

Abstract
Osteogenesis imperfecta (OI), also known as “brittle bone disease,” is a rare inherited genetic disorder characterized by bone fragility and often associated with short stature. The mutation in WNT1 causes autosomal recessive OI (AR‐OI) due to the key role of WNT/β‐catenin signaling in bone formation. WNT1 mutations cause phenotypes in OI of varying degrees of clinical severity, ranging from moderate to progressively deforming forms. The nucleotide change c.677C > T is one of the recurrent variants in the WNT1 alleles in Chinese AR‐OI patients. To explore the effects of mutation c.677C > T on WNT1 function, we evaluated the activation of WNT/β‐catenin signaling, cell proliferation, osteoblast differentiation, and osteoclast differentiation in WNT1c.677C>T, WNT1c.884C>A, and wild type WNT1 transfected into MC3T3‐E1 preosteoblasts. Plasmids containing wild type WNT1, WNT1c.677C>T, and WNT1c.884C>A cDNAs were constructed. Protein levels of phosphorylation at serine 9 of GSK‐3β (p‐GSK‐3β), GSK‐3β, nonphosphorylated β‐catenin (non‐p‐β‐catenin), and β‐catenin were detected with western blot. Cell proliferation was determined using MTS. BMP‐2 and RANKL mRNA and protein levels were detected by qPCR and western blot. Our results showed that WNT1c.677C>T failed to activate WNT/β‐catenin signaling and impaired the proliferation of preosteoblasts. Moreover, compared to wild type WNT1, WNT1c.677C>T downregulated BMP‐2 protein expression and was exhibited a diminished capacity to suppress the RANKL protein level. In conclusion, mutation c.677C > T hindered the ability of WNT1 to induce the WNT/β‐catenin signaling pathway and it affected the WNT/β‐catenin pathway which might potentially contribute to hampered bone homeostasis.
Keywords: AR‐OI, mutant WNT1, WNT/β‐catenin signaling pathway
1. INTRODUCTION
Osteogenesis imperfecta (OI) is a monogenic, heritable skeletal dysplasia that affects children and is characterized by increased bone fragility and decreased bone mass, leading to increased risk of fractures. Clinical manifestations of OI vary considerably among patients, from severe bone deformities or perinatal lethality to individuals with almost no fractures (Rauch & Glorieux, 2004). Approximately 90% of OI cases have an autosomal dominant pattern and are mostly caused by heterozygous mutations in COL1A1 and COL1A2, the genes encoding collagen type I α chain (Forlino, Cabral, Barnes, & Marini, 2011). Other dominant and recessive inherited forms of OI are rare and can be caused by mutations in genes involved in the posttranslational process and transportation of collagen, such as CRTAP, LEPRE1, PPIB, SERPINH1, FKBP10, SERPINF1, and BMP1 (Barbirato et al., 2015; Marini et al., 2017; Martinez‐Glez et al., 2012). Recently, several defects in new genes that are not primarily associated with collagen production have been identified as causative factors in OI, due to advances in high‐throughput sequencing. These noncollagenous OI‐causative genes are mostly implicated in osteoblast or osteoclast function and include such genes as SP7, WNT1, TMEM38B, and CREB3L1 (Cao, Zhang, & Zhang, 2019; Fiscaletti et al., 2018; Keller et al., 2018; Marini, Reich, & Smith, 2014).
WNT proteins are a family of 19 glycoproteins that play a vital role in developmental and regenerative processes in numerous tissues. Extracellular WNT ligands interact with Frizzled (Frz) receptor and lipoprotein receptor‐related protein 5 or 6 (LRP5/6) on cell membranes, leading to stabilization and nuclear accumulation of β‐catenin and subsequent transcriptional regulation of downstream genes (Baron & Rawadi, 2007; Takahashi, Maeda, Ishihara, Uehara, & Kobayashi, 2011). Numerous studies show that WNT/β‐catenin signaling is critical to bone formation and enhances osteoblast progenitor proliferation, osteoblast differentiation and survival (Baron & Kneissel, 2013), as well as indirectly repressing osteoclast differentiation and bone resorption through increased secretion of osteoprotegerin (OPG) (Kramer et al., 2010; Takahashi et al., 2011). In addition, the receptor activator of nuclear factor‐κB ligand (RANKL) is produced by osteoblasts, which can trigger the differentiation and maturation of osteoclasts by combining with RANK on the surface of preosteoclasts, and osteoprotegerin (OPG) prevents the interaction between RANK and RANKL by binding with RANKL, thus suppressing bone resorption and osteoclast differentiation (Ozer, Dagdelen, & Erbas, 2018). Moreover, WNTs participate in almost all aspects of bone health, including fetal skeletal development, bone mass accrual in childhood, and maintenance of bone homeostasis in adulthood (Baron & Kneissel, 2013).
WNT1 is a key ligand in bone. It was first found to be involved in early‐onset osteoporosis which is autosomal recessive OI (AR‐OI) in 2013. Laine et al. (2013) identified a heterozygous missense mutation c.652T > G (p.C218G) in dominantly inherited, early‐onset osteoporosis patients and a homozygous nonsense mutation c.884C > A (p.295*) in patients with severe AR‐OI. Subsequently, a growing number of studies have corroborated the association between WNT1 and AR‐OI, discovered novel mutations in WNT1, and expanded the OI spectrum. Somayyeh Fahiminiya et al. (2013) found that recessive inactivating mutations in WNT1 (homozygous missense mutation c.428G > T, homozygous frame shift deletion c.287‐300del, compound heterozygous for a frame shift insertion, and the missense mutations c.946‐949insAACA and c.1063G > T) are causes of moderate to severe OI. In 2016, Aldinger KA et al. (2016) reported a compound heterozygous mutation (c.184C > T and c.677C > T) in WNT1 in a Chinese child with severe AR‐OI accompanied by brain malformations. Recently, homozygous or compound heterozygous variants in WNT1 have been considered the major cause for OI patients of AR inheritance in East Asia (56%). The nucleotide change c.677C > T (p.Ser226Leu) is one of the recurrent variants in the WNT1 alleles in Chinese AR‐OI patients (Li et al., 2019). However, mechanism by which mutation c.677C > T causes AR‐OI is still unclear.
In this study, we constructed plasmids containing cDNAs of wild type WNT1 and WNT1 with the mutation c.677C > T (WNT1c.677C>T) and c.884C > A (WNT1c.884C>A). The WNT1c.884C>A mutation was used as the positive control group. We then evaluated the impact of WNT1c.677C>T on the WNT/β‐catenin signaling pathway, cell proliferation and differentiation of preosteoblasts to explore the effects of mutation c.677C > T on WNT1 function.
2. MATERIALS AND METHODS
2.1. Cell culture
The mouse preosteoblast cell line MC3T3‐E1 was obtained from the American Type Culture Collection (ATCC). The cells were cultured in an Alpha DMEM medium containing 10% fetal bovine serum (Gibco, Grand Island, NY, USA) and incubated at 37°C in a humidified atmosphere of 5% CO2.
2.2. Plasmid construction and transfection
Wild type and mutant WNT1 (WNT1, WNT1c.677C>T, and WNT1c.884c>A) cDNA was purchased from General Biosystems (Anhui, China) and cloned into the pcDNA3.1‐His vector. The inserts were verified by DNA sequencing. MC3T3‐E1 cells were transfected using Lipofectamin3000 (ThermoFisher, Waltham, MA, USA) according to the manufacturer's instructions. Briefly, the cells were seeded into a 6‐well plate 24 hours prior to transfection. Following this, 2 μg of each plasmid were transfected into the cells. After 72 hours, the cells were harvested for mRNA and protein expression analysis.
2.3. Quantitative real‐time PCR (qPCR)
Total RNA was isolated from cells using the TRIzol reagent (ThermoFisher, Waltham, MA, USA). The synthesis of cDNA was performed with the PrimeScript™ RT reagent Kit and gDNA Eraser (Takara, Tokyo, Japan). The qPCR was performed on a 7900HT Fast Realtime PCR system (Applied Biosystems) with GoTaq® qPCR Master Mix (Promega, Madison, WI, USA) following standard procedures. Thermal cycling conditions were as follows: 2 minutes at 50°C and 10 minutes at 95°C, followed by 40 cycles at 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 2 minutes. The housekeeping gene GAPDH was used as an internal control. The primers utilized in this study comprised the following: H‐WNT1 forward‐5′‐CGATGGTGGGGTATTGTGAAC‐3′; H‐WNT1 reverse‐5′‐CCGGATTTTGGCGTATCAGAC‐3′; Mouse‐BMP‐2 forward‐5′‐GGGACCCGCTGTCTTCTAGT; Mouse‐BMP‐2 reverse‐5′‐ TCAACTCAAATTCGCTGAGGAC; Mouse‐BMP‐4 forward‐5′‐ GAGCCATTCCGTAGTGCCAT; Mouse‐BMP‐4 reverse‐5′‐ACGACCATCAGCATTCGGTT; Mouse‐RANKL forward‐5′‐AGGCTGGGCCAAGATCTCTA; Mouse‐RANKL reverse‐5′‐GTCTGTAGGTACGCTTCCCG; GAPDH forward‐5′‐AGGTCGGTGTGAACGGATTTG‐3′; GAPDH reverse‐5′‐ TGTAGACCATGTAGTTGAGGTCA‐3′.
2.4. Western blot
The cells were lysed with RIPA buffer containing proteinase and phosphatase inhibitors (Sigma‐Aldrich, St. Louis, MO, USA). The concentration of protein was determined with the Pierce™ BCA Protein Assay Kit (ThermoFisher, Waltham, MA, USA), and equal amounts of protein were resolved by SDS‐PAGE on a 12% gel and transferred to a PVDF membrane. The membranes were blocked with 5% defatted milk for 1 hour and incubated with primary antibody anti‐WNT1 (ab15251, Abcam, Cambridge, UK), anti‐β‐catenin (sc7199, Santa Cruz, Dallas, TX, USA), anti‐GSK‐3β (ab32391, Abcam, Cambridge, UK), anti‐p‐GSK‐3β (9323s, Cell Signaling Technology, Beverly, MA, USA), anti‐non‐p‐β‐Catenin (19807T, Cell Signaling Technology, Beverly, MA, USA), anti‐BMP2 (18933‐1, Proteintech, Hubei, China), anti‐RANKL (ab45039, Abcam, Cambridge, UK), or anti‐GAPDH (60004‐1‐1 g, Proteintech, Hubei, China) overnight at 4°C. The membranes were then washed and incubated with specific secondary antibodies. Finally, the blots were visualized using chemiluminescence (ECL; Forevergen Biosciences Center, Guangzhou, China).
2.5. Cell viability test
The transfected cells were transferred into 96‐well plates and cultured for 24, and 48 hours at 37°C in a humidified 5% CO2 atmosphere. Cell viability was measured by assay (4, 5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2h‐tetrazolium (MTS) according to the standard protocol. The 10 μL MTS reagent (Promega) was added to each well and incubated with the cells for 4 hours. Absorbance was monitored using the plate reader at OD = 490 nm. All the samples were assayed in triplicate.
2.6. Statistical analysis
All experiments were repeated at least three times. Data are presented as means ± SD. A one‐way ANOVA analysis was carried out to evaluate the significant differences between groups. *p < .05 and *p < .01 were considered statistically significant.
3. RESULTS
3.1. Mutation c.677C > T impaired the capacity of WNT1 to induce the WNT/β‐catenin signaling pathway
Mutation c.677C > T in WNT1 is considered as a causative factor of AR‐OI. The NM number was NM_005430.4, the MAF was T = 0.000008/2 (GnomAD_exomes), and Rs number was rs1366948268. The nucleotide change c.677C > T in exon 4 affected the serine at position 226, which was highly conserved across species (Figure 1a). The mutation substitutes polar serine with the nonpolar hydrophobic amino acid leucine. In silico analysis predicted that the p.Ser226 Leu mutation was probably damaging (Figure 1b, c).
FIGURE 1.
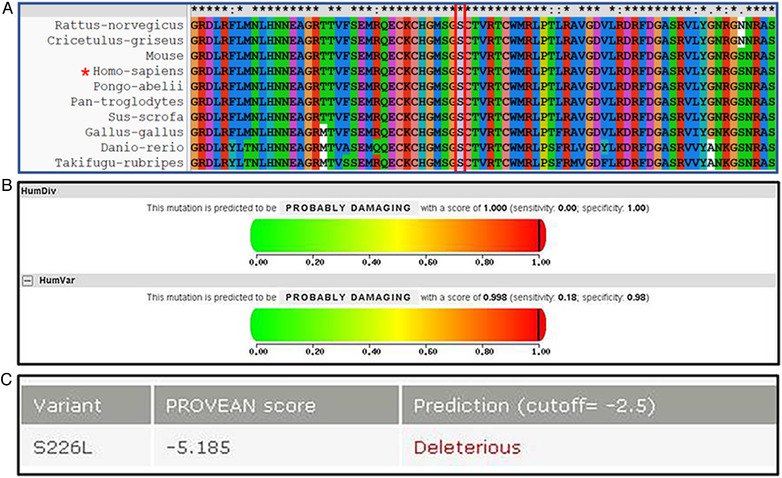
Analysis of amino acids at mutation sites of WNT1c.677C>T. (a) Protein sequence alignments of a fragment of WNT1 from diverse organisms, showing conservation of the S226 residue labeled in black frame. (b) In silico analysis of mutation c.677C > T with PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2/). (c) In silico analysis of mutation c.677C > T with SIFT (http://sift.jcvi.org/) [Color figure can be viewed at wileyonlinelibrary.com]
WNT1 with mutation c.884C > A has been found in AR‐OI patients, and this mutation shows an impaired capacity to induce canonical WNT signaling, their target genes, and mineralization in vitro (Laine et al., 2013). To explore the effect of c.667C > T mutation on WNT1 protein functions, wild type WNT1, WNT1c.677C>T, and WNT1c.884C>A plasmids were delivered into MC3T3‐E1 cells. As shown in Figure 2a, mRNAs of WNT1, WNT1c.677C>T, and WNT1c.884C>A were increased significantly in transfected cells compared to the vector group (**p < .01). Western blot results also indicated that transfections and the overexpression of wild type and mutant WNT1 were successful (Figure 2b), and WNT1 protein levels in the WNT1c.677C>T group were lower than those of wild type group (Figure 2b, c). WNT1 increased phosphorylation at serine 9 of GSK‐3β (p‐GSK‐3β) and nonphosphorylated β‐catenin (non‐p‐β‐catenin) level without a change in total GSK‐3β or β‐catenin protein levels (Figure 2b, d, e). In contrast with wild type WNT1, both WNT1c.677C>T and WNT1c.884C>A failed to alter, or only weakly altered the protein levels of p‐GSK‐3β and non‐p‐β‐catenin (Figure 2b, d, e).
FIGURE 2.
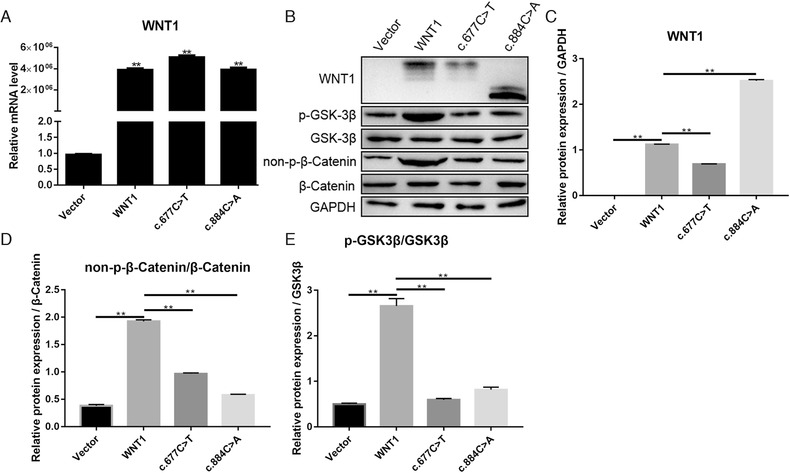
Expression level of genes in MC3T3‐E1 cells that were transfected with wild type WNT1, WNT1c.677C>T, WNT1c.884C>A, and vector. (a) At 72‐hour posttransfection, the WNT1 mRNA level in transfected cells was detected using quantitative real time PCR (qPCR). (b) WNT1, p‐GSK‐3β, GSK‐3β, non‐p‐β‐catenin and β‐catenin proteins in whole cells were detected using western blot. (c), (d), and (e) Semiquantitative analysis of proteins in graph b. **p < .01 represents the significance
3.2. WNT1c.677C>T suppressed the proliferation of preosteoblast cells
To investigate the role of mutation in preosteoblast proliferation, MTS assay was performed. As shown in Figure 3, wild type WNT1 significantly enhanced cell proliferation at 24, 48, and 72 hours, compared to the vector group. WNT1c.677C>T and WNT1c.884C>A failed to enhance the proliferation of preosteoblasts at 24, 48, and 72 hours, compared to wild type WNT1.
FIGURE 3.
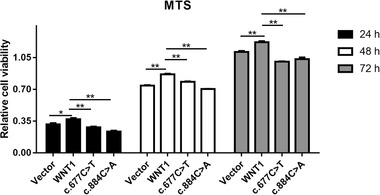
The viability of MC3T3‐E1 cells that were transfected with wild type WNT1, WNT1c.677C>T, WNT1c.884C>A, and vector for 24‐, 48‐, and 72‐hour posttransfection. *p < .05 and **p < .01 represent significant difference
3.3. Wnt1c.677C>T showed compromised capacity for enhancing osteoblast differentiation
Morphogenetic protein 4 (BMP‐4) is essential for osteoblast differentiation and osteogenesis (Wu et al., 2016). BMP‐2 is also one of the key extracellular signal molecules in bone tissue. BMP‐2 directs osteoblast differentiation and bone formation (Kaneko et al., 2000). The induction of WNT/β‐catenin signaling activates downstream gene expression, including bone morphogenetic protein 2 (BMP‐2) (Zhang et al., 2013). To evaluate the effects of mutant WNT1 on osteoblast differentiation, the mRNA expression levels of BMP‐2 and BMP‐4 were detected by qPCR. The mRNA expression level of BMP‐4 was lower in the WNT1c.677C>T mutation group than that of WNT1 wild type (Figure 4a). BMP‐2 protein level was also detected in treated cells. As expected, wild type WNT1 significantly elevated both mRNA and protein levels of BMP‐2. WNT1c.677C>T increased BMP‐2 mRNA, but failed to upregulate BMP‐2 protein. WNT1c.884C>A decreased BMP‐2 protein and mRNA (Figure 4b, d, e). The results indicated that WNT/β‐catenin signaling not only affected the BMP‐2 expression, but also might affect the BMP‐4 expression.
FIGURE 4.
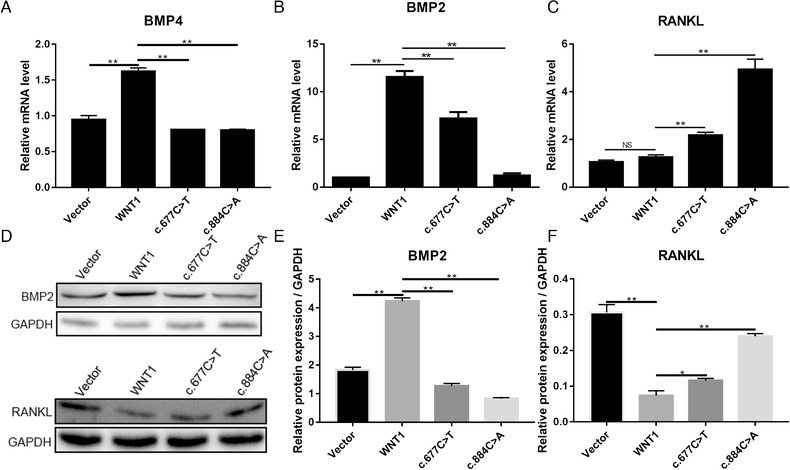
The effect of mutant WNT1 on osteoblast and osteoclast differentiation in MC3T3‐E1 cells was transfected with wild type WNT1, WNT1c.677C>T, WNT1c.884C>A, and vector. At 72‐hour posttransfection, the BMP4 (a), BMP2 (b), and RANKL (c) mRNA expression levels in transfected cells were detected using quantitative real time PCR (qPCR). (d) BMP2 and RANKL protein expression levels were detected using western blot. Semiquantitative analysis of BMP2 (e) and RANKL (f) proteins in graph d. *p < .05 and **p < .01 represent a significant difference; NG indicates no significance
RANKL, expressed by osteoblasts, is essential for osteoclast differentiation (Wright, McCarthy, Middleton, & Marshall, 2009). To explore the role of mutant WNT1 in osteoclast differentiation, qPCR and western blotting assays were performed to determine the mRNA and protein of RANKL in treated MC3T3‐E1 cells. RANKL mRNA expression level in MC3T3‐E1 transfected with wild type WNT1 was similar to that of MC3T3‐E1 treated with vector (Figure 4c). A decrease in RANKL protein was observed in cells transfected with normal WNT1 (Figure 4d). WNT1c.677C>T slightly increased RNAKL mRNA, but reduced RANKL protein expression, compared with vector group (Figure 4c, d, f). RANKL protein in the WNT1c.677C>T group was higher than in the normal WNT1 group (d, f). Our results indicated that WNT1c.677C>T failed to enhance osteoblast differentiation and showed diminished capacity for suppressing osteoclast differentiation.
4. DISCUSSION
OI is a genetic disease characterized by an increase in bone fragility and a decrease in bone mass (Moosa et al., 2019; Rauch & Glorieux, 2004). LRP5 gene mutations affecting osteoporosis‐pseudo glioma syndrome or high‐bone mass phenotype have been shown to be part of the typical WNT signaling mechanism (Baron & Kneissel, 2013). WNT ligands activate the canonical pathway through the formation of a complex of WNT, Frizzled, and LRP5 or LRP6. This complex promotes the phosphorylation of GSK‐3β at serine 9, which suppresses the kinase activity of GSK‐3β, leading to an increase in non‐p‐β‐catenin and the accumulation of non‐p‐β‐catenin in the nucleus. In the nucleus, β‐catenin interacts with T‐cell factor/lymphoid enhancer factor (TCF/LEF) family members to initiate the transcription of target genes (Baron & Rawadi, 2007). WNT/β‐catenin signaling plays an important role in skeletal development and homeostasis. Its effects begin during embryonic fetal development and continue throughout life (Liu, Kohlmeier, & Wang, 2008; Zhong, Ethen, & Williams, 2014). Mutations in genes associated with the WNT signaling pathway are involved in skeletal diseases. For instance, mutant LRP5 is associated with high‐bone mass (Roetzer et al., 2018) and osteoporosis‐pseudoglioma syndrome (Levasseur, Lacombe, & de Vernejoul, 2005), and defects in WNT1 cause early‐onset osteoporosis and OI (Laine et al., 2013). Currently, 16 variants of WNT1 are known to be involved in AR‐OI and early‐onset osteoporosis (Makitie, Kampe, Taylan, & Makitie, 2017). Interestingly, WNT1 mutations cause phenotypes in OI of varying degrees of clinical severity, ranging from moderate to progressively deforming forms (Fahiminiya et al., 2013). The nucleotide changes in c.506dupG, c.620G > A, and c.677C > T are recurrent variants in the WNT1 alleles in Chinese AR‐OI patients (Li et al., 2019).
WNT proteins consist of amino‐terminal and carboxy‐terminal domains (NTD and CTD). The NTD is composed of a cluster of α‐helices with 10 of the conserved cysteine residues forming five disulfide bridges, whereas the CTD is dominated by two β‐sheets and maintained by six disulfide bridges. NTD contains the thumb and palm regions, and CTD contains the forefinger (Janda, Waghray, Levin, Thomas, & Garcia, 2012). WNT proteins have the appearance of a hand with a thumb and forefinger protruding from a thicker palm region, reaching out to pinch the Frizzled receptor. Ser226 is located within the thumb region of NTD. The substitution of Ser by Leu likely disrupts the structure of the thumb, which is important for binding to the Frizzled receptor. The nonsense mutation c.884C < A results in a mutant WNT1 with a truncated 76‐amino‐acid at the C‐terminal end, the mutation occurs in the forefinger region. Destabilized interactions between WNT1 and Frizzled could impair the signal transduction of the canonical WNT pathway and fail to induce the accumulation of nuclear β‐catenin. Moreover, the substitution of Ser by Leu probably retains Frizzled and LRP5/6 binding but lacks the ability to induce signaling, leading to a dominant‐negative effect. Our results found that WNT1c.677C>T failed to stimulate the WNT/β‐catenin signaling pathway. Neither WNT1c.677C>T nor WNT1c.884C>A could induce p‐GSK‐3β and the accumulation of non‐p‐β‐catenin. Additionally, we found that WNT1c.677C>T and WNT1c.884C>A exhibited a dominant‐negative effect on the proliferation of preosteoblasts.
BMP‐4 is essential for osteoblast differentiation and osteogenesis (Wu et al., 2016). BMP‐2 is also one of the key extracellular signal molecules in bone tissue and an important member of the TGF‐β surperfamily that can direct osteoblast differentiation and osteogenesis (Blair et al., 2017). BMP‐2 can also induce specific transcriptional programs required for bone formation (Zhao et al., 2002). Numerous studies have demonstrated that BMP‐2 is essential to the differentiation of preosteoblastic cells into osteoblastic phenotypes. In 2013, Zhang et al. (2013) reported that the WNT/β‐catenin signaling pathway is an upstream activator of BMP2 expression in osteoblasts, and that β‐catenin directly stimulates BMP‐2 transcription through the Tcf/Lef response elements in the BMP‐2 promoter. Our results found that the mutation c.677C > T weakened the ability of WNT1 to induce BMP‐2 and BMP‐4 mRNA transcription, whereas c.884C > A nearly abolished the induction. Moreover, WNT1c.677C>T failed to significantly upregulate BMP‐2 protein, whereas WNT1c.884C>A decreased BMP‐2 protein. RANKL is an important ligand expressed by osteoblasts for osteoclast differentiation. RANKL stimulates osteoclast maturation and function through interactions with its receptor, RANK, expressed at the surface of osteoclasts (Yasuda et al., 1998). Elevated RANKL is observed in β‐catenin‐deficient osteoblasts, whereas adenomatous polyposis coli (Apc)‐deficient osteoblasts display a decreased expression of RANKL and constitutive activation of β‐catenin in the conditional Apc mutants resulted in dramatically increased bone deposition and a disappearance of osteoclasts (Glass et al., 2005; Holmen et al., 2005). Our data showed that WNT1 significantly decreased RANKL, compared with c.677C > T and c.884C > A groups, which is consistent with the results of other published studies.
5. CONCLUSIONS
The present study found that WNT1c.677C>T failed to activate WNT/β‐catenin signaling in preosteoblast cells and impaired the proliferation of preosteoblasts. Moreover, WNT1c.677C>T did not enhance BMP‐2 protein expression to promote osteoblast differentiation and exhibited a diminished capacity to suppress osteoclast differentiation by decreasing the RANKL protein level. The results of this study confirm that WNT1c.677C>T may play an important role in osteogenesis imperfecta.
FUNDING SOURCE
This work was supported by Dongguan Social Science and Technology Development Project (NO. 2018507150011653).
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Bashan Zhang was involved in the design and analysis of the experiment. Bashan Zhang, Rong Li, Wenfeng Wang, Xueming Zhou, Beijing Luo, Zinian Zhu, Xibo Zhang, Aijiao Ding contributed to the experiment.
Zhang B, Li R, Wang W, et al. The role of WNT1 mutant variant (WNT1) in osteogenesis imperfecta. Ann Hum Genet. 2020;84:447–455. 10.1111/ahg.12399
[The copyright line for this article was changed on 26 August 2020 after original online publication.]
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Aldinger, K. A. , Mendelsohn, N. J. , Chung, B. H. , Zhang, W. , Cohn, D. H. , Fernandez, B. , … Curry, C. J. (2016). Variable brain phenotype primarily affects the brainstem and cerebellum in patients with osteogenesis imperfecta caused by recessive WNT1 mutations. Journal of Medical Genetics, 53(6), 427–430. 10.1136/jmedgenet-2015-103476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbirato, C. , Trancozo, M. , Almeida, M. G. , Almeida, L. S. , Santos, T. O. , Duarte, J. C. , … Paula, F. (2015). Mutational characterization of the P3H1/CRTAP/CypB complex in recessive osteogenesis imperfecta. Genetics and Molecular Research [Electronic Resource], 14(4), 15848–15858. 10.4238/2015.December.1.36 [DOI] [PubMed] [Google Scholar]
- Baron, R. , & Kneissel, M. (2013). WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nature Medicine, 19(2), 179–192. 10.1038/nm.3074 [DOI] [PubMed] [Google Scholar]
- Baron, R. , & Rawadi, G. (2007). Wnt signaling and the regulation of bone mass. Current Osteoporosis Reports, 5(2), 73–80. [DOI] [PubMed] [Google Scholar]
- Blair, H. C. , Larrouture, Q. C. , Li, Y. , Lin, H. , Beer‐Stoltz, D. , Liu, L. , … Nelson, D. J. (2017). Osteoblast differentiation and bone matrix formation in vivo and in vitro. Tissue Engineering Part B: Reviews, 23(3), 268–280. 10.1089/ten.TEB.2016.0454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, Y. J. , Zhang, H. , & Zhang, Z. L. (2019). Novel mutations in the Wnt1, Tmem38b, P4hb, and Pls3 genes in four unrelated Chinese families with osteogenesis imperfecta. Endocrine Practice, 25(3), 230–241. 10.4158/EP-2018-0443 [DOI] [PubMed] [Google Scholar]
- Fahiminiya, S. , Majewski, J. , Mort, J. , Moffatt, P. , Glorieux, F. H. , & Rauch, F. (2013). Mutations in WNT1 are a cause of osteogenesis imperfecta. Journal of Medical Genetics, 50(5), 345–348. 10.1136/jmedgenet-2013-101567 [DOI] [PubMed] [Google Scholar]
- Fiscaletti, M. , Biggin, A. , Bennetts, B. , Wong, K. , Briody, J. , Pacey, V. , … Munns, C. F. (2018). Novel variant in Sp7/Osx associated with recessive osteogenesis imperfecta with bone fragility and hearing impairment. Bone, 110, 66–75. 10.1016/j.bone.2018.01.031 [DOI] [PubMed] [Google Scholar]
- Forlino, A. , Cabral, W. A. , Barnes, A. M. , & Marini, J. C. (2011). New perspectives on osteogenesis imperfecta. Nature Reviews Endocrinology, 7(9), 540–557. 10.1038/nrendo.2011.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass, D. A. , 2nd, Bialek, P., Ahn, J. D., Starbuck, M. , Patel, M. S. , Clevers, H. , … Karsenty, G. (2005). Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Developmental Cell, 8(5), 751–764. 10.1016/j.devcel.2005.02.017 [DOI] [PubMed] [Google Scholar]
- Holmen, S. L. , Zylstra, C. R. , Mukherjee, A. , Sigler, R. E. , Faugere, M. C. , Bouxsein, M. L. , … Williams, B. O. (2005). Essential role of beta‐catenin in postnatal bone acquisition. Journal of Biological Chemistry, 280(22), 21162–21168. 10.1074/jbc.M501900200 [DOI] [PubMed] [Google Scholar]
- Janda, C. Y. , Waghray, D. , Levin, A. M. , Thomas, C. , & Garcia, K. C. (2012). Structural basis of Wnt recognition by Frizzled. Science, 337(6090), 59–64. 10.1126/science.1222879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko, H. , Arakawa, T. , Mano, H. , Kaneda, T. , Ogasawara, A. , Nakagawa, M. , … Hakeda, Y. (2000). Direct stimulation of osteoclastic bone resorption by bone morphogenetic protein (BMP)‐2 and expression of BMP receptors in mature osteoclasts. Bone, 27(4), 479–486. [DOI] [PubMed] [Google Scholar]
- Keller, R. B. , Tran, T. T. , Pyott, S. M. , Pepin, M. G. , Savarirayan, R. , McGillivray, G. , … Byers, P. H. (2018). Monoallelic and biallelic CREB3L1 variant causes mild and severe osteogenesis imperfecta, respectively. Genetics in Medicine, 20(4), 411–419. 10.1038/gim.2017.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer, I. , Halleux, C. , Keller, H. , Pegurri, M. , Gooi, J. H. , Weber, P. B. , … Kneissel, M. (2010). Osteocyte Wnt/beta‐catenin signaling is required for normal bone homeostasis. Molecular and Cellular Biology, 30(12), 3071–3085. 10.1128/MCB.01428-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine, C. M. , Joeng, K. S. , Campeau, P. M. , Kiviranta, R. , Tarkkonen, K. , Grover, M. , … Makitie, O. (2013). WNT1 mutations in early‐onset osteoporosis and osteogenesis imperfecta. New England Journal of Medicine, 368(19), 1809–1816. 10.1056/NEJMoa1215458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levasseur, R. , Lacombe, D. , & de Vernejoul, M. C. (2005). LRP5 mutations in osteoporosis‐pseudoglioma syndrome and high‐bone‐mass disorders. Joint, Bone, Spine, 72(3), 207–214. 10.1016/j.jbspin.2004.10.008 [DOI] [PubMed] [Google Scholar]
- Li, L. , Mao, B. , Li, S. , Xiao, J. , Wang, H. , Zhang, J. , … Zhang, X. (2019). Genotypic and phenotypic characterization of Chinese patients with osteogenesis imperfecta. Human Mutation, 40(5), 588–600. 10.1002/humu.23718 [DOI] [PubMed] [Google Scholar]
- Liu, F. , Kohlmeier, S. , & Wang, C. Y. (2008). Wnt signaling and skeletal development. Cellular Signalling, 20(6), 999–1009. 10.1016/j.cellsig.2007.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makitie, R. E. , Kampe, A. J. , Taylan, F. , & Makitie, O. (2017). Recent discoveries in monogenic disorders of childhood bone fragility. Current Osteoporosis Reports, 15(4), 303–310. 10.1007/s11914-017-0388-6 [DOI] [PubMed] [Google Scholar]
- Marini, J. C. , Forlino, A. , Bachinger, H. P. , Bishop, N. J. , Byers, P. H. , Paepe, A. , … Semler, O. (2017). Osteogenesis imperfecta. Nature reviews Disease primers, 3, 17052 10.1038/nrdp.2017.52 [DOI] [PubMed] [Google Scholar]
- Marini, J. C. , Reich, A. , & Smith, S. M. (2014). Osteogenesis imperfecta due to mutations in non‐collagenous genes: Lessons in the biology of bone formation. Current Opinion in Pediatrics, 26(4), 500–507. 10.1097/MOP.0000000000000117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Glez, V. , Valencia, M. , Caparros‐Martin, J. A. , Aglan, M. , Temtamy, S. , Tenorio, J. , … Ruiz‐Perez, V. L. (2012). Identification of a mutation causing deficient BMP1/mTLD proteolytic activity in autosomal recessive osteogenesis imperfecta. Human Mutation, 33(2), 343–350. 10.1002/humu.21647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosa, S. , Yamamoto, G. L. , Garbes, L. , Keupp, K. , Beleza‐Meireles, A. , Moreno, C. A. , … Netzer, C. (2019). Autosomal‐recessive mutations in MESD cause osteogenesis imperfecta. American Journal of Human Genetics, 105(4), 836–843. 10.1016/j.ajhg.2019.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozer, F. F. , Dagdelen, S. , & Erbas, T. (2018). Relation of RANKL and OPG levels with bone resorption in patients with acromegaly and prolactinoma. Hormone and Metabolic Research, 50(7), 562–567. 10.1055/a-0630-1529 [DOI] [PubMed] [Google Scholar]
- Rauch, F. , & Glorieux, F. H. (2004). Osteogenesis imperfecta. Lancet, 363(9418), 1377–1385. 10.1016/s0140-6736(04)16051-0 [DOI] [PubMed] [Google Scholar]
- Roetzer, K. M. , Uyanik, G. , Brehm, A. , Zwerina, J. , Zandieh, S. , Czech, T. , … Klaushofer, K. (2018). Novel familial mutation of LRP5 causing high bone mass: Genetic analysis, clinical presentation, and characterization of bone matrix mineralization. Bone, 107, 154–160. 10.1016/j.bone.2017.12.002 [DOI] [PubMed] [Google Scholar]
- Takahashi, N. , Maeda, K. , Ishihara, A. , Uehara, S. , & Kobayashi, Y. (2011). Regulatory mechanism of osteoclastogenesis by RANKL and Wnt signals. Frontiers in Bioscience (Landmark Ed), 16, 21–30. [DOI] [PubMed] [Google Scholar]
- Wright, H. L. , McCarthy, H. S. , Middleton, J. , & Marshall, M. J. (2009). RANK, RANKL and osteoprotegerin in bone biology and disease. Current Reviews in Musculoskeletal Medicine, 2(1), 56–64. 10.1007/s12178-009-9046-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, L. A. , Wang, F. , Donly, K. J. , Baker, A. , Wan, C. , Luo, D. , … Chen, S. (2016). Establishment of immortalized BMP2/4 double knock‐out osteoblastic cells is essential for study of osteoblast growth, differentiation, and osteogenesis. Journal of Cellular Physiology, 231(6), 1189–1198. 10.1002/jcp.25266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda, H. , Shima, N. , Nakagawa, N. , Yamaguchi, K. , Kinosaki, M. , Mochizuki, S. , … Suda, T. (1998). Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis‐inhibitory factor and is identical to TRANCE/RANKL. PNAS, 95(7), 3597–3602. 10.1073/pnas.95.7.3597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, R. , Oyajobi, B. O. , Harris, S. E. , Chen, D. , Tsao, C. , Deng, H. W. , & Zhao, M. (2013). Wnt/beta‐catenin signaling activates bone morphogenetic protein 2 expression in osteoblasts. Bone, 52(1), 145–156. 10.1016/j.bone.2012.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, M. , Harris, S. E. , Horn, D. , Geng, Z. , Nishimura, R. , Mundy, G. R. , & Chen, D. (2002). Bone morphogenetic protein receptor signaling is necessary for normal murine postnatal bone formation. Journal of Cell Biology, 157(6), 1049–1060. 10.1083/jcb.200109012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, Z. , Ethen, N. J. , & Williams, B. O. (2014). WNT signaling in bone development and homeostasis. Wiley Interdisciplinary Reviews‐Developmental Biology, 3(6), 489–500. 10.1002/wdev.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.