

Abstract
Patients with systemic lupus erythematosus (SLE) display increased numbers of immature neutrophils in the blood, but the exact role of these immature neutrophils is unclear. Neutrophils that sediment within the peripheral blood mononuclear cell fraction after density centrifugation of blood are generally defined as low‐density neutrophils (LDNs). Far beyond antimicrobial functions, LDNs are emerging as decision‐shapers during innate and adaptive immune responses. Traditionally, neutrophils have been viewed as a homogeneous population. However, the various LDN populations identified in SLE to date are heterogeneously composed of mixed populations of activated mature neutrophils and immature neutrophils at various stages of differentiation. Controversy also surrounds the role of LDNs in SLE in terms of whether they are proinflammatory or polymorphonuclear myeloid‐derived suppressor cells. It is clear that LDNs in SLE can secrete increased levels of type I interferon (IFN) and that they contribute to the cycle of inflammation and tissue damage. They readily form neutrophil extracellular traps, exposing modified autoantigens and oxidized mitochondrial DNA, which contribute to autoantibody production and type I IFN signaling, respectively. Importantly, the ability of LDNs in SLE to perform canonical neutrophil functions is polarized, based on mature CD10+ and immature CD10− neutrophils. Although this field is still relatively new, multiomic approaches have advanced our understanding of the diverse origins, phenotype, and function of LDNs in SLE. This review updates the literature on the origin and nature of LDNs, their distinctive features, and their biologic roles in the immunopathogenesis and end‐organ damage in SLE.
Traditional concepts of SLE immunopathogenesis
Systemic lupus erythematosus (SLE) is an autoimmune disease of unknown etiology. Its incidence has been constant in recent decades, with ~4–5 per 100,000 people affected each year and a female:male bias peaking at 9:1 in adulthood 1, 2. However, the severity and mortality are higher in childhood‐onset and male SLE 3. It is one of the most common systemic autoimmune diseases, with a prevalence of 40–100 per 100,000 people in the US 4, 5. Common hypotheses about SLE immunopathogenesis suggest that environmental triggers, such as infectious agents, operate in the context of genetic and epigenetic influences, resulting in aberrations in antigen presentation, lymphoid signaling, apoptosis, epitope modification, and antigen and immune complex (IC) clearance 6, 7. Ordinarily, SLE is a polygenic disease, with >50 lupus susceptibility loci now identified 8, 9. Risk and severity are also associated with an increasing number and additive effect of susceptibility alleles 10. Given that SLE is an immunologic disease, it is not surprising that the top genetic associations with disease are those associated with immunity, including the HLA genes IRF5, IRF7, IRAK1, TNFAIP3, TNIP1, IFIH1, TYK2, and C1Q 11.
Traditionally, SLE has been considered a disease of perturbed adaptive immunity, due to the critical pathogenic roles of B cells and T cells 12, 13. Characteristic antinuclear antibodies (ANAs), such as anti–double‐stranded DNA, anti‐RNA, and anti‐RNA–associated proteins, are produced by autoreactive B cells through both extrafollicular pathways and germinal center reactions 14, 15. These ANAs form ICs with nuclear material released following excessive or inappropriate cell death processes. The subsequent IC deposition in tissues results in a proinflammatory loop, which involves the recruitment of multiple immune cells with cytokine release and the destruction of multiple organs 16, 17. Type I interferons (IFNs) comprise a family of cytokines that have been associated with SLE. They are divided into 5 classes (IFNα, IFNβ, IFNε, IFNκ, and IFNω), with IFNα being further categorized into 12 subtypes 18. IFNα was first described in the peripheral blood of SLE patients by Preble et al in 1982 19. Since then, an “IFN signature” has been identified in the peripheral blood mononuclear cells (PBMCs) of SLE patients 20, 21.
This up‐regulation of IFN‐regulated genes occurs in a subset of adult SLE patients and may reflect differences in genetics, environmental triggers, or disease severity 21. It is also now clear that although IFNα subtypes are important in SLE, additional type I IFNs, such as IFNβ and IFNκ, play key roles in disease 18, 22, 23. This interest in type I IFNs initiated a plethora of clinical trials with therapeutic targets directed at IFNα and type I IFN receptor, which have more recently been redirected toward those expressing the IFN signature 24. Neutrophils have generated interest for their role in the production of type I IFNs and for their proinflammatory and antiinflammatory functions in SLE 21, 25, 26, 27. Moreover, there is a burgeoning number of reports showing that they play an integral role in disease pathogenesis.
Neutrophil fundamentals
To understand dysfunction of any cell type, it is necessary to understand the underlying biology. Neutrophils are the most abundant immune cell type in the human peripheral blood, acting as the first responders during sterile and microbial insults 28. Chemotaxis and extravasation toward the potential pathogen are mediated through the activation of a number of surface receptors, including selectins and integrins, chemokine receptors, Fcγ receptors (FcγRs), and fMLP receptors. Neutrophils target microorganisms through the generation of reactive oxygen species (ROS) via respiratory burst, the release of bactericidal enzymes through degranulation, and phagocytosis, a process that engulfs the potentially pathogenic organism 26, 29, 30. More recently, NETosis has been described as a mechanism to trap potential pathogens within a network of expelled cell contents termed neutrophil extracellular traps (NETs) 29. Neutrophils also play a key role in eliminating opsonized bacteria and in antibody‐dependent cell‐mediated cytotoxicity 31, 32. They constitutively express a low‐affinity IgG receptor, FcγRIIa (CD32A) and the glycosyl phosphatidylinositol–linked FcγRIIIb (CD16B), which serves as a coreceptor 33, 34. Upon the crosslinking of FcγR in vitro, neutrophils activate, degranulate, produce ROS, and can trigger NETosis 33. In addition, neutrophils interact with the complement pathway to ensure clearance of invading pathogens, primarily through C5a 35.
The in vivo half‐life of human circulating neutrophils is 19–90 hours. This estimate is derived from the ratio of the number of neutrophils in the blood to the number of mitotic neutrophil precursors in the bone marrow 36, 37. Consistent with this relatively short lifespan, 50% of the bone marrow is devoted to neutrophil production, releasing ~5–10 × 1010 cells per day, which increases during an infection 38, 39. This lifespan increases following exposure to cytokines or other proinflammatory agents present in infected or inflamed tissue 40, 41.
Neutrophil heterogeneity is an important feature of immune pathophysiology, and therefore strategies for assessing neutrophil subsets based on their maturation have been proposed 28, 30. Maturation from committed proliferative neutrophil precursors (pre‐neutrophils) into nonproliferative immature neutrophils and then mature neutrophils can be detected, using changes in cell surface molecules 28, 30.
Low‐density neutrophils in SLE
In 1986, Hacbarth and Kajdacsy‐Balla were the first to describe the presence of “low buoyant density neutrophils” in the peripheral blood mononuclear cell (PBMC) preparations obtained from adult SLE patients 42 (Figure 1). This preparation remains common and involves the centrifugation of whole blood, mixed with media, layered over a polysaccharide or silica gradient with a density of ~1.077 gm/ml (e.g., Ficoll‐Paque, Histopaque‐1077, Percoll) for humans 43. Due to their relative high density, neutrophils end up below the Ficoll layer, on top of the erythrocyte fraction (often termed high‐density neutrophils [HDNs]). The PBMC fraction is found in the interphase between the Ficoll layer and the plasma 43. In this original study, the authors hypothesized that humoral factors in patient plasma induced activation of the neutrophils in vitro, causing degranulation and decreasing their buoyant density, leaving them to settle with the PBMCs as low‐density neutrophils (LDNs) 42. Supporting this idea, stimulation of whole blood with fMLP was shown to lead to an increase in the percentage of LDNs at the PBMC fraction in a dose‐dependent manner, representing up to 12.5% of the population 44, 45. Furthermore, the activation of neutrophils led to increased expression of CD66b, which is mobilized from intracellular granules to the cell membrane during degranulation, possibly contributing to the LDN increase 45.
Figure 1.
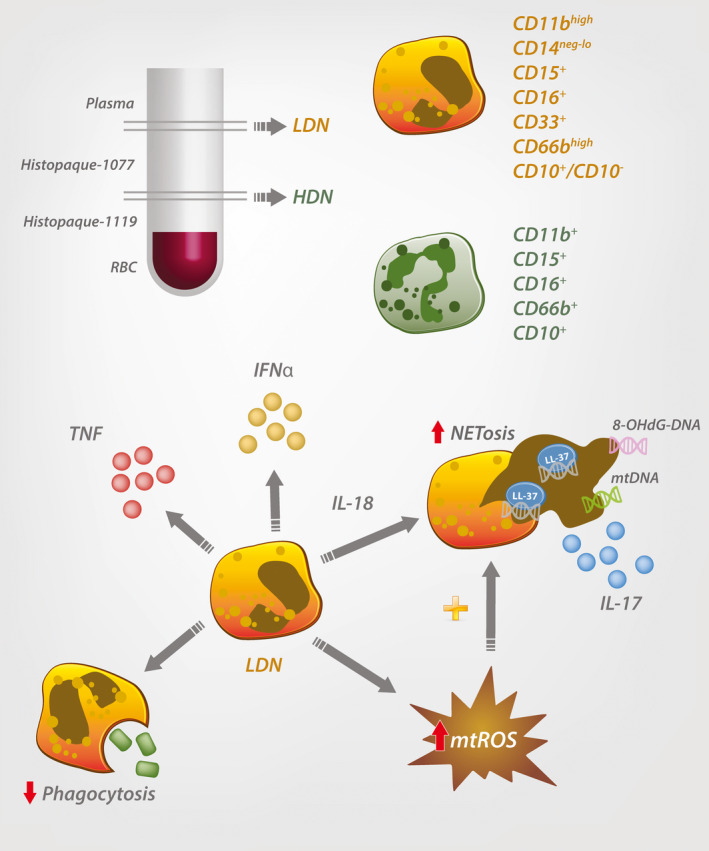
Phenotypic and biologic properties of low‐density neutrophils (LDNs) in systemic lupus erythematosus (SLE). SLE LDNs are found within the peripheral blood mononuclear cell fraction. CD11b, CD16, and CD66b are common markers to identify mature high‐density neutrophils (HDNs). However, LDNs comprise a heterogeneous population of CD10− pre‐neutrophils, immature neutrophils, and CD10+ mature neutrophils. SLE LDNs secrete increased levels of proinflammatory cytokines, have impaired phagocytosis, and enhanced neutrophil extracellular trap (NET) formation via mitochondrial reactive oxygen species (mtROS) production, leading to elaboration of oxidized mitochondrial DNA (mtDNA) and interleukin‐17 (IL‐17). RBC = red blood cell; TNF = tumor necrosis factor; IFNα = interferon‐α; 8‐OHdG = 8‐hydroxydeoxyguanosine.
In 2003, Bennett et al performed microarray analysis of PBMCs from pediatric SLE patients and identified high expression of neutrophil‐specific genes 20. This “granulocyte signature” was due to an increase of LDNs in the PBMC layer 20. This signature was not caused by steroid treatment as it was demonstrated in several newly diagnosed, untreated pediatric SLE patients 20. Moreover, intravenous pulse high‐dose glucocorticoid treatment extinguished the IFN signature without affecting the granulocyte signature in pediatric SLE PBMCs 20.
Among low‐density granulocytes, which encompass neutrophils, eosinophils, and basophils 26, 43, only LDNs have been described in SLE. The frequency of LDNs found contaminating the PBMC fraction is significantly higher in SLE patients, at ~17% of total PBMCs, compared to ~5% in healthy donor PBMCs 26, 46, 47 (Table 1). Neutrophils with low‐density features have also been described in other rheumatic diseases such as antineutrophil cytoplasmic antibody–associated vasculitis, myositis, primary antiphospholipid syndrome, and psoriasis 48, 49, 50, 51, 52, 53. In summary, LDNs form a contaminating granulocytic population in the PBMC layer of fractionated blood, which is increased in SLE.
Table 1.
Biologic and clinical characteristics of LDNs in SLEa
| Prevalence, % | HD 0.81–5.00; SLE 2.37–17.00 |
| Morphology | Promyelocytes, myelocytes, metamyelocytes, bands, and segmented neutrophils |
| Immunophenotype | All LDNs CD3−CD19−CD20−CD56− and CD11bhighCD14−/lowCD15+CD16+CD33+CD66bhigh; mature LDNs CD10+; immature LDNs CD10− |
| Functional properties | Increased TNF, IFNα, IL‐17+ NETs; decreased phagocytosis; decreased chemotactic activity; increased mtROS production; increased spontaneous NETosis with NET‐mtDNA release |
| Homeostatic/pathologic relevance | CD10+ LDNs associated with noncalcified plaque burden severity, vascular inflammation, and lower HDL cholesterol efflux capacity in SLE patients, and negatively correlated with renal function in white SLE patients; CD10− LDNs positively correlated with proteinuria in white SLE patients |
LDNs = low‐density neutrophils; SLE = systemic lupus erythematosus; HD = healthy donors; TNF = tumor necrosis factor; IFNα = interferon‐α; IL‐17 = interleukin‐17; NETs = neutrophil extracellular traps; mtROS = mitochondrial reactive oxygen species; mtDNA = mitochondrial DNA; HDL = high‐density lipoprotein.
SLE LDN surface markers, nuclear morphology, and activation markers
LDNs can be distinguished from monocytes in the PBMC layer by their high granularity and expression of surface molecules using flow cytometry 20, 26, 47 (Table 1). LDNs also lack major histocompatibility complex class II and the costimulatory molecule CD86 (B7.2), in contrast to monocytes, and express CD15, CD16b (FcγIIIb), CD33, and CD11b (Table 1). Comparatively, SLE LDNs express higher levels of the activation markers CD11b, CD66b, CD63, and CD107a, relative to autologous HDNs 26, 47. CD11c, CD31, granulocyte colony‐stimulating factor (G‐CSF) receptor, and granulocyte–macrophage colony‐stimulating factor receptor have also been found on their surface, in contrast to HDNs 54, 55. However, they do not differ with regard to surface L‐selectin (CD62L), which is a sensitive measure of activation 26.
More recently, 2 subpopulations of SLE LDNs which comprise immature CD10− and mature CD10+ neutrophils have been identified 46, 56 (Table 1). Similarly, morphologic analysis of SLE LDNs shows all stages of neutrophil development, including promyelocytes, myelocytes, metamyelocytes, bands, and segmented neutrophils 20, 46, 54, 56 (Table 1). Moreover, up to 60% of human SLE LDNs are mature, in contrast to HDNs, which consist of >90% mature cells 26, 54.
Genetics, epigenetics, and gene expression of SLE LDNs
Analyses of genomic instability have shown increased copy number alterations and microsatellite instability in SLE LDNs compared to matched HDNs from the same individuals 8. These somatic alterations are consistent with the notion of DNA strand break repair and a replication error–prone status, supporting a model whereby genomic damage contributes to the development of an abnormal population of neutrophils 8. Consistent with these findings, chronically elevated ROS, which often occurs in SLE, affects several aspects of DNA damage response and may lead to accumulation of such genomic changes 57, 58. An assessment of epigenetic accessibility by Coit et al also revealed a robust and consistent demethylation of IFN signature genes in SLE LDNs and HDNs compared to healthy donor HDNs 59.
A more stratified transposase‐accessible chromatin sequencing study by Mistry et al has shown that immature SLE LDNs have more open peaks compared to mature SLE LDNs, reflecting enhanced chromatin accessibility and suggesting increased gene activity 56. Similarly, an analysis of the transcriptome of SLE LDNs using bulk RNA sequencing showed increases in cell cycle progression genes in the CD10− population, confirming their immaturity 56. SLE LDNs also express higher messenger RNA (mRNA) levels for >200 genes relative to autologous or healthy donor HDNs, including serine proteases, bactericidal proteins, and other molecules involved in neutrophil regulation of inflammatory responses 60. Interestingly, type I IFN signaling transcripts, which include MX1, IFIT3, IFI44, and RSAD2, are increased only in mature CD10+ SLE LDNs and not in the immature CD10− population 54, 56. However, this may simply reflect the exposition time to the inflammatory milieu. Taken together, these data suggest that LDNs may be a mixed cell population consisting of mature cells, together with immature and activated/regulatory populations.
Cytokine profiles of SLE LDNs
Resting healthy donor HDNs, SLE HDNs, and LDNs show similar levels of cytokine release 26. However, phorbol myristate acetate–stimulated SLE LDNs secrete more tumor necrosis factor (TNF) and have higher levels of IFNα mRNA compared to autologous SLE and healthy donor HDNs 26 (Table 1). Furthermore, cultured supernatants from resting or stimulated SLE LDNs induce type I IFN–inducible genes, in contrast to HDNs from SLE patients or healthy donors, suggesting that they release Toll‐like receptor (TLR) ligands and/or IFNα itself 26. Although plasmacytoid dendritic cells provide the highest levels of IFNα upon stimulation, all other nucleated cells have the capacity to produce IFNα 25, 61. SLE LDN supernatants also induce IFNγ, TNF, and lymphotoxin α (LTα) from T cells, suggesting a proinflammatory phenotype of LDNs 47. However, more research is required to fully comprehend the capacity of LDNs in cytokine production.
SLE LDNs show a propensity for NETosis rather than phagocytosis
LDNs from SLE patients have a lower capacity for phagocytosis compared to autologous HDNs from SLE patients or healthy donors 26 (Table 1). This may be due to the relatively high proportion of immature cells, since CD10− SLE LDNs are less effective at phagocytosis compared to mature CD10+ SLE LDNs 56. Findings from a study by Lood et al suggest that this reduction may be due to an increase in the cleavage of FcγRIIa following activation by TLR‐7/8 ligands 62. In contrast, SLE LDNs show an increase in spontaneous NETosis and the release of self‐reactive material compared to autologous and healthy donor HDNs 27, 60, 63. Enhanced NETosis also results in comparatively higher secretions of interleukin‐17 (IL‐17), an important cytokine associated with T cell activation, particularly in autoimmunity 60, 64 (Table 1). The NETosis in SLE LDNs is, in part, due to an increase in mitochondrial ROS (mtROS) release 27, 65 (Figure 1 and Table 1). Importantly, mtROS production is sufficient for the generation of NETs even in the absence of functional NADPH oxidase 27. The oxidized mitochondrial DNA (mtDNA) derived from the LDN NETs is proinflammatory, activating stimulator of IFN genes to induce type I IFN signaling in target cells 27. Interestingly, mitochondrial stress alone can induce the release of mtDNA and induce IFN signaling. Inhibition of this process decreases NET formation and mtDNA release, reducing lupus‐like disease in a model system 66.
Additional cytokines, ligands, and pathways associated with NETosis have also been described in the pathogenesis of SLE. Lipopolysaccharide was described early on in the literature as a major stimulant of NETosis. However, in the absence of a bacterial infection, the specific stimulus in SLE remains unclear. IL‐18 has been shown as an effective stimulus of NET release by SLE LDNs in active lupus nephritis 67, 68. Lood et al have also demonstrated that RNP‐containing ICs can induce mitochondria mobilization, mtROS production, and the release of oxidized mtDNA 27. Similarly, van Dam et al demonstrated that SLE ICs are able to induce nonlytic NETosis in healthy donor HDNs via FcγR signaling, with rapid extrusion of NETs enriched with oxidized mtDNA 69. Therefore, ICs, mitochondrial dysfunction, and cytokines may all contribute to increased NETosis in SLE LDNs.
SLE LDNs and myeloid‐derived suppressor cells are distinct cell types
In 2007, Gabrilovic et al suggested the term “myeloid‐derived suppressor cells” (MDSCs), informed by the myeloid origin, immunosuppressive function, and systemic expansion of these cells in a cancer‐related context 70. MDSCs consist of 2 large groups of cells: polymorphonuclear MDSCs (PMN‐MDSCs) and monocytic MDSCs (M‐MDSCs) 71. PMN‐MDSCs are phenotypically and morphologically similar to neutrophils, while M‐MDSCs are similar to monocytes 71. Traditionally, PMN‐MDSCs are enriched in the low‐density PBMC fraction using density centrifugation methods, in a similar manner to LDNs 71. PMN‐MDSCs and SLE LDNs also express similar surface molecules, including CD11b+CD14−CD15+CD66b+, leading to a hypothesis that they are the same type of cells 26, 46, 70 (Table 2). However, an evaluation of the literature on PMN‐MDSCs, primarily from cancer research, and SLE LDNs also reveals differences between the neutrophil groups (Table 2). The lectin‐like oxidized low‐density lipoprotein receptor 1 was identified to distinguish PMN‐MDSCs from other neutrophils, although it is expressed on only one‐third of PMN‐MDSCs in cancer patients 72. Furthermore, it is expressed on LDNs from healthy donors and SLE patients at similar levels 47, eliminating its role in categorization.
Table 2.
Phenotypic, biochemical, molecular, and functional properties of SLE LDNs and PMN‐MDSCsa
| SLE LDNs | PMN‐MDSCs | |
|---|---|---|
| Physical characteristic | ||
| Density | Low | Low |
| Morphology | Promyelocytes, myelocytes, metamyelocytes, bands and segmented neutrophils | Metamyelocytes, bands, and segmented neutrophils |
| Surface marker | ||
| FSC | NA | High |
| CD11b | +++ | ++ |
| CD14 | −/+ | − |
| CD15 | +++ | +++ |
| CD66b | +++ | +++ |
| LOX‐1 | +/++/+++ | + |
| Biomarker | ||
| ROS | + | +++ |
| ARG1 | ++ | ++ |
| PD‐L1 | + | + |
| Immunometabolic status | ||
| ER stress | ++ | ++ |
| Functional test | ||
| Inhibition of T cell proliferation | No | Yes |
| Inhibition of IFNγ production | No | Yes |
Positive and negative signs indicate the level of expression of relevant markers in SLE LDNs or polymorphonuclear myeloid‐derived suppressor cells (PMN‐MDSCs). NA = not applicable; LOX‐1 = lectin‐like oxidized low‐density lipoprotein receptor 1; ROS = reactive oxygen species; ARG1 = arginase 1; PD‐L1 = programmed death ligand 1; ER = endoplasmic reticulum (see Table 1 for other definitions).
The hallmark of PMN‐MDSCs is their ability to suppress T cell function 72, which is contrary to findings from LDN studies in SLE (Table 2). Cultured SLE LDNs, or their supernatants, fail to suppress proliferation of activated healthy donor naive CD4+ T cells 47. Moreover, the addition of SLE LDNs enhances the production of IFNγ, TNF, and LTα by stimulated CD4+ T cells, in contrast to HDNs 47. This may be due to the comparative heterogeneous nature of SLE LDNs discussed above. The addition of predominantly immature CD10− SLE LDNs enhances T cell proliferation in vitro; however, isolated CD10+ LDNs can promote immunosuppression via a CD18‐mediated contact‐dependent arginase 1 release 46. Similarly, studies on immature CD10− LDNs from donors receiving recombinant human G‐CSF show increased T cell proliferation and IFNγ production 46, suggesting that maturity is a key factor in suppressive function. In contrast to SLE LDNs, morphologic examinations of PMN‐MDSCs in cancer patients showed segmented cells, and therefore maturity, in 8 of 9 studies 43. It is currently unknown whether LDNs from pediatric SLE patients are suppressive or proinflammatory.
Programmed death ligand 1 (PD‐L1) is expressed in MDSCs, and this may contribute to the success of recent anti–PD‐L1 immunotherapies used in cancer 70 (Table 2). Inhibition of this suppressive receptor, particularly on T cells, results in increased activation of the immune system and elimination of tumor cells. On MDSCs, inhibition of PD‐L1 may reduce their suppressive action. Interestingly, PD‐L1 has also been detected in LDNs from SLE patients 47. Further research is necessary to determine whether activating PD‐L1 will be an effective novel therapy in SLE through its suppression of the immune system.
LDNs are associated with organ damage in SLE
LDNs and premature cardiovascular disease
Cardiovascular disease (CVD) accounts for more than one‐third of all deaths in SLE patients 73. This is due to a plethora of factors, including autoantibodies, systemic inflammation, and endothelial injury, in addition to traditional CVD risk factors 74. A longer disease duration, higher damage index, and less aggressive immunosuppression are associated with increased heart disease, suggesting that immune dysregulation contributes to plaque progression and vascular complications, which are essential factors associated with CVD 73.
Endothelial dysfunction is an early central phase in the evolution of atherosclerosis and has been described as the intermediate link between CVD risk factors and the development of atherosclerosis 75. SLE LDNs have been associated with endothelial damage and abnormal endothelial proliferation in vitro 54. Furthermore, the reduced ability of SLE endothelial progenitor cells to differentiate into mature endothelial cells is mediated in part by IFNα produced by CD10+ SLE LDNs 26. Mature CD10+ LDNs have also been associated with noncalcified plaque burden severity and lower high‐density lipoprotein (HDL) cholesterol efflux capacity in SLE patients 56, 76 (Table 1). HDL cholesterol efflux capacity is the ability of HDL to promote efflux of cholesterol from macrophages and has a strong inverse correlation with CVD 77. In support of these findings, SLE LDNs have higher levels of genes associated with the regulation of vascular inflammation and noncalcified plaque burden, including AZU1, MPO, CTSG, PRTN3, ELANE, and DEFA3, compared to healthy donors 76 (Table 1).
In vitro culture of SLE LDNs with human umbilical vein endothelial cells results in increased endothelial cytotoxicity compared to autologous or healthy donor HDNs 26, 60. This cytotoxicity is dependent on NET formation and the externalization and activation of matrix metalloproteinases 26, 60. In support of these findings, administration of a potent antioxidant and ROS inhibitor, idebenone, inhibits NET formation and improves endothelial function in the MRL/lpr murine model of SLE 65. Collectively, these data suggest that LDNs contribute to the excess cardiovascular risk in SLE and that targeting mitochondrial dysfunction or LDNs directly may be a potential treatment strategy.
LDNs, cutaneous lupus, and lupus nephritis
Netting neutrophils have been observed in biopsy samples from patients with cutaneous LE and lupus nephritis 60, 78, 79. The majority of SLE patients with elevated levels of LDNs display clinical skin manifestations, including vasculitis 26. Furthermore, skin biopsies from SLE patients with several forms of cutaneous LE revealed neutrophil infiltration and the presence of NET‐DNA 60. Similarly, lupus nephritis has also been associated with impaired NET degradation, and NET‐DNA has been observed in kidney biopsy samples from lupus nephritis patients 80. The presence of NET‐mtDNA may also suggest increased LDN activity specifically 60, 79. Given the importance of the findings of these few studies, further investigations into the functional role of neutrophils in SLE are required.
Therapeutic targeting of LDNs in SLE
LDNs represent a novel therapeutic target in SLE. Identification of the pathogenic LDN subset may facilitate development of drugs that target selective neutrophil populations while preserving critical aspects of neutrophil‐mediated host defense 81. Medications that target key events in NET formation in LDNs or promote NET clearance may provide novel therapeutic strategies in SLE. These include medications already in use for treatment of SLE, as well as novel agents under investigation (Table 3).
Table 3.
Potential therapies targeting LDNs or NET formation in SLEa
| Drug | Mechanism of action | Effect on neutrophils/NETs | Effect on disease/clinical use |
|---|---|---|---|
| Chloroquine/hydroxychloroquine | Unknown | Chloroquine inhibits NETosis in SLE LDNs in vitro | Antimalarials used as first‐line treatment in SLE |
| Colchicine | Possibly via inhibition of tubulin polymerization | Inhibits spontaneous NETosis in Behçet's syndrome HDNs in vitro | Used to treat SLE pericarditis |
| Cyclosporine | Modulates calcium‐dependent signal transduction by calcineurin inhibition | Inhibits ionomycin‐ and IL‐8–induced NETosis in healthy donor HDNs in vitro | Used to treat membranous lupus nephritis |
| DNase I | Enzymatic degradation of DNA | Enzymatic degradation of NET‐DNA | Well tolerated in phase I study in 17 patients with lupus nephritis |
| Eculizumab | Monoclonal antibody against complement C5 to inhibit the cleavage of C5 to C5a and C5b, possibly via reduction of C5a‐primed neutrophils for NETosis | ANCA‐induced NETosis in C5a‐primed healthy donor HDNs in vitro | Used to treat SLE patients with thrombotic microangiopathy |
| Idebenone | Antioxidant that protects cells against ROS toxicity, improves mitochondrial physiology | Inhibits spontaneous NETosis in SLE LDNs but not SLE HDNs in vitro | Reduced disease activity and organ damage in lupus mouse models |
| Metformin | Unknown | Decreases NET‐DNA and NET‐mtDNA from healthy donor HDNs in vitro | Open‐label study showed reduced SLE flares and steroid‐sparing effect |
| N‐acetylcysteine | Free radical scavenger | Inhibits NETosis and free radical formation from healthy donor HDNs in vitro | Well tolerated in phase I study in 36 SLE patients, with reduction in ADHD Self‐Report Scale scores |
| Tofacitinib | JAK1 and JAK3 inhibitor | Decreases NETosis in bone marrow HDNs obtained from MLR/lpr mice treated with tofacitinib | Well tolerated in a phase Ib/IIa study in 30 SLE patients, with reduction in circulating LDNs in tofacitinib‐treated group |
| Rituximab (followed by belimumab) | Rituximab: monoclonal antibody against CD20; belimumab: monoclonal antibody against BAFF | Decreases spontaneous NETosis in SLE HDNs ex vivo by reducing autoantibodies | Rituximab followed by belimumab was safe in a phase II study, with clinical responses in patients with severe refractory SLE |
| Vitamin D | Unknown | 1,25(OH)2D3 decreases NETosis in SLE HDNs in vitro | Meta‐analysis of RCTs showed that vitamin D supplementation in SLE is safe and may improve fatigue |
HDNs = high‐density neutrophils; ANCA = antineutrophil cytoplasmic antibody; ADHD = attention deficit hyperactivity disorder; RCTs = randomized controlled trials (see Table 1 for other definitions).
Conclusions
Neutrophils play a fundamental role in protection against invading pathogens, but overactivity is associated with prolonged inflammation and tissue destruction. The discovery that they fractionate with the PBMC layer in SLE revolutionized concepts pertaining to their role in disease. The neutrophil‐related gene signatures in PBMCs have been associated with common comorbidities of SLE, including cardiovascular disease and lupus nephritis 76, 82, 83, 84, suggesting their critical role in pathogenesis. The physical reasons as to why the LDNs fractionate in this layer remains a target of investigation. The parallel emergence of LDNs and PMN‐MDSCs has complicated the field, given their similar phenotypes and densities 85. However, their different functional roles in immune suppression and activation demonstrate that they are not the same.
So far, findings indicate that LDNs are a heterogeneous population, often with a large proportion of immature cells. There are no definitive markers to identify this population for in‐depth characterization. Therefore, isolating LDNs remains a multistep and lengthy process, which limits what can be done because of the rapid apoptosis occurring ex vivo. An emerging area of research will seek to understand the interplay of LDNs, NETosis, and the adaptive immune system, based on existing data showing that LDNs can regulate T cell responses 16. While much has yet to be characterized, targeting LDNs in SLE remains a promising area for novel therapeutic intervention.
AUTHOR CONTRIBUTIONS
All authors drafted the article, revised it critically for important intellectual content, and approved the final version to be published.
No potential conflicts of interest relevant to this article were reported.
References
- 1. Jonsson H, Nived O, Sturfelt G, Silman A. Estimating the incidence of systemic lupus erythematosus in a defined population using multiple sources of retrieval. Br J Rheumatol 1990;29:185–8. [DOI] [PubMed] [Google Scholar]
- 2. Margery‐Muir AA, Bundell C, Nelson D, Groth DM, Wetherall JD. Gender balance in patients with systemic lupus erythematosus [review]. Autoimmun Rev 2017;16:258–68. [DOI] [PubMed] [Google Scholar]
- 3. Hersh AO, Trupin L, Yazdany J, Panopalis P, Julian L, Katz P, et al. Childhood‐onset disease as a predictor of mortality in an adult cohort of patients with systemic lupus erythematosus. Arthritis Care Res (Hoboken) 2010;62:1152–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self‐DNA–peptide complexes in systemic lupus erythematosus. Sci Transl Med 2011;3:73ra19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hayter SM, Cook MC. Updated assessment of the prevalence, spectrum and case definition of autoimmune disease [review]. Autoimmun Rev 2012;11:754–65. [DOI] [PubMed] [Google Scholar]
- 6. Wakeland EK, Liu K, Graham RR, Behrens TW. Delineating the genetic basis of systemic lupus erythematosus [review]. Immunity 2001;15:397–408. [DOI] [PubMed] [Google Scholar]
- 7. Scharer CD, Blalock EL, Mi T, Barwick BG, Jenks SA, Deguchi T, et al. Epigenetic programming underpins B cell dysfunction in human SLE. Nat Immunol 2019;20:1071–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Singh N, Traisak P, Martin KA, Kaplan MJ, Cohen PL, Denny MF. Genomic alterations in abnormal neutrophils isolated from adult patients with systemic lupus erythematosus. Arthritis Res Ther 2014;16:R165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vaughn SE, Kottyan LC, Munroe ME, Harley JB. Genetic susceptibility to lupus: the biological basis of genetic risk found in B cell signaling pathways [review]. J Leukoc Biol 2012;92:577–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morel L, Rudofsky UH, Longmate JA, Schiffenbauer J, Wakeland EK. Polygenic control of susceptibility to murine systemic lupus erythematosus. Immunity 1994;1:219–29. [DOI] [PubMed] [Google Scholar]
- 11. Ghodke‐Puranik Y, Niewold TB. Genetics of the type I interferon pathway in systemic lupus erythematosus. Int J Clin Rheumatol 2013;8: 10.2217/ijr.13.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Crispin JC, Kyttaris VC, Terhorst C, Tsokos GC. T cells as therapeutic targets in SLE [review]. Nat Rev Rheumatol 2010;6:317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dorner T, Jacobi AM, Lee J, Lipsky PE. Abnormalities of B cell subsets in patients with systemic lupus erythematosus. J Immunol Methods 2011;363:187–97. [DOI] [PubMed] [Google Scholar]
- 14. Blanco F, Kalsi J, Isenberg DA. Analysis of antibodies to RNA in patients with systemic lupus erythematosus and other autoimmune rheumatic diseases. Clin Exp Immunol 1991;86:66–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tipton CM, Fucile CF, Darce J, Chida A, Ichikawa T, Gregoretti I, et al. Diversity, cellular origin and autoreactivity of antibody‐secreting cell population expansions in acute systemic lupus erythematosus. Nat Immunol 2015;16:755–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Knight JS, Kaplan MJ. Lupus neutrophils: ‘NET’ gain in understanding lupus pathogenesis [review]. Curr Opin Rheumatol 2012;24:441–50. [DOI] [PubMed] [Google Scholar]
- 17. Wellmann U, Letz M, Herrmann M, Angermuller S, Kalden JR, Winkler TH. The evolution of human anti‐double‐stranded DNA autoantibodies. Proc Natl Acad Sci U S A 2005;102:9258–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ronnblom L, Leonard D. Interferon pathway in SLE: one key to unlocking the mystery of the disease. Lupus Sci Med 2019;6:e000270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Preble OT, Black RJ, Friedman RM, Klippel JH, Vilcek J. Systemic lupus erythematosus: presence in human serum of an unusual acid‐labile leukocyte interferon. Science 1982;216:429–31. [DOI] [PubMed] [Google Scholar]
- 20. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 2003;197:711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Crow MK. Type I interferon in the pathogenesis of lupus [review]. J Immunol 2014;192:5459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hamilton JA, Wu Q, Yang P, Luo B, Liu S, Li J, et al. Cutting edge: intracellular IFN‐β and distinct type I IFN expression patterns in circulating systemic lupus erythematosus B cells. J Immunol 2018;201:2203–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sarkar MK, Hile GA, Tsoi LC, Xing X, Liu J, Liang Y, et al. Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal‐derived interferon κ. Ann Rheum Dis 2018;77:1653–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med 2020;382:211–21. [DOI] [PubMed] [Google Scholar]
- 25. Decker P. Neutrophils and interferon‐ɑ‐producing cells: who produces interferon in lupus? Arthritis Res Ther 2011;13:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, et al. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol 2010;184:3284–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lood C, Blanco LP, Purmalek MM, Carmona‐Rivera C, de Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus‐like disease. Nat Med 2016;22:146–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Evrard M, Kwok IW, Chong SZ, Teng KW, Becht E, Chen J, et al. Developmental analysis of bone marrow neutrophils reveals populations specialized in expansion, trafficking, and effector functions. Immunity 2018;48:364–79.e8. [DOI] [PubMed] [Google Scholar]
- 29. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science 2004;303:1532–5. [DOI] [PubMed] [Google Scholar]
- 30. Ng LG, Ostuni R, Hidalgo A. Heterogeneity of neutrophils [review]. Nat Rev Immunol 2019;19:255–65. [DOI] [PubMed] [Google Scholar]
- 31. Pillinger MH, Abramson SB. The neutrophil in rheumatoid arthritis [review]. Rheum Dis Clin North Am 1995;21:691–714. [PubMed] [Google Scholar]
- 32. Ravetch JV, Kinet JP. Fc receptors [review]. Annu Rev Immunol 1991;9:457–92. [DOI] [PubMed] [Google Scholar]
- 33. Wang Y, Jonsson F. Expression, role, and regulation of neutrophil Fcɣ receptors [review]. Front Immunol 2019;10:1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Van de Winkel JG, Capel PJ. Human IgG Fc receptor heterogeneity: molecular aspects and clinical implications [review]. Immunol Today 1993;14:215–21. [DOI] [PubMed] [Google Scholar]
- 35. Camous L, Roumenina L, Bigot S, Brachemi S, Fremeaux‐Bacchi V, Lesavre P, et al. Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood 2011;117:1340–9. [DOI] [PubMed] [Google Scholar]
- 36. Pillay J, den Braber I, Vrisekoop N, Kwast LM, de Boer RJ, Borghans JA, et al. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood 2010;116:625–7. [DOI] [PubMed] [Google Scholar]
- 37. Lahoz‐Beneytez J, Elemans M, Zhang Y, Ahmed R, Salam A, Block M, et al. Human neutrophil kinetics: modeling of stable isotope labeling data supports short blood neutrophil half‐lives. Blood 2016;127:3431–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thieblemont N, Wright HL, Edwards SW, Witko‐Sarsat V. Human neutrophils in auto‐immunity. Semin Immunol 2016;28:159–73. [DOI] [PubMed] [Google Scholar]
- 39. Kaplan MJ. Role of neutrophils in systemic autoimmune diseases [review]. Arthritis Res Ther 2013;15:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kaplan MJ. Neutrophils in the pathogenesis and manifestations of SLE [review]. Nat Rev Rheumatol 2011;7:691–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chakravarti A, Rusu D, Flamand N, Borgeat P, Poubelle PE. Reprogramming of a subpopulation of human blood neutrophils by prolonged exposure to cytokines. Lab Invest 2009;89:1084–99. [DOI] [PubMed] [Google Scholar]
- 42. Hacbarth E, Kajdacsy‐Balla A. Low density neutrophils in patients with systemic lupus erythematosus, rheumatoid arthritis, and acute rheumatic fever. Arthritis Rheum 1986;29:1334–42. [DOI] [PubMed] [Google Scholar]
- 43. Pillay J, Tak T, Kamp VM, Koenderman L. Immune suppression by neutrophils and granulocytic myeloid‐derived suppressor cells: similarities and differences [review]. Cell Mol Life Sci 2013;70:3813–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schmielau J, Finn OJ. Activated granulocytes and granulocyte‐derived hydrogen peroxide are the underlying mechanism of suppression of T cell function in advanced cancer patients. Cancer Res 2001;61:4756–60. [PubMed] [Google Scholar]
- 45. Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, et al. Arginase I‐producing myeloid‐derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res 2009;69:1553–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marini O, Costa S, Bevilacqua D, Calzetti F, Tamassia N, Spina C, et al. Mature CD10+ and immature CD10– neutrophils present in G‐CSF-treated donors display opposite effects on T cells. Blood 2017;129:1343–56. [DOI] [PubMed] [Google Scholar]
- 47. Rahman S, Sagar D, Hanna RN, Lightfoot YL, Mistry P, Smith CK, et al. Low‐density granulocytes activate T cells and demonstrate a non‐suppressive role in systemic lupus erythematosus. Ann Rheum Dis 2019;78:957–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Grayson PC, Carmona‐Rivera C, Xu L, Lim N, Gao Z, Asare AL, et al. Neutrophil‐related gene expression and low‐density granulocytes associated with disease activity and response to treatment in antineutrophil cytoplasmic antibody–associated vasculitis. Arthritis Rheumatol 2015;67:1922–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, et al. Mast cells and neutrophils release IL‐17 through extracellular trap formation in psoriasis. J Immunol 2011;187:490–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yalavarthi S, Gould TJ, Rao AN, Mazza LF, Morris AE, Núñez‐Alvarez C, et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: a newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol 2015;67:2990–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Van den Hoogen LL, Fritsch‐Stork RD, van Roon JA, Radstake TR. Low‐density granulocytes are increased in antiphospholipid syndrome and are associated with anti–β2‐glycoprotein I antibodies: comment on the article by Yalavarthi et al [letter]. Arthritis Rheumatol 2016;68:1320–1. [DOI] [PubMed] [Google Scholar]
- 52. Teague HL, Varghese NJ, Tsoi LC, Dey AK, Garshick MS, Silverman JI, et al. Neutrophil subsets, platelets, and vascular disease in psoriasis. JACC Basic Transl Sci 2019;4:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang S, Shen H, Shu X, Peng Q, Wang G. Abnormally increased low‐density granulocytes in peripheral blood mononuclear cells are associated with interstitial lung disease in dermatomyositis. Mod Rheumatol 2017;27:122–9. [DOI] [PubMed] [Google Scholar]
- 54. Carmona‐Rivera C, Kaplan MJ. Low‐density granulocytes: a distinct class of neutrophils in systemic autoimmunity [review]. Semin Immunopathol 2013;35:455–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lakschevitz FS, Hassanpour S, Rubin A, Fine N, Sun C, Glogauer M. Identification of neutrophil surface marker changes in health and inflammation using high‐throughput screening flow cytometry. Exp Cell Res 2016;342:200–9. [DOI] [PubMed] [Google Scholar]
- 56. Mistry P, Nakabo S, O'Neil L, Goel RR, Jiang K, Carmona‐Rivera C, et al. Transcriptomic, epigenetic, and functional analyses implicate neutrophil diversity in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci U S A 2019;116:25222–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Srinivas US, Tan BW, Vellayappan BA, Jeyasekharan AD. ROS and the DNA damage response in cancer [review]. Redox Biol 2018;25:101084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lightfoot YL, Blanco LP, Kaplan MJ. Metabolic abnormalities and oxidative stress in lupus. Curr Opin Rheumatol 2017;29:442–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Coit P, Yalavarthi S, Ognenovski M, Zhao W, Hasni S, Wren JD, et al. Epigenome profiling reveals significant DNA demethylation of interferon signature genes in lupus neutrophils. J Autoimmun 2015;58:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol 2011;187:538–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kalliolias GD, Ivashkiv LB. Overview of the biology of type I interferons. Arthritis Res Ther 2010;12 Suppl 1:S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lood C, Arve S, Ledbetter J, Elkon KB. TLR7/8 activation in neutrophils impairs immune complex phagocytosis through shedding of FcgRIIA. J Exp Med 2017;214:2103–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gupta S, Chan DW, Zaal KJ, Kaplan MJ. A high‐throughput real‐time imaging technique to quantify NETosis and distinguish mechanisms of cell death in human neutrophils. J Immunol 2018;200:869–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Beringer A, Noack M, Miossec P. IL‐17 in chronic inflammation: from discovery to targeting [review]. Trends Mol Med 2016;22:230–41. [DOI] [PubMed] [Google Scholar]
- 65. Blanco LP, Pedersen HL, Wang X, Lightfoot YL, Seto N, Carmona‐Rivera C, et al. Improved mitochondrial metabolism and reduced inflammation following attenuation of murine lupus with coenzyme Q10 analog idebenone. Arthritis Rheumatol 2020;72:454–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kim J, Gupta R, Blanco LP, Yang S, Shteinfer‐Kuzmine A, Wang K, et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus‐like disease. Science 2019;366:1531–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kahlenberg JM, Carmona‐Rivera C, Smith CK, Kaplan MJ. Neutrophil extracellular trap‐associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol 2013;190:1217–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mende R, Vincent FB, Kandane‐Rathnayake R, Koelmeyer R, Lin E, Chang J, et al. Analysis of serum interleukin (IL)‐1β and IL‐18 in systemic lupus erythematosus. Front Immunol 2018;9:1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Van Dam LS, Kraaij T, Kamerling SW, Bakker JA, Scherer UH, Rabelink TJ, et al. Intrinsically distinct role of neutrophil extracellular trap formation in antineutrophil cytoplasmic antibody–associated vasculitis compared to systemic lupus erythematosus. Arthritis Rheumatol 2019;71:2047–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand‐Rosenberg S, et al. The terminology issue for myeloid‐derived suppressor cells. Cancer Res 2007;67:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Veglia F, Perego M, Gabrilovich D. Myeloid‐derived suppressor cells coming of age. Nat Immunol 2018;19:108–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea‐Torres K, et al. Lectin‐type oxidized LDL receptor‐1 distinguishes population of human polymorphonuclear myeloid‐derived suppressor cells in cancer patients. Sci Immunol 2016;1:aaf8943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lewandowski LB, Kaplan MJ. Update on cardiovascular disease in lupus [review]. Curr Opin Rheumatol 2016;28:468–76.27227346 [Google Scholar]
- 74. Yu HH, Chen PC, Yang YH, Wang LC, Lee JH, Lin YT, et al. Statin reduces mortality and morbidity in systemic lupus erythematosus patients with hyperlipidemia: a nationwide population‐based cohort study. Atherosclerosis 2015;243:11–8. [DOI] [PubMed] [Google Scholar]
- 75. Laclaustra M, Frangi AF, Frangi AG, Casasnovas JA, Cia P. Association of endothelial function and vascular data with LDL‐c and HDL‐c in a homogeneous population of middle‐aged, healthy military men: evidence for a critical role of optimal lipid levels. Int J Cardiol 2008;125:376–82. [DOI] [PubMed] [Google Scholar]
- 76. Carlucci PM, Purmalek MM, Dey AK, Temesgen‐Oyelakin Y, Sakhardande S, Joshi AA, et al. Neutrophil subsets and their gene signature associate with vascular inflammation and coronary atherosclerosis in lupus. JCI Insight 2018;3:e99276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Smith CK, Vivekanandan‐Giri A, Tang C, Knight JS, Mathew A, Padilla RL, et al. Neutrophil extracellular trap–derived enzymes oxidize high‐density lipoprotein: an additional proatherogenic mechanism in systemic lupus erythematosus. Arthritis Rheumatol 2014;66:2532–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Madhusoodanan J. Core concept: role player or cellular rubbish? Biologists debate the function of neutrophil extracellular traps. Proc Natl Acad Sci U S A 2017;114:13309–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang H, Li T, Chen S, Gu Y, Ye S. Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof‐of-concept trial of metformin. Arthritis Rheumatol 2015;67:3190–200. [DOI] [PubMed] [Google Scholar]
- 80. Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A 2010;107:9813–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Grayson PC, Schauer C, Herrmann M, Kaplan MJ. Neutrophils as invigorated targets in rheumatic diseases [review]. Arthritis Rheumatol 2016;68:2071–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Jourde‐Chiche N, Whalen E, Gondouin B, Speake C, Gersuk V, Dussol B, et al. Modular transcriptional repertoire analyses identify a blood neutrophil signature as a candidate biomarker for lupus nephritis. Rheumatology 2017;56:477–87. [DOI] [PubMed] [Google Scholar]
- 83. Banchereau R, Hong S, Cantarel B, Baldwin N, Baisch J, Edens M, et al. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell 2016;165:551–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Toro‐Dominguez D, Martorell‐Marugan J, Goldman D, Petri M, Carmona‐Saez P, Alarcon‐Riquelme ME. Stratification of systemic lupus erythematosus patients into three groups of disease activity progression according to longitudinal gene expression. Arthritis Rheumatol 2018;70:2025–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Moses K, Brandau S. Human neutrophils: their role in cancer and relation to myeloid‐derived suppressor cells. Semin Immunol 2016;28:187–96. [DOI] [PubMed] [Google Scholar]