is a transcription factor that stimulates c-myc expression through FUSE (2, 3). Yeast two-hybrid analysis has shown that FBP binds to a protein within the FBP-interacting repressor (FIR) and that FIR represses c-myc transcription by suppressing the TFIIH/P89/XPB helicase (P89; ref. 4). Cells from patients with xeroderma pigmentosum group B (XPB) and xeroderma pigmentosum group D (XPD) are defective in FIR repression, which suggests that P89 mutations impair c-myc transcriptional regulation by FIR and contribute to tumor development (5). Expression of FIRΔexon2, an FIR splice variant that lacks exon 2, may promote tumor development by disabling authentic FIR c-myc repression (6).
Cyclin-dependent kinase (Cdk)2 controls c-Myc-mediated suppression of cellular senescence (7). Cdk2 interacts with c-Myc at gene promoters and affects c-Myc-dependent regulation of genes that encode proteins controlling cellular senescence (8). c-Myc enhances the activity of cyclinE/Cdk2. c-Myc and cyclinE/Cdk2 physically interact, which results in c-Myc phosphorylation at Ser62 (9). This enables c-Myc-mediated regulation of senescence-associated genes, such as p21, pl6, BMI-1, and hTERT, and results in suppression of oncogene-induced senescence during cancer development (9).
Splicing factor 3b (SF3b) is a subcomplex of the U2 small nuclear ribonucleoprotein in the spliceosome. SAP155 is required for proper FIR expression and vice versa, and SAP155 knockdown or SF3b inhibition disrupts alternative FIR splicing and generates FIRΔexon2 (10). Of note, FIR/FIRΔexon2/SAP155 forms a complex and simultaneously interferes with the well-established functions of FIR or SAP155, namely c-myc transcription and alternative splicing in cancer cells (10). Accordingly, interaction between FIR/FIRΔexon2 and SAP155 bridges c-myc/c-Myc expression and cell cycling.
In cancers, cell-cycle arrest for complete DNA-damage repair is highly inefficient because expression of the Cip/Kip family is reduced; thus, cell-cycle progression is accelerated (11, 12). In this study, we examined the effect of altered FIR/FIRΔexon2/SAP155 expression on p27Kip1 (P27) and P89 expression. We found that FIR/FIRΔexon2/SAP155 interaction connects c-myc and cell-cycle regulation by integrating the expression of P89/FIR/FIRΔexon2 or P27/cdk2/cyclinE. The importance of the FIR/FIRΔexon2/SAP155 interaction is discussed as a possible target for a c-myc/cell-cycle checkpoint regulator in cancer development or DNA-damage repair for clinical applications.
Materials and Methods
Excised human tumor samples
Tissues from 34 primary colorectal cancer cases were surgically excised at Chiba University Hospital (Chiba, Japan). Tumor samples were obtained from tumor epithelium immediately after surgical excision, and corresponding nontumor epithelial samples were obtained 5 to 10 cm away from the tumor. Two pathologists microscopically confirmed that all tumor tissue samples were adenocarcinomas. All the excised tissues were immediately placed in liquid nitrogen and stored at −80°C until analysis. Written informed consent was obtained from each patient before surgery.
Plasmids
Full-length FIR cDNA was cloned into a pCGNM2 plasmid vector to introduce a hemagglutinin (HA)-tag at the amino terminus (HA—FIR). The human c-myc (a v-myc avian myelocytomatosis viral oncogene homolog) expression plasmid subcloned into pcDNA3.1/GS was purchased from Invitrogen. The plasmids were prepared by CsCl ultracentrifugation or using the Endofree Plasmid Maxi Kit (Qiagen), and their DNA sequences were verified by sequencing.
Cell cultures and plasmid transfections
HeLa and U2OS cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco—BRL) with 10% fetal calf serum (FCS). The cells were transfected using Lipofectamine Plus or Lipofectamine 2000 (Gibco-BRL) according to the manufacturer’s instructions. Cell cultures routinely used either 1 × 105 cells per well in 6-well plates overnight or 5 × 105 cells per 10 cm in dishes overnight.
Immunocytochemistry
Colorectal cancer tissues and their corresponding normal tissues were prepared for immunocytochemistry as described previously (6, 13).
Protein extraction and Western blotting
Cellular proteins were extracted with radioimmunoprecipitation assay buffer (1% w/w Nonidet P-40, 1% w/v sodium deoxycholate, 0.1% w/v SDS, 0.15 mol/L NaCl, 2 mmol/L EDTA, 1 mmol/L phenylmethylsulfonylfluoride) for 30 minutes on ice, after which cellular debris was removed by centrifugation. The supernatants (cell extracts) were analyzed by immunoblotting. Protein concentrations were determined (Bio-Rad), and proteins (10 mg per sample) were separated by SDS-PAGE with 4% to 12% gradient gels (NuPAGE) and then transferred onto nitrocellulose membranes. Alternatively, the protein extracts were separated by electrophoresis on a 7.5% to 15% Perfect NT Gel. Subsequently, the proteins were transferred to polyvinylidene fluoride membranes using a tank transfer apparatus (Bio-Rad). The membranes were blocked with 0.5% skimmed milk in PBS. Antigens were detected with enhanced chemiluminescence detection reagents (GE Healthcare). The primary and secondary antibodies are listed in Supplementary Table S1, and they were described previously (10). Band intensities were determined using Total Lab TL120 image analysis software.
MTS assay
Cells were cultured at 5 × 104 cells per well in flat-bottomed 96-well plates. After 3 days, CellTiter 96 Aqueous One Solution Reagent (Promega) was added to each well according to the manufacturer’s instructions. After culturing for 4 hours, cell viability was determined by measuring the absorbance at 490 nm using a Bio-Rad 550 plate reader (Bio-Rad).
Soft agar colony formation assay
The cells (2 × 103) were inoculated in 0.3% low-melting temperature agarose (FMC Bio Products) in DMEM supplemented with 10% FCS, and colonies were scored after incubating for 2 weeks.
Flow cytometry analysis
Cells were processed as described previously (6). To assess the effects of increased FIR levels on cell-cycle progression, the cells were transfected with HA-FIR and immunostained with anti-HA. The cells were also stained for DNA using 10 μg/mL of propidium iodide (PI). HA and PI levels in the cells were determined by flow cytometry. To determine the cell-cycle phase, 1 × 106 HeLa cells were washed twice with PBS and fixed with 70% ethanol, and nuclei were stained with PI in an RNase A solution (250 μg/mL). A total of 10,000 cells for each sample were analyzed by setting PI on the x-axis and HA on the y-axis using a FACSCanto II (Becton, Dickinson and Company).
Apoptosis detection
Apoptosis was detected using the APOPercentage apoptosis assay (Funakoshi Co., Ltd.) according to the manufacturer’s instructions. In brief, 100 μL of 0.4% gelatin was added to each well of a plate and allowed to settle for at least 10 minutes before adding 2 × 104 and 5 × 104 cells in 200 μL of culture medium into each well. The cells were incubated at 37°C in 5% CO2 until they reached confluence (approximately 24 hours). The incubation medium and gelatin were removed, and the cells were rinsed with fresh medium. Fresh culture medium (100 μL/well) containing 5 μL of APOPercentage Dye and the apoptotic inducer or inhibitor was added to the center of each well, and incubation was continued for 30 minutes. The culture medium/dye mixture was carefully removed with a syringe, and the cells were gently washed twice with 200 μL per well of PBS. The assay plate was immediately transferred to the stage of an inverted objective microscope, and at least 3 photomicrographs of a representative area of each well were obtained.
Reverse transcriptase PCR
Total RNA was extracted from HeLa cells using an RNeasy Mini Kit (Qiagen). cDNA was synthesized from total RNA using the first-strand cDNA Synthesis Kit for reverse transcriptase (RT)-PCR (Roche). Using cDNA as a template, FIR cDNA was amplified with suitable primers by RT-PCR. LightCycler software version 3.3 (Roche) was used for real-time RT-PCR analysis. Primers for RT-PCR and quantitative RT-PCR (qRT-PCR) of siRNAs were used in accordance with the manufacturers’ instructions (10).
siRNAs for FIR, SAP155, and cdk2
FIR, SAP155, and cdk2 siRNA duplexes were purchased from Sigma-Aldrich. The target sequences for FIR siRNA and SAP155 siRNA oligonucleotides were as reported previously (10), and the target sequences for cdk2 siRNA are listed in Supplementary Table S2. Transient siRNA transfection was carried out using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. The transfected cells were cultured for 72 hours at 37°C in a CO2 incubator.
FIR and FIRΔexon2 adenovirus vectors
FIR and FIRΔexon2 adenovirus vectors were prepared as described previously (10).
Spliceostatin A and bleomycin sulfate from Streptomyces verticillus.
Spliceostatin A (SSA), an SF3b inhibitor, was prepared as described previously (14). Bleomycin sulfate from S. verticillus was used according to the supplier’s instructions (B2434; Sigma-Aldrich).
Statistical analysis
P89 expression was compared between cancer tissues (T) and noncancer epithelium (N) by the Student t test and the Wilcoxon test.
Results
TFIIH/P89/ERCC3/XPB is activated in colon cancer tissues
FIR is believed to suppress c-myc through inhibiting DNA helicase activity of TFIIH/p89/ERCC3/XPB (P89; refs. 4, 15); therefore, P89 expression should be critical for c-myc regulation through FIR (4, 16). This study revealed that both total FIRs (authentic FIR and FIR splicing variants including FIRΔexon2) and P89 expression were significantly activated in colorectal cancer tissues (T) compared with corresponding nontumor tissues (N; Fig. 1A), and knockdown of FIR with siRNA suppressed P89 expression (Fig. 1B). How is the inhibition of P89 by FIR and the downregulation of P89 upon FIR knockdown reconciled? As our previous study showed that siRNA knockdown of FIR reduced SAP155 levels and vice versa (10), we examined whether SAP155 knockdown by siRNA affected P89 expression. Intriguingly, SAP155 knockdown by siRNA also suppressed P89 expression (Fig. 1C). In addition, immunohistochemistry showed that the expressions of total FIR, SAP155, and P89 were strongly interrelated in colon cancer tissues (Fig. 1D). These results support the hypothesis that FIR/FIRΔexon2 and SAP155 or their interaction is required, at least in part, for sustained P89 expression in HeLa cells and colon cancer tissues. Furthermore, while strong downregulation of FIR is observed even at the lowest concentrations of siRNA, the levels of SAP155 are apparently upregulated at these concentrations, whereas they are downregulated at higher siRNA concentrations (Figs. 1B). What is the explanation for these FIR’s dual effects to SAP155 expression? One explanation is that some feedback mechanism, mechanical- or morphologic-dependent protein interactions exist between FIR and SAP155. More detailed experiments are required to answer these questions. Patients’ characteristics, their tumor features, and disease prognosis were listed (Supplementary Table S3). As the number of patients was relatively small in this study, no significant or apparent correlation was made between the degree of upregulation of total FIRs expression and tumor features or disease prognosis.
Figure 1.
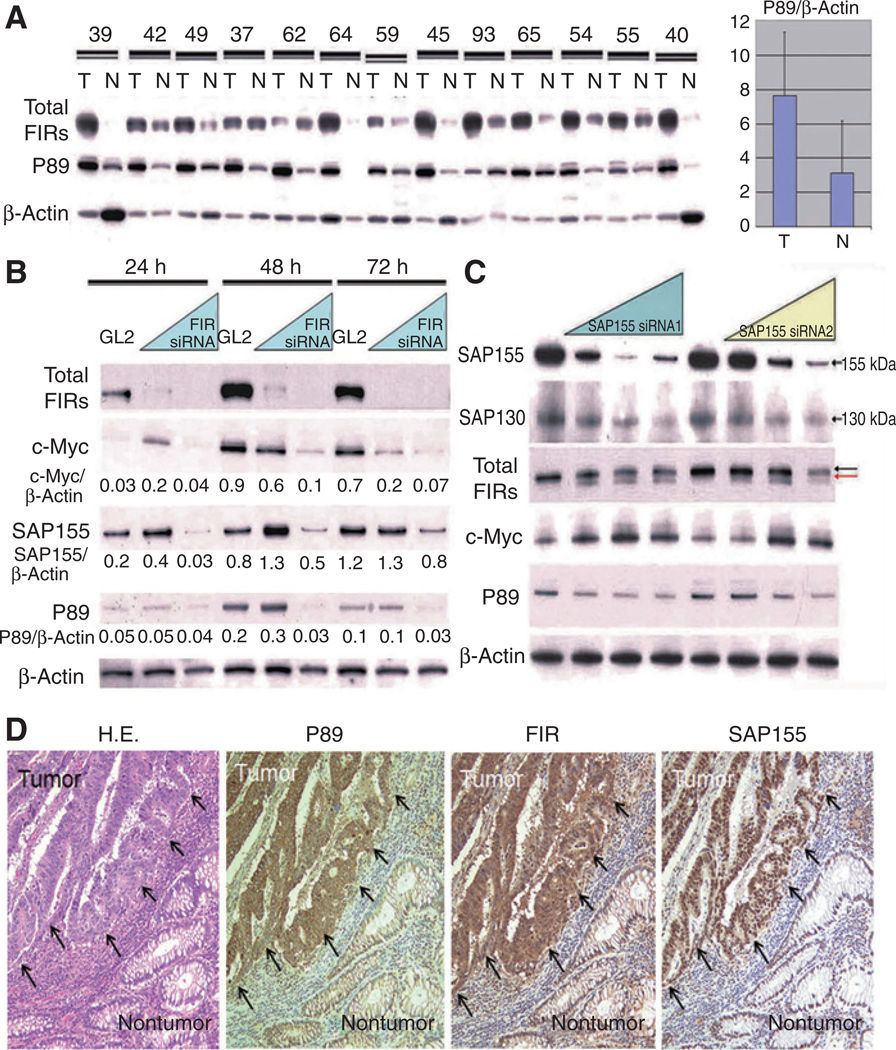
FIR represses endogenous c-Myc with wild-type TFIIH/p89/XPB/ERCC3. A, both total FIRs (authentic FIR and FIR splicing variants including FIRΔexon2) and P89 expression were higher in colon cancer tissues (T) than in corresponding noncancer tissues (N). P < 0.005 by the Wilcoxon test, and P < 0.01 by the t test. B, FIR knockdowns by siRNA (5 or 20 nmol/L) shows reduced c-Myc, SAP155, and P89 expression in HeLa cells. Each protein expression was indicated as to the ratio to β-actin. C, SAP155 siRNA (5, 10, or 25 nmol/L) suppressed P89 expression but increased c-Myc expression in HeLa cells, as well as shifting the FIR bands (arrows) and decreasing SAP130 expression. D, P89, total FIR, and SAP155 are more strongly expressed in excised human colorectal cancer tissues (arrows) than in normal colon mucosa. Hemotoxylin and eosin (H&E) staining of colorectal cancer tissues and normal colon mucosa is also shown.
Altered FIR expression suppresses P27Kip1/cyclinE expression
As a previous study showed that SF3b is required for proper P27 pre-mRNA splicing (14) and that SAP155 is a subunit of SF3b that forms a stable complex with FIR/FIRΔexon2 (10), we hypothesized that altered FIR expression influences P27 by modifying SAP155 expression. As expected, FIR knockdown by siRNA inhibited p27 pre-mRNA splicing and reduced the P27 expression level (Fig. 2A) with a decrease in cyclinE/phosphorylated-cdk2 (p-cdk2) expression (Fig. 2B). Although the 2 different siRNAs led to similar levels of FIR downregulation, their effects on P27 levels were very different, with siFIR1 leading to a modest reduction in P27, and siFIR2 leading to a much greater reduction (compare Fig. 2A, lanes 2 and 3 with lanes 4 and 5). There was no significant difference of SAP155 expression observed between siFIR1 and siFIR2 (data not shown). These results support the hypothesis that the effects on P27 are not simply or directly due to siFIR-mediated SAP155 downregulation (10, 14). Accordingly, mechanical or physical interaction between FIR and SAP155 is potentially pivotal for P27 pre-mRNA splicing rather than simply FIR or SAP155 expression. Enhanced c-Myc expression remarkably suppressed P27 and cdk2 expression, indicating that crosstalk exists between p27 and c-Myc expression (Fig. 2C). These results strongly indicated that altered FIR/FIRΔexon2/SAP155 expression affected cell cycling by altering P27 level.
Figure 2.
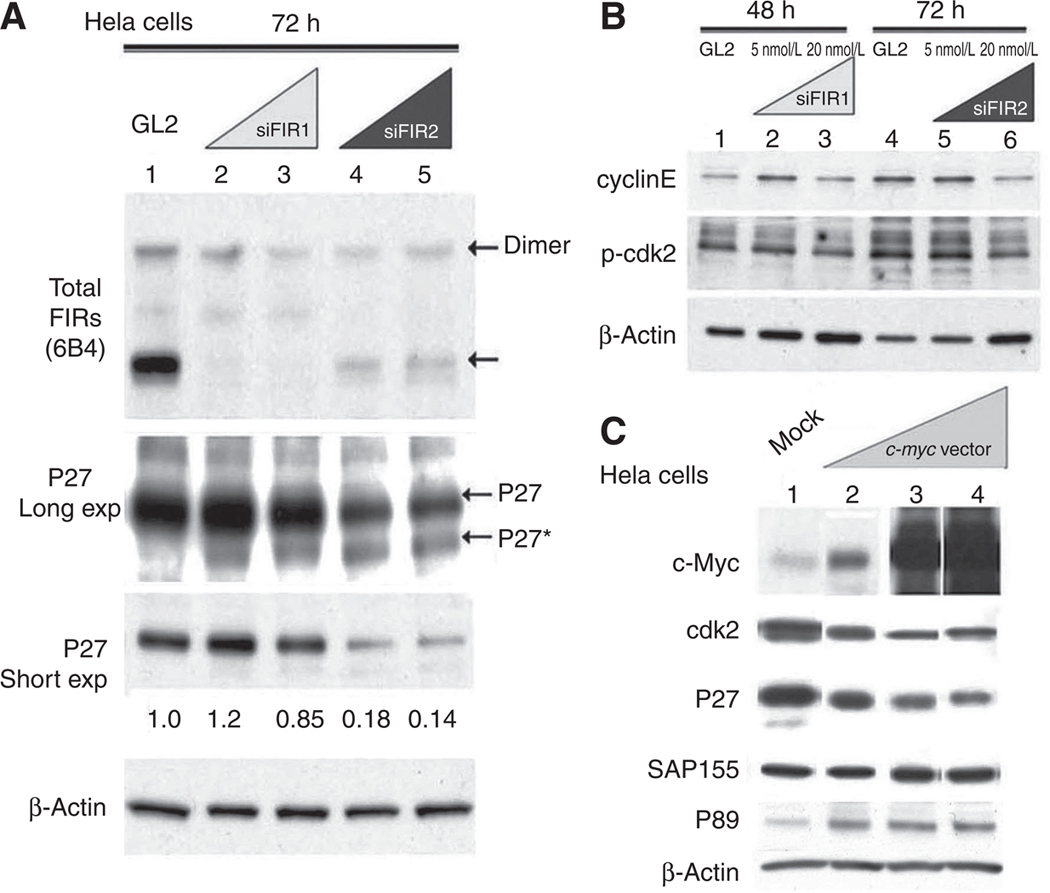
FIR knockdown by siRNA induces aberrant P27Kip1 splicing (P27*) with decreased p-cdk2/cyclinE expression. A, P27 and P27* were detected after FIR knockdown by siFIR1 and siFIR2 oligonucleotides. Lane 1: mock (20 nmol/LGL2 siRNA); lanes2 and 4: 5 nmol/L FIR siRNA; lanes 3 and 5: 20 nmol/L of FIR siRNA. After short exposure to film, P27 intensity with mock siRNA (GL2) was set at 1.0. B, p-Cdk2 and cyclinE expression was determined after FIR knockdown by siRNA. Lanes 1 and 4: mock (20 nmol/LGL2 siRNA); lanes2 and 5: 5 nmol/L FIR siRNA; lanes 3 and 6: 20 nmol/L FIR siRNA. C, c-Myc overexpression reduced P27, P89, and cdk2 expression. HeLa cells at 50% to 60% confluence were transfected with pcDNA3.1 vector alone (Mock, lane 1) or with pcDNA3.1/c-myc (1 ng, lane 2; 10 ng, lane 3; 100 ng, lane 4). The cells were harvested 48 hours after transfection and subjected to Western blotting. β-Actin served as an internal control.
SAP155 is required for proper pre-mRNA splicing of both FIR and P27
Altered P27 pre-mRNA splicing can be monitored by quantifying the ratio of the aberrant splicing variant of P27 (P27*) to P27 (14). The results in Fig. 3A indicated that SAP155 siRNA decreased P27 expression with increased c-Myc expression and FIR splicing alteration. In addition, the P27*/P27 ratio increased in accordance with the SAP155 siRNA concentration concomitant with cyclinE suppression (Fig. 3B) and decreased cyclinE expression (Fig. 3C). PUF60 and PUFΔexon2 mRNAs were detected by RT-PCR (Fig. 3C). Cdk2 siRNA suppressed cyclinE, but not FIR, SAP155, or c-Myc (Fig. 3D). As reported previously (10), total FIR protein was shifted to a lower molecular weight, suggesting that SAP155 is required for FIR splicing (Fig. 3A and C, bottom). Together, these results indicated that SAP155, a subunit of SF3b, is required for proper pre-mRNA splicing of both P27 and FIR.
Figure 3.
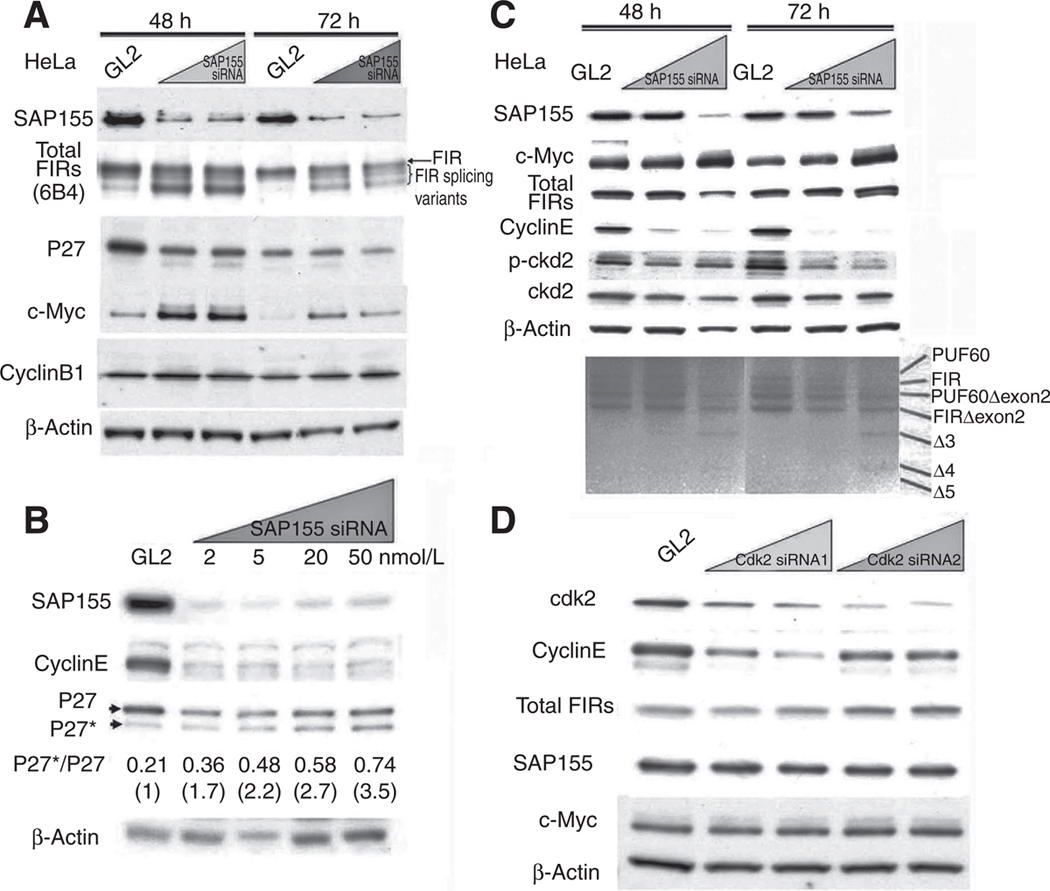
SAP155 knockdown by siRNA reduces both FIR and P27Kip1 expression and increases their alternative splicing variants. A, SAP155 knockdown by siRNA (5 or 20 nmol/L for 48 or 72 hours) show reduced P27 expression. Some of the FIR protein is shifted to a lower molecular weight, indicating that SAP155 is required for FIR splicing. B, SAP155 knockdowns by siRNA for 48 hours inhibited P27 pre-mRNA splicing in a dose-dependent manner, as indicated by the P27*/P27 ratio. The P27*/P27 ratios were 0.21 (GL2 siRNA), 0.36 (2 nmol/L SAP155 siRNA), 0.48 (5 nmol/L), 0.58 (20 nmol/L), and 0.74 (50 nmol/L). When the P27*/P27 ratio for GL2 was set at 1, the P27*/P27 ratios were 1.7 (2 nmol/L SAP155 siRNA), 2.2 (5 nmol/L), 2.7 (20 nmol/L), and 3.5 (50 nmol/L). CyclinE expression also decreased with SAP155 siRNA silencing. C, FIR, cyclinE, p-cdk2, and cdk2 expression was suppressed, and RT-PCR shows that FIR splicing variants were increased by SAP155 knockdown by siRNA in HeLa cells. A novel FIR splicing variant, Δ5, was detected by RT-PCR. D, Cdk2 knockdown by siRNA decreased cyclinE expression but not FIR or SAP155 expression.
SSA, an SF3b inhibitor, alters P27/cdk2/cyclinE expression in HeLa cells
Because SSA, a natural SF3b inhibitor, affected the P27*/P27 expression ratio, cdk2/cyclinE expression should be altered by SSA treatment. SSA treatment altered P27* expression (Fig. 4A) and increased c-Myc expression (Fig. 4B), as reported previously (10, 14). SSA treatment decreased p-cdk2/cyclinE expression in HeLa cells (Fig. 4B). Collectively, these results indicate that SAP155 is pivotal for cdk2/cyclinE expression, possibly by regulating proper P27 pre-mRNA splicing, and SF3b inhibition by SSA or SAP155 siRNA affected both P27 and FIR pre-mRNA splicing. Thus, the FIR/FIRΔexon2/SAP155 interaction links cell-cycle progression and cell growth by modulating P27/cdk2/cyclinE and c-Myc expression.
Figure 4.

SSA, a SF3b inhibitor, induces P27* and reduces p-cdk2/cyclinE expression. A, HeLa cells were treated with SSA (100 ng/ml) for 0,1,6, and 24 hours. P27Kip1 and its splicing variant P27* were detected by Western blotting using anti-P27Kip1 antibody. P27* expression was higher than authentic P27 following SSA treatment. Schematic constructs of P27 and P27* are shown. SSA inhibited P27 pre-mRNA alternative splicing, and P27* was generated using a stop codon within intron 1 (between exon 1 and exon 2). B, the expression of c-Myc, total cdk2, phosphorylated cdk2 (Phos-cdk2), cyclinE, and cyclinB1 were examined after SSA treatment. β-Actin served as an internal control.
SAP155 siRNA inhibits HeLa cell growth and HA-FIR arrests cells at G1
Because SSA inhibition of SF3b function has cytotoxic effects, SAP155 siRNA should induce significant cell damage. As expected, SAP155 siRNA induced significant cytotoxicity with apoptosis in HeLa cells (Fig. 5A). SAP155 siRNA significantly reduced cell viability as assessed by MTS assay (Fig. 5B, left) and inhibited colony formation in a soft agar gel assay (Fig. 5B, right). These results indicated that proteins regulated by SAP155 engage in cell survival, growth, and migration. To assess the effect of FIR on cell cycling, HA-FIR was transfected to U2OS cells. Interestingly, HA-FIR-transfected cells were blocked in G1 (Fig. 5C). c-Myc downregulation arrests cells at Gx, independent ofP27 expression (17, 18). Ad-FIR suppressed both cyclinE and P27, but did not alter cdk2 expression (Supplementary Fig. S1). These results suggest that c-Myc downregulation may have a more significant effect on cell growth and survival independent of P27.
Figure 5.

SAP155 siRNA inhibits cell growth concomitant with cell-cycle disruption. A, SAP155 siRNA (20 nmol/L) treatment for 48-hours induced apoptosis as determined by the APOPercentage apoptosis assay™. B, SAP155 siRNA treatment significantly decreased cell viability as determined by the MTS assay (left). SAP155 siRNA inhibited colony formation in a soft agar gel assay (top right). GL2 siRNA was used as a control (bottom right). C, U2OS cells were transfected with HA-FIR and immunostained with anti-HA. DNA was stained with PI. The HA- and PI-stained cells were analyzed by flow cytometry. At 24 hours after transfection, 71.9% of the HA-positive cells were in G0/G1, whereas the cell-cycle profile of HA-negative cells was indistinguishable from that of untransfected cells. Both untransfected cells and HA-negative transfected cells had 30% to 32% of cells in G1.
FIR is required for endogenous P89 expression
A previous report showed that P89 coimmunoprecitated with FIR (4). Therefore, the FIR/FIRΔexon2/SAP155 interaction significantly affects proper P89 expression. As siRNA against SAP155 suppresses FIR expression (10), we examined whether decreased P89 expression due to SAP155 siRNA was rescued by Ad-FIR or Ad-FIRΔexon2. Ad-FIR, but not Ad-FIRΔexon2, restored expression of P89 compromised by SAP155 knockdown (Figs. 1C and 6A, arrows), indicating that endogenous P89 expression is at least partly sustained by authentic FIR expression. However, the fold-increase of FIR did not differ significantly between treatments with GL2 and SAP155 siRNAs (Fig. 6A, arrows); thus, the rescue by FIR was related to the FIR/SAP155 interaction rather to an ectopic effect of FIR overexpression alone (Fig. 6A, arrows). Although elevated FIR suppresses c-Myc and induces apoptosis (6), FIR is paradoxically activated in cancer cells with elevated c-Myc (Fig. 1A). One explanation for this may be that the spliced variant of FIR, FIRΔexon2, competes with authentic FIR for FUSE binding in cancers (10). Another explanation is that FIR/FIRΔexon2 forms a sustained complex with SAP155 in cancers; thus, rapid, efficient, and effective responses to DNA damage could be possible by integrating c-Myc and P27 regulation. Therefore, bleomycin, a DNA-damaging and apoptosis-inducing chemical (19, 20), was used to survey the cellular responses of c-Myc and P27. Bleomycin-induced DNA damage was accompanied by suppression of total FIR, c-Myc, P89, and P27 expression (Fig. 6B). These results indicate that FIR/FIRΔexon2/SAP155 interaction is independently critical for both c-myc transcription, in part via P89 expression, and for cell-cycle regulation via P27 expression.
Figure 6.

Endogenous P89 expression suppressed by SAP155 siRNA is rescued by Ad-FIR. A, HeLa cells were treated with SAP155 siRNAat day 0 in a 6-well plate with GL2 siRNA as a control. On day 1, the cells were transfected with Ad-FIR or Ad-FIRΔexon2. The cells were harvested on day 2, and the SAP155, P89, and total FIR expression levels were determined by Western blotting. Lanes 1 and 4: Ad-GFP; lanes 2 and 5: Ad-FIR; lanes 3 and 6: Ad-FIRΔexon2. Ad-FIR, but not Ad-FIRΔexon2, rescued P89 suppression by SAP155 siRNA (compare arrows in lane 2 to lane 1 and lane 5 to lane 4, respectively). The ratio of P89 expression of lane 2 to lane 1 was 1.4, the same of lane 5 to lane 4 was 1.9. B, HeLa cells were treated with the DNA-damaging chemical bleomycin. Lane 1: mock; lanes 2–5: 0, 10, 50, and 100 ng/mL of bleomycin, respectively. DNA damage was indicated by γH2AX activation.
Discussion
Recently, we reported on the physical association of FIR, FIRΔexon2, and SAP155, the antagonistic effects of FIR and FIRΔexon2 on c-myc transcription, and the coordinated regulation of FIR, SAP155, and c-Myc in colorectal cancer (10). In this report, we elaborated on these previous findings by evaluating the effects of SAP155 and FIR isoforms on the expression of P27, cdk2/cyclinE, and P89. This study provides novel information on a regulatory circuit, which is important for cell growth and is of potential relevance for colorectal cancer progression.
FIR/FIRΔexon2/SAP155 complex formation potently disturbs alternative FIR pre-mRNA and changes the ratio of FIRΔexon2/FIR mRNA expression. Thus, this process is a novel bona, fide molecular switch for c-myc gene expression (10). Our study further shows the following: (i) FIR/FIRΔexon2/SAP155 is required for both proper P27 expression and its pre-mRNA splicing; (ii) FIR/FIRΔexon2/SAP155 is required for sustained P89 expression in colon cancers; and (iii) overexpressed HA-FIR arrests U2OS cells at G1. In addition, bleomycin, a DNA-damaging and apoptosis-inducing chemical, suppressed total FIR, c-Myc, P89, and P27 expression, and thus, (iv) the FIR/FIRΔexon2/SAP155 complex links c-myc transcription and P27 regulation (Fig. 7). The FIR system suppresses c-myc and SAP155 regulates P27 pre-mRNA splicing in noncancer cell (Fig. 7, top). In contrast, the sustained FIR/FIRΔexon2/SAP155 complex disturbs c-myc regulation and P27 pre-mRNA splicing (Fig. 7, bottom). FIRΔexon2 may possibly compete with FIR for FUSE binding as a trans-acting element; however, FIR is involved in sustained P89 expression, probably as a cis-acting element (Fig. 7, bottom).
Figure 7.

A scheme for the effects of FIR/FIRΔexon2/SAP155 interactions on c-myc transcription and cell-cycle regulator P27/cdk2/cyclinE expression. The FIR/FIRΔexon2/SAP155 interaction links c-myc and P27Kip1 expression, and integrates cell growth, cell proliferation, and cell-cycle control. FIR suppresses c-myc and P27Kip1 suppresses cdk2/cyclinE, which arrests cells in the G1 phase (top), but sustained expression of the FIR/FIRΔexon2/SAP155 complex accelerates cell entry into the S-phase (bottom). In S-phase or proliferating cells, sustained expression of FIR/SAP155 or the FIRΔexon2/SAP155 complex disrupts the well-established functions of FIR for c-myc transcriptional repression and SAP155 splicing activity. In addition, FIRΔexon2competeswith FIR for FUSE binding; therefore, the c-myc gene is activated in cancer. Because SAP155 is required for proper P27Kip1 pre-mRNA splicing, sustained expression of the FIR/SAP155 complex or mechanical change of FIR-SAP155 interaction inhibits P27Kip1 pre-mRNA splicing by disrupting SF3b function.
These results raise the question of what cellular events FIR/FIRΔexon2/SAP155 monitors by integrating c-Myc, cell cycling, and splicing. One possibility is that the FIR/FIRΔexon2/SAP155 interaction is involved in DNA-damage repair responses, because some DNA repair proteins coimmunoprecipitate with FIR (10), and bleomycin-induced DNA damage significantly reduced total FIR, P89, and P27 expression (Fig. 6B). At this point, c-myc expression should be tightly constrained when cells respond to certain stimulations (16).
When DNA damage occurs in cells, c-Myc and cell-cycle regulation should be monitored by certain integrated systems because cell fate is determined by the extent of DNA damage. For example, the double-strand break repair proteins Ku86 and DNA-PKcs coimmunoprecipitate with FIR, indicating that FIR and Ku86/DNA-PKcs may form a complex (unpublished data). In cancer cells, DNA damage appears to accumulate without complete repair because the threshold for a cell-cycle checkpoint is lowered compared with that in normal cells. Thus, c-myc expression and cell-cycle regulation should be integrated or monitored as a cell-fate switch for responding to DNA damage by undergoing DNA repair, apoptosis, or transformation (21).
In undeviating regulation conditions, as occurs in normal cells, c-Myc responds to multiple intracellular feedback mechanisms to be constrained within physiologic bounds. P27 suppresses cyclinE expression and arrests cells in G1 (22). c-Myc represses the transcription of key growth control and differentiation genes, including p21, p27, and growth-arrest and DNA-damage proteins (23, 24). In addition, p27 reportedly acts as a barometer for targeted therapies, because reduced p27 levels and p27 mislocalization have potential prognostic and therapeutic implications (25). However, under dysregulated conditions, as seen in cancer cells, the threshold for these checkpoints, such as DNA damage repair during cell cycle, is avoided because cell-cycle suppressors, such as the p21Cip1/p27 family of CDK inhibitors, are reduced (11, 12, 26, 27). Elucidation of the mechanisms of the FIR/SAP155 interaction promises to reveal the relation between c-myc expression, tumor progression, differentiation, and other signaling systems such as DNA-damage responses.
Therefore, inhibiting or dissociating the FIR/FIRΔexon2/SAP155 interaction is potentially a promising target for cancer therapy. A sendai virus vector encoding FIR showed synergistic effects with cisplatin on malignant mesothelioma (28). Furthermore, SF3b is a cancer treatment target (14, 29). Since the FIR/FIRΔexon2/SAP155 complex is induced in colorectal cancers and disrupts proper FIR pre-mRNA splicing, FIR splicing variants may be novel cancer screening markers (30, 31). Thus, the crosstalk between SAP155 and FIR/FIRΔexon2 is a candidate mechanism or sensor for monitoring cell-cycle checkpoints, because c-Myc executes cell decisions, including proliferation, differentiation, or apoptosis, by controlling a variety of candidate targets whose products are involved in multiple metabolic and regulatory pathways (32, 33).
Supplementary Material
Acknowledgments
The authors thank Dr. Naohito Nozaki of the Department of Biochemistry and Molecular Biology, Kanagawa Dental College (Yokosuka, Kanagawa) for preparing FIR monoclonal antibodies.
Grant Support
This study was supported in part by Grant-in-Aid 18591453 for priority areas in cancer research from the Ministry of Education, Science, Sports, and Culture of Japan and “Seed Finding Programs” and “Mini-Feasibility Study Project” of the JST (Japan Science and Technology) Agency (to K. Matsushita), and a Grant-in-Aid for Scientific Research (S) from the Japan Society for the Promotion of Science (to M. Yoshida).
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
M. Yoshida is employed (other than primary affiliation; e.g., consulting) as a senior program officer in Research Center for Science Systems, JSPS and has honoraria from speakers’ bureau from Johns Hopkins University. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Avigan MI, Strober B, Levens D.Afar upstream element stimulates c-myc expression in undifferentiated leukemia cells. J Biol Chem 1990;265:18538–45. [PubMed] [Google Scholar]
- 2.Duncan R, Bazar L, Michelotti G, Tomonaga T, Krutzsch H, Avigan M, et al. A sequence-specific, single-strand binding protein activates the far upstream element of c-myc and defines a new DNA-binding motif. Genes Dev 1994;8:465–80. [DOI] [PubMed] [Google Scholar]
- 3.Bazar L, Meighen D, Harris V, Duncan R, Levens D, Avigan M. Targeted melting and binding of a DNA regulatory element by a transactivator of c-myc. J Biol Chem 1995;270:8241–8. [DOI] [PubMed] [Google Scholar]
- 4.Liu J, He L, Collins I, Ge H, Libutti D, Li J, et al. The FBP interacting repressor targets TFIIH to inhibit activated transcription. Mol Cell 2000;5:331–41. [DOI] [PubMed] [Google Scholar]
- 5.Liu J, Akoulitchev S, Weber A, Ge H, Chuikov S, Libutti D, et al. Defective interplay of activators and repressors with TFIH in xeroderma pigmentosum. Cell 2001;104:353–63. [DOI] [PubMed] [Google Scholar]
- 6.Matsushita K, Tomonaga T, Shimada H, Shioya A, Higashi M, Matsubara H, et al. An essential role of alternative splicing of c-myc suppressor FIR in carcinogenesis. Cancer Res 2006;66:1409–17. [DOI] [PubMed] [Google Scholar]
- 7.Campaner S, Doni M, Hydbring P, Verrecchia A, Bianchi L, Sardella D, et al. Cdk2 suppresses cellular senescence induced by the c-myc oncogene. Nat Cell Biol 2010;12:54–9. [DOI] [PubMed] [Google Scholar]
- 8.Hydbring P, Bahram F, Su Y, Tronnersjö S, Högstrand K, von der Lehr N, et al. Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proc Natl Acad Sci USA 2010;107:58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hydbring P, Larsson LG. Tipping the balance: Cdk2 enables Myc to suppress senescence. Review. Cancer Res 2010;70:6687–91. [DOI] [PubMed] [Google Scholar]
- 10.Matsushita K, Kajiwara T, Tamura M, Satoh M, Tanaka N, Tomonaga T, et al. SAP155-mediated splicing of FUSE-binding protein-interacting repressor (FIR) serves as a molecular switch for c-myc gene expression. Mol Cancer Res 2012;10:787–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsushita K, Kobayashi S, Kato M, Itoh Y, Okuyama K, Sakiyama S, et al. Reduced messenger RNA expression levelofp21 CIP1 inhuman colorectal carcinoma tissues and its association with p53 gene mutation. Int J Cancer 1996;69:259–64. [DOI] [PubMed] [Google Scholar]
- 12.Starostina NG, Kipreos ET. Multiple degradation pathways regulate versatile CIP/KIP CDK inhibitors. Trends Cell Biol 2012;22:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsushita K, Takenouchi T, Shimada H, Tomonaga T, Hayashi H, Shioya A, et al. Strong HLA-DR antigen expression on cancer cells relates to better prognosis of colorectal cancer patients: Possible involvement of c-myc suppression by interferon-g in situ. Cancer Sci 2006;97:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat Chem Biol 2007;3:576–83. [DOI] [PubMed] [Google Scholar]
- 15.Weber A, Liu J, Collins I, Levens D. TFIIH operates through an expanded proximal promoter to fine-tune c-myc expression. Mol Cell Biol 2005;25:147–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, Chen QM. Far upstream element binding protein 1: a commander of transcription, translation and beyond. Review. Oncogene. 2012. August 27. doi: 10.1038/onc.2012.350. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walter KA, Hossain MA, Luddy C, Goel N, Reznik TE, Laterra J. Scatter factor/hepatocyte growth factor stimulation of glioblastoma cell cycle progression through G(1)is c-Myc dependent and independent of p27 suppression, Cdk2 activation, or E2F1-dependent transcription. Mol Cell Biol 2002;22:2703–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benaud CM, Dickson RB. Adhesion-regulated G-i cell cycle arrest in epithelial cells requires the downregulation of c-Myc. Oncogene 2001;20:4554–67. [DOI] [PubMed] [Google Scholar]
- 19.Hamilton RF Jr, Li L, Felder TB, Holian A. Bleomycin induces apoptosis in human alveolar macrophages. Am J Physiol 1995;269:318–25. [DOI] [PubMed] [Google Scholar]
- 20.Vernole P, Tedeschi B, Caporossi D, Maccarrone M, Melino G, Annicchiarico-Petruzzelli M. Induction of apoptosis by bleomycin in resting and cycling human lymphocytes. Mutagenesis 1998;13: 209–15. [DOI] [PubMed] [Google Scholar]
- 21.Alexander R, Beggs JD. Cross-talk in transcription, splicing and chromatin: who makes the first call? Biochem Soc Trans 2010;38: 1251–56. [DOI] [PubMed] [Google Scholar]
- 22.Morris TA, DeLorenzo RJ, Tombes RM. CaMK-II inhibition reduces cyclin D1 levels and enhances the association of p27kip1 with Cdk2 to cause G1 arrest in NIH 3T3 cells. Exp Cell Res 1998;240: 218–27. [DOI] [PubMed] [Google Scholar]
- 23.Gartel AL, Shchors K. Mechanisms of c-myc-mediated transcriptional repression of growth arrest genes. Exp Cell Res 2003;283:17–21. [DOI] [PubMed] [Google Scholar]
- 24.Si J, Yu X, Zhang Y, DeWille JW. Myc interacts with Max and Miz1 to repress C/EBP delta promoter activity and gene expression. Mol Cancer 2010;9:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wander SA, Zhao D, Slingerland JM. p27: a barometer of signaling deregulation and potential predictor of response to targeted therapies. Review. Clin Cancer Res 2011;17:12–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Philipp-Staheli J, Payne SR, Kemp CJ. p27(Kip1): regulation and function of a haploinsufficient tumor suppressor and its misregulation in cancer. Exp Cell Res 2001;264:148–68. [DOI] [PubMed] [Google Scholar]
- 27.Borriello A, Caldarelli I, Bencivenga D, Criscuolo M, Cucciolla V, Tramontano A, et al. p57(Kip2) and cancer: time for a critical appraisal. Mol Cancer Res 2011;9:1269–84. [DOI] [PubMed] [Google Scholar]
- 28.Kitamura A, Matsushita K, Takiguchi Y, Shimada H, Tomonaga T, Matsubara H, et al. Synergistic effect of non-transmissible Sendai virus vector encoding the c-myc suppressor FUSE-binding protein-interacting repressor plus cisplatin in treatment of malignant pleural mesothelioma. Cancer Sci 2011;102:1366–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y, Shimizu H, Uesugi M, et al. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat Chem Biol 2007;3:570–5. [DOI] [PubMed] [Google Scholar]
- 30.Kajiwara T, Matsushita K, Itoga S, Tamura M, Tanaka N, Tomonaga T, et al. SAP155-mediated c-myc suppressor FBP-interacting repressor splicing variants are activated in colon cancer tissues. Cancer Sci 2013;104:149–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsushita K,Tomonaga T, Kajiwara T, Shimada H, Itoga S, Hiwasa T, et al. c-myc suppressor FBP-interacting repressor for cancer diagnosis and therapy. Front Bios 2009;14:3401–8. [DOI] [PubMed] [Google Scholar]
- 32.Dang CV, Resar LM, Emison E, Kim S, Li Q, Prescott JE, et al. Function of the c-Myc oncogenic transcription factor. Exp Cell Res 1999;235:63–77. [DOI] [PubMed] [Google Scholar]
- 33.Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012;151:68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.