

Abstract
Background
Inhibition of the programmed cell death protein 1 (PD‐1) pathway has demonstrated clinical benefit in metastatic urothelial cancer (mUC); however, response rates of 15% to 26% highlight the need for more effective therapies. Bruton tyrosine kinase (BTK) inhibition may suppress myeloid‐derived suppressor cells (MDSCs) and improve T‐cell activation.
Methods
The Randomized Phase 2 Trial of Acalabrutinib and Pembrolizumab Immunotherapy Dual Checkpoint Inhibition in Platinum‐Resistant Metastatic Urothelial Carcinoma (RAPID CHECK; also known as ACE‐ST‐005) was a randomized phase 2 trial evaluating the PD‐1 inhibitor pembrolizumab with or without the BTK inhibitor acalabrutinib for patients with platinum‐refractory mUC. The primary objectives were safety and objective response rates (ORRs) according to the Response Evaluation Criteria in Solid Tumors, version 1.1. Secondary endpoints included progression‐free survival (PFS) and overall survival (OS). Immune profiling was performed to analyze circulating monocytic MDSCs and T cells.
Results
Seventy‐five patients were treated with pembrolizumab (n = 35) or pembrolizumab plus acalabrutinib (n = 40). The ORR was 26% with pembrolizumab (9% with a complete response [CR]) and 20% with pembrolizumab plus acalabrutinib (10% with a CR). The grade 3/4 adverse events (AEs) that occurred in ≥15% of the patients were anemia (20%) with pembrolizumab and fatigue (23%), increased alanine aminotransferase (23%), urinary tract infections (18%), and anemia (18%) with pembrolizumab plus acalabrutinib. One patient treated with pembrolizumab plus acalabrutinib had high MDSCs at the baseline, which significantly decreased at week 7. Overall, MDSCs were not correlated with a clinical response, but some subsets of CD8+ T cells did increase during the combination treatment.
Conclusions
Both treatments were generally well tolerated, although serious AE rates were higher with the combination. Acalabrutinib plus pembrolizumab did not improve the ORR, PFS, or OS in comparison with pembrolizumab alone in mUC. Baseline and on‐treatment peripheral monocytic MDSCs were not different in the treatment cohorts. Proliferating CD8+ T‐cell subsets increased during treatment, particularly in the combination cohort. Ongoing studies are correlating these peripheral immunome findings with tissue‐based immune cell infiltration.
Keywords: immunotherapy, programmed cell death receptor 1, protein kinase inhibitor, urologic neoplasms
Short abstract
In this randomized phase 2 study of metastatic urothelial cancer, a combination of pembrolizumab and a Bruton tyrosine kinase inhibitor (acalabrutinib) does not improve clinical outcomes in comparison with pembrolizumab alone. Comprehensive flow cytometry is used to evaluate circulating immune cells during treatment.
Introduction
Urothelial cancer is common, with more than 80,000 new cases and 17,000 deaths estimated in the United States in 2019. 1 As with many cancer types, immune evasion is one of the key hallmarks of urothelial cancer. 2 Monoclonal antibodies that block negative regulators of T cells such as programmed cell death protein 1 (PD‐1) amplify immune responses, and over the past 5 years, the treatment landscape for metastatic urothelial cancers (mUCs) has expanded to include multiple standard‐of‐care PD‐1 and programmed cell death ligand 1 (PD‐L1) inhibitors. 3
PD‐1 and PD‐L1 inhibitors are able to block immunosuppressive signals and improve the anticancer activity of effector T cells. 4 However, in late‐phase clinical studies of PD‐1 inhibitors in patients with mUC in the chemotherapy‐refractory setting, objective response rates (ORRs) are low (15%‐26%). 5 , 6 , 7 , 8 In the Keynote 045 study, a phase 3 trial that randomized platinum‐refractory patients with mUC to receive either pembrolizumab, a PD‐1 inhibitor, or chemotherapy, pembrolizumab improved median overall survival (OS; 10.3 vs 7.4 months; hazard ratio, 0.73; 95% confidence interval [CI], 0.59‐0.91; P = .002) 4 and was established as a preferred immunotherapy regimen for the second‐line treatment of platinum‐refractory mUC. 3 Pembrolizumab had an ORR of 21.1% in that trial, and it seemed to be durable in some patients (duration of response range, 1.6‐15.6 months; median not reached). 4 Therefore, approximately 80% of patients with platinum‐refractory mUC do not have an objective response to pembrolizumab monotherapy, and the effect of PD‐1 inhibition alone may be improved by adding treatments that target other suppressive signals in the tumor microenvironment.
Myeloid cells in the tumor microenvironment, particularly myeloid‐derived suppressor cells (MDSCs), can release cytokines such as tumor growth factor β, interleukin 10, interleukin 1b, nitric oxide, and indolamine 2,3‐dioxygenase, and this can lead to T‐cell suppression and an immunosuppressive environment necessary for tumor cell growth. 9 , 10 , 11 Circulating MDSCs have been correlated with poor outcomes in several solid tumors, including metastatic melanoma 12 and urothelial cancer. 13 Mechanisms to suppress MDSCs in the tumor microenvironment include promoting MDSC differentiation into mature myeloid cells, preventing MDSC infiltration, depleting MDSCs, and inhibiting MDSC activation. Bruton tyrosine kinase (BTK) has been shown as a mechanism for MDSC growth in the tumor microenvironment, 14 and this suggests a role for BTK inhibition in lowering MDSCs and, therefore, reducing T‐cell suppression. 15 , 16 , 17
Interestingly, studies examining circulating MDSCs in patients treated with PD‐1 inhibition have shown that alterations in the myeloid cell compartment correlate with clinical outcomes. Specifically, patients with tumor progression had proportionally higher circulating MDSC levels and a high myeloid gene signature. 13 , 18 , 19 , 20 Recent preclinical results show that elevated MDSC levels are responsible for this lack of response and that elimination of MDSCs may lead to increased efficacy with PD‐1 inhibition. 21 , 22 In addition, BTK‐dependent activation of mast cells, myeloid cells, and other immunocytes in peritumoral inflammatory stroma has been shown to sustain the complex microenvironment needed for solid tumor maintenance. 23 , 24 Taken together, these findings suggest that BTK inhibition may offer an attractive strategy for treating solid tumors by enhancing the antitumor effects of PD‐1–directed monotherapy.
Acalabrutinib is a highly selective and potent BTK inhibitor that has shown significant clinical responses in chronic lymphocytic leukemia 25 and mantle cell lymphoma. 26 To assess the activity of acalabrutinib as an adjunct to PD‐1–directed therapy, we performed a randomized phase 2 trial of pembrolizumab alone or combined with acalabrutinib in patients with platinum‐resistant mUC. Because of the important hypothesis that BTK inhibition would inhibit suppressive myeloid cells, a key exploratory endpoint was the characterization of peripheral MDSCs at the baseline and during treatment.
Materials and Methods
Study Design and Participants
This trial was the Randomized Phase 2 Trial of Acalabrutinib and Pembrolizumab Immunotherapy Dual Checkpoint Inhibition in Platinum‐Resistant Metastatic Urothelial Carcinoma (RAPID CHECK; also known as ACE‐ST‐005 and Keynote 143; ClinicalTrials.gov identifier NCT02351739). This was a randomized, open‐label, multicenter phase 2 trial that recruited patients from 15 sites across the United States (Table 1). Eligible patients had mUC or a mixed histology (percentage unspecified) of the bladder or upper urinary tract, including the ureters and renal pelvis; had progression during or after (within 1 year of) 1 or more platinum‐based chemotherapy regimens; were 18 years old or older; had an Eastern Cooperative Oncology Group performance status of 0 or 1; and had measurable disease according to the Response Evaluation Criteria in Solid Tumors, version 1.1 (RECIST 1.1). Archival tumor tissue was collected, or a fresh biopsy was obtained if archival tissue was not available.
Table 1.
Baseline Clinical Characteristics
| Characteristic | Pembrolizumab (n = 35) | Pembrolizumab + Acalabrutinib (n = 40) |
|---|---|---|
| Age, median (range), y | 65 (46‐96) | 68 (36‐83) |
| Male sex, No. (%) | 28 (80) | 29 (73) |
| ECOG PS, No. (%) | ||
| 0 | 13 (37) | 12 (30) |
| 1 | 20 (57) | 28 (70) |
| 2 | 2 (6) | 0 |
| Disease stage IV, No. (%) | 35 (100) | 40 (100) |
| No. of prior therapies, No. (%) | ||
| 1 | 15 (43) | 21 (53) |
| 2 | 13 (37) | 9 (23) |
| ≥3 | 7 (20) | 10 (25) |
| Prior therapies, No. (%) | ||
| Platinum combination a | 31 (89) | 35 (88) |
| Cisplatin | 28 (80) | 25 (63) |
| Carboplatin | 11 (31) | 18 (45) |
| Taxane | 6 (17) | 7 (18) |
| MVAC | 5 (14) | 8 (20) |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; MVAC, methotrexate, vinblastine, Adriamycin (doxorubicin), and cisplatin; PS, performance status.
Platinum combination: cisplatin, carboplatin, oxaliplatin, and nedaplatin.
Patients were excluded if they had known central nervous system metastases; significant cardiovascular disease within 6 months of screening; ongoing immunosuppressive therapy or active autoimmune disease; a history of interstitial lung disease or pneumonitis; prior exposure to BTK inhibitors or antibodies directed against PD‐1, PD‐L1, or PD‐L2; concurrent warfarin use; inadequate hematologic values (including an absolute neutrophil count < 1.5 × 109/L, a platelet count < 100 × 109/L, and a hemoglobin level < 8.0 g/dL); or inadequate renal function (creatinine clearance < 30 mL/min).
The institutional review board or independent ethics committee approved the protocol at each study site. The study was performed in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines. All patients who were enrolled in this study provided written informed consent to participate.
Procedures
Patients were randomized 1:1 to receive either pembrolizumab or pembrolizumab and acalabrutinib. Pembrolizumab was dosed at 200 mg intravenously every 3 weeks, and acalabrutinib was given at 100 mg by mouth twice daily. A dose reduction was required for treatment‐related dose‐limiting toxicities (defined as grade 4 vomiting or diarrhea; grade 3 nausea, vomiting, or diarrhea lasting longer than 72 hours; other toxicities higher than grade 3; or a dose delay due to toxicity for longer than 21 days) or other intolerable adverse events (AEs); dose‐reduction levels for acalabrutinib included 100 mg by mouth daily or 50 mg by mouth twice daily. Study treatments were limited to 24 months from the date of the first dose for patients who were tolerating treatment and not progressing. Patients with disease progression on pembrolizumab monotherapy were allowed to cross over to the combination cohort.
An interim safety analysis was planned for dose‐limiting toxicities after 6 patients were treated in the combination cohort. Patients with disease progression in the pembrolizumab monotherapy cohort were allowed to cross over to pembrolizumab and acalabrutinib if they were deemed to be good candidates by the treating physician, with discussion with medical monitors. The severity of treatment‐emergent AEs was assessed with the Common Terminology Criteria for Adverse Events, version 4.03. Responses were determined by investigator assessment using the RECIST 1.1 criteria.
Archival tissue specimens were obtained, and immunohistochemistry (IHC) for PD‐L1 expression was performed with the PD‐L1 IHC 22C3 pharmDx assay (Agilent, Carpinteria, California). The combined positive score (CPS) of the number of staining tumor and immune cells with respect to total tumor cells was used, and PD‐L1 positivity was defined as at least 1 cell per 100 tumor cells staining for PD‐L1 (PD‐L1 CPS > 1).
Outcomes
The primary objectives of this study were to characterize the safety profile of the acalabrutinib and pembrolizumab combination in patients with platinum‐resistant mUC and to determine the ORR for pembrolizumab and acalabrutinib versus pembrolizumab monotherapy. Secondary objectives included progression‐free survival (PFS) and OS for each treatment cohort. PFS was defined as the time from the start of treatment until disease progression or death from any cause and was estimated with Kaplan‐Meier methods. OS was defined as the time from enrollment until death and was also estimated with Kaplan‐Meier methods.
We also report here a key exploratory objective of immune profiling for changes in peripheral MDSCs based on the hypothesis that BTK inhibition would lower the MDSC population. Other prespecified exploratory endpoints, including full immune profiling of peripheral blood, tissue‐based analysis for T‐cell infiltration, and RNA sequencing for inflammatory expression signatures, are ongoing.
Multidimensional Immune Cell Profiling
Peripheral blood mononuclear cells (PBMCs) were collected at the baseline and during treatment (weeks 4, 7, and 10). Multidimensional flow cytometry was performed on PBMCs in 3 panels (tumor‐reactive T cells, exhaustion, and regulatory T cells/MDSCs) for the following markers: CD3, CD4, CD8, PD‐1, PD‐L1, PD‐L2, B7‐H3, CTLA‐4, ICOS, LAG3, TIM3, Ki‐67, CD45RA, CCR7, CD38, CD14, HLA‐DR, CD16/CD56, CD19/CD20, CD25, CD127, CD39, and CCR4 (Supporting Fig. [Link], [Link], [Link]). Monocytic MDSCs were defined as cells that were lineage‐negative (negative for CD3, CD56, CD16, CD19, and CD20) and CD14+/HLA‐DRlow.
Statistical Analysis
The sample size of this randomized 2‐arm trial was determined with a Z test for normal approximation of the binomial distribution (based on a 1‐sided α value of 0.10 and 80% power) to detect the hypothesized difference in the ORR (from 18% in the pembrolizumab cohort to 40% in the combination pembrolizumab and acalabrutinib cohort). The final sample size was 37 patients in each cohort. All enrolled patients who received at least 1 dose of the study drug were included in the safety and intention‐to‐treat analysis sets. Descriptive statistics were used to summarize the safety and efficacy data. The disease control rate was defined as the proportion of patients who achieved stable disease (SD), a partial response (PR), or a complete response (CR), and the ORR was defined as the proportion of patients who achieved a PR or a CR. Kaplan‐Meier methods were used to estimate PFS and OS. Common censoring rules were used for PFS and OS.
For the multidimensional flow cytometry analysis, the mean of the relative cell subset frequency (plus or minus the standard deviation) was calculated and associated with clinical responses (best RECIST 1.1 response). Wilcoxon rank sum was used to compare treatment arms and was based on best disease progression. The Benjamini‐Hochberg method was used to correct for multiple testing with a false discovery rate of 0.1 or less. The clinical trial was registered at ClinicalTrials.gov, number NCT02351739.
Role of the Funding Source
Acerta Pharma (a member of the AstraZeneca group) sponsored and designed the study in collaboration with a subgroup of investigators and Merck & Co, Inc (Kenilworth, New Jersey), and it managed the clinical trial database, including the oversight of data collection and data analysis. The corresponding author had full access to all the data in the study and wrote the final manuscript for publication.
Results
Clinical Trial Outcomes
Patients (n = 78) were enrolled between May 28, 2015 and January 26, 2016. In total, 75 patients were treated with at least 1 dose of the study drug; 35 patients were randomized to receive pembrolizumab monotherapy, and 40 patients received pembrolizumab and acalabrutinib (Supporting Fig. 2). Nine patients who progressed after pembrolizumab monotherapy crossed over to receive the combination of pembrolizumab and acalabrutinib.
The baseline characteristics for all treated patients are listed in Table 1. All patients had metastatic disease at the baseline, and they had a median of 2 prior lines of therapy without prior immune checkpoint inhibitor therapy.
The primary efficacy endpoint of ORR (PR and CR) was 25.7% (95% CI, 12.5%‐43.3%) in the pembrolizumab cohort and 20.0% (95% CI, 9.1%‐35.7%) in the pembrolizumab and acalabrutinib cohort. The disease control rate (SD, PR, and CR) was 45.7% (95% CI, 28.8%‐63.4%) in the pembrolizumab cohort and 47.5% (95% CI, 31.5%‐63.9%) in the pembrolizumab and acalabrutinib cohort (Table 2). The median PFS was 1.6 months (95% CI, 1.4‐4.2 months) in the pembrolizumab cohort and 2.2 months (95% CI, 1.4‐3.5 months) in the pembrolizumab and acalabrutinib cohort. The median OS was 11.4 months (range, 5.7‐21.1 months) for the pembrolizumab cohort and 6.3 months (range, 3.6‐12.3 months) for the pembrolizumab and acalabrutinib cohort (Fig. 1).
Table 2.
Objective Response Rate and Disease Control Rate in the Clinical Trial
| Pembrolizumab (n = 35) | Pembrolizumab + Acalabrutinib (n = 40) | |
|---|---|---|
| Best response, No. (%) | ||
| CR | 3 (8.6) | 4 (10) |
| PR | 6 (17.1) | 4 (10) |
| SD | 7 (20) | 11 (27.5) |
| PD | 15 (42.9) | 15 (37.5) |
| Missing/unknown | 4 (11.4) | 6 (15) |
| Objective response rate (CR + PR), No. (%) | 9 (25.7) | 8 (20) |
| 95% CI, % | 12.5‐43.3 | 9.1‐35.7 |
| Disease control rate (CR + PR + SD), No. (%) | 16 (45.7) | 19 (47.5) |
| 95% CI, % | 28.8‐63.4 | 31.5‐63.9 |
Abbreviations: CI, confidence interval; CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease.
Figure 1.
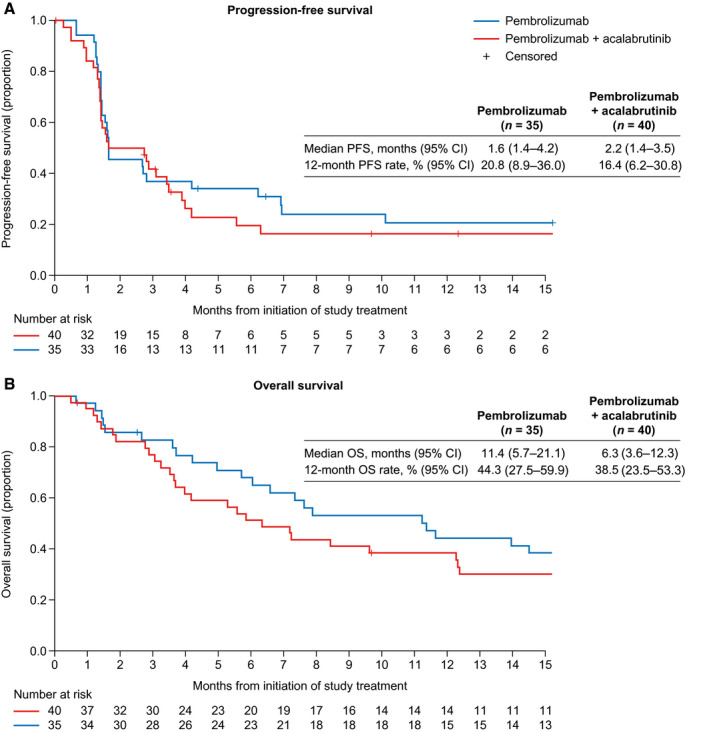
(A) PFS and (B) OS by treatment cohort. CI indicates confidence interval; OS, overall survival; PFS, progression‐free survival.
Adverse Events
Of the 75 patients randomized and treated with study medication, 34 (97.1%) treated with pembrolizumab and 39 (97.5%) treated with pembrolizumab and acalabrutinib experienced any AE. Nineteen patients (54.3%) in the pembrolizumab arm and 30 patients (75.0%) in the pembrolizumab and acalabrutinib arm experienced a grade 3/4 AE (Table 3). Thirty patients (85.7%) in the pembrolizumab arm and 21 patients (52.5%) in the pembrolizumab and acalabrutinib arm experienced an AE related to pembrolizumab only. The most frequent pembrolizumab alone–related AEs in the pembrolizumab and acalabrutinib arm were fatigue, diarrhea, increased alanine aminotransferase (ALT), and increased aspartate aminotransferase (AST). Seven patients (20.0%) in the pembrolizumab cohort and 9 patients (22.5%) in the pembrolizumab and acalabrutinib cohort experienced a grade 3/4 AE related to pembrolizumab only, as assessed by the investigator. The most frequent grade 3/4 pembrolizumab‐related AEs experienced by patients in the pembrolizumab and acalabrutinib cohort were increased ALT, increased AST, and maculopapular rash.
Table 3.
Treatment‐Related AEs
| AEs | Pembrolizumab (n = 35), No. (%) | Pembrolizumab + Acalabrutinib (n = 40), No. (%) |
|---|---|---|
| Any AEs related to acalabrutinib | ||
| Grade 1/2 | — | 11 (27.5) |
| Grade 3/4 | — | 8 (20.0) |
| Most frequent AEs related to acalabrutinib a | ||
| Fatigue | — | 6 (15.0) |
| Nausea | — | 2 (5.0) |
| Vomiting | — | 2 (5.0) |
| Anemia | — | 2 (5.0) |
| Decreased appetite | — | 2 (5.0) |
| Diarrhea | — | 2 (5.0) |
| Headache | — | 2 (5.0) |
| Decreased platelet count | — | 2 (5.0) |
| Any AEs related to pembrolizumab | ||
| Grade 1/2 | 23 (65.7) | 11 (27.5) |
| Grade 3/4 | 7 (20) | 9 (22.5) |
| Grade 5 | 0 | 1 (2.5) |
| Most frequent AEs related to pembrolizumab a | ||
| Fatigue | 12 (34.3) | 6 (15.0) |
| Decreased appetite | 6 (17.1) | 2 (5.0) |
| Diarrhea | 3 (8.6) | 5 (12.5) |
| Hypothyroidism | 6 (17.1) | 1 (2.5) |
| Rash, maculopapular | 4 (11.4) | 3 (7.5) |
| ALT increase | 2 (5.7) | 5 (12.5) |
| Rash | 4 (11.4) | 3 (7.5) |
| AST increase | 1 (2.9) | 5 (12.5) |
| Nausea | 4 (11.4) | 2 (5.0) |
| Pruritus | 3 (8.6) | 2 (5.0) |
| Vomiting | 3 (8.6) | 2 (5.0) |
| Anemia | 2 (5.7) | 2 (5.0) |
| Dry skin | 2 (5.7) | 2 (5.0) |
| Peripheral edema | 1 (2.9) | 3 (7.5) |
| Abdominal pain | 1 (2.9) | 2 (5.0) |
| Arthralgia | 2 (5.7) | 1 (2.5) |
| Cough | 0 | 3 (7.5) |
| Dry mouth | 2 (5.7) | 1 (2.5) |
| Dyspnea | 0 | 3 (7.5) |
| Infusion‐related reaction | 2 (5.7) | 1 (2.5) |
| Pneumonia | 2 (5.7) | 1 (2.5) |
| Pyrexia | 2 (5.7) | 1 (2.5) |
| Blood alkaline phosphatase increase | 0 | 2 (5.0) |
Abbreviations: AE, adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase.
All grades; 2 or more patients.
Nineteen patients (47.5%) treated with pembrolizumab and acalabrutinib experienced an AE related to acalabrutinib only. The most frequent acalabrutinib‐related AEs in the pembrolizumab and acalabrutinib cohort included fatigue, nausea, vomiting, anemia, decreased appetite, diarrhea, headache, and decreased platelet count. A total of 8 patients (20.0%) in the pembrolizumab and acalabrutinib cohort experienced a grade 3/4 AE related to acalabrutinib only. The most frequent grade 3/4 acalabrutinib‐related AEs in the pembrolizumab and acalabrutinib cohort were fatigue, anemia, diarrhea, decreased platelet count, and dyspnea.
Twenty‐six patients (65.0%) in the pembrolizumab and acalabrutinib arm experienced an AE related to pembrolizumab and acalabrutinib. The most frequent AEs related to both were fatigue, increased ALT, and diarrhea. A total of 8 patients (20.0%) in the pembrolizumab and acalabrutinib cohort experienced a grade 3/4 AE related to acalabrutinib, most frequently increased fatigue (10.0%), with anemia, diarrhea, a decreased platelet count, and exertional dyspnea each experienced by 1 patient (2.5%).
Over the course of the study, 8 patients (22.9%) in the pembrolizumab cohort and 16 patients (40.0%) in the pembrolizumab and acalabrutinib cohort discontinued treatment because of a treatment‐emergent AE. In addition, 55 patients (73.3%) died: 24 patients in the pembrolizumab cohort (including 7 patients after they had crossed over to the combination cohort) and 31 patients in the pembrolizumab and acalabrutinib cohort. Of these patients, 48 (64.0%) died of disease progression. Two patients (2.7%) died in relation to AEs (one from pneumonia within 30 days of the last dose and the other from sepsis more than 30 days after the last dose; both were in the pembrolizumab and acalabrutinib cohort).
Fifteen patients (42.9%) in the pembrolizumab cohort and 23 patients (57.5%) in the pembrolizumab and acalabrutinib cohort experienced a serious AE (Supporting Table 1); the most frequent serious AEs occurring in ≥5% of patients in either cohort included acute kidney injury (5.7% in the pembrolizumab cohort vs 12.5% in the pembrolizumab and acalabrutinib cohort), sepsis (2.9% vs 7.5%), urinary tract infection (5.7% vs 5.0%), hematuria (2.9% vs 5.0%), nausea (5.7% vs 0%), pneumonia (5.7% vs 2.5%), and ascites (0% vs 5.0%).
Eight patients (22.9%) in the pembrolizumab cohort and 16 patients (40.0%) in the pembrolizumab and acalabrutinib cohort experienced treatment‐emergent AEs that led to study medication discontinuation. AEs that led to treatment discontinuation in ≥5% of the patients in the pembrolizumab cohort and the pembrolizumab and acalabrutinib cohort included increased ALT (0% in the pembrolizumab cohort vs 7.5% in the pembrolizumab and acalabrutinib cohort), increased AST (0% vs 7.5%), pneumonia (5.7% vs 2.5%), colitis (0% vs 5.0%), fatigue (0% vs 5.0%), and vomiting (0% vs 5.0%).
Crossover Patients
There were 9 patients who crossed over to combination therapy after disease progression on pembrolizumab monotherapy. These 9 patients had a median duration of treatment with pembrolizumab of 2.9 months (range, 1.64‐13.14 months) before entering the crossover treatment cohort. After crossing over to the combination treatment, they had a median duration of treatment with pembrolizumab of 1.4 months (range, 0.7‐2.8 months) and with acalabrutinib of 1.6 months (range, 0.8‐3.3 months); 78% of these crossover patients (7 of 9) died of disease progression more than 30 days after the last dose of treatment, and 22% of these patients (2 of 9) experienced increased transaminases after the crossover that led to dose discontinuation.
PD‐L1 Analysis
Of 60 evaluable tumors, 49 (81.7%) were PD‐L1 positive by Dako IHC 22C3 testing. Twenty‐two of these patients were treated with pembrolizumab, and 27 patients were treated with pembrolizumab and acalabrutinib. The ORR was 22.7% (5 of 22) in patients treated with pembrolizumab and 22.2% (6 of 27) in patients treated with pembrolizumab and acalabrutinib.
Peripheral Immune Cell Profiling
Acalabrutinib, through its BTK inhibition, was hypothesized to exert anti–solid tumor responses predominantly through inhibition of immunosuppressive MDSCs. To test this hypothesis, we evaluated peripheral circulating populations of MDSCs (defined as lineage‐negative, HLA‐DRlow, CD14+; Supporting Fig. 1A). Baseline MDSC populations did not correlate with best clinical responses with the treatment cohorts combined (Fig. 2A). Nine of the 75 patients (12%) had >20% MDSCs (as a proportion of the monocyte population) at the baseline (Fig. 2A). In addition, monitoring changes in MDSCs over time at weeks 4, 7, and 10 did not correlate with the best objective response for either treatment cohort (Fig. 2B and Supporting Fig. 3).
Figure 2.
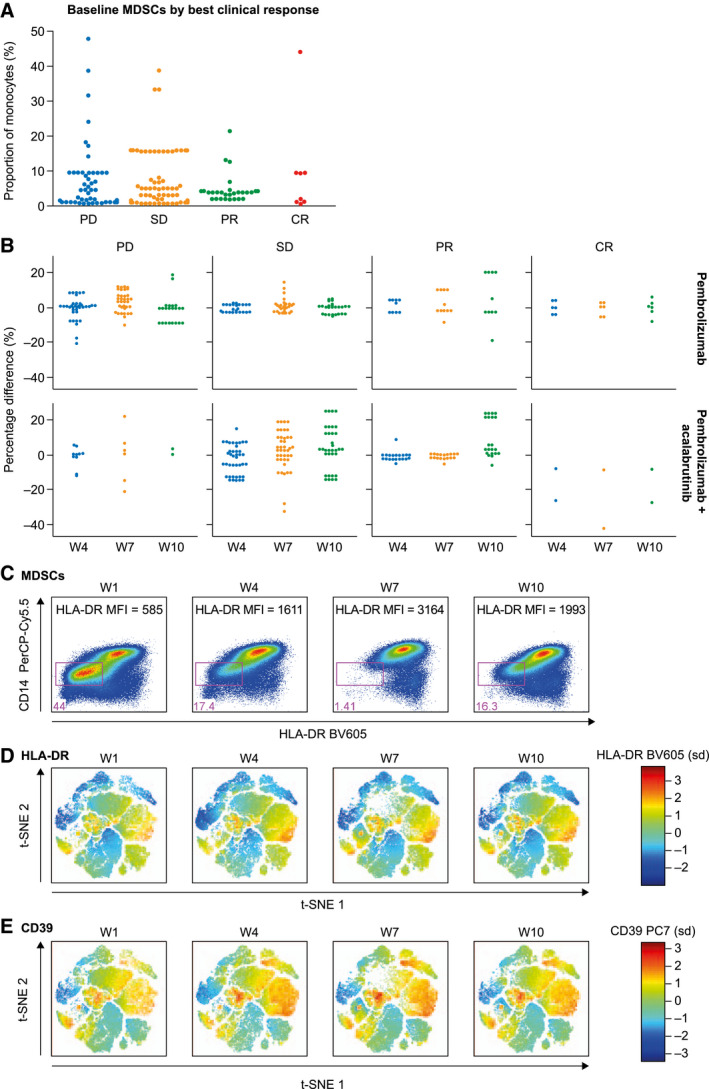
(A) Baseline monocytic MDSCs based on clinical responses and (B) changes during treatment in comparison with the baseline based on the treatment cohort and the best clinical response. (C) Peripheral MDSCs of an exceptional responder to pembrolizumab and acalabrutinib depicted by flow cytometry and (D,E) t‐SNE plots by HLA‐DR and CD39, respectively. CR indicates complete response; MFI, median fluorescent intensity; MDSC, myeloid‐derived suppressor cell; PerCP‐Cy5.5, PerCP (Peridinin chlorophyll)‐Cy5.5; PD, progressive disease; PR, partial response; SD, stable disease; t‐SNE, t‐distributed stochastic neighbor embedding.
MDSCs in an Exceptional Responder
One of the outlier patients for high baseline monocytic MDSCs (also positive for PD‐L1) was an exceptional responder to the combination treatment of pembrolizumab and acalabrutinib. This patient's monocytes included 44% MDSCs at the baseline, which decreased significantly at week 4 to 17% and then at week 7 to 1.4% (Fig. 2C). At week 10, this population of MDSCs seemed to increase slightly to 16.3%. t‐distributed stochastic neighbor embedding plots were generated to visualize these cells on the basis of HLA‐DR and CD39 expression (Fig. 2D,E).
Baseline Cellular Subsets With Clinically Relevant Markers
In evaluating monocyte subsets with clinically relevant cell‐surface markers, we found that subsets of monocytes expressing LAG3, PD‐L2, B7‐H3, and CTLA‐4 were generally lower at the baseline for patients with CRs than those with a PR, SD, or progressive disease. However, these differences in cellular subsets were not statistically significant between clinical response groups. There was a trend toward a difference between clinical response groups in CD4+ T cells co‐expressing CTLA‐4 and B7‐H3 as well as both PD‐1 and CTLA‐4. Differences in CD4+ T cells expressing CD28 and ICOS between response groups were not statistically significant when we separated out all response groups (Supporting Table 2), but CD4+ T cells expressing ICOS were different when we compared patients with disease control (CR, PR, or SD) and those with disease progression (Supporting Table 2 and Supporting Fig. 4G).
Cellular Subset Change on Treatment
When comparing peripheral cellular subsets between week 4 and the baseline for patients with disease control (CR, PR, or SD) and those with disease progression, we found several proliferating (Ki‐67+) T‐cell populations that had statistically significant increases in patients with a clinical benefit versus progressive disease (Supporting Table 3). These included CD4+ T cells lacking PD‐1 and both CD8+ T cells not expressing CD28 and those not expressing PD‐1. CD8+ T cells as well as subsets of CD8+ T cells not expressing CD28 but expressing TIM3 or expressing both PD‐1 and TIM3 had increases on treatment in patients with clinical disease control versus progressive disease (Supporting Table 3).
In evaluating changes between the baseline and week 7 and comparing the extremes of disease response (CR vs progressive disease), we found regulatory T cells that expressed CCR4 and HLA‐DR as well as regulatory T cells that expressed CD39 and HLA‐DR to be statistically significant (but not after Benjamin‐Hochberg correction; Fig. 3 and Supporting Table 4). A comparison between treatment arms showed that the difference between week 7 and the baseline for a population of PD‐L1–positive monocytes was statistically significant (Supporting Table 4). Finally, several subsets of circulating CD8+ T cells changed from the baseline to week 7 and were different on the basis of the treatment cohort. These included CD8+ T cells expressing CTLA‐4, expressing PD‐1, double positive for CD39 and HLA‐DR, double positive for CD39 and Ki‐67, double positive for PD‐1 and TIM3, and negative for CD28 but positive for ICOS (Fig. 3 and Supporting Table 4).
Figure 3.
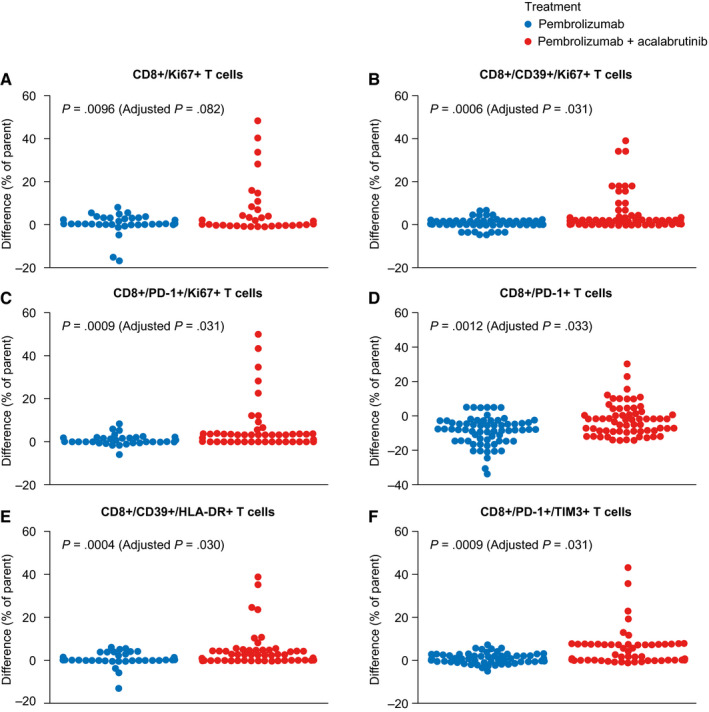
T‐cell subsets that increased from the baseline to week 7 in patients treated with the combination of pembrolizumab and acalabrutinib. These included (A) CD8 T cells, (B) CD8/CD39+ T cells, and (C) CD8/PD‐1 T cells. CD8 cells expressing (D) PD‐1, (E) CD39/HLA‐DR, and (F) PD‐1/TIM3 also increased in patients treated with the combination of pembrolizumab and acalabrutinib. PD‐1 indicates programmed cell death protein 1.
Discussion
Overall, this randomized study of acalabrutinib added to pembrolizumab found that it did not improve clinical outcomes in comparison with pembrolizumab monotherapy in mUC. Therefore, in contrast to the initial hypothesis, BTK inhibition with acalabrutinib did not significantly augment the activity of pembrolizumab. This study was performed before standard‐of‐care access to atezolizumab or pembrolizumab; none of the patients treated in the trial had previous exposure to any PD‐1 or PD‐L1 inhibition. In this context, ORRs of 25.7% for pembrolizumab monotherapy and 20% for pembrolizumab and acalabrutinib cohorts were well within the expected ORR range for immunotherapy‐naive patients. Comparing toxicities, we found that the main differences between the 2 treatment cohorts included increases in fatigue (all grades, 85% in the combination cohort vs 48.6% in the monotherapy cohort) as well as transaminitis (grade 3/4 elevated ALT, 22.5% vs 0%; grade 3/4 elevated AST, 10% vs 0%). Overall, 22.9% of the patients in the pembrolizumab cohort and 40.0% in the pembrolizumab and acalabrutinib cohort discontinued treatment because of a treatment‐emergent AE. Notably, among the 9 patients who crossed over from pembrolizumab to pembrolizumab and acalabrutinib, 2 (22%) had increases in transaminases that led to treatment discontinuation. The hepatic microenvironment holds a rich balance of infiltrating T cells and macrophages, which may have an impact on immune‐mediated hepatotoxicity. 27 Hypothetically, if BTK inhibition potentiates immune‐mediated damage in the liver, then future studies of combination BTK and PD‐1 inhibition should specifically monitor for hepatic dysfunction. These differences in the toxicity profile may not be particular to acalabrutinib but should be considered when future trials are performed with BTK inhibition combined with PD‐1 or PD‐L1 inhibition.
Most patients in this trial were considered PD‐L1 positive per a CPS > 1, and the ORR for PD‐L1–positive patients (22% in either cohort) was similar to the overall ORR. Since this study was conducted, the definition of PD‐L1 positivity has changed to a CPS > 10; therefore, the rates of PD‐L1–positive patients were higher in this trial than other subsequent, larger trials in mUC. 5 , 6 , 7 , 8 In this study, PD‐L1 positivity, defined by a CPS > 1, was not predictive for a treatment response.
The complex interactions of tumor and immune cells can be characterized through an evaluation of both the immune cells in the tumor microenvironment 28 , 29 and those in the circulation as part of the immunome. 13 , 30 , 31 The peripheral immunome has been shown to correlate with clinical responses to vaccine therapy in metastatic breast and prostate cancer 30 and with immune checkpoint therapy in patients treated with immune checkpoint inhibitors. 13 , 31 Comprehensive immune profiling of PBMCs via flow cytometry has been shown previously to be a way of characterizing many cellular subsets during treatment with avelumab, although circulating subsets of T cells or MDSCs did not correlate with a clinical benefit. 31 In mUC, prior work has shown that peripheral MDSC populations expressing PD‐L1 or PD‐1 correlated with the type of immune checkpoint inhibitor that the patient received. Specifically, PD‐L1–positive monocytic MDSCs decreased on treatment with PD‐L1–targeting therapy (atezolizumab or avelumab), and PD‐1–expressing monocytic and immature MDSCs decreased on treatment with PD‐1–targeting therapy (pembrolizumab). 13 Another recent study showed the correlation of lower circulating MDSC levels in the neoadjuvant setting and pathologic CR at the time of cystectomy. 32
In our study, the absolute MDSCs at the baseline or changes on treatment were not sufficient as biomarkers to predict a treatment response. In 1 exceptional responder to the combination of pembrolizumab and acalabrutinib, MDSCs were significantly elevated at the baseline and did decrease substantially from the baseline to week 7. Further exploration may be indicated for patients with high baseline MDSCs measured by flow cytometry, but with high baseline MDSCs (>20%) in only 12% of our patient cohort, such a study would have limited patient accrual.
Recently, increases in proliferating circulating CD8+ T cells between the baseline and week 1 were shown to be associated with a clinical benefit in patients with metastatic thymic epithelial tumors treated with pembrolizumab. 33 Similarly, in our study, several subsets of circulating CD8+ T cells increased between the baseline and week 4 for patients who derived disease control (CR, PR, or SD) versus progression of disease. When we compared treatment arms in our study, patients treated with the combination of pembrolizumab and acalabrutinib had increased proliferating CD8+ T cells that expressed CD39 and proliferating CD8+ T cells that expressed PD‐1. In the pembrolizumab and acalabrutinib treatment cohort, T cells that co‐expressed CD39/HLA‐DR and those that co‐expressed PD‐1/TIM3 also increased between the baseline and week 7. Future studies will include evaluating circulating T‐cell subsets at earlier time points to determine immunological changes at week 1 and week 2.
CD39 (ENTPD1), a cell‐surface ectonucleotidase mediating ATP hydrolysis and generating extracellular adenosine, has been characterized as a feature of regulatory T cells, 34 , 35 , 36 tumor‐associated macrophages, 37 and MDSCs, 38 , 39 , 40 which are important in T‐cell suppression in the tumor microenvironment. Our finding of increasing populations of CD39‐expressing peripheral T cells over time during treatment with pembrolizumab and acalabrutinib (Fig. 3B,E) suggests that these T cells may be pushed out of the tumor microenvironment and into the circulation as a consequence of treatment. These findings have yet to be correlated with an ongoing analysis of tissue‐based infiltrating subsets of T cells and monocytes. CD39 positivity was confirmed on this post hoc biomarker analysis, and further research is needed to understand its clinical utility.
In conclusion, this randomized phase 2 clinical trial of patients with mUC treated with pembrolizumab or pembrolizumab and acalabrutinib did not show improved responses with the combination. Although this trial did not show clinical efficacy, at least 1 other ongoing trial is combining ibrutinib with pembrolizumab and with other chemotherapies in this disease setting (NCT02599324) to evaluate whether the addition of BTK and interleukin 2–inducible T‐cell kinase inhibition with PD‐1 inhibition will have greater tumor efficacy. On the basis of the hypothesis that BTK inhibition with acalabrutinib would suppress MDSC populations, peripheral monocytic MDSC characterization was performed in this study, and it did not show differences in either baseline or on‐treatment MDSC levels in all patients, regardless of treatment cohort. One exceptional responder to pembrolizumab and acalabrutinib had an increased proportion of MDSCs at the baseline that did decrease during treatment and nadired at week 7. Proliferating CD8+ T‐cell subsets also increased during treatment, particularly in the pembrolizumab and acalabrutinib cohort. The standard for evaluating the tumor microenvironment remains direct analysis of the tumor tissue. Ongoing studies are correlating these current peripheral immunome findings with those of the tumor microenvironment from tissue‐based analyses. Future studies are needed to carefully subtype T cells and MDSCs to evaluate their clinical utility as circulating, real‐time biomarkers for monitoring patients treated with immune checkpoint inhibitors.
Funding Support
This study was supported by Acerta Pharma, a member of the AstraZeneca group. Editorial support was provided by Oxford PharmaGenesis (Oxford, United Kingdom) and was funded by Acerta Pharma.
Conflict of Interest Disclosures
Tian Zhang reports research funding (to Duke) from AbbVie, Acerta, AstraZeneca, Janssen, Merck & Co, Inc, Merrimack, Novartis, OmniSeq, Pfizer, PGDx, Mirati, Regeneron, and Seattle Genetics; consulting/speaking for Exelixis, Genentech/Roche, Sanofi Aventis, and Genomic Health; consulting for AstraZeneca, Bristol‐Myers Squibb, Foundation Medicine, Janssen, Merck, Pfizer, Pharmacyclics, Amgen, Seattle Genetics, MJH Life Sciences, and Sanofi Aventis; and stock ownership in/employment by Archimmune Therapeutics and Capio Biosciences (spouse). Michael R. Harrison reports research funding (to Duke) from Acerta Pharma, Astellas Pharma, Bristol‐Myers Squibb, Clovis Oncology, Exelixis, Genentech, Janssen, Merck & Co, Inc, and Pfizer; personal fees from Bristol‐Myers Squibb and Pfizer; and consulting/speaking for AstraZeneca, Bayer, Exelixis, Fujifilm, Genentech, and Janssen. Peter H. O'Donnell reports stock and other ownership interest in Allergan and PrescriptIQ; honoraria from Astellas Pharma, AstraZeneca, Atheneum Partners, Genentech/Roche, the Harrison Consulting Group, the Dedham Group, Schlesinger Associates, FirstWorld Publication, Health Advances, Inovio Pharmaceuticals, Janssen Biotech, Kantar Health, Merck & Co, Inc, OncLive, Parexel, Quintiles, and Seattle Genetics; a consulting or advisory role with Merck; research funding (to the University of Chicago) from Acerta Pharma, AstraZeneca/MedImmune, Boehringer Ingelheim, Bristol‐Myers Squibb, Genentech/Roche, Janssen, Merck & Co, Inc, and Seattle Genetics; being named a coinventor on a pending patent for a genomic prescribing system for medication; travel/accommodation expenses from Genentech/Roche, Janssen, Merck & Co, Inc, and Seattle Genetics/Astellas Pharma; and other from Advance Medical, Janssen, Nektar, and the National Institutes of Health. Ajjai S. Alva reports consulting/advisory roles for Merck & Co, Inc, and AstraZeneca and research funding (to the University of Michigan Medical Center) from Clovis Oncology, Merck & Co, Inc, Bristol‐Myers Squibb, AstraZeneca, Bayer, Progenics, Janssen, Genentech, Esanik, Ionis, and Prometheus. Noah M. Hahn reports research funding (to Johns Hopkins) from Acerta Pharma, Astex Pharmaceuticals, AstraZeneca, Bristol‐Myers Squibb, Genentech/Roche, Incyte, Inovio, Merck & Co, Inc, Pieris, Principia Biopharma, and Seattle Genetics; consulting for AstraZeneca, Bristol‐Myers Squibb, Champions Oncology, CicloMed, Ferring, Genentech/Roche, GlaxoSmithKline, Health Advances, Incyte, Inovio, Janssen, Merck & Co, Inc, Mirati Therapeutics, Pieris, Principia Biopharma, Seattle Genetics, TARIS Biomedical, and TransMed/Inteliquet; and honoraria from Bladder Cancer Academy and PierView. Leonard J. Appleman reports research funding (to the University of Pittsburgh) from Acerta Pharma, Agensys, Astellas Pharma, AVEO, Bayer, Bristol‐Myers Squibb, Calithera Biosciences, Eisai, Exelixis, Genentech/Roche, Inovio Pharmaceuticals, Lilly, Merck & Co, Inc, Novartis, Peloton Therapeutics, Pfizer, Amgen, Tokai, and Seattle Genetics. John M. Burke reports consulting for AbbVie, AstraZeneca, Bayer, Celgene/Juno, Gilead/Kite, Roche/Genentech, Seattle Genetics, and Tempus Labs; speaking for Seattle Genetics; and personal fees from Adaptive Biotechnologies, Verastem, and Bristol‐Myers Squibb. Matthew I. Milowsky reports a consulting or advisory role for BioClin Therapeutics; has received research funding from Acerta Pharma, Astellas Pharma, AstraZeneca, Bristol‐Myers Squibb, Clovis Oncology, Incyte, Inovio, Merck & Co, Inc, Pfizer, Roche/Genentech, Seattle Genetics, Mirati Pharmaceuticals, Boehringer Ingelheim, Constellation Pharmaceuticals, Jounce Therapeutics, Syndax, Innocrin Pharma, Medimmune, Cerulean Pharma, and X4 Pharmaceuticals; has received an honorarium (paid to his institution) from Asieris; and has received travel expenses from Roche/Genentech. Amir Mortazavi reports working on advisory boards for Debiopharm Group and Seattle Genetics and research funding from Acerta Pharma, Astellas Pharma, Bristol‐Myers Squibb, Genentech, Merck & Co, Inc, Mirati Therapeutics, Novartis, Roche, and Seattle Genetics. Neal Shore reports research funding/consulting fees from Amgen, Astellas, AstraZeneca, Bayer, Bristol‐Myers Squibb, Dendreon, Ferring, Genentech, Janssen, Merck & Co, Inc, Myovant, Nymox, Pfizer, Sanofi‐Genzyme, and Tolmar. Guru P. Sonpavde is on advisory boards for Amgen, Astellas/Agensys, AstraZeneca, Bayer, Bristol‐Myers Squibb, Eisai, EMD Serono, Exelixis, Genentech, Janssen, Merck & Co, Inc, Novartis, Pfizer, and Sanofi; reports consulting for Bicycle Therapeutics, the National Comprehensive Cancer Network, and Dava Oncology; has received research support for his institution from Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Celgene, Janssen, Merck & Co, Inc, Onyx‐Amgen, Pfizer, and Sanofi; reports lecture fees for continuing medical education from Clinical Care Options; is an author for UpToDate; is on steering committees for AstraZeneca, Astellas, Bristol‐Myers Squibb, Bavarian Nordic, QED, and Debiopharm Group; reports travel support for investigator meetings from Bristol‐Myers Squibb; and is a speaker for OncLive, Physician Education Resource, and Research to Practice. Emmett V. Schmidt is an employee of Merck Sharp & Dohme Corp, a subsidiary of Merck & Co, Inc, and is a stockholder in Merck & Co, Inc. Bojena Bitman is an employee of Acerta Pharma, a member of the AstraZeneca group. Veerendra Munugalavadla is an employee of Acerta Pharma, a member of the AstraZeneca group, and owns stock in AstraZeneca and Gilead Sciences. Raquel Izumi is an employee of Acerta Pharma, a member of the AstraZeneca group; owns stock in AstraZeneca; and has a patent for acalabrutinib. Priti Patel is an employee of Acerta Pharma, a member of the AstraZeneca group, and owns stock in AstraZeneca. Daniel J. George reports research funding (to Duke) from Acerta Pharma, Astellas Pharma, Bayer, Bristol‐Myers Squibb, Calithera, Dendreon, Exelixis, Innocrin, Janssen, Novartis, Pfizer, Sanofi, and Seattle Genetics; consulting and speaking for Bayer, Exelixis, and Sanofi; consulting for Astellas, AstraZeneca, Bristol‐Myers Squibb, Capio Biosciences, Flatiron, Innocrin, Janssen, Merck & Co, Inc, Merck, Sharp & Dohme, Michael J Hennessey Associates, Modra Pharmaceuticals, Myovant Sciences, Vizuri Health Sciences, Physician Education Resource, and Pfizer; honoraria from Ipsen and UroGPO; honoraria and travel support from UroToday; membership on steering committees for Bristol‐Myers Squibb, Nektar Therapeutics, and the National Cancer Institute; and other from American Association for Cancer Research, Axess Oncology, and Millennium Medical Publishing. The other authors made no disclosures.
Author Contributions
Tian Zhang: Clinical investigation, coordination of the investigation, drafting of the manuscript, design of the clinical trial, review and revision of the manuscript, and approval of the final manuscript. Michael R. Harrison: Clinical investigation, review and revision of the manuscript, and approval of the final manuscript. Peter H. O'Donnell: Clinical investigation, review and revision of the manuscript, and approval of the final manuscript. Ajjai S. Alva: Clinical investigation, review and revision of the manuscript, and approval of the final manuscript. Noah M. Hahn: Clinical investigation, review and revision of the manuscript, and approval of the final manuscript. Leonard J. Appleman: Clinical investigation, review and revision of the manuscript, and approval of the final manuscript. Jeremy Cetnar: Clinical investigation, review and revision of the manuscript, and approval of the final manuscript. John M. Burke: Clinical investigation, review and revision of the manuscript, and approval of the final manuscript. Mark T. Fleming: Clinical investigation, review and revision of the manuscript, and approval of the final manuscript. Matthew I. Milowsky: Clinical investigation, review and revision of the manuscript, and approval of the final manuscript. Amir Mortazavi: Clinical investigation, review and revision of the manuscript, and approval of the final manuscript. Neal Shore: Clinical investigation, review and revision of the manuscript, and approval of the final manuscript. Guru P. Sonpavde: Clinical investigation, review and revision of the manuscript, and approval of the final manuscript. Emmett V. Schmidt: Review and revision of the manuscript and approval of the final manuscript. Bojena Bitman: Clinical trial statistical analysis, review and revision of the manuscript, and approval of the final manuscript. Veerendra Munugalavadla: Interpretation of the data, drafting of the manuscript, review and revision of the manuscript, and approval of the final manuscript. Raquel Izumi: Design of the clinical trial, review and revision of the manuscript, and approval of the final manuscript. Priti Patel: Sponsor's responsible medical officer, review and revision of the manuscript, and approval of the final manuscript. Janet Staats: Translational research, review and revision of the manuscript, and approval of the final manuscript. Cliburn Chan: Translational research, review and revision of the manuscript, and approval of the final manuscript. Kent J. Weinhold: Translational research, review and revision of the manuscript, and approval of the final manuscript. Daniel J. George: Clinical investigation, design of the clinical trial, review and revision of the manuscript, and approval of the final manuscript.
Supporting information
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Zhang T, Harrison MR, O'Donnell PH, Alva AS, Hahn NM, Appleman LJ, Cetnar J, Burke JM, Fleming MT, Milowsky MI, Mortazavi A, Shore N, Sonpavde GP, Schmidt EV, Bitman B, Munugalavadla V, Izumi R, Patel P, Staats J, Chan C, Weinhold KJ, George DJ. A randomized phase 2 trial of pembrolizumab versus pembrolizumab and acalabrutinib in patients with platinum‐resistant metastatic urothelial cancer. Cancer. 2020:126:4485–4497. 10.1002/cncr.33067
See editorial on pages 4446‐50, this issue.
The last 2 authors contributed equally to this article.
We thank all patients, families, and caregivers who participated in this study.
This trial is registered at ClinicalTrials.gov (NCT02351739).
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics 2019. CA Cancer J Clin. 2019;69:7‐34. [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 3. National Comprehensive Cancer Network . Bladder Cancer (Version 5.2018). Accessed December 29, 2018. https://www.nccn.org/professionals/physician_gls/pdf/bladder.pdf
- 4. Chen DS, Mellman I. Elements of cancer immunity and the cancer‐immune set point. Nature. 2017;541:321‐330. [DOI] [PubMed] [Google Scholar]
- 5. Bellmunt J, de Wit R, Vaughn DJ, et al. Pembrolizumab as second‐line therapy for advanced urothelial cancer. N Engl J Med. 2017;376:1015‐1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Plimack ER, Bellmunt J, Gupta S, et al. Safety and activity of pembrolizumab in patients with locally advanced or metastatic urothelial cancer (KEYNOTE‐012): a non‐randomised, open‐label, phase 1b study. Lancet Oncol. 2017;18:212‐220. [DOI] [PubMed] [Google Scholar]
- 7. Rosenberg JE, Hoffman‐Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum‐based chemotherapy: a single‐arm multicenter phase 2 trial. Lancet. 2016;387:1909‐1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sharma P, Callahan MK, Bono P, et al. Nivolumab monotherapy in recurrent metastatic urothelial carcinoma (CheckMate 032): a multicenter, open‐label, two‐stage, multi‐arm, phase 1/2 trial. Lancet Oncol. 2016;17:1590‐1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu Y, Wei G, Cheng WA, et al. Targeting myeloid‐derived suppressor cells for cancer immunotherapy. Cancer Immunol Immunother. 2018;67:1181‐1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Molina‐Cerrillo J, Alonso‐Gordoa T, Gajate P, Grande E. Bruton's tyrosine kinase (BTK) as a promising target in solid tumors. Cancer Treat Rev. 2017;58:41‐50. [DOI] [PubMed] [Google Scholar]
- 11. Wesolowski R, Markowitz J, Carson WE. Myeloid derived suppressor cells—a new therapeutic target in the treatment of cancer. J Immunother Cancer. 2013;1:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jordan KR, Amaria RN, Ramirez O, et al. Myeloid‐derived suppressor cells are associated with disease progression and decreased overall survival in advanced stage melanoma patients. Cancer Immunol Immunother. 2013;62:1711‐1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tseng A, Diaz‐Montero CM, Rayman PA, et al. Immunological correlates of response to immune checkpoint inhibitors in metastatic urothelial carcinoma. Target Oncol. 2018;13:599‐609. [DOI] [PubMed] [Google Scholar]
- 14. Schmidt U, Boucheron N, Unger B, Ellmeier W. The role of Tec family kinases in myeloid cells. Int Arch Allergy Immunol. 2004;134:65‐78. [DOI] [PubMed] [Google Scholar]
- 15. Gunderson AJ, Kaneda MM, Tsujikawa T, et al. Bruton tyrosine kinase–dependent immune cell cross‐talk drives pancreas cancer. Cancer Discov. 2016;6:270‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Monu NR, Frey AB. Myeloid‐derived suppressor cells and anti‐tumor T cells: a complex relationship. Immunol Invest. 2012;41:595‐613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stiff A, Trikha P, Wesolowski R, et al. Myeloid‐derived suppressor cells express Bruton's tyrosine kinase and can be depleted in tumor‐bearing hosts by ibrutinib treatment. Cancer Res. 2016;76:2125‐2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti–PD‐L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558‐562. [DOI] [PubMed] [Google Scholar]
- 19. Weide B, Martens A, Zelba H, et al. Myeloid‐derived suppressor cells predict survival of patients with advanced melanoma: comparison with regulatory T‐cells and NY‐ESO‐1– or melan‐A–specific T cells. Clin Cancer Res. 2014;20:1601‐1609. [DOI] [PubMed] [Google Scholar]
- 20. Meyer C, Cagnon L, Costa‐Nunes CM, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. 2014;63:247‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Highfill SL, Cui Y, Giles AJ, et al. Disruption of CXCR2‐mediated MDSC tumor trafficking enhances anti‐PD1 efficacy. Sci Transl Med. 2014;6:237ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim K, Skora AD, Li Z, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid‐derived cells. Proc Natl Acad Sci U S A. 2014;111:11774‐11779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ponader S, Chen SS, Buggy JJ, et al. The Bruton tyrosine kinase inhibitor PCI‐32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119:1182‐1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de Rooij MF, Kuil A, Geest CR, et al. The clinically active BTK inhibitor PCI‐32765 targets B‐cell receptor‐ and chemokine‐controlled adhesion and migration in chronic lymphocytic leukemia. Blood. 2012;119:2590‐2594. [DOI] [PubMed] [Google Scholar]
- 25. Byrd JC, Harrington B, O'Brien S, et al. Acalabrutinib (ACP‐196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:323‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang M, Rule S, Zinzani PL, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE‐LY‐004): a single arm, multicentre, phase 2 trial. Lancet. 2018;391:659‐667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Suzman DL, Pelosof L, Rosenberg A, Avigan MI. Hepatotoxicity of immune checkpoint inhibitors: an evolving picture of risk associated with a vital case of immunotherapy agents. Liver Int. 2018;38:976‐987. [DOI] [PubMed] [Google Scholar]
- 28. Fridman WH, Pages F, Sautes‐Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12:298‐306. [DOI] [PubMed] [Google Scholar]
- 29. Bindea G, Mlecnik B, Tosolini M, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782‐795. [DOI] [PubMed] [Google Scholar]
- 30. Farsaci B, Donahue RN, Grenga I, et al. Analyses of pretherapy peripheral immunoscore and response to vaccine therapy. Cancer Immunol Res. 2016;4:755‐765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Donahue RN, Lepone LM, Grenga I, et al. Analyses of the peripheral immunome following multiple administrations of avelumab, a human IgG1 anti–PD‐L1 monoclonal antibody. J Immunother Cancer. 2017;5:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ornstein MC, Diaz‐Montero CM, Rayman P, et al. Myeloid‐derived suppressors cells (MDSC) correlate with clinicopathologic factors and pathologic complete response (pCR) in patients with urothelial carcinoma (UC) undergoing cystectomy. Urol Oncol. 2018;36:405‐412. [DOI] [PubMed] [Google Scholar]
- 33. Kim KH, Cho J, Ku BM, et al. The first‐week proliferative response of peripheral blood PD‐1+ CD8+ T‐cells predicts the response to anti–PD‐1 therapy in solid tumors. Clin Cancer Res. 2019;25:2144‐2154. [DOI] [PubMed] [Google Scholar]
- 34. Borsellino G, Kleinewietfeld M, Di Mitri D, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225‐1232. [DOI] [PubMed] [Google Scholar]
- 35. Boer MC, van Meijgaarden KE, Bastid J, Ottenhoff TH, Joosten SA. CD39 is involved in mediating suppression by Mycobacterium bovis BCG‐activated human CD8(+) CD39(+) regulatory T‐cells. Eur J Immunol. 2013;43:1925‐1932. [DOI] [PubMed] [Google Scholar]
- 36. Gupta PK, Godec J, Wolski D, et al. CD39 expression identifies terminally exhausted CD8+ T‐cells. PLoS Pathog. 2015;11:e1005177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Montalban Del Barrio I, Penski C, Schlahsa L, et al. Adenosine‐generating ovarian cancer cells attract myeloid cells which differentiate into adenosine‐generating tumor associated macrophages—a self‐amplifying, CD39‐ and CD73‐dependent mechanism for tumor immune escape. J Immunother Cancer. 2016;4:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Limagne E, Euvrard R, Thibaudin M, et al. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX‐bevacizumab drug treatment regimen. Cancer Res. 2016;76:5241‐5252. [DOI] [PubMed] [Google Scholar]
- 39. Li L, Wang L, Li J, et al. Metformin‐induced reduction of CD39 and CD73 blocks myeloid‐derived suppressor cell activity in patients with ovarian cancer. Cancer Res. 2018;78:1779‐1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev. 2017;276:121‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material