

Abstract
Adaptivity is an essential trait of life. One type of adaptivity is the reconfiguration of a functional system states by correlating sensory inputs. We report polymer transformers, which can adaptively reconfigure their composition from a state of a mixed copolymer to being enriched in either monomer A or B. This is achieved by embedding and hierarchically interconnecting two chemically fueled activation/deactivation enzymatic reaction networks for both monomers via a joint activation pathway (network level) and an AB linker monomer reactive to both A and B (species level). The ratio of enzymes governing the individual deactivation pathways (our external signals) control the enrichment behavior in the dynamic state. The method shows high programmability of the reconfigured state, rejuvenation of transformation cycles, and quick in situ adaptation. As a proof‐of‐concept, we showcase this dynamic reconfiguration for colloidal surface functionalities.
Keywords: adaptive materials, polymer chemistry, supramolecular chemistry, systems chemistry
A strategy, called polymer transformers, is developed to adaptively reconfigure functional polymer states with the possibility for rejuvenation and reconfiguration into different outputs in new cycles. This is achieved by interdigitating two ATP‐powered enzymatic reaction networks of dynamically polymerizing monomer mixtures into a joint system.
Signal correlation is a key function in living organisms to realize adaptation and evolution.1 For example, stem cells can differentiate into different lineages by correlating mechanical, topographical, and (bio)chemical sensory inputs using (bio)chemical reaction networks.2 Induced pluripotent stem cells can even regenerate from a mature state and then transform to different functions.3 Although these abilities for adaptive reconfiguration of functional states are of formidable complexity, an abstraction allows to define one subset of adaptive behavior in synthetic systems. This is the ability to form distinct functional outcomes depending on the strength of different signals in complex sensory landscapes, and to be able to reconfigure the outcome quickly and potentially repeatedly.
For future synthetic materials systems, it is important to develop methods to endow systems with capacities for adaptivity and reconfiguration so that they can be transformed into other desired functions when meeting different environments. Dynamically self‐assembling systems with controlled subunit exchange are a favorable starting point as dynamics provide an inherent capacity for fast reorganization. This is seen in the dissipative self‐assembly of the microtubules in cells, which are in a dynamic flux‐like state so that the cytoskeleton can quickly reconfigure into new spatial patterns.4 Synthetic out‐of‐equilibrium systems fueled by chemical energy have been targeted for a range of systems,5, 6, 7, 8, 9, 10, 11, 12 but structural dynamics, which is a key for quick reconfiguration, has thus far only been carefully shown in limited systems.13, 14, 15, 16
Inspired by the abilities for reconfiguration of living systems, we report an abstracted chemical systems approach, termed polymer transformers, which allows the reconfiguration of functional material states by embedding the underlying species into two interdigitated enzymatic reaction networks (iERNs). The initial material state can transform into other states following certain signal conditions, return to the initial state, and then be reconfigured to a different state. Enzymes are used as signaling units to decisively influence the outcome of iERNs. Small DNA‐based monomers, ME, MN, ML, that dynamically polymerize into long polymer strands using ERNs of an ATP‐powered T4 DNA ligase‐mediated polymerization and concurrent restriction/cutting by restriction endonucleases (REases), constitute the (co)polymers for reconfiguration (Figure 1). DNA monomers are models for synthetic polymers, as they can advantageously be manipulated by enzymes as signal processors in a highly defined fashion.17 Fluorophores attached to ME and MN describe a first functional output on a polymer level.
Figure 1.
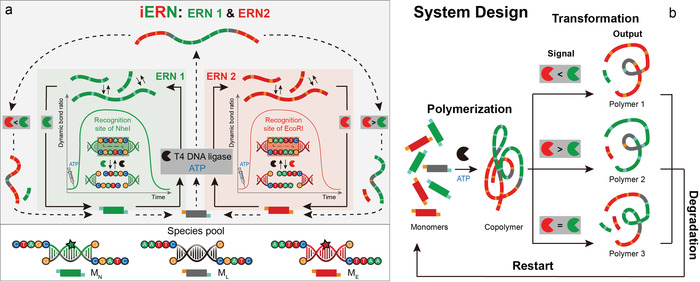
a) iERN and b) polymer transformers reconfiguring their functional state via iERN.
The interdigitation of the iERNs occurs on two levels: Individually, MN and ME can operate in their own ERN1 and ERN2, respectively. When mixed into one system, a connection on the ERN level occurs, because the ligase is jointly used. Upon addition of the heterolinker ML, the interconnection occurs additionally on the building block, and, hence, on the structural level (Figure 1 a; Supporting Information, Figure S1). Previously, for simple, single‐species ERNs of this type, we confirmed that i) the degree of ligation in the dynamic steady state (DySS) plateau depends on the ratio of ligase/REase, ii) structural dynamics (constant bond shuffling and subunit exchange) occurs, and iii) the lifetimes depend on the ATP concentration.16, 18, 19
The adaptive transformation will operate as follows (Figure 1 b): First, ligase (black) and ATP polymerize the three monomers into a mixed copolymer, serving as a homogenous initial material state. Afterwards, we add two REases (green, red) as signals with a certain ratio to dynamize the system. This leads to a transformation into different copolymer types that enrich selected monomers and sort out the other, depending on the kinetic balance of iERN as instructed by the REases ratio. The quick reconfiguration is allowed by the dynamics of the system, which is in constant balance between ligation and cutting. As ATP is consumed, all copolymers degrade into the initial monomers owing to cleavage by REases. After heat inactivation of the enzymes, a new round of transformations can start.
We first identified two REases, EcoRI and NheI, that can be operated at similar activities (Supporting Information, Figure S2). Before targeting the transformation, we elucidate the kinetics of the ATP‐fueled transient growth of ME and MN as a function of the REase ratio in a joint reaction. ML is absent, causing individual homopolymerizations. Since ME and MN share the ligase, the system is mainly controlled by the activity of the REases. Figure 2 a–d displays time‐dependent agarose gel electrophoresis (AGE) of transient polymerizations at different ratios of EcoRI and NheI, which can be quantified as mass‐weighted average chain length distributions in base pairs ( ; Figure 2 e–h, details of analysis in Ref. [16] and the Supporting Information; please see definitions of enzyme activity U in the Supporting Information). When EcoRI cutting activity is higher than NheI (Figure 2 a), red ME can only grow to oligomers (ca. 90 bp), while green MN reaches a longer polymer state (ca. 260 bp). This is due to more frequent cleavage by EcoRI, causing a lower degree of ligation and hence a shortened . Reduction of [EcoRI] to 8.0 and 5.0 U μL−1 (constant [NheI]=1.0 U μL−1) first doubles the ME chain length to ca. 180 bp and then allows ME to reach similar levels as MN (ca. 350 bp). An inversion, meaning high of MN and low of ME, is obtained by inverting the REase ratio (Figures 2 d,h). Both networks, ERN1 and ERN2, are clearly connected via the activation pathway (shared ligase; Figure 2 a–c,e–g). Upon reduction of [EcoRI] from 10 to 5 U μL−1, both of ME and MN increase noticeably. This is because the ligase is less required in ligating the ME sequences at lower [EcoRI], and hence more available for ligating longer MN chains. These results confirm that tuning the cutting kinetics with different REase ratios enables two DNA polymers to be simultaneously grown to a tunable in the presence of shared ligase. Yet, first level interdigitation effects by joint use of the activation reaction need to be considered.
Figure 2.
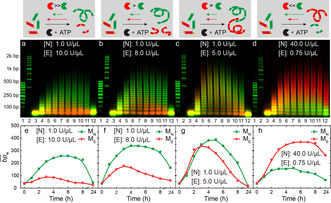
REase cutting kinetics govern transient homopolymerizations. a)–d) Time‐dependent polymerizations and e–h) developments of with different ratios of EcoRI ([E]) and NheI ([N]) when ME and MN shared the ligase and ATP. Lane assignment (individual channels in Figure S3): 1. fluorescent 50 bp DNA ladder; 2. fluorescent 200 bp DNA ladder; 3. 0 h; 4. 1 h; 5. 2 h; 6. 3 h; 7. 4 h; 8. 5 h; 9. 6 h; 10. 7 h; 11. 8 h; 12. 24 h.
We next target distinct reconfiguration by incorporating the heterolinker ML. This further interdigitates the two ERNs on a species and structural level, and, critically, forces copolymer formation. An ATP‐driven ligase‐mediated polymerization of [ME]/[MN]/[ML]=3:3:1 gives a mixed, static copolymer (Pinitial; Lane 5 in Figures 3 a–d). Subsequently, we demonstrate its transformation by adding various ratios of EcoRI and NheI, aiming to enrich or deplete the copolymer structure from one of its species. Figure 3 a shows that a high NheI activity transforms the orange Pinitial to a polymer containing almost exclusive red fluorescence. This means that the balance in iERN kinetics reconfigures the copolymer to be mainly composed of ME, while MN are mostly sorted out as monomers, because the excessive cutting frequency leads to a low degree of ligation. The overall lower values of the dynamic structures after adding the REases compared to Pinitial originate from entering the DySS polymerization with a ligase/REase balance. Using a REase balance with a stronger EcoRI activity, the situation can be inverted into enrichment in MN (green polymers; Figure 3 b). Also, a balance with a mixed copolymer structure can be obtained for similar cutting frequency of both REases (Figure 3 c). A symmetric reduction of the REase concentrations, while maintaining their ratio, leads to higher , because lower overall [REase] causes higher degree of ligation (Figures 3 c,g/d,h).
Figure 3.
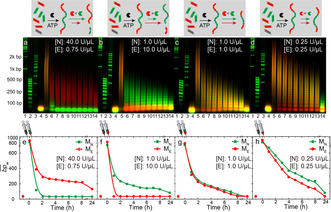
REase ratios and concentrations govern transformations. a)–d) Time‐dependent polymerizations and e)–h) developments of with different ratios/concentrations of EcoRI and NheI when containing ME, MN, and ML. Lane assignment (individual channels in the Supporting Information, Figure S4): 1. fluorescent 1k bp DNA ladder; 2. fluorescent 50 bp DNA ladder; 3. fluorescent 200 bp DNA ladder; 4. before ligation; 5. after 16 h ligation; adding REases: 6. 1 h; 7. 2 h; 8. 3 h; 9. 4 h; 10. 5 h; 11. 6 h; 12. 7 h; 13. 8 h; 14. 24 h.
These results confirm that this transformation process is governed by the kinetics of REases. Appropriate tuning allows to reconfigure one state (fully mixed copolymer with green and red fluorescence) into a new state with different constitutions and controllable material compositions (largely one type of sorted homopolymer with red/green fluorescence). We also find that more heterolinkers ML or less total monomer concentration can cause a shorter chain length (Supporting Information, Figures S5, S6). Although fluorescence is used as a model function, it is obvious that conceptually other physical properties can be changed.20
Next, we integrated the concept into a series to consecutively transform the same system and sort out different enriched states from an originally mixed Pinitial (Figures 4 a,b). In Round 1, after obtaining Pinitial, a REase mixture with more EcoRI was added for dynamization. As expected, red ME is sorted out and a green polymer enriched in MN appears. This degrades over time due to ATP consumption. Before Round 2, heating to 85 °C (1 h) was used to inactivate all enzymes. We began Round 2 with ligase and ATP to reobtain Pinitial followed by dynamization with REases containing less EcoRI, leading to a reconfigured polymer enriched in red ME. For Round 3, after generating Pinitial, the system is transformed back to the state of Round 1, although the activities of the enzymes have a slight loss owing to the accumulation of glycerol of repeated enzyme addition (Supporting Information, Figure S8).
Figure 4.
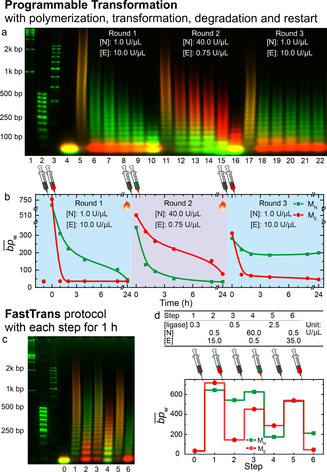
Programmable transformation and FastTrans protocol. Time‐dependent polymerizations and developments of of a),b) programmability and c),d) FastTrans protocol. Lane assignment for (a) (individual channels in the Supporting Information, Figure S7): 1. fluorescent 1k bp DNA ladder; 2. fluorescent 50 bp DNA ladder; 3. fluorescent 200 bp DNA ladder; 4. before ligation; Round 1: 5. after 16 h ligation; enzyme heating deactivation and adding REases: 6. 1 h; 7. 2 h; 8. 4 h; 9. 8 h; 10. 24 h; Round 2: 11. after 16 h ligation; enzyme heating deactivation and adding REases: 12. 1 h; 13. 2 h; 14. 4 h; 15. 8 h; 16. 24 h; Round 3: 17. after 16 h ligation; enzyme heating deactivation and adding REases: 18. 1 h; 19. 2 h; 20. 4 h; 21. 8 h; 22. 24 h. Lane assignment for (c) (individual channels in the Supporting Information, Figure S9): 0. before ligation; 1–6. Step 1–6.
We further developed a FastTrans protocol for rapid and in situ transformation, in which each step finishes in only one hour and where the enzyme inactivation is omitted (Figures 4 c,d). By adding the enzymes consecutively, a homogeneous polymer with little sorting is generated at the start, as well as for later steps by ligase (re)supply (Steps 1,3,5), whereas the sorting to enriched green MN polymers (Steps 2,6), or red ME polymers (Step 4) requires the indicated REase ratios to favor restriction appropriately. Although the approach is less effective owing to enzyme accumulation, the in situ transformations with sorting into preferred polymers of one color/monomer and short oligomers of the other color/monomer and re‐homogenization are clearly visible in AGE and length evaluation. This approach would be beneficial when handling temperature‐sensitive components, where no heating rejuvenation can be done.
To demonstrate a broader applicability, we showcase the reconfiguration of the surface functionality on colloids as important soft matter model systems21 (Figure 5; Supporting Information, Figures S10–S12). After growing Pinitial chains on streptavidinylated beads using biotinylated DNA primers, we incubated these beads with different REase ratios to adaptively dynamize the surface‐grown polymers. When exposed to a high EcoRI activity, the initial colloid surface with a mixed red and green fluorescence reconfigures to one with stronger green MN and low red ME (Figures 5 a,b). The color channel‐specific intensity distributions across a particle clearly show the pronounced changes and underscore that the surface coating has strongly sorted out ME and is enriched in MN. The inverse result with more ME on the colloidal surface is obtained for higher NheI activity (Figure 5 c,f). When realizing similar activities of EcoRI and NheI, a surface layer containing comparable ME and MN is present (Figure 5 d,g). The reconfiguration on the surface can be obtained because the iERN dynamizes the whole polymer structure and leads to constant bond shuffling and unit exchange.
Figure 5.
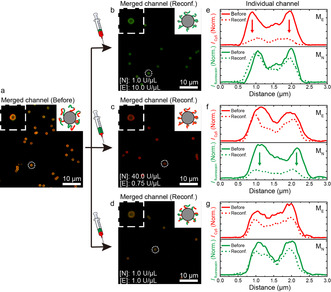
Transformation of colloidal surface functions. Confocal fluorescence images and fluorescence intensity changes for polymer transformation on the bead surfaces a) before and with b),e) stronger EcoRI, c),f) stronger NheI, and d),g) similar enzymatic activity for 3 h.
In summary, we introduced iERNs to demonstrate a simple and efficient concept, polymer transformers, that can adaptively reconfigure their state from mixed copolymers to sorted out polymer structures that are enriched in one monomer. REases which act on the individual species are used as signals for the adaptive response. The heterolinker (and to a lesser extent the joint use of the activating reaction) is the important component to interdigitate both otherwise independent ERN‐based steady state structures. Driving the system in an ATP‐driven DySS with control over the subunit exchange allows to implement high levels of dynamics and fine‐tune the sorting out phenomena. Using this method, we can obtain various polymers with different compositions from the same starting material by simply tuning the REase ratio because of the enzymatically controlled process. The method is robust and allows repeated rejuvenation and several consecutive transformation cycles, even with a FastTrans protocol to achieve the in situ transformation in only one hour. As a first proof‐of‐concept application, we showcased reconfiguration of colloid surface functions. This method will promote concepts for adaptive materials design and assist the systems chemistry‐based development of more intelligent materials design and applications, such as robotics or self‐regulation systems. On a conceptual level, we believe that related approaches may be feasible in supramolecular chemistry, or classical polymer science, which however requires to improve regulatory networks in these systems.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy—EXC‐2193/1‐390951807 via “Living, Adaptive and Energy‐Autonomous Materials Systems” (livMatS). We acknowledge support by the European Research Council starting Grant (TimeProSAMat) Agreement 677960. Open access funding enabled and organized by Projekt DEAL.
M. Sun, J. Deng, A. Walther, Angew. Chem. Int. Ed. 2020, 59, 18161.
References
- 1. Walther A., Adv. Mater. 2020, 32, 1905111. [Google Scholar]
- 2. De Luca M., Aiuti A., Cossu G., Parmar M., Pellegrini G., Robey P. G., Nat. Cell Biol. 2019, 21, 801–811. [DOI] [PubMed] [Google Scholar]
- 3. Rowe R. G., Daley G. Q., Nat. Rev. Genet. 2019, 20, 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cheeseman I. M., Desai A., Nat. Rev. Mol. Cell Biol. 2008, 9, 33–46. [DOI] [PubMed] [Google Scholar]
- 5. Boekhoven J., Hendriksen W. E., Koper G. J., Eelkema R., van Esch J. H., Science 2015, 349, 1075. [DOI] [PubMed] [Google Scholar]
- 6. Merindol R., Walther A., Chem. Soc. Rev. 2017, 46, 5588–5619. [DOI] [PubMed] [Google Scholar]
- 7. Mattia E., Otto S., Nat. Nanotechnol. 2015, 10, 111–119. [DOI] [PubMed] [Google Scholar]
- 8. Rieß B., Grötsch R. K., Boekhoven J., Chem 2020, 6, 552–578. [Google Scholar]
- 9. van Rossum S. A. P., Tena-Solsona M., van Esch J. H., Eelkema R., Boekhoven J., Chem. Soc. Rev. 2017, 46, 5519–5535. [DOI] [PubMed] [Google Scholar]
- 10. Singh N., Formon G. J. M., De Piccoli S., Hermans T. M., Adv. Mater. 2020, 32, 1906834. [DOI] [PubMed] [Google Scholar]
- 11. Te Brinke E., Groen J., Herrmann A., Heus H. A., Rivas G., Spruijt E., Huck W. T. S., Nat. Nanotechnol. 2018, 13, 849–855. [DOI] [PubMed] [Google Scholar]
- 12. Kundu P. K., Samanta D., Leizrowice R., Margulis B., Zhao H., Borner M., Udayabhaskararao T., Manna D., Klajn R., Nat. Chem. 2015, 7, 646–652. [DOI] [PubMed] [Google Scholar]
- 13. van Ravensteijn B. G. P., Hendriksen W. E., Eelkema R., van Esch J. H., Kegel W. K., J. Am. Chem. Soc. 2017, 139, 9763–9766. [DOI] [PubMed] [Google Scholar]
- 14. Ogden W. A., Guan Z., ChemSystemsChem 2020, 2, e1900030. [Google Scholar]
- 15. Heuser T., Weyandt E., Walther A., Angew. Chem. Int. Ed. 2015, 54, 13258–13262; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13456–13460. [Google Scholar]
- 16. Heinen L., Walther A., Sci. Adv. 2019, 5, eaaw0590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Luo D., Mater. Today 2003, 6, 38–43. [Google Scholar]
- 18. Deng J., Walther A., J. Am. Chem. Soc. 2020, 142, 685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deng J., Bezold D., Jessen H., Walther A., Angew. Chem. Int. Ed. 2020, 59, 12084–12092; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 12182–12190. [Google Scholar]
- 20. Sun X., Chwatko M., Lee D. H., Bachman J. L., Reuther J. F., Lynd N. A., Anslyn E. V., J. Am. Chem. Soc. 2020, 142, 3913–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lin H., Lee S., Sun L., Spellings M., Engel M., Glotzer S. C., Mirkin C. A., Science 2017, 355, 931. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary