

Abstract
Exfoliation syndrome (XFS) is an age-related disease defined by the deposition of aggregated fibrous material (XFM) in the peri-cellular space. Principal morbidity occurs in the eye, where XFM accumulates on the anterior ocular tissues. GWAS have found that certain genetic variants of lysyl oxidase-like 1 (LOXL1), a matrix cross-linking enzyme that is required for elastic fiber formation confer risk for the development of XFS, but are not a single causative factor as many genetically affected individuals do not develop XFS or subsequent glaucoma (XFG). We have found that XFG cells display defects in lysosomes, microtubules, autophagy, and mitochondria resembling defects found in cells from age-related syndromes, such as the main neurodegenerative diseases. In the majority of these diseases, the determining cellular factor is a protein containing intrinsically disordered regions (IDRs) and displaying a high propensity for aggregation. We have found that in XFG patient-derived cells, LOXL1 protein is actively subjected to autophagic clearance, suggesting that LOXL1 is undergoing aggregation. In silico analysis demonstrates that LOXL1’s first 369 aa constitute an IDR with the highest disorder probability peak centering around the known risk positions. Experimentally, we have found over-expression of either unmodified LOXL1 or fluorescent chimeras preserving the well-structured N-terminus cause copious intracellular aggregation and that aggregation wanes when the high IDR peaks are deleted. Overall, our work suggests that XFS/G results from the aggregation of the LOXL1 protein coupled with a reduction of cellular proteostasis capabilities in aging, resulting in a chronic build-up of LOXL1-containing protein aggregates.
1. Introduction
The age-dependent Exfoliation Syndrome (XFS) is a systemic disease characterized by the formation of aggregated protein deposits called Exfoliation Material (XFM) in all tissues that can synthesize elastin fibers (Nazarali, Damji, & Damji, 2018). While there are reports showing XFS health impact in multiple organs, in particular in peri-cardiac vessels (Anastasopoulos, Founti, & Topouzis, 2015; Prince & Ritch, 1986), the most significant morbidity occurs in the eye. Flaky material accumulates on the ciliary body, iris, lens, and trabecular meshwork. When glaucoma develops in the presence of these aggregates, it is referred to as Exfoliation Glaucoma (XFG)(Aboobakar, Johnson, Stamer, Hauser, & Allingham, 2016; Henry et al., 1987; Ritch & Schlotzer-Schrehardt, 2001; Ritch, Schlotzer-Schrehardt, & Konstas, 2003). XFG displays a marked resistance to classical pharmacological treatments for primary open-angle glaucoma (POAG) and induces a much faster vision loss. The XFG contribution to the incidence of open-angle glaucoma shows a strong dependence on the ethnic background, geography, and environmental variables (Pasquale et al., 2014). It constitutes between 20 and 60% of glaucoma cases in many regions of the world, including several Scandinavian countries, Russia, Iran, Ethiopia, and the South African Bantu tribe (Bialasiewicz, Wali, Shenoy, & Al-Saeidi, 2005; Forsius, 1988; Konstas & Ringvold, 2018; Ringvold, 1996), and overall, it accounts for 20%–25% of the estimated 70 million worldwide glaucoma sufferers. Because the earliest onset of POAG is around age 40, but for XFG is around age 60, both XFG prevalence and its fractional contribution to total glaucoma can be projected to increase as worldwide lifespan extends. For instance, in rural Finland XFS prevalence in the 60–69, 70–79 and 80 + age cohorts are, 14.2%, 21.9% and 34.7%, respectively. Projecting these numbers forward, yields a >90% probability of XFS incidence for a presently hypothetical, 100–110 cohort (Krause et al., 1988).
2. Macroscopic, structural and biochemical features of XFG
Whereas elsewhere the XFS material will manifest as extracellular deposits within an extracellular matrix not accessible to the naked eye, the accumulations in the eye occur within the aqueous space of the eye, resulting in their easy recognition as white flaky deposits (Fig. 1-A and B). Ultrastructural studies have shown that the material originates from the surface of tissues facing the posterior aqueous chamber. In the ciliary body, putative aggregated material has been visualized at invaginations of the aqueous facing, basolateral membrane of the non-pigmented epithelial layer (Schlotzer-Schrehardt, 2009). The pigment-rich pseudostratified epithelium covering the inner surface of the iris may also secrete XFM. Whether XFM is made there or not, one of the more dramatic manifestations of XFG is the release of pigment granules from these cells, indicating that XFG causes cellular stress. Yet, the most visible accumulations occur at the equator of the crystalline lens. The cellular source of these deposits remains to be elucidated. Since the permeability of the human lens capsule has an MW cut off of < 170 Kd (Kastner et al., 2013; Lee, Vroom, Fishman, & Bent, 2006) this XFM may arrive at the lens from the CB-Iris carried up by the aqueous flow. Nevertheless, ultrastructural studies suggest that the deposit may be generated by the lens cells and transferred through the capsule by an impaling-type mechanical force. The glaucoma-promoting effect of XFS may be related to the iris epithelium release of pigment granules, which carried away by the fluid flow to the outflow facility, deposit there and thereby increase outflow resistance elevating the intraocular pressure. This secondary consequence of XFS may combine with a putative effect on the integrity of the lamina cribrosa, the protective cushion for the retinal neurons at the optic nerve head; XFM accumulation there may weaken the lamina resulting in damage to the optic nerve fibers at lower pressures than those needed to elicit the damage in POAG. Ultrastructural examination (atomic force microscopy) reveals features of organized structures within XFM. Short beaded stretches crosslink at variable angles (Fig. 2A). These structures do not resemble the structure of fibrillin fibrils (Fig. 2B).
Fig. 1. (A) Schematic description of the cellular components of the anterior segments of the eye in relation to aqueous flow.
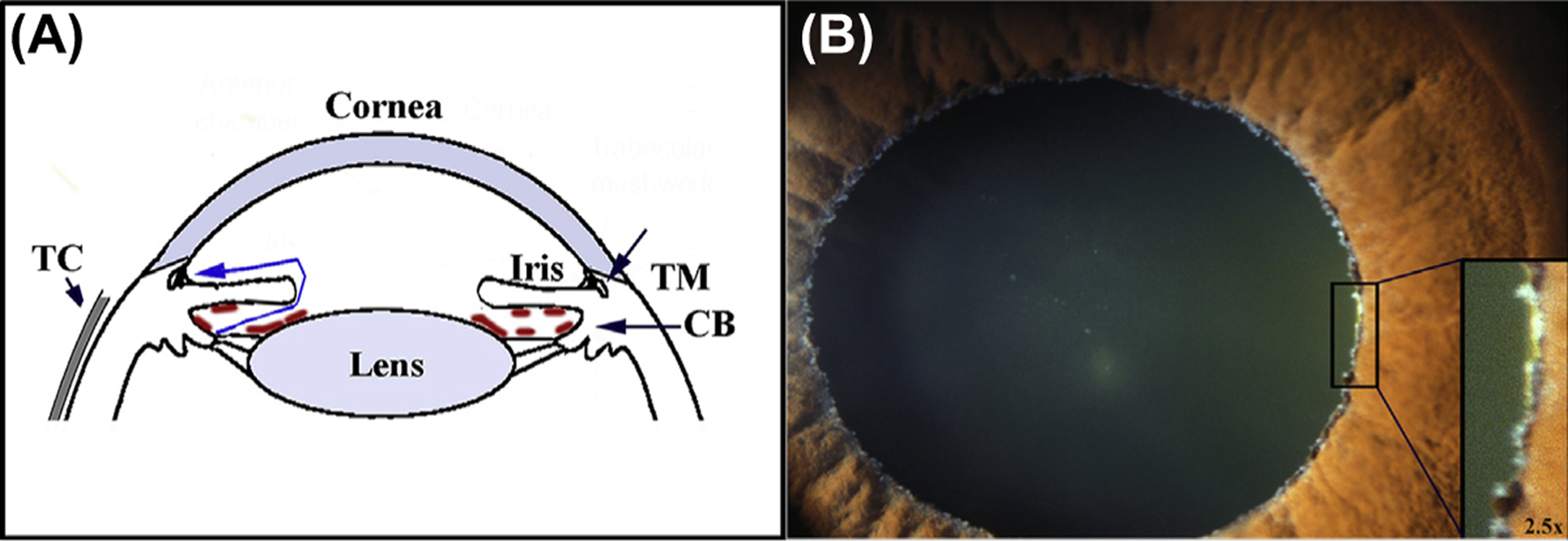
Fluid (blue line) is generated at the inner surface of the epithelium of the ciliary body CB) in the posterior aqueous chamber. From there it flows through the space between the iris and lens into the anterior aqueous chamber to exit the eye through the trabecular meshwork (TM) also referred to as the outflow facility. Increased, abnormal flow resistance of the facility causes elevation of the intraocular pressure (IOP). The increased pressure lead to the development of glaucoma, loss of retinal visual function due to retinal neural death. In individuals affected by exfoliation syndrome (XSF), proteinaceous material (shown tan marks in sketch) forms or deposits on the surface of the epithelial facing the posterior aqueous chamber, including the equator of the lens. The deposition is frequently associated with a type of glaucoma, exfoliation glaucoma, characterized by drug-unresponsive IOP increases and rapid progression of glaucoma. The Tenon’s capsule (TC) is a thin, elastic collagenous lining covering the sclera. (B) Flaky white deposits in the equator of the lens. The pupil has been dilated.
Fig. 2.

(A) XFM structure. (B) Fibrillin microfibrils.
At the biochemical level, mass spectra proteomic studies have shown that XFM incorporates an inordinate number of different proteins. The initial analysis identified, fibrillin-1, microfibril-associated glycoprotein, and clusterin/ApoJ and elastin, between other components (Ronci, Sharma, Martin, Craig, & Voelcker, 2013; Schlotzer-Schrehardt, 2009; Sharma et al., 2009, 2018). The first two proteins are logical candidates to contribute to a fibrous structure. The presence of Clusterin/ApoJ is unsurprising; this protein is secreted by cells when a proximal extracellular aggregate forms in an attempt to dissociate it. Other more unexpected components such as basement membrane and blood proteins may merely become sequestered in the forming meshwork. LOXL1, the main subject of this review was identified as an XFM component only after its relationship to XFG was indicated by the genomic wide association studies (GWAS; described below). Since LOXL1’s primary function is to catalyze the weaving of elastic fibers from the tropoelastin monomers through its deaminase activity, the presence of both elastin and LOXL1 peptides in the aggregates suggests the involvement of the elaborate extracellular process for elastin weaving in aggregate formation (Fig. 3). Supporting this hypothesis, it has been recently shown that mutations in fibulin 5, the scaffold protein that spatially positions LOXL1 for tropoelastin deamination, fosters the formation of extracellular aggregates (Padhy, Kapuganti, Hayat, Mohanty, & Alone, 2019).
Fig. 3. Schematic description of elastic fiber development.
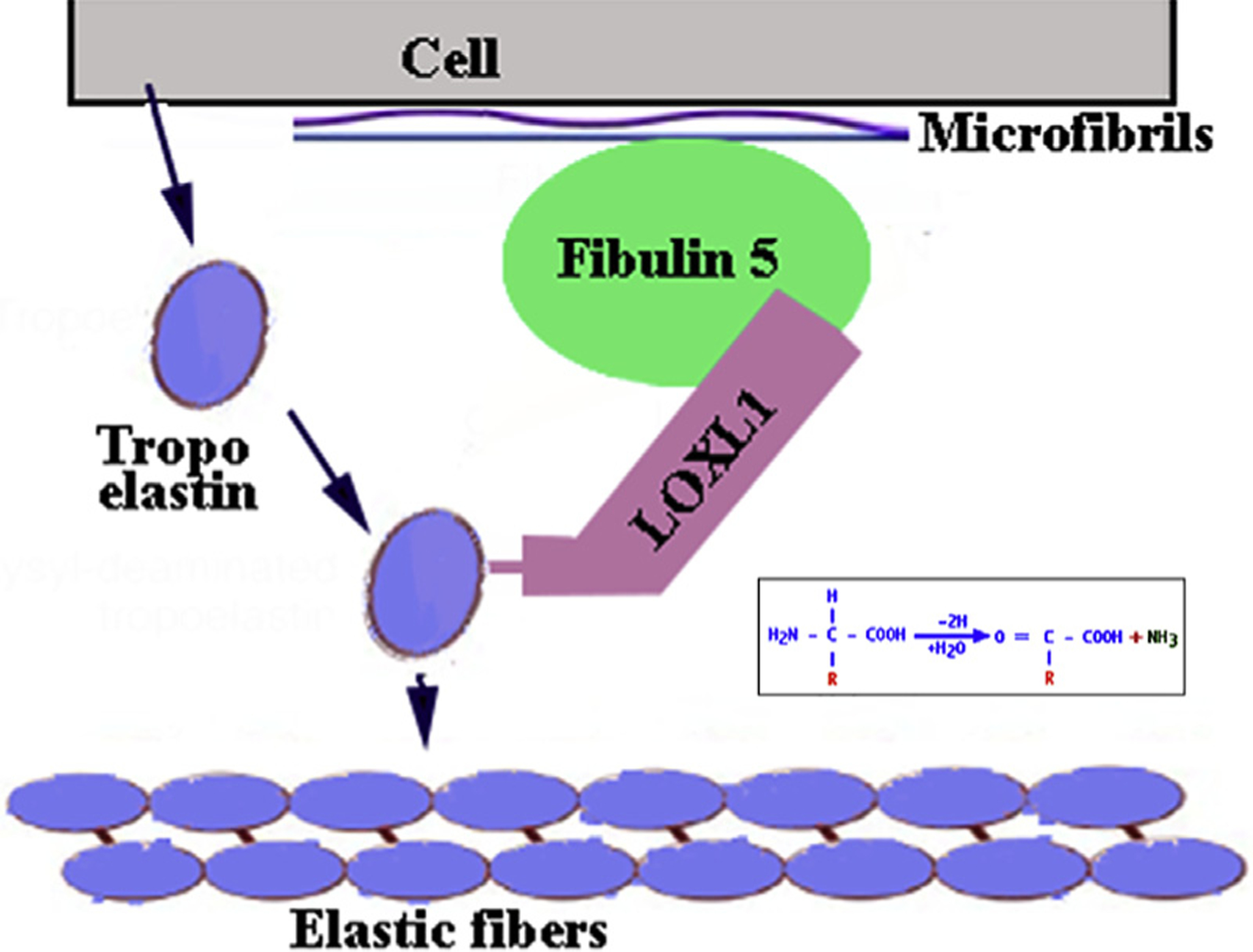
At the peri-cellular space the adapter protein Fibulin 5 anchoring at Fibrillin 1, binds the deaminase LOXL1 setting it in the proper spatial configuration to cause deamination of secreted Tropoelastin monomers. The reactive aldehyde groups allow homologous polymerization of Tropoelastin to form elastic fibers.
3. LOXL1 variants and XFG risk
A major advance toward the elucidation of the molecular drivers of XFG was provided by a GWAS that found that in the Swedish population the XFG risk was markedly affected by three sequence variants in LOXL1. One risk variant occurs in a long non-coding (Inc) region of the LOXL1 gene. The other two SNPs are CDS variants involving arginine (R) or leucine at the amino acid position (aa) 141 and glycine (G) or aspartic acid (D) at aa 153 (Hauser et al., 2015; Malukiewicz et al., 2011; Thorleifsson et al., 2007). The two risk-linked variants, R141 and G153 are not rare; in most world populations 153G incidence is about 70%. The strong statistical link results from the fact that in most studies including both regional LOXL1-focused genetic investigation and a very large worldwide GWAS (Abu-Amero et al., 2010; Chen et al., 2010; de Juan-Marcos et al., 2016; Hewitt et al., 2008; Thorleifsson et al., 2007) the incidence of G153 in XFS patients is 99% or 100 % (Fan et al., 2008). Similar numbers, though less accentuated, support the link of 141R to XFG. These links have been challenged by confounding results obtained with two high XFS prevalence populations. In the Japanese high risk is conferred by leucine occupation of aa 141, whereas in the Bantu, high XFS risk is associated with aspartic acid at aa 151 (Chen et al., 2010; Hayashi, Gotoh, Ueda, Nakanishi, & Yoshimura, 2008; Rautenbach, Bardien, Harvey, & Ziskind, 2011; Williams et al., 2010). Given that presently no high XFS prevalence population in which the variants at either of these two positions display risk indifference has been identified, seeing these divergent observations as voiders of the presumptive LOXL1 variant-XFS link will not be prudent. Rather, a more fitting explanation is that the CDS variant risk reflects the interactions of these two sites with other proteins or other locations within LOXL1, which in turn, contain variants alternatively facilitating or obstructing, the process of XFM formation. This conjecture fits the facts a) that the Japanese and the Bantu, which end having no variant commonality for risk, happens to be the two populations at both ends of the human genomic clustering map (Fig. 4) and b) the uniqueness of the genomic variant complement in the Bantu is suggested by the fact that, intriguingly the Bantu is the only population in the world where the G/D ratio at aa 153 deviates from a universal 70/30 % ratio (Rautenbach et al., 2011; Williams et al., 2010). More recently, a functional variant in the LOXL1 locus was identified immediately upstream of the long noncoding RNA LOXL1-AS1, that changes promotor activity and thus the level of its expression. Investigations into LOXL1-AS1’s function indicate involvement in the maintenance of the extracellular matrix and other pathways integral to the pathophysiology of XFG. (Hauser et al., 2015). Future studies will test the impact of LOXL1-AS1 on the development of Exfoliation Glaucoma.
Fig. 4. Human genetic clustering.
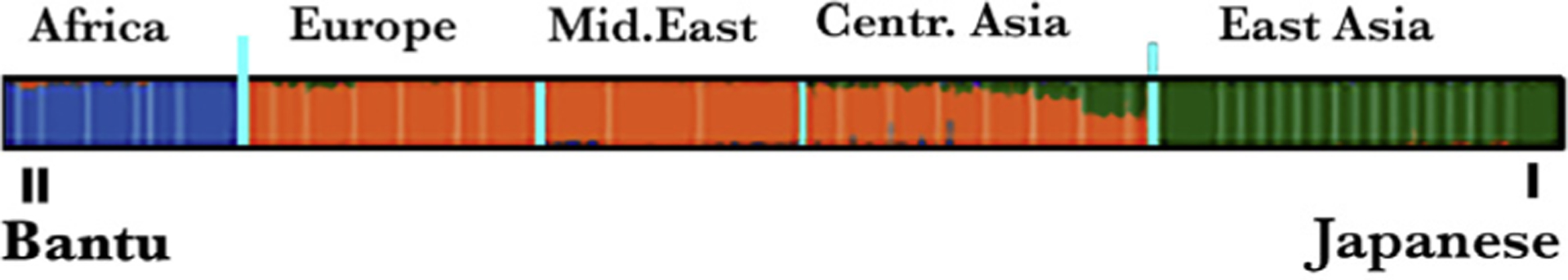
The South African Bantu and Japanese sit at the opposite sides of the spectrum of the differences in expression of genetic variants. One representative gene cluster, of several studied, is shown.
4. LOXL1 autophagy and LOXL1 in XFG patient cells
For studies based on actual disease, presently the only available option consists of Tenon’s capsule tissue obtained from trabeculectomies (glaucoma surgery to relieve intraocular pressure) of XFG patients and the tenon fibroblasts (XFG-TF) that can be cultured out of these minute biopsies (Kottler et al., 2005; Zenkel et al., 2011). While the tenon tissue is not intraocular, the systemic nature of the syndrome and the fact that the samples derive from individuals with glaucoma and documentable XFM accumulations, make XFG-TFs a useful conduit to understanding the drivers of XFG. Studies were based on the comparison against TF derived from non-XFG patients, either primary open-angle glaucoma (POAG) or non-glaucoma (NG) donors (Aboobakar et al., 2016; Bernstein, Ritch, & Wolosin, 2018; Want et al., 2016; Wolosin, Ritch, & Bernstein, 2018). Our studies have shown that XFG-TFs are larger than POAG and NG-TFs, proliferate at a slower rate and fail to organize in parallel bundles typical of cultured fibroblasts. Furthermore, XFG-TFs display the multiple abnormalities and dysfunctions observed in cells from the main neurodegenerative diseases and other age-related aggregopathies, including, impeded autophagic flux, disturbed LC3 expression, engorged and mislocalized lysosomes and endosomes, and decaying mitochondria (Bernstein et al., 2018; Want et al., 2016; Wolosin et al., 2018). A model for the formation of toxic intracellular aggregations leading to larger extracellular aggregates comes from Alzheimer’s. For a long time, it was assumed that neuronal death was caused by bulky extracellular amyloid aggregations. Nonetheless, it is now understood that the toxicity is caused by small intracellular aggregations (Iadanza, Jackson, Hewitt, Ranson, & Radford, 2018; Jeong, 2017; Tam & Pasternak, 2012). As graphically indicated in Fig. 5, Alzheimer’s extracellular deposits seem to result from the export of intracellular aggregates as a cell survival mechanism. It has been speculated that the intracellular aggregate toxicity reflects hindrances to cellular autophagy (Cruz, Kumar, Yuan, Arikkath, & Batra, 2018; Olzscha et al., 2011). The same possibility exists for XFG. In terms of LOXL1, we have found that in the XFG-derived fibroblasts LOXL1 accumulates in particles of larger size compared to controls and it is bound and exported by a chaperone that exports only misfolded proteins (unpublished). Additionally, when autophagy is blocked with the inhibitors Spautin-1 and Bafilomycin the amount of LOXL1 protein detected increases in XFG-TFs but not in POAG cells suggesting that LOXL1 is processed in the autophagy pathway that engulfs aggregates, in XFG-TFs only (Bernstein et al., 2018).
Fig. 5. Macroautophagy.
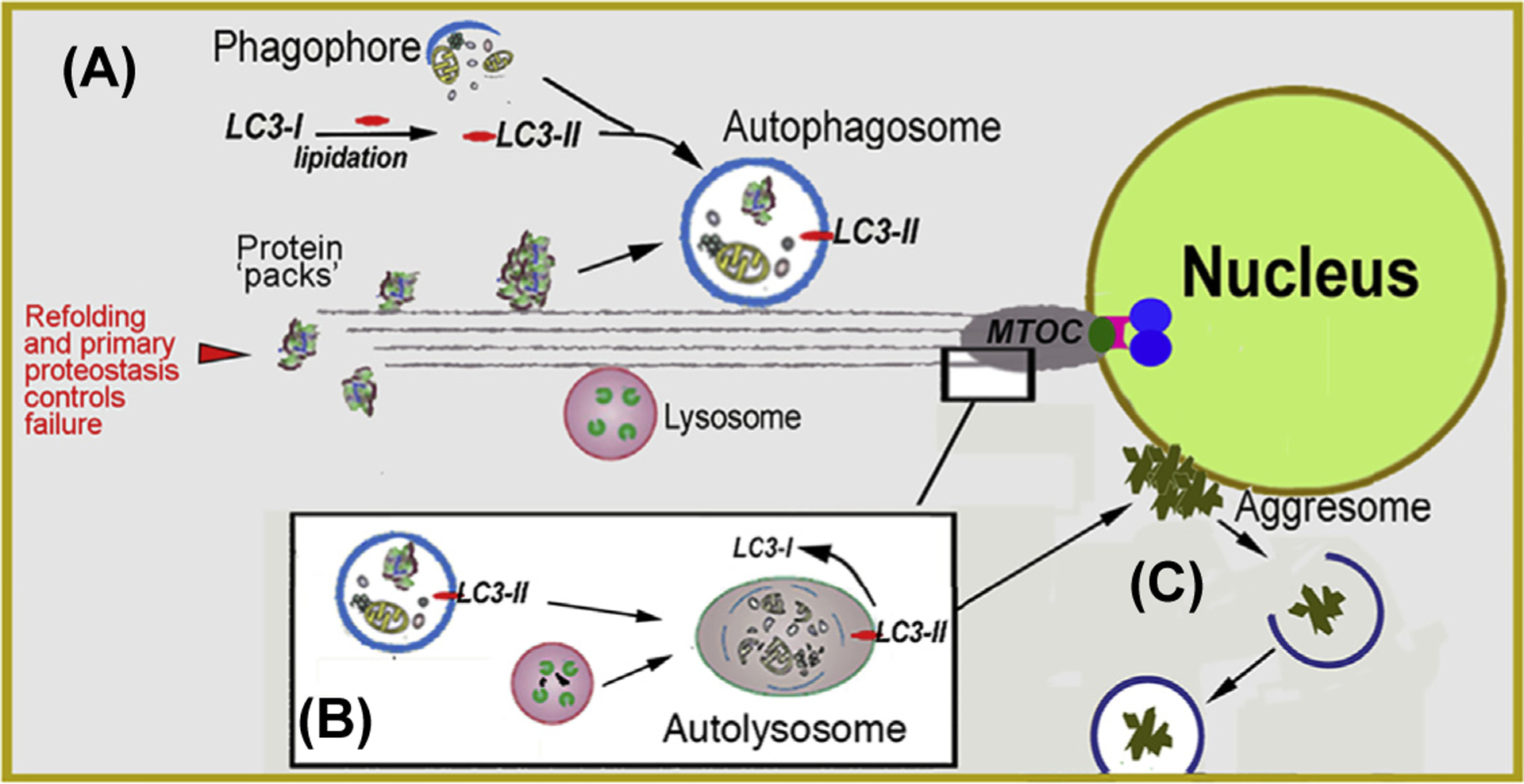
(A) Biogenesis of autophagosomes from precursor phagophores within the cytosol is orchestrated by the lipidated form of LC3 (LC3-II). Autophagosomes engulf ‘macroscopic’ cellular detritus, naturally decaying organelles, in particular, mitochondria (i.e., mitophagy) and misfolded or denatured proteins that have resisted the primary protein folding quality control mechanisms. (B) Autophagosomes fuse with lysosomes at the microtubule organizing center (MTOC). (C) Detritus that resist the combined proteostatic processes may accumulate in body inclusions, (e.g., Lewy body in senile dementia) categorized alternatively as aggresomes. If as demonstrated for tau protein propagation in Alzheimer’s and a-synuclein protein in Parkinson’s, then the aggregates may propagate to adjacent cells by particulate phagocytosis or endocytosis.
5. LOXL1 and intrinsic disorder
Given the observations described in the previous section on LOXL1 phenomena in XFG-TFs, it is plausible the LOXL1’s role in XFG parallels the role played by proteins in other age-related aggregopathies, namely as a germinal factor for XFM genesis. Extensive studies across the spectrum of age-related aggregopathies have established that they are usually mediated by one or more proteins containing intrinsically disordered regions (IDR) s. The IDR aggregation propensity reflects their ability to transiently exist, or dynamically adapt, undesirable hydrophobic β-sheet conformations of low solubility that can ‘stack’(De Simone et al., 2012; Martinelli, Lopes, John, Carlini, & Ligabue-Braun, 2019; Uversky, 2016) or even transfer the denatured conformation to an otherwise healthy protein, in a prion-like manner (Frost & Diamond, 2010; Kumar & Udgaonkar, 2019; Vaquer-Alicea & Diamond, 2019). This potential for unchecked propagation of hydrophobic denaturation represents a serious danger to the cell from a number of different perspectives, in particular for their interference with microtubule transport processes (Clark, Yeaman, Blizzard, Chuckowree, & Dickson, 2016; Cruz et al., 2018; Dubey, Ratnakaran, & Koushika, 2015; Olzscha et al., 2011). Incipient, small aggregates are the most toxic elements. The copious extracellular amyloid brain deposits found in cadaver brains in Alzheimer’s and probably, the large inclusion bodies observed in other neurodegenerative diseases, result in part from aging of the protecting and lytic mechanisms that neutralize aggregation within a cell (Fig. 5).
The study of proteins with extended disordered regions is challenging, they easily aggregate and cannot be crystallized. As a result, a large number of prediction-based systems providing disorder prediction (probability) that use a variety of different computational methods have been established (Atkins, Boateng, Sorensen, & McGuffin, 2015). Utilization of several of these servers to analyze the LOXL1 sequence uniformly yielded a prediction of disorder for most of the LOXL1 N-terminus extending up to amino acid (aa) 324. The results generated by one of the most advanced predictors, PdDOS, is shown in Fig. 6A. The most notable feature is the presence of two peaks of near > 95 % probability, centered around aa 80 and 155, respectively. The latter peak encompasses the two CDS variants associated with XFG risk. The C-terminus, containing the enzymatic function of LOXL1 (DP < 0.5), is fully structured. Intriguingly, all other members of the LOXL family are fully structured proteins (Fig. 6B) and BlastP analysis the human 1–324 IDR sequence against the GeneBank proteomic collections (eukaryotic and prokaryotic) yields high conservation across the LOXL1 mammalian spectrum but no significant homology to any other protein. This feature suggests that LOXL1 may play be involved in unique complex cellular interactions (Uversky, 2016).
Fig. 6. IDR in LOXL proteins.
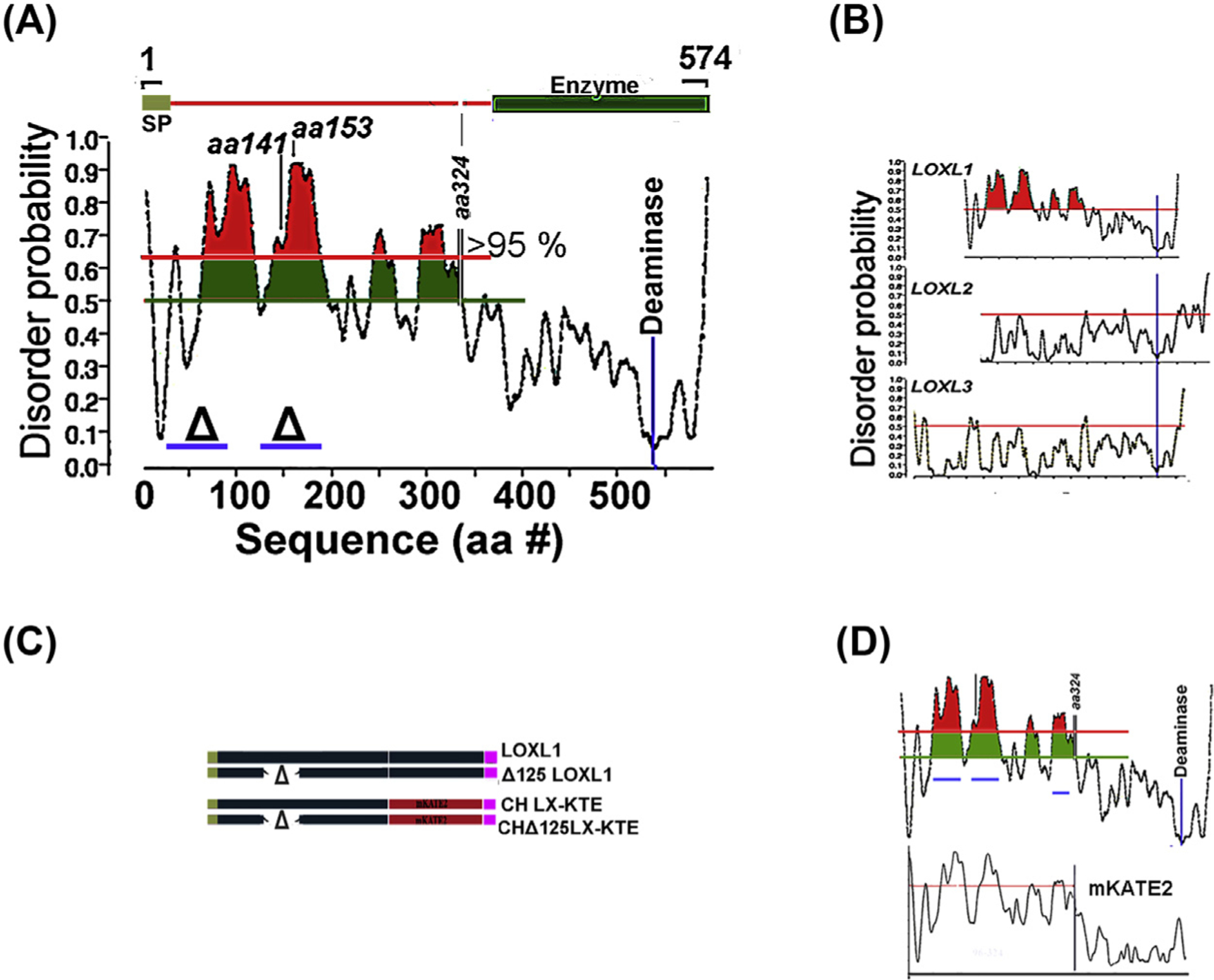
(A) Order/disorder probability prediction in the LOXL1 sequence. The zones where prediction confidence is > 95% is highlighted in red and the location for the enzymatic activity is indicated. (B) Comparison of order/disorder in LOXL family. (C) Comparison of order/disorder pattern in parent LOXL1 sequence and its mKATE2 chimera. (D) Examples of transgene LOXL1 constructs and their equivalent chimeras used to test aggregation propensity in overexpression assays.
6. The role of the IDR in LOXL1 aggregation
To examine whether the predicted disorder in LOXL1 confers to this protein an aggregation propensity, we employed lentivirus transduction of immortalized fibroblasts to generate, after antibiotic selection, cell sublines with high expression from a CMV promoter of either the whole LOXL1 sequence or sequences in which the aa stretches 28–95 (Δ28) or 125–185 (Δ125) were deleted (blue bars in Fig. 6A). Additionally, the structured enzymatic C-terminus of LOXL1, starting from aa 324, was replaced by the highly structured monomeric (m), red fluorescent protein mKATE2. The polypeptides were designed without tags or with either 6xHis (HIS) or FLAG C-terminus tags (Fig. 6C, schematic of Δ125). Flow cytometry demonstrated that all mKATE2 cells lines had a comparable mean level of fluorescence and Gaussian intensity distributions. Selected results from the over-expression studies are shown in Fig. 7. Over expression of whole LOXL1 creates small aggregates when grown in FBS-containing media (Fig. 7A) However, when cells are stressed with growth in supplemented-serum free media (SSFM, Fig. 7B) this phenotype is exacerbated, ring-like vesicular structures are apparent. In both cases the Δ 28 and Δ 125 have reduced aggregate formation compared to the whole LOXL1. Interestingly, the center of the ring-like LOXL1-staining structures in Fig. 7B can be identified with a protein aggregation-specific dye (Proteostat, Fig. 7B, bottom). Like LOXL1-HIS, images of the chimera Ch LX-KTE, demonstrate the same ring-like structures (Fig. 7C). A plausible explanation for the LOXL1-Proteostat staining profiles (and similarly, the mKATE2 fluorescence patterns) is that the center of large vesicles is undergoing intense aggregation of LOXL1 or CH LX-KTE and, while initially reflected in increased fluorescence at the center (LOXL1-binding secondary Ab or mKATE fluorescence), eventually inhibits (quenches) fluorophore function. As in the case of the parent LOXL1 sequences the Δ versions of CH LX KTE showed only enlarged vesicular structures distributed through the cytosol (Fig. 7C). In Fig. 7D, the LX-KTE fluorescent images are set in the cellular context by the staining of the actin filaments with FITC-phalloidin.
Fig. 7. Aggregation in overexpressed LOXL1 and homolog mKATE2 sequences.
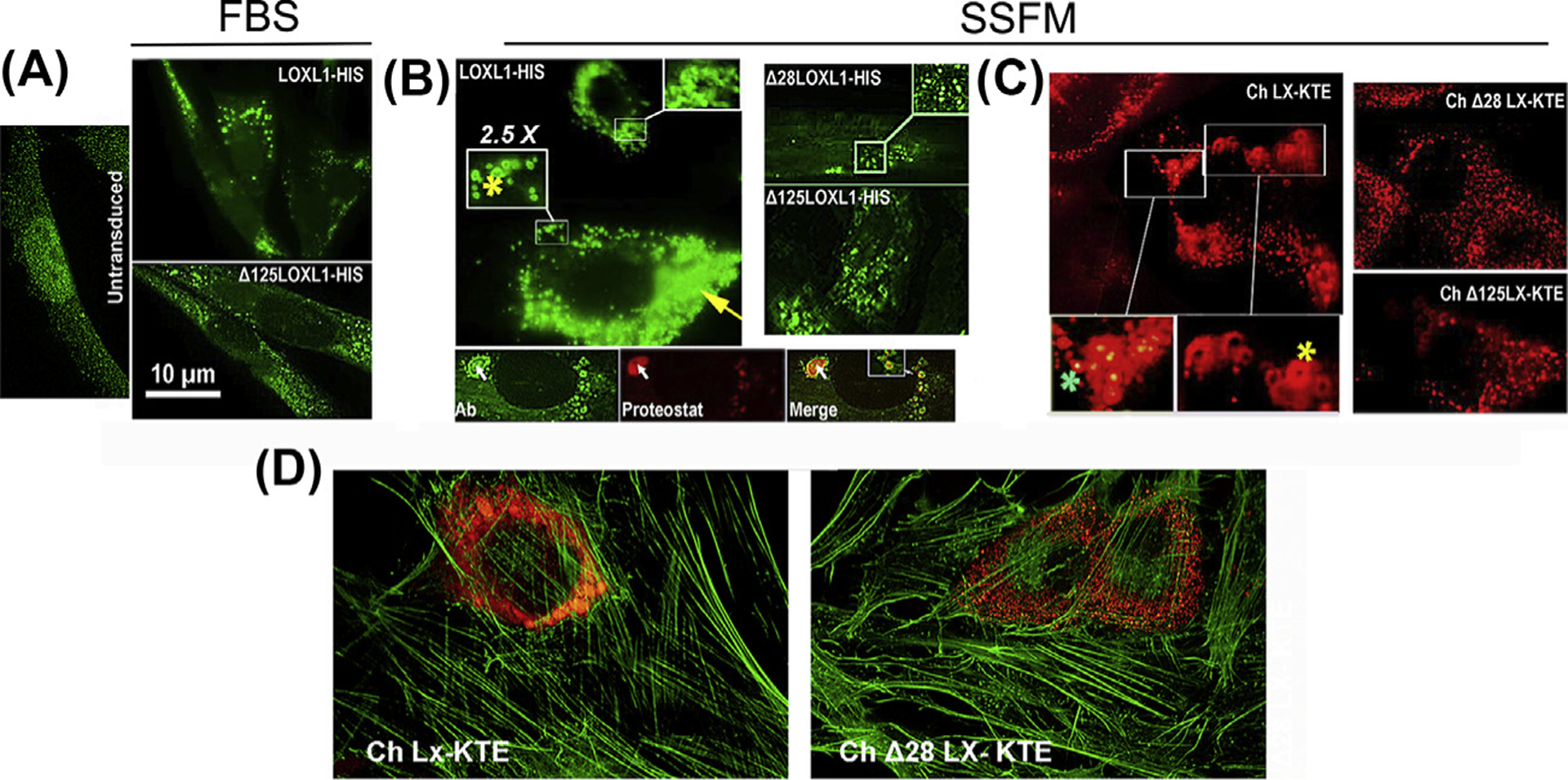
Representative images of BJ5a cells transduced with the indicated constructs. LOXL1 constructs were stained with an N-terminus anti-LOXL1 monoclonal Ab (H11 clone). Chimeras are red fluorescent. (A) Cultures in FBS-containing media. Left. Untransduced cells. Top. Cells transduced with LOXL1-HIS. Enlarged vesicles accumulated near the nuclei are the most common feature of cells expressing high levels of the transgene. Bottom. Typical stain profile for the Δ125–185 LOXL1 version. Vesicles are smaller and distribute throughout the cytosol. (B) Left top. The same transduction with LOXL1-HIS cDNA in supplemented-serum free media (SSFM). In addition to engorged vesicles (yellow arrow), very large particles and ring-shaped profiles (yellow asterisks) are frequent. Bottom 3-frame composite. Anti-LOXL1- Proteostat dual stain. Whereas the LOXL1 Ab stains the periphery of particles generating ring-shaped fluorescence, the aggregated-protein dye (Proteostat) labels the center of the same particles (white arrow). Right. LOXL1 with deleted domains lack this grossly vesicular phenotype. (C) The fluorescence patterns of the mKATE2 chimeras. Left. Ch LX-KTE fluorescence. Large vesicles and ring-like profiles (yellow asterisk) and engorged vesicles. There are also vesicles where the fluorescence intensity is higher at the center, rather than absent or lower (green asterisk). Right. The fluorescence of Δ versions. There are no aggregation profiles. (D) Micrographs of cells transduced with whole sequence (1–324) mKATE2 chimera and a chimera with a 28–95 aa deletion (Δ28) and counterstained with FITC-phalloidin.
7. Summary
The aggregation of the LOXL1 protein coupled with age-related decline in cellular proteostasis capabilities may result in a build-up of LOXL1-containing protein aggregates. In silico and experimental work demonstrate LOXL1’s propensity to aggregate and provide a road map to identify the loci that determine or facilitate the initiation of aggregation. This latter knowledge could be instrumental in the design of strategies to neutralize aggregation without the need to fully understand the process of fibrous growth.
Acknowledgments
This work was supported by The MYS Family US Charitable Foundation, Inc., Research to Prevent Blindness, The Bright Focus Foundation, The Glaucoma Foundation, and a donation from Barbie and Tony Mayer (AMB and JMW), NIH-NEI R01 EY024942 and Lions District 20-Y (AMB) and NIH-NEI R01 EY018748 (JMW).
References
- Aboobakar IF, Johnson WM, Stamer WD, Hauser MA, & Allingham RR (2016). Major review: Exfoliation syndrome; advances in disease genetics, molecular biology, and epidemiology. Experimental Eye Research, 154, 88–103. 10.1016/j.exer.2016.11.011. [DOI] [PubMed] [Google Scholar]
- Abu-Amero KK, Osman EA, Dewedar AS, Schmidt S, Allingham RR, & Al-Obeidan SA (2010). Analysis of LOXL1 polymorphisms in a Saudi Arabian population with pseudoexfoliation glaucoma. Molecular Vision, 16, 2805–2810. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/21197115. [PMC free article] [PubMed] [Google Scholar]
- Anastasopoulos E, Founti P, & Topouzis F (2015). Update on pseudoexfoliation syndrome pathogenesis and associations with intraocular pressure, glaucoma and systemic diseases. Current Opinion in Ophthalmology, 26(2), 82–89. 10.1097/ICU.0000000000000132. [DOI] [PubMed] [Google Scholar]
- Atkins JD, Boateng SY, Sorensen T, & McGuffin LJ (2015). Disorder prediction methods, their applicability to different protein targets and their usefulness for guiding experimental studies. International Journal of Molecular Sciences, 16(8), 19040–19054. 10.3390/ijms160819040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein AM, Ritch R, & Wolosin JM (2018). Exfoliation syndrome: A disease of autophagy and LOXL1 proteopathy. Journal of Glaucoma. 10.1097/IJG.0000000000000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialasiewicz AA, Wali U, Shenoy R, & Al-Saeidi R (2005). Patients with secondary open-angle glaucoma in pseudoexfoliation (PEX) syndrome among a population with high prevalence of PEX. Clinical findings and morphological and surgical characteristics. Ophthalmologe, Der, 102(11), 1064–1068. 10.1007/s00347-005-1226-2. [DOI] [PubMed] [Google Scholar]
- Chen H, Chen LJ, Zhang M, Gong W, Tam PO, Lam DS, et al. (2010). Ethnicity-based subgroup meta-analysis of the association of LOXL1 polymorphisms with glaucoma. Molecular Vision, 16, 167–177. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/20142848. [PMC free article] [PubMed] [Google Scholar]
- Clark JA, Yeaman EJ, Blizzard CA, Chuckowree JA, & Dickson TC (2016). A case for microtubule vulnerability in amyotrophic lateral sclerosis: Altered dynamics during disease. Frontiers in Cellular Neuroscience, 10, 204 10.3389/fncel.2016.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz E, Kumar S, Yuan L, Arikkath J, & Batra SK (2018). Intracellular amyloid beta expression leads to dysregulation of the mitogen-activated protein kinase and bone morphogenetic protein-2 signaling axis. PLoS One, 13(2), e0191696 10.1371/journal.pone.0191696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simone A, Kitchen C, Kwan AH, Sunde M, Dobson CM, & Frenkel D (2012). Intrinsic disorder modulates protein self-assembly and aggregation. Proceedings of the National Academy of Sciences of the United States of America, 109(18), 6951–6956. 10.1073/pnas.1118048109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey J, Ratnakaran N, & Koushika SP (2015). Neurodegeneration and microtubule dynamics: Death by a thousand cuts. Frontiers in Cellular Neuroscience, 9, 343 10.3389/fncel.2015.00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan BJ, Pasquale L, Grosskreutz CL, Rhee D, Chen T, DeAngelis MM, … Wiggs JL (2008). DNA sequence variants in the LOXL1 gene are associated with pseudoexfoliation glaucoma in a U.S. clinic-based population with broad ethnic diversity. BMC Medical Genetics, 9, 5 10.1186/1471-2350-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsius H (1988). Exfoliation syndrome in various ethnic populations. Acta Ophthalmologica, (Suppl. 184), 71–85. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/2853925. [DOI] [PubMed] [Google Scholar]
- Frost B, & Diamond MI (2010). Prion-like mechanisms in neurodegenerative diseases. Nature Reviews Neuroscience, 11(3), 155–159. 10.1038/nrn2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser MA, Aboobakar IF, Liu Y, Miura S, Whigham BT, Challa P, … Allingham RR (2015). Genetic variants and cellular stressors associated with exfoliation syndrome modulate promoter activity of a lncRNA within the LOXL1 locus. Human Molecular Genetics, 24(22), 6552–6563. 10.1093/hmg/ddv347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H, Gotoh N, Ueda Y, Nakanishi H, & Yoshimura N (2008). Lysyl oxidase-like 1 polymorphisms and exfoliation syndrome in the Japanese population. American Journal of Ophthalmology, 145(3), 582–585. 10.1016/j.ajo.2007.10.023. [DOI] [PubMed] [Google Scholar]
- Henry JC, Krupin T, Schmitt M, Lauffer J, Miller E, Ewing MQ, et al. (1987). Long-term follow-up of pseudoexfoliation and the development of elevated intraocular pressure. Ophthalmology, 94(5), 545–552. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/3601370. [DOI] [PubMed] [Google Scholar]
- Hewitt AW, Sharma S, Burdon KP, Wang JJ, Baird PN, Dimasi DP, … Craig JE (2008). Ancestral LOXL1 variants are associated with pseudoexfoliation in Caucasian Australians but with markedly lower penetrance than in Nordic people. Human Molecular Genetics, 17(5), 710–716. 10.1093/hmg/ddm342. [DOI] [PubMed] [Google Scholar]
- Iadanza MG, Jackson MP, Hewitt EW, Ranson NA, & Radford SE (2018). A new era for understanding amyloid structures and disease. Nature Reviews Molecular Cell Biology. 10.1038/s41580-018-0060-8. [DOI] [PubMed] [Google Scholar]
- Jeong S (2017). Molecular and cellular basis of neurodegeneration in Alzheimer’s disease. Molecules and Cells, 40(9), 613–620. 10.14348/molcells.2017.0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Juan-Marcos L, Escudero-Dominguez FA, Hernandez-Galilea E, Cabrillo-Estevez L, Cruz-Gonzalez F, Cieza-Borrella C, … Gonzalez-Sarmiento R (2016). Association of lysyl oxidase-like 1 gene polymorphisms in pseudoexfoliation syndrome and pseudoexfoliation glaucoma in a Spanish population. Ophthalmic Genetics, 37(1), 25–30. 10.3109/13816810.2014.921316. [DOI] [PubMed] [Google Scholar]
- Kastner C, Lobler M, Sternberg K, Reske T, Stachs O, Guthoff R, et al. (2013). Permeability of the anterior lens capsule for large molecules and small drugs. Current Eye Research, 38(10), 1057–1063. 10.3109/02713683.2013.803288. [DOI] [PubMed] [Google Scholar]
- Konstas AGP, & Ringvold A (2018). Epidemiology of exfoliation syndrome. Journal of Glaucoma, 27(Suppl. 1), S4–S11. 10.1097/IJG.0000000000000908. [DOI] [PubMed] [Google Scholar]
- Kottler UB, Junemann AG, Aigner T, Zenkel M, Rummelt C, & Schlotzer-Schrehardt U (2005). Comparative effects of TGF-beta 1 and TGF-beta 2 on extracellular matrix production, proliferation, migration, and collagen contraction of human Tenon’s capsule fibroblasts in pseudoexfoliation and primary open-angle glaucoma. Experimental Eye Research, 80(1), 121–134. 10.1016/j.exer.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Krause U, Alanko HI, Karna J, Miettinen R, Larmi T, Jaanio E, … Takala J (1988). Prevalence of exfoliation syndrome in Finland. Acta Ophthalmologica, (Suppl. 184), 120–122. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/2853908. [DOI] [PubMed] [Google Scholar]
- Kumar H, & Udgaonkar JB (2019). Mechanistic approaches to understand the prion-like propagation of aggregates of the human tau protein. Biochimica et Biophysica Acta - Proteins and Proteomics. 10.1016/j.bbapap.2019.04.004. [DOI] [PubMed] [Google Scholar]
- Lee CJ, Vroom JA, Fishman HA, & Bent SF (2006). Determination of human lens capsule permeability and its feasibility as a replacement for Bruch’s membrane. Biomaterials, 27(8), 1670–1678. 10.1016/j.biomaterials.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Malukiewicz G, Lesiewska-Junk H, Linkowska K, Mielnik M, Grzybowski T, & Sulima N (2011). Analysis of LOXL1 single nucleotide polymorphisms in Polish population with pseudoexfoliation syndrome. Acta Ophthalmologica, 89(1), e64–66. 10.1111/j.1755-3768.2010.02083.x. [DOI] [PubMed] [Google Scholar]
- Martinelli AHS, Lopes FC, John EBO, Carlini CR, & Ligabue-Braun R (2019). Modulation of disordered proteins with a Focus on neurodegenerative diseases and other pathologies. International Journal of Molecular Sciences, 20(6). 10.3390/ijms20061322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarali S, Damji F, & Damji KF (2018). What have we learned about exfoliation syndrome since its discovery by John Lindberg 100 years ago? British Journal of Ophthalmology, 102(10), 1342–1350. 10.1136/bjophthalmol-2017-311321. [DOI] [PubMed] [Google Scholar]
- Olzscha H, Schermann SM, Woerner AC, Pinkert S, Hecht MH, Tartaglia GG, … Vabulas RM (2011). Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell, 144(1), 67–78. 10.1016/j.cell.2010.11.050. [DOI] [PubMed] [Google Scholar]
- Padhy B, Kapuganti RS, Hayat B, Mohanty PP, & Alone DP (2019). De novo variants in an extracellular matrix protein coding gene, fibulin-5 (FBLN5) are associated with pseudoexfoliation. European Journal of Human Genetics. 10.1038/s41431-019-0482-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquale LR, Jiwani AZ, Zehavi-Dorin T, Majd A, Rhee DJ, Chen T, … Levkovitch-Verbin H (2014). Solar exposure and residential geographic history in relation to exfoliation syndrome in the United States and Israel. JAMA Ophthalmology, 132(12), 1439–1445. 10.1001/jamaophthalmol.2014.3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince AM, & Ritch R (1986). Clinical signs of the pseudoexfoliation syndrome. Ophthalmology, 93(6), 803–807. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/3737125. [DOI] [PubMed] [Google Scholar]
- Rautenbach RM, Bardien S, Harvey J, & Ziskind A (2011). An investigation into LOXL1 variants in black South African individuals with exfoliation syndrome. Archives of Ophthalmology, 129(2), 206–210. 10.1001/archophthalmol.2010.349. [DOI] [PubMed] [Google Scholar]
- Ringvold A (1996). Epidemiology of glaucoma in northern Europe. European Journal of Ophthalmology, 6(1), 26–29. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/8744847. [DOI] [PubMed] [Google Scholar]
- Ritch R, & Schlotzer-Schrehardt U (2001). Exfoliation syndrome. Survey of Ophthalmology, 45(4), 265–315. pii:S0039–6257(00)00196-X. [DOI] [PubMed] [Google Scholar]
- Ritch R, Schlotzer-Schrehardt U, & Konstas AG (2003). Why is glaucoma associated with exfoliation syndrome? Progress in Retinal and Eye Research, 22(3), 253–275. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/12852486. [DOI] [PubMed] [Google Scholar]
- Ronci M, Sharma S, Martin S, Craig JE, & Voelcker NH (2013). MALDI MS imaging analysis of apolipoprotein E and lysyl oxidase-like 1 in human lens capsules affected by pseudoexfoliation syndrome. Journal of Proteomics, 82, 27–34. 10.1016/j.jprot.2013.01.008. [DOI] [PubMed] [Google Scholar]
- Schlotzer-Schrehardt U (2009). Molecular pathology of pseudoexfoliation syndrome/glaucoma–new insights from LOXL1 gene associations. Experimental Eye Research, 88(4), 776–785. 10.1016/j.exer.2008.08.012. [DOI] [PubMed] [Google Scholar]
- Sharma S, Chataway T, Burdon KP, Jonavicius L, Klebe S, Hewitt AW, … Craig JE (2009). Identification of LOXL1 protein and Apolipoprotein E as components of surgically isolated pseudoexfoliation material by direct mass spectrometry. Experimental Eye Research, 89(4), 479–485. 10.1016/j.exer.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Sharma S, Chataway T, Klebe S, Griggs K, Martin S, Chegeni N, … Craig JE (2018). Novel protein constituents of pathological ocular pseudoexfoliation syndrome deposits identified with mass spectrometry. Molecular Vision, 24, 801–817. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/30713420. [PMC free article] [PubMed] [Google Scholar]
- Tam JH, & Pasternak SH (2012). Amyloid and Alzheimer’s disease: Inside and out. The Canadian Journal of Neurological Sciences, 39(3), 286–298. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/22547507. [DOI] [PubMed] [Google Scholar]
- Thorleifsson G, Magnusson KP, Sulem P, Walters GB, Gudbjartsson DF, Stefansson H, … Stefansson K (2007). Common sequence variants in the LOXL1 gene confer susceptibility to exfoliation glaucoma. Science, 317(5843), 1397–1400. 10.1126/science.1146554. [DOI] [PubMed] [Google Scholar]
- Uversky VN (2016). Paradoxes and wonders of intrinsic disorder: Complexity of simplicity. Intrinsically Disordered Proteins, 4(1), e1135015 10.1080/21690707.2015.1135015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquer-Alicea J, & Diamond MI (2019). Propagation of protein aggregation in neurodegenerative diseases. Annual Review of Biochemistry, 88, 785–810. 10.1146/annurev-biochem-061516-045049. [DOI] [PubMed] [Google Scholar]
- Want A, Gillespie SR, Wang Z, Gordon R, Iomini C, Ritch R, … Bernstein AM (2016). Autophagy and mitochondrial dysfunction in tenon fibroblasts from exfoliation glaucoma patients. PLoS One, 11(7), e0157404 10.1371/journal.pone.0157404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SE, Whigham BT, Liu Y, Carmichael TR, Qin X, Schmidt S, … Allingham RR (2010). Major LOXL1 risk allele is reversed in exfoliation glaucoma in a black South African population. Molecular Vision, 16, 705–712. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/20431720. [PMC free article] [PubMed] [Google Scholar]
- Wolosin JM, Ritch R, & Bernstein AM (2018). Is autophagy dysfunction a key to exfoliation glaucoma? Journal of Glaucoma, 27(3), 197–201. 10.1097/IJG.0000000000000606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenkel M, Krysta A, Pasutto F, Juenemann A, Kruse FE, & Schlotzer-Schrehardt U (2011). Regulation of lysyl oxidase-like 1 (LOXL1) and elastin-related genes by pathogenic factors associated with pseudoexfoliation syndrome. Investigative Ophthalmology & Visual Science, 52(11), 8488–8495. 10.1167/iovs.11-8361. [DOI] [PubMed] [Google Scholar]