

Abstract
Background
Genome‐wide association studies have identified over 100 single‐nucleotide polymorphisms (SNPs) associated with prostate cancer (PrCa), and polygenic risk scores (PRS) based on their combined genotypes have been developed for risk stratification. We aimed to assess the contribution of PRS to PrCa risk in a large multisite study.
Methods
The sample included 1972 PrCa cases and 1919 unaffected controls. Next‐generation sequencing was used to assess pathogenic variants in 14 PrCa‐susceptibility genes and 72 validated PrCa‐associated SNPs. We constructed a population‐standardized PRS and tested its association with PrCa using logistic regression adjusted for age and family history of PrCa.
Results
The mean age of PrCa cases at diagnosis and age of controls at testing/last clinic visit was 59.5 ± 7.2 and 57.2 ± 13.0 years, respectively. Among 1740 cases with pathology data, 57.4% had Gleason score ≤ 6, while 42.6% had Gleason score ≥ 8. In addition, 39.6% cases and 20.1% controls had a family history of PrCa. The PRS was significantly higher in cases than controls (mean ± SD: 1.42 ± 1.11 vs 1.02 ± 0.76; P < .0001). Compared with men in the 1st quartile of age‐adjusted PRS, those in the 2nd, 3rd, and 4th quartile were 1.58 (95% confidence interval [CI]: 1.31‐1.90), 2.36 (95% CI: 1.96‐2.84), and 3.98 (95% CI: 3.29‐4.82) times as likely to have PrCa (all P < .0001). Adjustment for family history yielded similar results. PRS predictive performance was consistent with prior literature (area under the receiver operating curve = 0.64; 95% CI: 0.62‐0.66).
Conclusions
These data suggest that a 72‐SNP PRS is predictive of PrCa, supporting its potential use in clinical risk assessment.
Keywords: polygenic, prostate cancer
1. INTRODUCTION
Prostate cancer (PrCa) is one of the most heritable human cancers, with 57% of the variation in disease risk attributable to genetic factors. 1 PrCa risk assessment has been primarily based on family history (FH) and screening of rare pathogenic/likely pathogenic variants (RPVs). Germline RPVs in cancer susceptibility genes have been observed in 8% to 12% of men with localized PrCa, 2 and up to 20% of men with advanced or metastatic disease. 3 , 4 , 5 , 6 In addition to rare RPVs, common genetic variations also contribute to PrCa risk. 7 Genome‐wide association studies (GWAs) have identified more than 100 single‐nucleotide polymorphisms (SNPs) associated with increased PrCa susceptibility. While each individual SNP contributes only modestly to this susceptibility, their combined effects are estimated to account for 33% of the familial risk of PrCa. 8 It has further been shown that a polygenic risk score (PRS) based on a combination of SNP genotypes (a) can be consistently estimated across a variety of populations and geographic regions, (b) may have substantial predictive value for PrCa risk stratification, and (c) has predictive performance superior to that based on FH alone. 9 , 10 , 11 , 12 , 13
In a comparison of self‐reported FH and PRS as two measures of inherited risk for PrCa in several study populations, Sun et al 10 observed substantial variation in the proportion of positive FH but similar mean PRS among participating sites of the REDUCE trial. Despite utilization of the same protocol to obtain FH, sites in Eastern and Western Europe reported 4.2% and 10.9% of men with PrCa‐affected first‐ or second‐degree relatives, respectively, while the proportion in North American was 22.8%; similar variation was also observed among sites within the same country. The FH‐associated relative risks (RRs) for PrCa among these three regions ranged from 1.20 to 1.91. Recent studies in men of European ancestry from the PRACTICAL consortium and others have reported RR for PrCa associated with FH to be as high as 2.5. 14 , 15 In contrast, RRs associated with a 33‐SNP PRS were relatively homogenous among REDUCE study sites, ranging from 1.69 to 1.89. PRS‐associated RRs recently reported by PRACTICAL and others have ranged from 1.74 to 1.86, despite differences in the number of SNPs included in each PRS model. 14 , 15 Moreover, Sun et al 10 reported superior predictive performance of the PRS compared with FH alone, and demonstrated that the addition of FH information to the PRS model did not further improve prediction of PrCa. Taken together, the consistent risk estimates associated with PRSs across multiple studies, and its ability to more accurately identify men who develop PrCa, provide evidence supporting its potential use in clinical risk assessment.
In this report, we examine the extent to which a 72‐SNP PRS is predictive of PrCa in men who tested negative for RPVs in PrCa‐susceptibility genes. We also assessed whether the contribution of overall genetic risk represented by the PRS varied by age and/or FH and estimated absolute lifetime risk for PrCa to age 85.
2. MATERIALS AND METHODS
2.1. Patient population
This is a case‐control validation study in which 4327 patients were ascertained from three study sites: Johns Hopkins University Hospital (JHH: 1725 cases and 541 controls), Ambry Genetics (AG: 448 cases and 1229 controls), and NorthShore University HealthSystem's Genomic Health Initiative (NSGHI: 384 controls), and were eligible for study inclusion if they were male, self‐reported European ancestry, and greater than or equal to 18 years of age at PrCa diagnosis (JHH cases), genetic testing (AG cases and controls), or last clinic visit (JHH and NSGHI controls). All sites submitted samples as permitted by an applicable Institutional Review Board or under HHS regulations (45 CFR 46.1019b)(4). JHH cases were patients undergoing radical prostatectomy for the treatment of clinically localized PrCa in 2002 to 2015 and were included if disease was organ‐confined and Gleason score ≤6 or ≥8, as determined upon pathological evaluation of the prostatectomy specimen. Only the high‐ and low‐grade PrCa JHH cases were included because they were previously curated to assess the association of RPVs in cancer susceptibility genes with PrCa. Men who were undergoing screening for PrCa at The Johns Hopkins Hospital and the Johns Hopkins University Applied Physics Laboratory (Columbia, MD) during the same time period were asked to participate as control subjects, as described in Zheng et al. 16 Briefly, blood samples for preparation of DNA, serum prostate‐specific antigen (PSA) levels, digital rectal examination results, and demographic information were available for these subjects. History of additional non‐PrCa primaries was not available. A total of 541 men met our inclusion criteria as JHH control subjects for this study: European American ancestry by self‐report, normal digital rectal examination, PSA level less than 4.0 ng/mL, and older than 55 years. AG cases and controls were men referred for multigene panel testing in 2017 to 2019. Among AG cases, 104 patients had Gleason ≥8, and 59 patients had Gleason ≤6, and 230 patients had no pathology information. AG controls were unaffected with PrCa at the time of testing; 53% of AG controls had a personal history of at least one non‐PrCa primary. NSGHI controls had a minimum of 1 year of clinical history available in the electronic health record (EHR) and were excluded if any ICD‐9/10 diagnosis of cancer was present at any time in the EHR. PrCa‐specific FH information was available for the majority of cases (n = 1877) and controls from AG and JHH (n = 1467). Men who tested positive for RPVs in any PrCa‐susceptibility gene (ATM, BRCA1, BRCA2, CHEK2, EPCAM, HOXB13, NBN, PALB2, MLH1, MSH2, MSH6, PMS2, RAD51D, TP53) were excluded from further analysis (n = 358).
2.2. SNP selection
SNPs that met the following criteria were selected and added to the next‐generation sequencing (NGS) library for genetic testing panel of PrCa genes: (a) reported with genome‐wide significance in more than one GWA analysis, based on a sample size of more than 500 cases and more than 500 controls in any population; (b) are not strongly correlated; (c) effects are race‐/ethnicity‐specific (ie, only SNPs that meet (a) and (b) in a specific population were included). For this study, we selected SNPs reported in studies of individuals of North European ancestry. 8 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 We identified 72 SNPs meeting these criteria, of which 3 could not be directly genotyped and were substituted by tag SNPs, with pairwise r 2 = 1.0 and distance ranging from 519 to 8504 bp between each proxy and originally reported SNP (Table S1).
2.3. Molecular analysis and quality control
Sequencing quality for Illumina NextSeq 500 were monitored during the sequencing run and include visualization of intensity vs cycle plots, and cluster intensity over the duration of the run. Other quality metrics that were evaluated for the entire sequencing run upon completion of sequencing and demultiplexing of the samples include metrics for percent perfect index reads, percent of bases ≥Q30, and overall mean quality score. Samples passing sequencing quality metrics (>85% perfect index, >75% bases >Q30, and mean Qscore >30) were input into a proprietary NGS data‐processing pipeline in a parallelized fashion, starting with alignment of sequencing reads to human reference genome build (GRCh37/hg19), followed by variant and genotype calling on the panel genes and all SNP positions. Additionally, NGS coverage was evaluated at all sites for every sample, and any samples with more than 20 sites at no or low coverage (<20X) were excluded from genotype calling (n = 64; 1.5% of samples sequenced) and downstream statistical analysis.
NGS data were examined to assess missing rates for each sample, and each SNP. Samples were excluded if more than seven SNPs were missing due to bioinformatics quality control thresholds (n = 14; 0.3% of samples passing bioinformatics quality control [QC]). SNP calls were checked for consistency with publicly available databases (GRCh37/hg19; Ensembl release 91 29 ) and literature‐reported reference and risk alleles. We compared SNP allele frequencies among control subjects to those available in the gnomAD non‐Finnish European population to ensure consistency with the reference population. Hardy‐Weinberg equilibrium (HWE) was assessed for all SNPs among controls using R package HardyWeinberg. 30 To assess the assumption of SNP effects consistent with a multiplicative model, we examined all possible pairwise SNP × SNP interactions associated with PrCa using logistic regression, with a 1‐df likelihood ratio test for the interaction; P values were false discovery rate‐corrected for multiple testing. We additionally tested for higher order SNP interactions using logic regression. 31
2.4. Statistical analysis
Based on the 72 established PrCa‐risk SNPs, we computed a population‐standardized PRS for each individual using an approach consistent with prior literature. 12 Using previously published estimates of the per‐allele odds ratios (ORs) and risk allele frequencies (p) for each SNP, and assuming independent and additive risks on the log OR scale, we computed the unscaled population average risk for autosomal SNPs as:
and for sex chromosome SNPs as:
Adjusted risk values were then calculated as:
for the three genotypes defined by the number of risk alleles: 0, 1, or 2, respectively. Missing genotypes were imputed with a population average risk of 1.0. The adjusted risk values for each SNP were then multiplied to estimate the overall PRS‐associated risk as the population‐standardized PRS for each individual based on his/her observed genotypes.
Logistic regression models were used to estimate the ORs for PrCa by quartile of the PRS, with the 1st quartile category (<25th percentile) as the reference. ORs for PrCa were also estimated for the following percentiles of the distribution of the PRS, assuming 25th to 75th percentile as the reference: less than 1st, 1st to 10th, 10th to 25th, 25th to 75th, 75th to 90th, 90th to 99th, greater than 99th percentile. Sensitivity analyses of PRS‐PrCa association were performed for JHH and AG samples separately, as well as for Gleason ≥8 and Gleason ≤6 groups separately. A continuous PRS was tested for association with FH, defined as a binary indicator for the presence of greater than or equal to one first‐ or second‐degree relatives with PrCa, using logistic regression. To determine whether the effect of the PRS varied by other patient characteristics such as age at testing/last clinic visit (for JHH PrCa cases, age at diagnosis was used) and/or FH, we used a 1‐df likelihood ratio test to assess evidence for interaction between PRS and each characteristic, by comparing models with and without a multiplicative interaction term. To further assess whether the PRS was associated with low‐ or high‐risk PrCa, we tested for association between PRS and Gleason score among PrCa cases, with and without adjustment for diagnosis age; PRS association with diagnosis age was also tested among cases. Area under the receiver operating curve (AUROC) was computed using R package pROC. R (v.3.5.1) and used for all statistical analyses; all statistical tests were two‐sided, and P< .05 were considered nominally statistically significant.
Lifetime risk to age 85 years was estimated based on PRS‐specific ORs, population‐based age‐specific PrCa incidences rates, and both all‐cause and PrCa‐specific mortality rates, to account for competing causes of death. Population rates were based on non‐Hispanic white males in the United States (SEER, www.seer.cancer.gov 32 , 33 ). Absolute risk was estimated with ABSRISK v.1.0. 34
3. RESULTS
3.1. Patient characteristics
A total of 4327 patient samples underwent NGS for the present study. After the assessment of bioinformatics QC and inclusion/exclusion criteria, data from 3891 patients (1972 PrCa cases and 1919 controls) were available for analysis. The mean age at diagnosis for cases and at testing or last clinic visit for controls was 59.5 ± 7.2 and 57.2 ± 13.0 years, respectively. Detailed patient characteristics by site were included in Table S2.
3.2. SNP characteristics
The mean SNP call rate (the proportion of individuals whose genotype was successfully determined) was 99.8% ± 0.7% (range: 94.7%‐100.0%). SNP risk allele frequencies among controls ranged from 3.4% to 94.6% and were consistent with the gnomAD non‐Finnish European population (range: 3.1%‐94.3%; mean ± SD absolute difference among SNPs: 1.6% ± 1.5%; P = .98). Among the 69 SNPs located on autosomal chromosomes, none deviated significantly from HWE (all P ≥ .001); the three SNPs located on the X chromosome are not expected to meet classic HWE in an all‐male population. 35 Consistent with the findings of previous large studies, we did not detect any significant pairwise or high‐order interactions among the SNPs after multiple testing correction. 14
3.3. PRS association with PrCa
The sum of the risk alleles across the 72 SNPs was approximately normally distributed among cases (range: 49‐83) and controls (range: 41‐81) (mean ± SD risk allele count: 64.7 ± 5.4 vs 62.2 ± 5.3; P < .0001; Figure 1). The mean ± SD PRS was significantly higher for cases vs controls (1.42 ± 1.11 vs 1.02 ± 0.76; P < .0001). The continuous PRS was significantly associated with PrCa risk, with an OR per standard deviation of the PRS of 1.72 (95% CI: 1.59‐1.88). Compared with men in the 1st quartile of PRS, those in the 2nd, 3rd, and 4th quartile were 1.60 (95% CI: 1.33‐1.92), 2.32 (95% CI: 1.93‐2.79), and 3.85 (95% CI: 3.19‐4.65) times as likely to have PrCa (Table 1). Maximum AUROC for PRS discrimination of cases and controls was reached at a threshold of PRS = 0.96, corresponding to a PPV = 0.61 and NPV = 0.60 (AUROC = 0.64; 95% CI: 0.62‐0.66; Figure 2). Adjustment for age resulted in nearly identical PRS ORs per quartile and model discriminatory performance (AUROC = 0.64) compared with the unadjusted model. Further adjustment for FH resulted in slight attenuation of PRS ORs per quartile, but similar AUROC (0.64), which was superior to the model of FH alone (AUROC = 0.60; 95% CI: 0.58‐0.61). Results were similar when stratified by site (JHH and AG; Table S3). In the fully adjusted model, both FH and PRS quartile were significantly associated with PrCa (all P < .001), implying that each factor independently contributes to disease risk. The small reduction in PRS effect estimates was driven primarily by the strong relationship between FH and PrCa; the PRS was not associated with FH among cases (P = .05) or controls (P = .47). Moreover, we observed no significant interactions between the PRS and age (P = .64), or FH (P = .90). Consistent with the findings of others, PRS‐associated risk for PrCa did not vary by increasing age or FH (Table 2).
Figure 1.
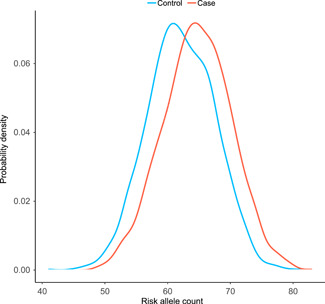
Distribution of the sum of risk alleles across 72 single‐nucleotide polymorphisms, for cases (red) compared with controls (blue). Probability density on the y axis represents the proportion of cases and controls, respectively, with a given risk allele count (x axis). The mean ± standard deviation risk allele count in cases vs controls: 64.7 ± 5.4 vs 62.2 ± 5.3; P < .0001 [Color figure can be viewed at wileyonlinelibrary.com]
Table 1.
Odds ratios (ORs) and 95% confidence intervals (CIs) for PrCa per quartile of PRS
| Unadjusted | Adjusted for age | Adjusted for age and FHa | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PRS quartile or covariate | PRS threshold | Case, n | Control, n | OR | 95% CI | P value | OR | 95% CI | P value | OR | 95% CI | P value |
| ≤25% | <0.62 | 337 | 636 | 1.00 | … | … | 1.00 | … | … | 1.00 | … | … |
| 25%‐50% | 0.62‐0.96 | 446 | 527 | 1.60 | 1.33‐1.92 | <.001 | 1.58 | 1.31‐1.90 | <.001 | 1.58 | 1.29‐1.95 | <.001 |
| 50%‐75% | 0.96‐1.53 | 536 | 436 | 2.32 | 1.93‐2.79 | <.001 | 2.36 | 1.96‐2.84 | <.001 | 2.18 | 1.77‐2.68 | <.001 |
| >75% | >1.53 | 653 | 320 | 3.85 | 3.19‐4.65 | <.001 | 3.98 | 3.29‐4.82 | <.001 | 3.85 | 3.10‐4.80 | <.001 |
| Age | 1.03 | 1.03‐1.04 | <.001 | 1.06 | 1.05‐1.07 | <.001 | ||||||
| FH | 2.68 | 2.26‐3.17 | <.001 | |||||||||
Abbreviations: FH, family history; PrCa, prostate cancer; PRS, polygenic risk scores.
FH is defined as the presence or absence of at least one first‐ or second‐degree relative with prostate cancer. Adjustment for FH resulted in reduced sample size and therefore altered PRS quartile thresholds, because once samples were removed, the distribution is altered. Analyses including FH included 3344 (86%) of 3891 subjects meeting all inclusion/exclusion criteria and missing less than or equal to seven SNPs; a total of 1877 (95%) cases and 1467 (76%) controls were included.
Figure 2.
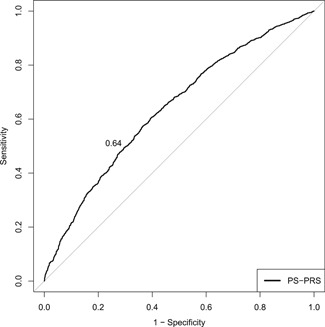
AUROC for the accuracy of the PRS in distinguishing between prostate cancer cases and controls (AUROC = 0.64, 95% CI: 0.62‐0.66). AUROC, area under the receiver operating curve; CI, confidence interval; PRS, polygenic risk scores
Table 2.
Odds ratios (ORs) and 95% confidence intervals (CIs) per standard deviation of PRS in the presence or absence of family history of PrCa, compared with Al Olama et al 14 and Schumacher et al 15
Examining age‐ and FH‐adjusted effects stratified by PRS percentiles on a finer scale, we observed monotonic increasing ORs with increasing PRS compared with the 25th to 75th percentile range (median risk as the reference) (Table 3). Importantly, the ORs above and below the median risk were comparable to and had 95% CIs overlapping with two other large studies: a 25‐SNP PRS and 147‐SNP PRS assessed in 40 414 and 72 729 PrCa cases and controls of European ancestry, respectively. 14 , 15 We also note that men with high PRS (≥90th percentile) included 83 controls, of whom 64 (77%) were negative for PrCa FH.
Table 3.
Odds ratios (ORs) and 95% confidence intervals (CIs) for PrCa per PRS percentile, compared with Al Olama et al 14 and Schumacher et al 15
| Present studya (72 SNPs) | Al Olama et alb (25 SNPs) | Schumacher et alc (147 SNPs) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PRS threshold | Case, n | Control, n | P value | OR | 95% CI | OR | 95% CI | OR | 95% CI | |
| <1% | <0.24 | 10 | 24 | .003 | 0.30 | 0.13‐0.64 | 0.14 | 0.08‐0.24 | 0.15 | 0.11‐0.20 |
| 1‐10% | 0.24‐0.43 | 107 | 194 | <.001 | 0.43 | 0.32‐0.56 | 0.41 | 0.36‐0.47 | 0.35 | 0.32‐0.37 |
| 10‐25% | 0.43‐0.63 | 222 | 279 | <.001 | 0.64 | 0.52‐0.79 | 0.63 | 0.57‐0.70 | 0.54 | 0.51‐0.57 |
| 25‐75% | 0.63‐1.56 | 937 | 735 | … | 1.00 | … | 1.00 | … | 1.00 | … |
| 75‐90% | 1.56‐2.29 | 349 | 152 | <.001 | 1.86 | 1.48‐2.33 | 1.68 | 1.54‐1.83 | 1.74 | 1.67‐1.82 |
| 90‐99% | 2.29‐4.97 | 222 | 79 | <.001 | 2.31 | 1.74‐3.10 | 2.31 | 2.09‐2.56 | 2.69 | 2.55‐2.82 |
| ≥99% | >4.97 | 30 | 4 | .001 | 5.76 | 2.20‐19.78 | 4.24 | 3.24‐5.53 | 5.71 | 5.04‐6.08 |
Abbreviations: PrCa, prostate cancer; PRS, polygenic risk scores; SNP, single‐nucleotide polymorphism.
ORs for the present study were estimated for percentiles of a population‐standardized PRS, adjusted for age and family history. Family history was defined as the presence or absence of greater than or equal to one first‐ or second‐degree family member with a personal history of PrCa. ORs for some percentiles of the PRS may be unstable due to a very small sample size (eg, ≥99%).
ORs shown for Al Olama et al 14 were estimated for percentiles of a weighted PRS, adjusted for age and family history. Family history was defined as the presence or absence of greater than or equal to one first‐ or second‐degree family member with a personal history of PrCa. Despite model differences, the present study ORs for PrCa risk are similar to those estimated by Al Olama et al, except for the ≥99% category.
ORs shown for Schumacher et al 15 were estimated for percentiles of a weighted PRS, and are not adjusted for age or family history. Despite model differences, the present study ORs for PrCa risk are similar to those estimated by Schumacher et al, except for the ≥99% category.
3.4. PRS associations with high‐ vs low‐risk PrCa and age of onset
Among 1740 cases with high‐ or low‐risk Gleason scores (Table S2), the PRS was associated with PrCa in both groups (Table S4). Interestingly, ORs for the low‐risk group were slightly higher than those estimated for the high‐risk group, although 95% CI were largely overlapping. There was no association between PRS and high‐ vs low‐risk PrCa (adjusted for diagnosis age: OR = 1.00; 95% CI: 0.91‐1.10; unadjusted: OR = 0.95; 95% CI: 0.87‐1.04); analysis of JHH cases alone resulted in similar estimates (adjusted for diagnosis age: OR = 0.98; 95% CI: 0.88‐1.08; unadjusted: OR = 0.93; 95% CI: 0.84‐1.03). However, the PRS was associated with diagnosis age (P < .0001). On average, cases were 0.7 (±0.3) years younger per quartile of increasing PRS. Men with a PRS in the less than 1st percentile were on average, 65.4 ± 5.7 years of age at diagnosis, while those with a PRS in the greater than or equal to 99th percentile were 55.7 ± 7.6 years, respectively (Table S5).
3.5. PRS‐associated lifetime risk
The mean ± SD lifetime risk to age 85 estimated for all cases vs. controls based on PRS‐associated risk was 11.6% ± 8.3% vs. 8.4% ± 6.1%. By PRS percentile, cumulative lifetime risk to age 85 for men in the less than 1st percentile of the PRS was 1.6% and increased with increasing PRS percentile to 44.2% for men in the greater than 99th percentile. Estimates of lifetime risk were similar for men with and without an FH of PrCa, which ranged from 1.8% to 48.1% and from 1.7% to 40.1% for the former and latter, respectively (Figure 3). Cumulative risk from ages 25 to 45 was close to 0% for all PRS percentiles, as PrCa incidence is extremely low in this age range. Risk by PRS percentile began to diverge at ~50 years of age, the age that most men are first screened for PrCa in the clinical setting.
Figure 3.
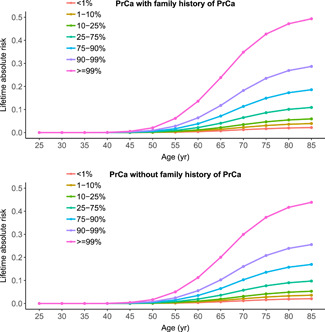
Cumulative lifetime (absolute) risk for prostate cancer by percentiles of PRS, stratified by family history (yes/no). PrCa, prostate cancer; PRS, polygenic risk scores [Color figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
We examined the performance of a 72‐SNP PRS in a population of men who were negative for rare RPVs in reported PrCa‐susceptibility genes. Our results demonstrate that the relative and lifetime risks of PrCa associated with the PRS for these men are similar to those reported previously in larger and/or broader populations. We also found that the predictive performance of the PRS, while modest, outperformed that of FH alone, as is consistent with prior literature.
Al Olama et al 14 from PRACTICAL, an international collaboration of nearly 80 PrCa studies conducted across multiethnic populations, evaluated the predictive performance of a 25‐SNP PRS in 40 414 men of European ancestry. Recently, Schumacher et al 15 reported results from the PrCa OncoArray project, which aimed to identify additional loci associated with PrCa and assessed a 147‐SNP PRS in 72 729 men of European ancestry. Investigators from both studies reported a strong association between PRS and PrCa risk, with an OR per PRS standard deviation of 1.74 (95% CI: 1.70‐1.78) and 1.86 (95% CI: 1.83‐1.89), respectively. Similarly, we estimated an OR of 1.72 (95% CI: 1.59‐1.88) for association with PrCa per PRS standard deviation. After adjusting for age and presence of PrCa FH, Al Olama et al reported that men with a PRS in the highest 10% of the distribution were 2.31 (95% CI: 2.09‐2.56) times as likely to develop PrCa as those with a median‐risk PRS (25th‐75th percentile of PRS distribution). Schumacher et al reported an OR of 2.69 (95% CI: 2.55‐2.82) for this same percentile category of PRS relative to the median‐risk category. Consistent with these findings, our age‐ and FH‐adjusted PRS‐associated risk for men in the 90th to 99th PRS percentile compared with the median range was 2.31 (95% CI: 1.74‐3.10). As observed in a prior and this study, the PRS and FH were independently associated with PrCa, and the PrCa risk associated with the PRS did not vary by the presence or absence of FH. This observation and recently increasing evidence suggested that PRS captures genetic risk components not quantified by FH and RPVs. 36
Compared with FH, which often suffers from incomplete information and wide variation, the PRS demonstrated superior predictive performance in this and a prior study. 10 Further adjustment of the PRS model for age and/or FH did not improve its discriminatory performance, which implies that age and/or FH do not add discriminatory power above that already accounted for by the PRS. Several other large studies have reported similar observations, with AUROCs for the PRS alone ranging from 0.59 to 0.62, 10 , 12 , 37 and AUROCs for the joint model of PRS and FH ranging from 0.60 to 0.64. 10 Notably, our findings suggested that a large proportion (77%) of unaffected men in the greater than or equal to 90th PRS percentile who were PrCa FH‐negative may have a high PrCa risk that might otherwise go unrecognized by FH assessment alone.
We observed an association between PRS and diagnosis age among PrCa cases, consistent with the findings of Schumacher et al that PRS effects are greater in magnitude for early‐onset PrCa (≤55 vs >55 years). 15 Also consistent with our findings, Schumacher et al found no association between the PRS and disease aggressiveness (OR [95% CI] for low‐/intermediate‐risk vs high‐risk PrCa: 1.89 [1.86‐1.93] vs 1.88 [1.83‐1.93]). In a large‐scale analysis of PrCa cases, ~85% of whom had clinically defined aggressive disease, Siebert et al 38 observed association between a 54‐SNP PRS and PrCa diagnosis age. 38
Absolute risks to age 85 reported by Al Olama et al ranged from 1.5% to 35.0% across the percentiles of PRS distribution for men without an FH of PrCa, and from 3.7% to 65.8% for men with a positive FH. 14 We observed similar lifetime risks in men without an FH and lower lifetime risks in men with an FH. Given the observed divergence of lifetime risk by PRS at approximately age 50 and the fact that 50 years is the age that most men are initially screened for PrCa, an approach combining clinical assessment with PRS profiling for estimation of lifetime risk may be useful for clinical risk assessment.
While our study validates a PRS that may be utilized to assess PrCa risk, we acknowledge its limitations. Our case‐control study is based on genotyping of retrospectively collected samples and limited clinical information available from three different sites. Selection bias may potentially confound our findings, although sensitivity analyses demonstrated that PRS results and inferences were comparable between sites. This PRS is based on men of European ancestry, and generalizing it to other races/ethnicities requires large‐scale GWAs and validation in samples of non‐European ethnicity. Lastly, as our study is based largely on retrospective data, it did not include estimates of cost to assess the PRS in the clinical setting, which would need to be addressed in a future clinical utility study.
5. CONCLUSIONS
This analysis suggests that a 72‐SNP PRS is predictive of PrCa and may be used to identify unaffected individuals at high risk of developing PrCa. Large‐scale population‐based studies are needed for assessing PRS association with prognosis. Additional studies are needed to assess PRS association with clinical features, validation in non‐European populations, interaction with high‐ and moderate‐penetrance PrCa predisposition genes, and clinical utility.
CONFLICT OF INTERESTS
MG, KW, ZS, JW, SLZ, BTH, WI, and JX declare that there are no conflict of interests. All other authors are employed by and receive a salary from Ambry Genetics.
AUTHOR CONTRIBUTIONS
MHB, JX, BTH, WI, and SL contributed to the concept and design. MHB, SL, HL, JC, RH, SG, MG, KW, ZS, JW, and SLZ contributed to the acquisition, analysis and interpretation of data. MHB drafted the manuscript; MHB, SL, JX, BTH, WI, HL, H‐ML, and BTD contributed to the critical revision of the manuscript.
Supporting information
Supporting information
Black MH, Li S, LaDuca H, et al. Validation of a prostate cancer polygenic risk score. The Prostate. 2020;80:1314–1321. 10.1002/pros.24058
A relevant whitepaper is published as a Scientific Poster on Ambry Genetics' website: https://www.ambrygen.com/file/material/view/985/AmbryScore_Prostate_WhitePaper_0.
REFERENCES
- 1. Mucci LA, Hjelmborg JB, Harris JR, et al. Familial risk and heritability of cancer among twins in Nordic countries. JAMA. 2016;315(1):68‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marshall CH, Fu W, Wang H, Baras AS, Lotan TL, Antonarakis ES. Prevalence of DNA repair gene mutations in localized prostate cancer according to clinical and pathologic features: association of Gleason score and tumor stage. Prostate Cancer Prostatic Dis. 2019;22(1):59‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA‐repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375(5):443‐453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mandelker D, Zhang L, Kemel Y, et al. Mutation detection in patients with advanced cancer by universal sequencing of cancer‐related genes in tumor and normal DNA vs guideline‐based germline testing. JAMA. 2017;318(9):825‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Giri VN, Hegarty SE, Hyatt C, et al. Germline genetic testing for inherited prostate cancer in practice: implications for genetic testing, precision therapy, and cascade testing. Prostate. 2019;79:333‐339. [DOI] [PubMed] [Google Scholar]
- 6. Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;162(2):454. [DOI] [PubMed] [Google Scholar]
- 7. Xu J, Labbate CV, Isaacs WB, Helfand BT. Inherited risk assessment of prostate cancer: it takes three to do it right. Prostate Cancer Prostatic Dis. 2020;23(1):59‐61. [DOI] [PubMed] [Google Scholar]
- 8. Al Olama AA, Kote‐Jarai Z, Berndt SI, et al. A meta‐analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nat Genet. 2014;46(10):1103‐1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Helfand B, Kearns J, Conran C, Xu J. Clinical validity and utility of genetic risk scores in prostate cancer. Asian J Androl. 2016;18(4):509‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sun J, Na R, Hsu FC, et al. Genetic score is an objective and better measurement of inherited risk of prostate cancer than family history. Eur Urol. 2013;63(3):585‐587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liss MA, Xu J, Chen H, Kader AK. Prostate genetic score (PGS‐33) is independently associated with risk of prostate cancer in the PLCO trial. Prostate. 2015;75(12):1322‐1328. [DOI] [PubMed] [Google Scholar]
- 12. Conran C, Na R, Chen H, et al. Population‐standardized genetic risk score: the SNP‐based method of choice for inherited risk assessment of prostate cancer. Asian J Androl. 2016;18(4):520‐524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen H, Liu X, Brendler CB, et al. Adding genetic risk score to family history identifies twice as many high‐risk men for prostate cancer: results from the Prostate Cancer Prevention trial. Prostate. 2016;76(12):1120‐1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Al Olama AA, Benlloch S, Antoniou AC, et al. Risk analysis of prostate cancer in PRACTICAL, a multinational consortium, using 25 known prostate cancer susceptibility loci. Cancer Epidemiol Biomarkers Prev. 2015;24(7):1121‐1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schumacher FR, Al Olama AA, Berndt SI, et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nature Genet. 2018;50(7):928‐936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zheng SL, Sun J, Cheng Y, et al. Association between two unlinked loci at 8q24 and prostate cancer risk among European Americans. JNCI. 2007;99(20):1525‐1533. [DOI] [PubMed] [Google Scholar]
- 17. Al Olama AA, Kote‐Jarai Z, Giles GG, et al. Multiple loci on 8q24 associated with prostate cancer susceptibility. Nat Genet. 2009;41(10):1058‐1060. [DOI] [PubMed] [Google Scholar]
- 18. Eeles RA, Kote‐Jarai Z, Al Olama AA, et al. Identification of seven new prostate cancer susceptibility loci through a genome‐wide association study. Nat Genet. 2009;41(10):1116‐1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eeles RA, Kote‐Jarai Z, Giles GG, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008;40(3):316‐321. [DOI] [PubMed] [Google Scholar]
- 20. Eeles RA, Olama AAA, Benlloch S, et al. Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nat Genet. 2013;45(4):385‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gudmundsson J, Sulem P, Gudbjartsson DF, et al. Genome‐wide association and replication studies identify four variants associated with prostate cancer susceptibility. Nat Genet. 2009;41(10):1122‐1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gudmundsson J, Sulem P, Manolescu A, et al. Genome‐wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007;39(5):631‐637. [DOI] [PubMed] [Google Scholar]
- 23. Gudmundsson J, Sulem P, Rafnar T, et al. Common sequence variants on 2p15 and Xp11.22 confer susceptibility to prostate cancer. Nat Genet. 2008;40(3):281‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hoffmann TJ, Van Den Eeden SK, Sakoda LC, et al. A large multiethnic genome‐wide association study of prostate cancer identifies novel risk variants and substantial ethnic differences. Cancer Discov. 2015;5(8):878‐891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kote‐Jarai Z, Olama AAA, Giles GG, et al. Seven prostate cancer susceptibility loci identified by a multi‐stage genome‐wide association study. Nat Genet. 2011;43(8):785‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schumacher FR, Berndt SI, Siddiq A, et al. Genome‐wide association study identifies new prostate cancer susceptibility loci. Hum Mol Genet. 2011;20(19):3867‐3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thomas G, Jacobs KB, Yeager M, et al. Multiple loci identified in a genome‐wide association study of prostate cancer. Nat Genet. 2008;40(3):310‐315. [DOI] [PubMed] [Google Scholar]
- 28. Yeager M, Orr N, Hayes RB, et al. Genome‐wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39(5):645‐649. [DOI] [PubMed] [Google Scholar]
- 29. Zerbino DR, Achuthan P, Akanni W, et al. Ensembl 2018. Nucleic Acids Res. 2018;46(D1):D754‐D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Graffelman J, Camarena JM. Graphical tests for Hardy‐Weinberg equilibrium based on the ternary plot. Hum Hered. 2008;65(2):77‐84. [DOI] [PubMed] [Google Scholar]
- 31. Schwender H, Ickstadt K. Identification of SNP interactions using logic regression. Biostatistics. 2008;9(1):187‐198. [DOI] [PubMed] [Google Scholar]
- 32. Fay MP, Pfeiffer R, Cronin KA, Le C, Feuer EJ. Age‐conditional probabilities of developing cancer. Stat Med. 2003;22(11):1837‐1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fay MP. Estimating age conditional probability of developing disease from surveillance data. Popul Health Metr. 2004;2(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dupont WD, Plummer WD Jr. Understanding the relationship between relative and absolute risk. Cancer. 1996;77(11):2193‐2199. [DOI] [PubMed] [Google Scholar]
- 35. Graffelman J, Weir BS. Testing for Hardy‐Weinberg equilibrium at biallelic genetic markers on the X chromosome. Heredity. 2016;116(6):558‐568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lambert SA, Abraham G, Inouye M. Towards clinical utility of polygenic risk scores. Hum Mol Genet. 2019;28(R2):R133‐R142. [DOI] [PubMed] [Google Scholar]
- 37. Aly M, Wiklund F, Xu J, et al. Polygenic risk score improves prostate cancer risk prediction: results from the Stockholm‐1 cohort study. Eur Urol. 2011;60(1):21‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Seibert TM, Fan CC, Wang Y, et al. Polygenic hazard score to guide screening for aggressive prostate cancer: development and validation in large scale cohorts. BMJ. 2018;360:j5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information