

Abstract
Tumor cell‐surface markers are usually overexpressed or mutated protein receptors for which spatiotemporal regulation differs between and within cancers. Single‐molecule fluorescence imaging can profile individual markers in different cellular contexts with molecular precision. However, standard single‐molecule imaging methods based on overexpressed genetically encoded tags or cumbersome probes can significantly alter the native state of receptors. We introduce a live‐cell points accumulation for imaging in nanoscale topography (PAINT) method that exploits aptamers as minimally invasive affinity probes. Localization and tracking of individual receptors are based on stochastic and transient binding between aptamers and their targets. We demonstrated single‐molecule imaging of a model tumor marker (EGFR) on a panel of living cancer cells. Affinity to EGFR was finely tuned by rational engineering of aptamer sequences to define receptor motion and/or native receptor density.
Keywords: aptamers, cell-surface receptors, live-cell imaging, PAINT, single-molecule tracking
Localization and tracking of individual membrane receptors on living cells was achieved upon transient binding of aptamer probes through minimally invasive solution diffusion. Probe affinity was modulated by rational engineering of the aptamer sequence. Detection of single molecules is linked to binding affinity, so probes with different affinity provide readouts on different properties, such as diffusive dynamics and receptor density levels.
Introduction
Altered cancer cell behavior often is due to the occurrence of mutations or overexpression of certain cell‐surface receptors, known as tumor cell‐surface markers. Single‐molecule imaging techniques are widely used to visualize these markers and to evaluate relevant biological features, such as receptor diffusion, and homotypic or heterotypic receptor‐receptor interactions.1 Single molecule localization microscopy (SMLM) provides static mapping of receptors with sub‐diffraction resolution,2 typically obtained after chemical fixation of cells. Single molecule tracking (SMT) measures receptor dynamics by tracking the motion of sparse biomolecules on living cells.3, 4 In both SMLM and SMT, labeling strategies are essential to directly assess native receptor behavior, and should ideally provide selective labeling with minimal alterations of both endogenous receptor behavior and cell conditions.5, 6
Considerable efforts have been made to overcome traditional labeling approaches based on bulky labels, e.g., antibodies (Abs) or overexpression of protein‐reporter fusions. Alternative affinity probes such as nanobodies,7, 8 affimers9 or aptamers10, 11 have been proposed. Among them, aptamers are produced via chemical or enzymatic synthesis in identical copies with precise dye stoichiometry and have been exploited to perform quantitative nanoscale imaging.10, 11, 12 However, poor compatibility with some chemical fixation protocols has been reported in some cases, which led to incomplete epitope recognition.11 This might pose considerable limitations in the routine use of aptamers or other fixative‐sensitive labels, since the endogenous availability of biomolecules can be reduced. Live‐cell imaging represents a valid alternative to define receptor behavior without altering the native status of target biomolecules due to fixative methods.13, 14 In live‐cell imaging, receptor mapping is hardly achievable at the nanoscale because of receptor motion, but single‐molecule dynamics is efficiently assessed. Moreover, cell surface distributions of receptors can still be observed on an intermediate scale (100 nm–1 μm), provided that the target biomolecules are sampled with sufficient labeling density.
Assessment of single‐molecule dynamics at high labeling density can be achieved with photobleaching‐insensitive approaches like universal PAINT (uPAINT). In this imaging method, living cells are directly exposed to a low concentration of dye‐labeled affinity probes that freely diffuse in solution and are imaged only upon binding of their target receptors, allowing tracking of molecules with unprecedented density.15 However, uPAINT still exploits rather invasive labels such as Abs or receptor‐activating endogenous ligands,6, 16 while less‐invasive implementations (e.g., use of Fab fragments17) are still limited. In addition, the slow dissociation rates of these high‐affinity probes hampers their turnover,15 leading to incomplete replenishment of photobleached probes with fresh ones. Photobleached labels thus remain tightly associated to their target receptors, which in turn cannot be further imaged. DNA duplexes with programmable hybridization kinetics were successfully exploited to promote faster exchange of fluorescent reporters.11, 18, 19 However, this strategy still requires a high affinity probe to label target receptors and expose a DNA docking strand for the hybridization with dye‐conjugated DNA strands diffusing in solution. In contrast, the use of inherently modular affinity probes that bind target receptors selectively and reversibly with minimal perturbation of their native function remains unexplored.
Here we introduce the use of aptamers as reversible, highly specific, and non‐perturbing affinity probes that are directly applied to living cells for single‐molecule imaging of cell‐surface receptors. SMT of Epidermal Growth Factor Receptor (EGFR) molecules was performed using a well‐described high‐affinity aptamer.20 Then, we rationally engineered the aptamer sequence to generate a variant with reduced affinity and faster dissociation rate from EGFR. This reversible probe allowed photobleaching‐insensitive detection of EGFR density levels on living cancer cells that was combined with SMT to obtain high‐density diffusion maps with no perturbation of EGFR endogenous behavior.
Results and Discussion
Principles of Single‐Molecule PAINT Imaging Using Aptamers as Probes
Aptamer designs include an extended 3′‐end “tail” that efficiently anneals with an “anti‐tail” oligonucleotide bearing an organic dye (Figure 1 a) to form a stable complex with precise 1:1 aptamer:fluorophore stoichiometry. The design is modular and can be applied to multiple aptamers as previously demonstrated,21, 22 and each aptamer‐tail can be directly conjugated to virtually any fluorophore of choice. Aptamer probes were incubated with living cells that expressed endogenous receptors. Low concentrations were used (pm to nm) ensuring low labeling density in each image frame, which is necessary for reliable detection of single‐molecule binding events. In addition, total‐internal‐reflection (TIR) illumination was employed to selectively excite only probes located on basal cell surface, significantly reducing fluorescence background from freely‐diffusing aptamers. Following the principles of PAINT, aptamers were detected and localized only upon binding of their target receptors (Figure 1 b). Tracking the motion of individual aptamer‐receptor complexes on the cell surface is performed until aptamers dissociate from their target receptors or leave the illumination plane or until dye photobleaching. However, because bound and unbound probes are both present, continuous labeling of receptors and constant localizations rate could be achieved upon binding of fresh aptamers to cell‐surface receptors, thus overcoming dissociation and photobleaching.
Figure 1.
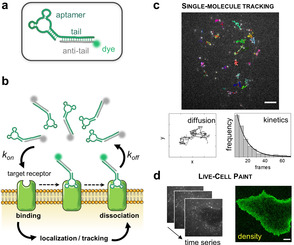
a) The fluorescently conjugated aptamer probe. The aptamer tail (green) is annealed to its complementary anti‐tail (grey) bearing a fluorophore (green sphere). b) Schematic of live‐cell imaging using aptamers. Thanks to TIR illumination, only aptamers (green fluorophores) that bind cell‐surface receptors are selectively excited and detected. In contrast, freely diffusing aptamers (grey fluorophores) are not observed. Transient binding of aptamers to target receptors enables single‐molecule imaging on nearly unperturbed living cells. c) Single‐molecule tracking was performed using sub‐nanomolar concentrations of probes (0.05–0.20 nm) to study receptor diffusion and aptamer binding kinetics. Examples of single EGFR trajectories on the surface of an A431 cell are shown (top image). Scale bar: 5 μm. X‐Y coordinates of a representative trajectory are also displayed. Diffusive status of a receptor can be studied by analysis of thousands of trajectories. Additionally, aptamer binding kinetics can be assessed by analyzing the distribution of trajectory durations, which follows a single‐exponential decay (black line). d) Membrane receptor densities are obtained by live‐cell PAINT using low nm concentrations (1–20 nm). Time‐lapse sequences are recorded (on the left), then a PAINT image (on the right) is reconstructed. Scale bar: 5 μm.
Two main readouts were obtained: (i) motion of individual receptors can be tracked, enabling measurement of molecular diffusion and extrapolation of the rates of fluorophore photobleaching and aptamer dissociation from their target receptors (calculated from analysis of trajectories duration) (Figure 1 c); (ii) localizations of individual receptors in every acquisition frame can be merged to reconstruct PAINT images (Figure 1 d). Although PAINT images cannot provide a quantitative receptor mapping on the nanoscale (because diffusing receptors can appear in different locations in multiple frames), they define localization densities that are proportional to the endogenous levels of cell‐surface receptors, and also determine receptor spatial distribution on a mesoscale (40–300 nm).23
The density of labeled cell‐surface receptors and affinity of aptamer probes are crucial to determine the type of readout attainable. Low labeling density (up to a few tens of labeled cell‐surface molecules) and high‐affinity aptamers are required to perform optimal SMT, in which molecules are tracked for long time with minimal overlap of trajectories. In contrast, high labeling density and efficient exchange of aptamer probes on the target are preferred for acquisition of PAINT images since a higher number of cell‐surface molecules (up to a few hundreds) are localized in every frame. Thus, degree of receptor labeling and type of readout measured can be controlled by tuning the affinity of aptamers.
Tracking Single EGFR Molecules with the MinE07 Aptamer
SMT analysis was performed on A431 cells, a human epidermoid carcinoma cell line that expresses exceptionally high levels of EGFR24, 25 (Table S1). A photostable dye, Atto647N,15, 16, 17 was conjugated to anti‐EGFR RNA aptamer MinE07,26, 27 which binds EGFR with high affinity (K D≈1 nm and apparent K D of 25–60 nm). Live A431 cells were exposed to 0.05 nm aptamer and imaged under moderate illumination (≈200 W cm−2) to obtain low labeling density and relatively long tracking time, thanks to a discontinuous irradiation (Video S1). The distributions of diffusion coefficient (D) values (Figure 2 a) calculated from ≈6000 single molecule trajectories indicates one main population of mobile receptors (D≈10−1 μm2 s−1) and a second population that can be considered immobile (D≈10−3 μm2 s−1). The latter was previously found to be associated with clathrin‐coated pits,28, 29 suggesting that these immobile EGFR molecules were being internalized via clathrin‐mediated endocytosis. Our measurements are nicely consistent with previous results obtained on transfected EGFR labeled with fluorescent proteins.29 Additionally, different EGFR diffusive states could be discriminated by SMT analysis (Figure 2 b,c and Figure S2). Similar distribution of D values was also obtained using the same MinE07 aptamer, but conjugated with AlexaFluor488 (AF488) (Figure S1 and Supplementary Notes), a dye with reduced affinity for phospholipidic bilayers than Atto647N,30, 31 demonstrating that results are negligibly affected by dye‐driven interactions.
Figure 2.
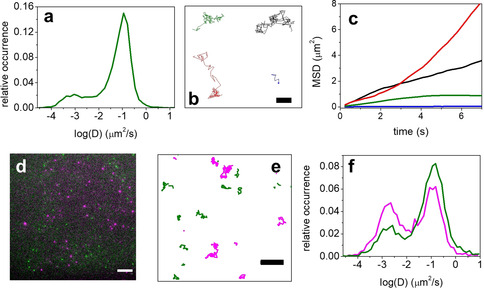
a–c) SMT of EGFR on living A431 cells exposed to 0.05 nm of anti‐EGFR aptamer (MinE07) conjugated to Atto647N. a) Distribution of measured D values for MinE07 is shown in green. b) Examples of selected long trajectories for each of these four diffusive states are shown: Brownian (black), confined (green), directed (red), and immobilization (blue). Scale bar: 2 μm. c) Corresponding mean‐square displacement (MSD) plots for four different types of EGFR motion. d–f) Two‐color single‐molecule tracking on living A431 cells exposed simultaneously to 0.05 nm AF488‐conjugated MinE07 (EGFR, green) and 0.20 nm Atto647N‐Waz aptamer (TfR, magenta). d) Representative example of one acquisition frame is displayed. Scale bar: 5 μm. e) Examples of trajectories of single EGFR (green) or TfR (magenta) on the cell surface. Scale bar: 2 μm. f) Distributions of D values calculated for the two channels (EGFR in green and TfR in magenta).
Simultaneous tracking of EGFR and TfR with MinE07 and Waz aptamers
Simultaneous SMT was performed for EGFR and a second tumor cell‐surface marker, Transferrin Receptor (TfR, CD71), on A431 cells. The Waz aptamer binds human TfR with an apparent K D in the range 200–400 nm.32, 33 A431 cells were co‐incubated with both AF488‐MinE07 and Atto647N‐Waz and excited using 488 nm and 647 nm lasers. Because EGFR is expressed on A431 cells at higher levels than TfR34, 35, 36 (Table S1), and MinE07 and Waz possess different K D values toward their respective target receptors, concentrations of the two aptamers were adjusted to 0.05 nm MinE07 and 0.20 nm Waz to achieve comparable numbers of labeled receptors in both AF488 and Atto647N channels (Video S2). Figure 2 d,e display representative examples of frame acquisition with dual‐color imaging and the corresponding trajectories obtained by SMT. Comparable numbers of trajectories for both channels were analyzed to calculate D values and their distribution (Figure 2 f). Both receptors showed mobile (D≈10−1 μm2 s−1) and immobile (D≈10−3 μm2 s−1) populations, albeit with different subpopulation ratios of these two diffusive states (Figure 2 f). EGFR exhibited a clear prevalence of mobile molecules, as observed in single‐color imaging (Figure 2 a and Figure S1). In contrast, mobile and immobile molecules were similar for TfR, consistent with previous reports.37, 38 As noted above for EGFR,28 recent work found immobile TfR to be located in proximity of clathrin‐coated pits, suggesting association with clathrin‐mediated endocytosis.37 Our findings show that high‐affinity aptamer probes, such as MinE07, are well‐suited for SMT of endogenous membrane protein receptors and for multi‐target tracking using spectrally distinct dyes.
Evaluation of the MinE07 Aptamer for SMT and PAINT
As a consequence of MinE07’s slow dissociation from EGFR (≈1×10−3 s−1,39 corresponding to t1/2≈10 min), tracking time is primarily limited by the photobleaching rate of the dye (occurring in the range of seconds). Low labeling density (up to a few tens of molecules per frame) enabled tracking receptors over seconds, as in Figure 2, while avoiding trajectory overlap. Because EGFR displays an asymmetric distribution on the cell surface and is enriched on the cell periphery,40, 41, 42 the labeling of sparse receptors was indispensable to efficiently track single EGFR at the edges of A431 cells without causing an overlap of trajectories. To achieve low labeling density and perform optimal SMT, sub‐nanomolar concentrations of MinE07 were used. Unfortunately, when using this range of concentrations, the influx rate of fresh probe is quite slow and does not fully overcome the photobleaching rate of the dye. As a result, long trajectories are recorded, but the number of tracked EGFRs decreases during acquisition time.
Overall, the high affinity and slow dissociation off‐rate of MinE07 make this aptamer ideal for applications of SMT, since target EGFRs are tracked for long time, but it shows limitations when probe concentration is increased to nanomolar range for live‐cell PAINT. In fact, to achieve simultaneous localization of multiple EGFRs (hundreds) in every acquisition frame, living A431 cells were exposed to 3 nm of MinE07 and continuous irradiation was used at relatively high power (≈1 kW cm−2). Then, single‐molecule localizations were merged to reconstruct the final PAINT images (Figure 3 a and S10a), estimating levels of cell‐surface EGFRs and their localizations. Multiple artifacts (yellow arrows) are evident in the reconstructed image of Figure 3 a, which consist of unrealistic “blacked‐out‐areas” located in close proximity to the basolateral edges of the cell. Such aberrantly irregular distribution of EGFR in PAINT images is related to the presence of multiple emitting probes located simultaneously at sub‐diffraction distance, and thereby not recognized as single molecules during the analysis procedure (Figure S10a and c).
Figure 3.
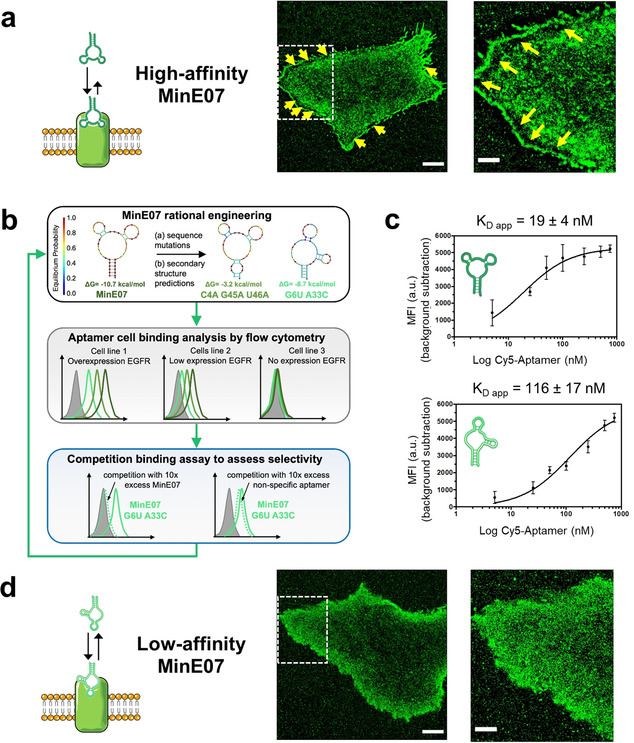
a) Reconstructed PAINT images of living A431 cells incubated with 3 nm MinE07 (high‐affinity MinE07). Scale bars: 5 μm (left) and 2 μm (right). Yellow arrows indicate aberrant irregular distribution of PAINT localizations at the edges of the cell. The image on the right shows a zoomed‐in view of the area within the dashed square. b) Workflow for aptamer engineering . Variant sequences were designed to destabilize the minimum free energy (MFE) secondary structure of MinE07 (top panel) and/or to promote alternative conformations, as shown for the MFE structure exhibited by MinE07_G6U/A33C (top panel, see also Figure S3). For each MinE07 variant, flow cytometry was used to assess binding of three cell lines that display differential expression of EGFR. Results and experimental details are reported in Figure S4‐S6. To assess retention of selectivity, candidate sequences were further tested in competition binding assays (Figure S7). c) Apparent dissociation constants (K D app) on HeLa cells were determined by flow cytometry using increasing concentrations of Cy5‐labeled anti‐EGFR aptamers (MinE07 and MinE07_G6U/A33C). Plots of median fluorescence intensity (MFI) versus aptamer concentration and apparent K D values are shown for MinE07 (top) and MinE07_G6U/A33C (bottom). Representative flow cytometry curves for all aptamer samples are shown in Figure S8. d) Reconstructed PAINT images of living A431 cells incubated with 3 nm MinE07_G6U/A33C (low‐affinity MinE07). Scale bars: 5 μm (left) and 2 μm (right). The image on the right shows a zoomed‐in view of the area within the dashed square.
This systematic undercounting is related to the long association lifetime of MinE07 to EGFR (t 1/2≈10 min39), which leads to a substantial lack of probe turnover during acquisition time (≈8 min). In fact, because of the high‐affinity, MinE07 probes tend to immediately overload areas of the cell membrane with higher EGFR levels, such as basolateral edges of A431 cells. Then, photobleaching of probes occurs during acquisition time, but MinE07 remains stably associated with EGFRs, thus preventing effective probe turnover. As a result, the localization rate during acquisition is unsteady. Emitters are initially very crowded on the cell surface and often over‐crowded at the cell edges, producing artifacts in PAINT images. Then, the density of emitting probes continuously decreases, due to photobleaching, without reaching a stable localization rate (Figure S10a and S10e), because of the inefficient replacement of photobleached probes.
Rational Engineering of a Low‐Affinity EGFR Aptamer
To overcome the limitations related to the high affinity of MinE07 aptamer when performing PAINT imaging, we generated MinE07 variants with lower affinity and faster off‐rates than MinE07 (Figure 3 b). First, MinE07 variants were designed to destabilize the MinE07 secondary structure and/or promote alternative conformations (Table S2 and Figure S3). Second, binding to EGFR‐expressing cells was tested by flow cytometry for a set of seven MinE07 variants. Only for flow cytometry, aptamers were conjugated with Cy5, and were incubated at increasing concentrations with three human cell lines that differentially express EGFR: HeLa (moderate levels43, 44), HEK293T (low levels25) and Ramos (EGFR‐negative45) (Table S1). MinE07 and all aptamer variants displayed a clear dose‐dependent staining of HeLa cells (Figure S4) relative to the low background observed with a control RNA aptamer that binds human CD4 (CD4BA),46, 47 a cell‐surface receptor expressed on normal and cancerous T cells but not on the employed solid tumor cell lines. Aptamer variants MinE07_G6U, MinE07_G6U/U46A, and MinE07_G6U/A33C showed reduced binding to HeLa cells (Figure S4), but dose‐dependent cell staining was clearly evident in both HeLa and (to a lesser extent) HEK293T cells (Figure S5 and S6). In contrast, no significant binding to the EGFR‐negative Ramos cells was observed for MinE07 or any of its variants (Figure S5 and S6). To rule out other potential mechanisms of obtaining false positive signals, two variants were further tested in competition binding assays against either MinE07 or CD4BA (Figure S7). As expected, binding of each MinE07 variant was completely inhibited upon incubation of HeLa cells with 10‐fold molar excess of MinE07, while retention of binding was observed when an excess of CD4BA was used as non‐specific competitor (Figure S7).
Finally, binding affinity to EGFR displayed on HeLa cells was quantitatively determined for MinE07_G6U/A33C, as this aptamer showed the lowest cell staining among all MinE07 variants. The apparent K D (K D app) for MinE07 (19±4 nm) indicated 6‐fold higher affinity than for MinE07_G6U/A33C (116±17 nm) (Figure 3 c and Figure S8), consistent with the reduced cell staining observed in Figure S4–S6. These results demonstrate that aptamer‐dependent cell binding is due to the presence of EGFR on the cell surface, that the magnitude of staining correlates with the reported levels of EGFR expression, and that the rationally engineered aptamer variants exhibit reduced binding of EGFR‐expressing cells relative to MinE07 while retaining the same selectivity.
Binding and dissociation kinetics of MinE07_G6U/A33C were assessed at single‐molecule level as a complement to the ensemble‐average flow cytometry measurements. Single‐molecule trajectory durations were measured at low labeling density under discontinuous excitation and were modulated on different sampling frequencies. Photobleaching linearly decreased at lower sampling frequencies, allowing the extrapolation of the dissociation rate constant to zero frequency (in absence of photobleaching).48 For both MinE07 and MinE07_G6U/A33C probes, photobleaching was considerably faster than dissociation (Figure S9). Dissociation off‐rate (k off) was too low for MinE07 to be reliably estimated with this method, consistent with a reported value of ≈1×10−3 s−1 from surface plasmon resonance.39 In contrast, k off≈4×10−2 s−1 for MinE07_G6U/A33C could be estimated, which corresponds to a dissociative half‐life of ≈15–20 seconds (Figure S9). This value is compatible with multiple association/dissociation events between EGFR and aptamer during the acquisition time of a PAINT image (minutes), enabling efficient probe turnover. Overall, estimation of the aptamer dissociation rate and ensemble measurements of apparent K D demonstrate that MinE07 (hereinafter “high‐affinity MinE07”) displayed high‐affinity and a slow off‐rate from EGFR that resembles a “catch‐and‐hold” behavior. In contrast, MinE07_G6U/A33C (hereinafter “low‐affinity MinE07”) displayed a “catch and release” behavior as a consequence of its faster off‐rate, appropriate for an efficient probe exchange on the targets over a time range of minutes.
Low‐Affinity MinE07 Outperforms High‐Affinity MinE07 as a Probe for PAINT
The faster off‐rate of low‐affinity MinE07 makes it an ideal probe when high labeling density is required as for live‐cell PAINT. When living A431 cells were incubated with 3 nm of low‐affinity MinE07 to reconstruct PAINT images (Figure 3 d), the artifacts seen for high‐affinity MinE07 were not observed. Due to its faster dissociation rate, this low‐affinity aptamer did not over‐accumulate in areas with high EGFR density, and individual EGFR molecules could be accurately localized (Figure 3 d and Figure S10b). Additionally, a steady single‐molecule localization rate can be achieved during acquisition time. Cell‐surface emitters are crowded in the initial frames of acquisition (roughly 700 frames, which are discarded during the reconstruction analysis of the PAINT images), and a fast decrease in localization rate occurs within this initial phase, due to photobleaching. Then, the density of emitting probes on the surface reaches a steady state. This leads to a constant localization rate throughout the rest of acquisition time (Figure S10), which is due to the effective replacement of photobleached probes on target receptors. Notably, these features are distinctive of photobleaching‐insensitive methodologies and allow the acquisition of PAINT images, where the number of localizations is proportional to endogenous EGFR levels on the basal membrane of living cells, while avoiding persistent or invasive EGFR staining.
Live‐Cell PAINT Defines EGFR Densities on the Surfaces of a Set of Cancer Cells
Because of the superior performance for the acquisition of PAINT images, low‐affinity MinE07 was used to qualitatively compare EGFR density on two breast cancer cells lines that express different levels of EGFR: MDA‐MB‐231 cells (moderate EGFR levels24, 49) and MCF‐7 cells (very low EGFR levels49, 50, 51) (Table S1). A431 cell line was used as a reference, and CD4BA was used as non‐targeting control aptamer. Time‐lapse sequences were recorded using 7 nm Atto647N‐conjugated aptamer probes and reconstructed PAINT images were used to calculate the localization density on the basal cell surface (Figure 4). Representative PAINT images are shown in Figure 4 a–c. There is a clear difference in EGFR localization densities among the three cell lines, which correlated with previously reported levels of EGFR. Separately, direct STORM (dSTORM)52 imaging was performed on the same cell lines to give an independent evaluation of the relative EGFR densities using a well‐established single‐molecule imaging approach. Unlike PAINT, dSTORM imaging was performed after standard chemical fixation of cells and EGFR immunostaining with a signal‐amplification system based on primary anti‐EGFR Abs and AF647‐labeled secondary Abs. The relative differences in EGFR localization densities between the cell lines obtained with dSTORM (Figure S11) closely resembled those of Figure 4. This confirms that the expected trend of cell‐surface EGFR levels on these cell lines is correctly reported by live‐cell PAINT, although differences of imaging methodology precludes a quantitative comparison.
Figure 4.
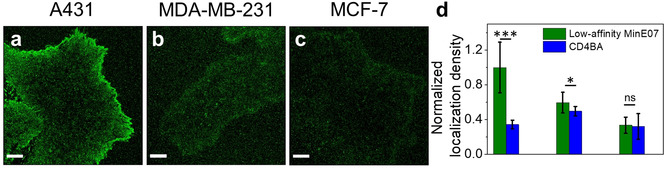
a–c) Representative reconstructed PAINT images obtained upon incubation of living cells with Atto647N‐conjugated low‐affinity MinE07 (7 nm). Scale bars 5 μm. d) Normalized localization densities on the membrane are reported for A431, MDA‐MB‐231 and MCF‐7 cells upon incubation with 7 nm low‐affinity MinE07 (green) or CD4BA (blue). Means and standard deviations are reported in histograms. Data were obtained from 10 different cells in each of two independent experiments. Statistical analysis was performed with non‐parametric Mann‐Whitney test: ***: p<0.001; **: p<0.05; *: p<0.1; ns: not statistically significant.
A431 cells displayed the highest localization densities (≈450 localizations per μm2), with a very high number of localizations concentrated at the basolateral edges of the cells (Figure 4 a). Labeling of EGFR on MDA‐MB‐231 cells (Figure 4 b) was moderate (≈250 localizations per μm2) compared to A431 cells but still detectable above background. In contrast, low‐affinity MinE07 showed minimal localizations on the cell surfaces of MCF‐7 cells (Figure 4 c) that was comparable to those obtained with aptamer CD4BA (Figure S12). Importantly, PAINT images were obtained on unperturbed living cancer cells, showing very different expression of EGFR (in the case of MCF‐7, similar to heathy epithelial cells50, 51, 53) and in absence of staining protocols that artificially amplify the signal. As for similar approaches based on reversible binders, contribution of non‐specific interactions affects the sensitivity for the detection of low amount of receptors. Strategies to improve the sensitivity of this methodology are extensively discussed in Supplementary Notes.
Simultaneous Mapping of EGFR Dynamics and Density on the Cell Surface
The optimal performance of low‐affinity MinE07 for single‐molecule imaging at high labeling density enables to simultaneously detect EGFR density by live‐cell PAINT (Figure 5 a) and to track single‐molecule motions using the same data set. Exposing living MDA‐MB‐231 cells to 17 nm of aptamer probes under continuous TIR irradiation, a high number of trajectories (between 50 000 and 70 000, with a duration rarely exceeding 1 s) could be measured with negligible overlaps and negligible contribution of bulk fluorescence, allowing prompt calculation of D and other dynamic parameters. Distribution of D values still showed two main populations of cell‐surface EGFRs (Figure S13), as seen for A431 cells (in Figure 2 a), but with a slight prevalence of nearly immobile molecules, which is likely due to increased static interactions with coverslip surface at higher probe concentrations (Figure S14). Thanks to the rapid exchange of bound and unbound low‐affinity MinE07 molecules to target receptors, the number of trajectories detected was not affected by photobleaching but was only dependent on the acquisition time. As a consequence, high trajectory density (≈50 per μm2, Figure S15a) was easily detected, in agreement with previously reported values obtained by uPAINT.15 In addition, a density map of trajectory distributions on the whole cell membrane area was calculated by binning the mass center of each trajectory with an arbitrary pixel size (Figure 5 b). Notably, because of this efficient sampling, specific subpopulations of molecules, corresponding to a statistically relevant number (thousands) of trajectories could be mapped on the cell surface (Figure 5 c–e). Trajectories obtained from the tracking analysis on MDA‐MB‐231 cells were first filtered according to values of their D or distance, then were spatially localized to obtain a 2D density map of the selected cell‐surface molecules. Importantly, when MDA‐MB‐231 cells were incubated with CD4BA, we observed significantly reduced values of trajectory density and different distributions of D values (Figure S15).
Figure 5.
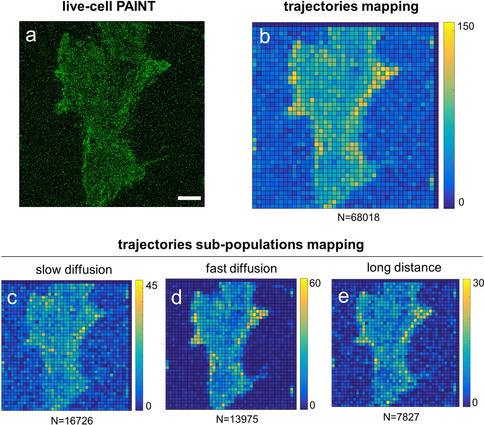
a,b) Reconstructed PAINT image (a), and density map of trajectories (>5 frames) detected by SMT (b) obtained upon incubation of living MDA‐MB‐231 cells with low‐affinity MinE07 aptamer (17 nm). Scale bars: 5 μm. c–e) Density maps of sub‐populations of trajectories were obtained after applying a filtering process (to the same field of view shown in a–c) to select molecules with (c) slow diffusion (2×10−3 μm2 s−1<D<2×10−2 μm2 s−1), (d) fast diffusion (10−1 μm2 s−1<D<1 μm2 s−1), or (e) long distance covered (1.3 μm<d<1.7 μm). Colors indicate the number of trajectories centered within each pixel of the map (size 1 μm). The color scale is reported on the bar on the right of each image, while the total number of trajectories in the map is at the bottom.
This combined SMT and live‐cell PAINT approach appears to constitute a suitable tool to investigate complex cell membrane processes occurring on the mesoscale, such as confinement of membrane components that typically appears as a relatively loose, heterogeneous, and highly dynamic spatial organization of molecules rather than a static and well‐defined structure.54 Similar combined analysis has been recently employed to study the dynamics of mesoscale confinement of CD44 receptor, bearing genetically‐encoded SNAP tag for labeling.55
In our approach, tracking of EGFRs at high labeling density is achieved using “silent labels”: small, non‐agonistic MinE07 probes, which compete with EGF but still allow formation of EGFR dimers (see Figure SN3 in Supplementary Notes) without triggering its autophosphorylation and signaling.26 Thus, these “silent labels” are suitable to monitor either basal EGFR motion and confinement in mesoscale domains or the ligand‐independent EGFR signaling. Examples of potential uses of high‐ and low‐affinity MinE07 as “silent labels” are reported in Supplementary Notes.
Conclusion
Our findings demonstrate that small, reversible aptamer probes with tunable affinity for EGFR can be exploited for the accurate tracking of individual EGFR motion or for the imaging and comparison of EGFR density levels on the membranes of several cancer cell lines. In our approach, unlike most of the current single‐molecule imaging methods, PAINT was performed directly on living cells and did not require persistent labeling of target molecules with genetically encoded tags or bulky, invasive probes such as Abs or native ligands. We demonstrated SMT of EGFR at low labeling density using a high affinity aptamer (MinE07), achieving tracking of receptor motion on the cell surface over relatively long times (seconds), essentially limited by the fluorophore photobleaching rate. Additionally, simultaneous SMT of two receptors (EGFR and TfR) was obtained using two different aptamers (MinE07 and Waz) conjugated with spectrally distinguishable dyes. In a complementary approach, live‐cell PAINT was used to localize single EGFRs at high labeling density on the cell surface and compare native expression levels of EGFR on different cell types. Because photobleaching insensitive PAINT imaging relies on transient binding kinetics and fast turnover of probes on the target, affinity probes with fast dissociation rates are required. Thus, we exploited a relatively easy and quick aptamer engineering strategy to generate anti‐EGFR aptamers (MinE07 variants) with lower affinity that retained the same selectivity as their parental sequence (high‐affinity MinE07). Importantly, a low‐affinity MinE07 aptamer, which displayed a faster dissociation from EGFR, significantly improved the performance of live‐cell PAINT. Finally, we showed that using this low‐affinity aptamer, live‐cell PAINT measurements obtained at high labeling density could be combined with SMT. Several features of individual cell‐surface receptors can be measured with statistically relevant sampling by this combined analysis, such as speed, distance and duration of individual trajectories, and trajectory density. Thus, behavior of single receptors can be mapped at a single‐cell level and using small, non‐invasive, “silent” affinity probes. Moreover, spatial organization of defined receptor subpopulations can be identified based on diffusive parameters. These results highlight the multiple possibilities offered by specifically tailored imaging probes with tunable affinity, such as aptamers. Overcoming traditional methodologies for imaging of cell‐surface receptors based on persistent labeling might open the way to the identification of multidimensional cancer‐associated surface protein signatures.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was financially supported by the Netherlands Science Organization (Grant 192.028) through the VIDI program, the Ministerio de Economía y Competitividad/Agencia Estatal de Investigación (AEI) (EURONANOMED/Acciones de Programación Conjunta Internacional PCIN‐2016‐025), the European Union's Horizon 2020 research and innovation program under the European Research Council grant ERC‐StG‐757397 (NANOSTORM) and the Marie Sklodowska‐Curie grant agreement 765497 (THERACAT), the “La Caixa” Foundation, the Generalitat de Catalunya (2017 SGR 01536), the MU Life Sciences Center (LSC) ‐ Early Concept Grant (ECG) 2019 for Innovative Collaborative Research involving Post‐Doctoral Researchers, and the UM Research and Creative Works Strategic Investment Program grant.
P. Delcanale, D. Porciani, S. Pujals, A. Jurkevich, A. Chetrusca, K. D. Tawiah, D. H. Burke, L. Albertazzi, Angew. Chem. Int. Ed. 2020, 59, 18546.
Contributor Information
Prof. Dr. Donald H. Burke, Email: burkedh@missouri.edu.
Prof. Dr. Lorenzo Albertazzi, Email: L.Albertazzi@tue.nl.
References
- 1. Stone M. B., Shelby S. A., Veatch S. L., Chem. Rev. 2017, 117, 7457–7477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sauer M., Heilemann M., Chem. Rev. 2017, 117, 7478–7509. [DOI] [PubMed] [Google Scholar]
- 3. Manzo C., Garcia-Parajo M. F., Rep. Prog. Phys. 2015, 78, 124601. [DOI] [PubMed] [Google Scholar]
- 4. Marchetti L., Bonsignore F., Gobbo F., Amodeo R., Calvello M., Jacob A., Signore G., Schirripa Spagnolo C., Porciani D., Mainardi M., Beltram F., Luin S., Cattaneo A., Proc. Natl. Acad. Sci. USA 2019, 116, 21563–21572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fornasiero E. F., Opazo F., BioEssays 2015, 37, 436–451. [DOI] [PubMed] [Google Scholar]
- 6. Cognet L., Leduc C., Lounis B., Curr. Opin. Chem. Biol. 2014, 20, 78–85. [DOI] [PubMed] [Google Scholar]
- 7. Ries J., Kaplan C., Platonova E., Eghlidi H., Ewers H., Nat. Methods 2012, 9, 582–584. [DOI] [PubMed] [Google Scholar]
- 8. Mikhaylova M., Cloin B. M. C., Finan K., van den Berg R., Teeuw J., Kijanka M. M., Sokolowski M., Katrukha E. A., Maidorn M., Opazo F., Moutel S., Vantard M., Perez F., van Bergen en Henegouwen P. M. P., Hoogenraad C. C., Ewers H., Kapitein L. C., Nat. Commun. 2015, 6, 7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schlichthaerle T., Eklund A. S., Schueder F., Strauss M. T., Tiede C., Curd A., Ries J., Peckham M., Tomlinson D. C., Jungmann R., Angew. Chem. Int. Ed. 2018, 57, 11060–11063; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 11226–11230. [Google Scholar]
- 10. Opazo F., Levy M., Byrom M., Schäfer C., Geisler C., Groemer T. W., Ellington A. D., Rizzoli S. O., Nat. Methods 2012, 9, 938–939. [DOI] [PubMed] [Google Scholar]
- 11. Strauss S., Nickels P. C., Strauss M. T., Jimenez Sabinina V., Ellenberg J., Carter J. D., Gupta S., Janjic N., Jungmann R., Nat. Methods 2018, 15, 685–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ta H., Keller J., Haltmeier M., Saka S. K., Schmied J., Opazo F., Tinnefeld P., Munk A., Hell S. W., Nat. Commun. 2015, 6, 7977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stanly T. A., Fritzsche M., Banerji S., García E., Bernardino de la Serna J., Jackson D. G., Eggeling C., Biol. Open 2016, 5, 1343–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pereira P. M., Albrecht D., Culley S., Jacobs C., Marsh M., Mercer J., Henriques R., Front. Immunol. 2019, 10, 675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Giannone G., Hosy E., Levet F., Constals A., Schulze K., Sobolevsky A. I., Rosconi M. P., Gouaux E., Tampé R., Choquet D., Cognet L., Biophys. J. 2010, 99, 1303–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Winckler P., Lartigue L., Giannone G., De Giorgi F., Ichas F., Sibarita J.-B., Lounis B., Cognet L., Sci. Rep. 2013, 3, 2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harwardt M.-L. I. E., Young P., Bleymüller W. M., Meyer T., Karathanasis C., Niemann H. H., Heilemann M., Dietz M. S., FEBS Open Bio 2017, 7, 1422–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schueder F., Stein J., Stehr F., Auer A., Sperl B., Strauss M. T., Schwille P., Jungmann R., Nat. Methods 2019, 16, 1101–1104. [DOI] [PubMed] [Google Scholar]
- 19. Spahn C., Hurter F., Glaesmann M., Karathanasis C., Lampe M., Heilemann M., Angew. Chem. Int. Ed. 2019, 58, 18835–18838; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 19011–19014. [Google Scholar]
- 20. Cheng S., Jacobson O., Zhu G., Chen Z., Liang S. H., Tian R., Yang Z., Niu G., Zhu X., Chen X., Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 948–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Porciani D., Tedeschi L., Marchetti L., Citti L., Piazza V., Beltram F., Signore G., Mol. Ther. Nucleic Acids 2015, 4, e235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Porciani D., Cardwell L. N., Tawiah K. D., Alam K. K., Lange M. J., Daniels M. A., Burke D. H., Nat. Commun. 2018, 9, 2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kusumi A., Fujiwara T. K., Morone N., Yoshida K. J., Chadda R., Xie M., Kasai R. S., Suzuki K. G. N., Semin. Cell Dev. Biol. 2012, 23, 126–144. [DOI] [PubMed] [Google Scholar]
- 24. Schaefer J. M., Barth C. W., Davis S. C., Gibbs S. L., J. Biomed. Opt. 2019, 24, 026002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Derer S., Bauer P., Lohse S., Scheel A. H., Berger S., Kellner C., Peipp M., Valerius T., J. Immunol. 2012, 189, 5230–5239. [DOI] [PubMed] [Google Scholar]
- 26. Li N., Nguyen H. H., Byrom M., Ellington A. D., PLoS ONE 2011, 6, e20299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.“Avidity Effects of MinE07, an Anti-EGFR Aptamer, on Binding to A431 Cells”: V. Avutu, The University of Texas at Austin, 2010.
- 28. Ibach J., Radon Y., Gelléri M., Sonntag M. H., Brunsveld L., Bastiaens P. I. H., Verveer P. J., PLoS ONE 2015, 10, e0143162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim D.-H., Kim D.-K., Zhou K., Park S., Kwon Y., Jeong M. G., Lee N. K., Ryu S. H., Chem. Sci. 2017, 8, 4823–4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hughes L. D., Rawle R. J., Boxer S. G., PLoS ONE 2014, 9, e87649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zanetti-Domingues L. C., Tynan C. J., Rolfe D. J., Clarke D. T., Martin-Fernandez M., PLoS ONE 2013, 8, e74200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kratschmer C., Levy M., Mol. Ther. Nucleic Acids 2018, 10, 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maier K. E., Jangra R. K., Shieh K. R., Cureton D. K., Xiao H., Snapp E. L., Whelan S. P., Chandran K., Levy M., Mol. Ther. Nucleic Acids 2016, 5, e321. [DOI] [PubMed] [Google Scholar]
- 34. Inoue T., Cavanaugh P. G., Steck P. A., Brünner N., Nicolson G. L., J. Cell. Physiol. 1993, 156, 212–217. [DOI] [PubMed] [Google Scholar]
- 35. Schonhorn J. E., Akompong T., Wessling-Resnick M., J. Biol. Chem. 1995, 270, 3698–3705. [DOI] [PubMed] [Google Scholar]
- 36. Warren R. A., Green F. A., Enns C. A., J. Biol. Chem. 1997, 272, 2116–2121. [DOI] [PubMed] [Google Scholar]
- 37. Fujiwara T. K., Iwasawa K., Kalay Z., Tsunoyama T. A., Watanabe Y., Umemura Y. M., Murakoshi H., Suzuki K. G. N., Nemoto Y. L., Morone N., Kusumi A., MBoC 2016, 27, 1101–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. De Brabander M., Nuydens R., Geerts H., Hopkins C. R., Cell Motil. Cytoskeleton 1988, 9, 30–47. [DOI] [PubMed] [Google Scholar]
- 39. Melancon M. P., Zhou M., Zhang R., Xiong C., Allen P., Wen X., Huang Q., Wallace M., Myers J. N., Stafford R. J., Liang D., Ellington A. D., Li C., ACS Nano 2014, 8, 4530–4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chibalina M. V., Poliakov A., Kendrick-Jones J., Buss F., Traffic 2010, 11, 1290–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Malliri A., Symons M., Hennigan R. F., Hurlstone A. F. L., Lamb R. F., Wheeler T., Ozanne B. W., J. Cell Biol. 1998, 143, 1087–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chung I., Akita R., Vandlen R., Toomre D., Schlessinger J., Mellman I., Nature 2010, 464, 783–787. [DOI] [PubMed] [Google Scholar]
- 43. Berkers J. A. M., van Bergen en Henegouwen P. M. P., Boonstra J., J. Biol. Chem. 1991, 266, 922–927. [PubMed] [Google Scholar]
- 44. Capuani F., Conte A., Argenzio E., Marchetti L., Priami C., Polo S., Di Fiore P. P., Sigismund S., Ciliberto A., Nat. Commun. 2015, 6, 7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hayes G. M., Chinn L., Cantor J. M., Cairns B., Levashova Z., Tran H., Velilla T., Duey D., Lippincott J., Zachwieja J., Ginsberg M. H., van der Horst E. H., Int. J. Cancer 2015, 137, 710–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Davis K., Nucleic Acids Res. 1998, 26, 3915–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wheeler L. A., Trifonova R., Vrbanac V., Basar E., McKernan S., Xu Z., Seung E., Deruaz M., Dudek T., Einarsson J. I., Yang L., Allen T. M., Luster A. D., Tager A. M., Dykxhoorn D. M., Lieberman J., J. Clin. Invest. 2011, 121, 2401–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guignet E. G., Segura J.-M., Hovius R., Vogel H., ChemPhysChem 2007, 8, 1221–1227. [DOI] [PubMed] [Google Scholar]
- 49. Moiseeva E. P., Heukers R., Manson M. M., Carcinogenesis 2007, 28, 435–445. [DOI] [PubMed] [Google Scholar]
- 50. Reilly R. M., Kiarash R., Sandhu J., Lee Y. W., Cameron R. G., Hendler A., Vallis K., Gariépy J., J. Nucl. Med. 2000, 41, 903–911. [PubMed] [Google Scholar]
- 51. Ding L., Tian C., Feng S., Fida G., Zhang C., Ma Y., Ai G., Achilefu S., Gu Y., Theranostics 2015, 5, 378–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. van de Linde S., Löschberger A., Klein T., Heidbreder M., Wolter S., Heilemann M., Sauer M., Nat. Protoc. 2011, 6, 991–1009. [DOI] [PubMed] [Google Scholar]
- 53. Smith L. M., Birrer M. J., Stampfer M. R., Brown P. H., Cancer Res. 1997, 57, 3046. [PubMed] [Google Scholar]
- 54. Kusumi A., Tsunoyama T. A., Hirosawa K. M., Kasai R. S., Fujiwara T. K., Nat. Chem. Biol. 2014, 10, 524–532. [DOI] [PubMed] [Google Scholar]
- 55. Sil P., Mateos N., Nath S., Buschow S., Manzo C., Suzuki K. G. N., Fujiwara T., Kusumi A., Garcia-Parajo M. F., Mayor S., MBoC 2019, mbc.E18-11-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary