

Abstract
Mutations in LMNA encoding lamin A/C and EMD encoding emerin cause cardiomyopathy and muscular dystrophy. Lmna null mice develop these disorders and have a lifespan of 7–8 weeks. Emd null mice show no overt pathology and have normal skeletal muscle but with regeneration defects. We generated mice with germline deletions of both Lmna and Emd to determine the effects of combined loss of the encoded proteins. Mice without lamin A/C and emerin are born at the expected Mendelian ratio, are grossly normal at birth but have shorter lifespans than those lacking only lamin A/C. However, there are no major differences between these mice with regards to left ventricular function, heart ultrastructure or electrocardiographic parameters except for slower heart rates in the mice lacking both lamin A/C and emerin. Skeletal muscle is similarly affected in both of these mice. Lmna+/− mice also lacking emerin live to at least 1 year and have no significant differences in growth, heart or skeletal muscle compared to Lmna+/− mice. Deletion of the mouse gene encoding lamina-associated protein 1 leads to prenatal death; however, mice with heterozygous deletion of this gene lacking both lamin A/C and emerin are born at the expected Mendelian ratio but had a shorter lifespan than those only lacking lamin A/C and emerin. These results show that mice with combined deficiencies of three interacting nuclear envelope proteins have normal embryonic development and that early postnatal defects are primarily driven by loss of lamin A/C or lamina-associated polypeptide 1 rather than emerin.
Introduction
The nuclear envelope is comprised of the nuclear membranes, nuclear lamina and nuclear pore complexes. The nuclear lamina is a network of intermediate filament proteins called lamins that polymerize to form a meshwork of filaments primarily associated with the inner nuclear membrane (1–6). In mammals, three genes encode nuclear lamins, including LMNA (Lmna in mice), which encodes lamin A and lamin C (lamin A/C) that are expressed in most terminally differentiated somatic cells (7,8). Approximately 80 integral proteins concentrate in the inner nuclear membrane in interphase cells, several of which bind to lamin A/C (9). Among these are emerin, encoded by EMD (Emd in mice) and lamina-associated polypeptide 1 (LAP1), encoded by TOR1AIP1 (Tor1aip1 in mice) (10–17). (We refer to Tor1aip1 as Lap1 in the rest of this paper.) In addition to binding to lamin A/C, emerin and LAP1 interact with each other (18).
Emery–Dreifuss muscular dystrophy (EDMD) is classically characterized by early contractures of the elbows, Achilles tendons and postcervical muscles, slowly progressive muscle wasting and weakness with a humeroperoneal distribution and dilated cardiomyopathy with conduction system defects (19,20). Mutations in EMD cause X-linked EDMD (OMIM # 310300) and related myopathies with dilated cardiomyopathy (11,21–23). In all of these cases, the dilated cardiomyopathy with conduction defects is the main life-threatening feature. Almost all pathogenic EMD mutations lead to lack of emerin expression (13,14,24,25). Autosomal dominant mutations and very infrequent compound heterozygous mutations in LMNA also cause EDMD (OMIM # 181350 and OMIM # 616516, respectively) as well as related disorders with dilated cardiomyopathy and variable skeletal muscle involvement (26–32). These include mutations that lead to haploinsufficiency of lamin A/C (26,29). Mutations in TOR1AIP1 have also been linked to muscular dystrophy and cardiomyopathy (33–35) (OMIM # 617072). The similar phenotypes caused by mutations in EMD, LMNA and TORI1AIP1 suggest that emerin, lamin A/C and LAP1 function in the same ‘pathway’ in striated muscle.
Phenotypes of mice with deletions of genes encoding nuclear envelope proteins differ however in respects to those in humans with mutations in homologous genes. Lmna−/− mice develop muscular dystrophy and dilated cardiomyopathy and die at ~7–8 weeks of age (36,37). These mice express no lamin A/C but do express low levels of a truncated variant (36,38). Their heterozygous Lmna+/− siblings have essentially normal lifespans and develop a relatively mild cardiomyopathy with conduction abnormalities at older ages (36,39). Mice that express only lamin C but no lamin A, which are generated by alternative splicing of the RNA encoded by Lmna, are also overtly normal (40).
Mice with germline deletion of Emd encoding emerin have essentially normal phenotypes with only gene expression abnormalities during skeletal muscle regeneration and minimal motor and cardiac atrioventricular conduction defects (41,42). This has severely limited the utility of mice lacking emerin as a small animal model of X-linked EDMD. To overcome this limitation, we generated mice with selective depletion of LAP1 from skeletal muscle at embryonic day (E) 17.5, which when crossed to mice with germline deletion of Emd leads to more severe myopathy than those with loss of LAP1 alone (18). These mice provide a model in which emerin loss of function contributes to overt skeletal muscle pathology with features of EDMD. However, deletion of its interacting protein LAP1 has variable pathological effects itself depending on the nature of the genetic alteration. Lap1−/− mice with germline depletion of LAP1 exhibit perinatal lethality, suggesting a critical role for the protein in the development of other organs including the brain, whereas Lap1+/− mice are essentially normal (17). Depletion of LAP1 selectively from skeletal muscle at embryonic day (E) 8.5 leads to postnatal skeletal muscle hypotrophy whereas deletion at E17.5 leads to a muscular dystrophy phenotype (18,43). Selective depletion of LAP1 from cardiomyocytes generates a relatively mild cardiomyopathy with left ventricular systolic dysfunction (44).
Mice with homozygous germline deletion of both Emd and Lmna are reportedly born at the expected frequency and have body masses similar to Lmna−/− mice up to 3 weeks of age (45). However, growth and survival beyond 3 weeks as well as postnatal cardiac and skeletal muscle structure and function have not been reported in the literature. We therefore undertook an analysis of mice with combined deletions of these genes, focusing on striated muscle. We also examined the effect of Emd deletion on Lmna+/− mice to determine if loss of emerin function contributes to skeletal or cardiac muscle pathology at 1 year of age. In addition, we examined how haploinsufficiency of Lap1 effects mice with deficiencies of lamin A/C and emerin.
Results
Mice with germline deletions of Lmna and Emd are born at expected Mendelian ratios
We performed a series of crosses of mice, all on the C57BL/6J background, to generate animals with various germline deletions in Lmna and Emd. To generate mice that were Lmna+/+, Lmna+/− or Lmna−/− combined with hemizygous (male) and homozygous (female) deletion of Emd, we first crossed Lmna+/+;Emd−/− mice to Lmna+/−;Emd+/y mice to obtain Lmna+/+;Emd+/−, Lmna+/+; Emd−/y, Lmna+/−;Emd+/− and Lmna+/−;Emd−/y mice. We then intercrossed the Lmna+/−;Emd+/− and Lmna+/−;Emd−/y offspring to obtain mice of 12 different genotypes, including Lmna+/−;Emd−/− and Lmna+/−;Emd−/y. We intercrossed mice of these two genotypes to generate male and female mice with germline deficiency of Emd combined with no, heterozygous or homozygous deletion of Lmna (Supplementary Material, Fig. S1A). We did not analyze female mice with heterozygous deletion Emd (analogous to human ‘X-linked EDMD carriers’). To obtain male and female mice that were Lmna+/+, Lmna+/− or Lmna−/− with a normal complement of Emd, we crossed Lmna+/−;Emd+/+ to Lmna+/−;Emd+/y mice (Supplementary Material, Fig. S1B).
Both male and female offspring of crosses between Lmna+/−;Emd−/− and Lmna+/−;Emd−/y mice were born at the expected Mendelian ratios as determined by χ2 tests (Fig. 1A). These offspring included Lmna−/−;Emd−/y and Lmna−/−;Emd−/− mice, confirming a previous report with smaller numbers that mice with germline deficiencies of both Lmna and Emd were born at expected frequencies (45). Male and female offspring of crossings between Lmna+/−;Emd+/+ and Lmna+/−;Emd+/y mice were also born at the expected Mendelian ratios (Fig. 1B). This also confirmed a previous report (36).
Figure 1.
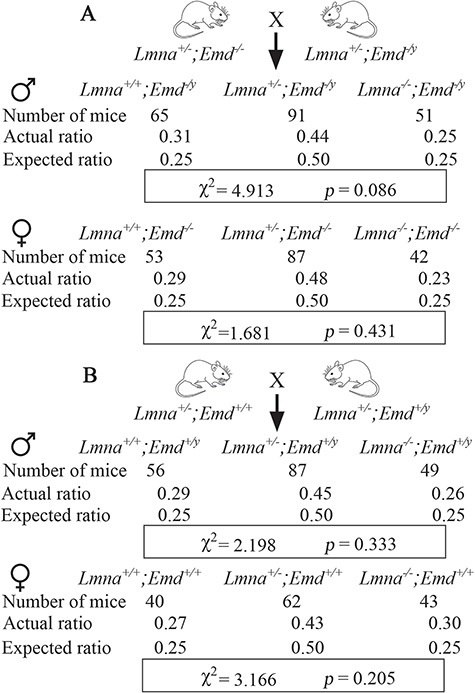
Mice with deletions in Lmna and Emd, including those with total germline deficiencies of both, are born at expected Mendelian ratios. (A) Offspring of crosses between Lmna+/−;Emd−/− and Lmna+/−;Emd−/y mice (see Supplementary Material, Fig. S1A). Top: numbers of mice born, actual ratios and expected ratios for male mice that were Lmna+/+, Lmna+/− or Lmna−/− combined with hemizygous deletion of Emd (Emd−/y). Bottom: numbers of mice born, actual ratios and expected ratios for female mice that were Lmna+/+, Lmna+/− or Lmna−/− combined with homozygous deletion of Emd (Emd−/−). χ2 and P-value for goodness of fit determination are shown in a rectangular box at the bottom of the results of male and female offspring. Standard χ2 value at α = 0.05 with 2 degrees of freedom is 5.991, which is higher than the calculated χ2 values. (B) Offspring of crosses between Lmna+/−;Emd+/+ and Lmna+/−;Emd+/y mice (see Supplementary Material, Fig. S1B). Top: numbers of mice born, actual ratios and expected ratios for male mice that were Lmna+/+, Lmna+/− or Lmna−/− with normal Emd (Emd+/y). Bottom: numbers of mice born, actual ratios and expected ratios for female mice that were Lmna+/+, Lmna+/− or Lmna−/− with normal Emd (Emd+/+). χ2 and P-value for goodness of fit determination are shown in a rectangular box at the bottom of the results for male and female offspring. Standard χ2 value at α = 0.05 with 2 degrees of freedom is 5.991, which is higher than the calculated χ2 values.
Postnatal growth and survival of mice with deletions of Lmna and Emd
At birth and within the first few days of life, there were no readily visible differences in the body sizes of mice of any genotype examined. By 3 weeks of age, mice with germline depletion of both lamin A/C and emerin (Lmna−/−;Emd−/y) and those with depletion of only lamin A/C (Lmna−/−;Emd+/y) were both visibly smaller than wild-type (Lmna+/+;Emd+/y) mice (Fig. 2A). Male mice lacking emerin (Lmna+/+;Emd−/y), lacking one Lmna allele (Lmna+/−;Emd+/y) or lacking both emerin and one Lmna allele (Lmna+/−;Emd−/y) had postnatal increases in body mass with age indistinguishable from wild-type (Lmna+/+;Emd+/y) mice, whereas those with homozygous Lmna depletion (Lmna−/−;Emd+/y) or homozygous Lmna deletion plus Emd deletion (Lmna−/−;Emd−/y) demonstrated a failure to thrive (Fig. 2B). Similarly, female mice lacking both Lmna alleles (Lmna−/−;Emd+/+) or both Lmna alleles and both Emd alleles (Lmna−/−;Emd−/−) had a failure to thrive whereas those with heterozygous deletion of Lmna (Lmna+/−;Emd+/+), homozygous deletion of Emd (Lmna+/+;Emd−/−) or heterozygous deletion of Lmna and lacking both Emd alleles (Lmna+/−;Emd−/−) all had increases in body mass with age similar to wild-type (Lmna+/+;Emd+/+) mice (Fig. 2C). Only mice with deletion of Lmna or deletion of both Lmna and Emd had significantly reduced median survivals compared to wild-type mice. When compared to each other, male mice with depletion of both lamin A/C and emerin (Lmna−/−;Emd−/y) had a statistically significant shorter median lifespan than those lacking only both Lmna alleles (Lmna−/−;Emd+/y) (Fig. 2D). Similarly, female mice lacking both lamin A/C and emerin (Lmna−/−;Emd−/−) also had a significantly shorter median lifespan than those lacking only both Lmna alleles (Lmna−/−;Emd+/+) (Fig. 2E). There were no significant differences in the median survivals between male Lmna−/−;Emd+/y and female Lmna−/−;Emd+/+ mice or between male Lmna−/−;Emd−/y and female Lmna−/−;Emd−/− mice. These data indicate that while prenatal development appears grossly normal, total germline deletion of Lmna and Emd combined leads to earlier death than deletion of only Lmna.
Figure 2.
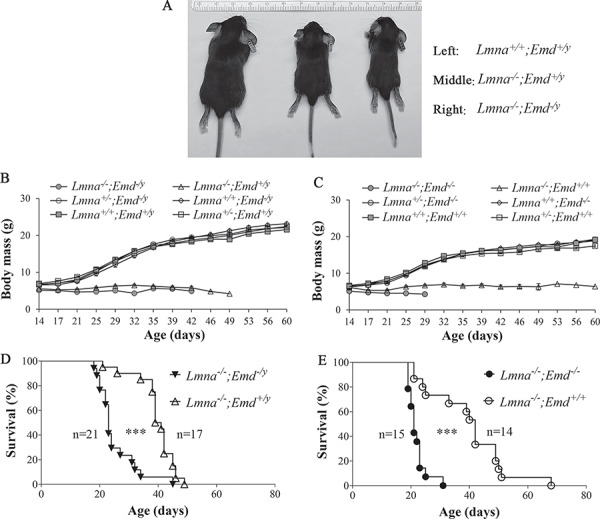
Size, body mass and survival of mice with deletions of Lmna and Emd. (A) photographs of Lmna+/+;Emd+/y mice (left), Lmna−/−;Emd+/y mice (middle) and Lmna−/−;Emd−/y mice (right) at 3 weeks of age. Ruler at top shows mm gradations. (B) Body masses (means ± standard errors) of male Lmna+/+;Emd+/y (n = 10), Lmna+/−;Emd+/y (n = 12), Lama−/−;Emd+/y (n = 21), Lmna−/−;Emd−/y (n = 14), Lmna+/−;Emd−/y (n = 13) and Lmna+/+;Emd−/y (n = 10) mice versus age. (C) Body masses (means ± standard errors) of female Lmna+/+;Emd+/+ (n = 10), Lmna+/−;Emd+/+ (n = 12), Lmna−/−;Emd+/+ (n = 15), Lmna−/−;Emd−/− (n = 12), Lmna+/−;Emd−/− (n = 12) and Lmna+/+;Emd−/− (n = 10) mice versus age. (D) Kaplan–Meier survival plots for male Lmna−/−;Emd−/y and Lmna−/−;Emd+/y mice. ***P < 0.001 for difference between Lmna−/−;Emd−/y and Lmna−/−;Emd+/y mice. (E) Kaplan–Meier survival plots for female Lmna−/−;Emd−/− and Lmna−/−;Emd+/+ mice. ***P < 0.001 for difference between Lmna−/−;Emd−/− and Lmna−/−;Emd+/+ mice. In (D) and (E), numbers of mice (n) in each group are indicted in the figure.
Lmna +/− mice of both sexes, Emd−/y mice and Emd−/− mice have all been reported to have similar survival to wild-type mice up to approximately a year (39,42). One report in the literature showed that Lmna+/−;Emd−/y and Lmna+/−;Emd−/− had normal body masses at 3 weeks of age (45). However, we are not aware of any longer-term follow-up of these mice. We therefore followed five Lmna+/−;Emd−/− mice and five Lmna+/−;Emd−/y mice for 1 year and all survived. At 1 year of age, the body mass of Lmna+/−;Emd−/y mice was indistinguishable from that of Lmna+/+;Emd+/y mice, Lmna+/−;Emd+/y mice and Lmna+/+;Emd−/y mice (Supplementary Material, Fig. S2A). Similarly, body masses of female Lmna+/+;Emd+/+ mice, Lmna+/−;Emd+/+ mice, Lmna+/−;Emd−/− mice and Lmna+/+;Emd−/− mice were not significantly different (Supplementary Material, Fig. S2B). Because there was no apparent sexual dimorphism in growth and survival between each genotype, in subsequent experiments male and female mice were analyzed in an aggregated manner (Emd+/y and Emd+/+ combined are referred to as Emd+ and Emd−/y and Emd−/− combined are referred to as Emd−).
Nuclear envelope protein expression in hearts of mice with deletions of Lmna and Emd
We performed immunoblotting to examine the expression of selected nuclear envelope proteins in hearts of mice with different combinations of Lmna and Emd deletions. We selected the heart because it is the most uniformly affected organ in humans with striated muscle disease caused by LMNA and EMD mutations. At 3 weeks of age, there was no difference in the expression levels of LAP1 (an integral inner nuclear membrane protein), lamin B1 (lamina component), Nup98 (a nuclear pore complex protein) or SUN2 (another integral inner nuclear membrane protein) in protein extracts of mouse hearts of any genotype examined, with lamin A/C and emerin undetectable or reduced as expected in mice with homozygous, heterozygous or hemizygous germline deletions of Lmna or Emd (Fig. 3A). Relatively low quantities (compared to lamin A) of a truncated lamin A variant with an apparent molecular mass of approximately 54 kDa, which was previously described (38), were also detected in protein extracts from hearts of mice that were homozygous for Lmna deletion, with even lower quantities detected in hearts of mice heterozygous for Lmna deletion (Fig. 3B). At 1 year of age, expression of LAP1, lamin B1, Nup98 and SUN2 in the hearts was the same in mice of all genotypes that lived to that long (Supplementary Material, Fig. S3).
Figure 3.
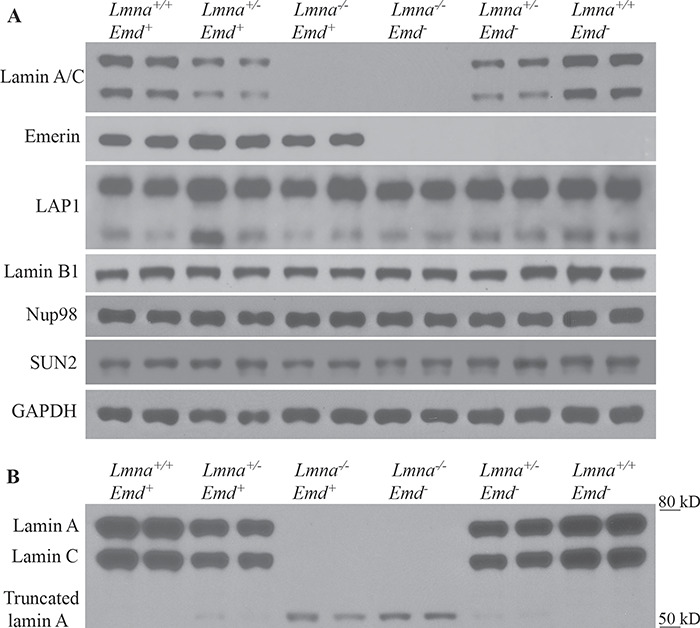
Immunoblots for selected nuclear envelope proteins in hearts of mice with deletions of Lmna and Emd. (A) Protein extracts from hearts of 3-week-old mice of indicated genotypes (two mice of each genotype) were probed with antibodies against lamin A/C, emerin, LAP1, lamin B1, Nup98, SUN2 or glyceraldehyde 3-phosphate dehydrogenase (GAPDH; loading control). In the blot probed with antibodies against LAP1, the upper band corresponds to the LAP1A and LAP1B isoforms and the lower band the LAP1C isoform. (B) Protein extracts from the mouse hearts of indicated genotypes (two mice of each genotype) were probed with anti-lamin A/C antibodies and exposed for 2 more minutes on X-ray film to show a truncated lamin A with apparent molecular mass of approximately 54 kDa in Lmna−/− mice and in lower quantities in Lmna+/− mice. Migrations of molecular mass standards are shown at the right of the blot.
Heart function in mice with deletions of Lmna and Emd
LMNA and EMD mutations in humans cause dilated cardiomyopathy (11,21–23,26–32). Lmna−/− mice develop slightly dilated left ventricles compared to wild-type mice at 2 weeks of age when diameters are normalized for body mass and a frank cardiomyopathy with left ventricular dilatation and decreased left ventricular fractional shortening (FS) by 4–6 weeks of age (37). Emd null mice do not develop dilated cardiomyopathy (41,42). We therefore performed experiments to determine if combined deletion of Lmna and Emd exacerbate cardiomyopathy that is subtly detectable in Lmna−/− mice at 2 weeks of age. We also analyzed Lmna+/−;Emd− mice at 1 year of age to determine if they develop a more severe dilated cardiomyopathy than Lmna+/− mice.
We performed M-mode echocardiography on Lmna+/+;Emd+, Lmna+/−;Emd+, Lmna−/−;Emd+, Lmna−/−;Emd−, Lmna+/−;Emd− and Lmna+/+;Emd− mice at 2 weeks of age (Fig. 4A). There was no significant difference in left ventricular end-diastolic diameter (LVEDD) between any of the mutant and wild-type genotypes (Fig. 4B). The left ventricular end-systolic diameter (LVESD) of Lmna−/−;Emd− was significantly larger than that of Lmna+/−;Emd− but there were no other significant differences between genotypes (Fig. 4C). When normalized to body mass, the ventricular diastolic and systolic diameters of Lmna−/−;Emd+ and Lmna−/−;Emd− mice were both significantly larger than those of wild-type mice but not different from each other (data not shown). The calculated left ventricular FS of Lmna−/−;Emd− mice was significantly lower than that of wild-type mice; however, there was no statistically significant difference between Lmna−/−;Emd+ and Lmna−/−;Emd− mice (Fig. 4D).
Figure 4.
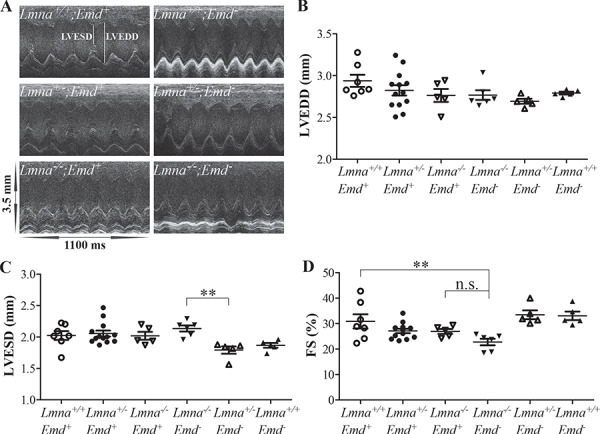
Echocardiography of mice with deletions of Lmna and Emd at 2 weeks of age. (A) Representative M-mode transthoracic echocardiographic tracings of mice of indicated genotypes. LVESD and LVEDD are indicted in the upper left tracing. (B) LVEDD for mice of genotypes indicated. (C) LVESD for mice of genotypes indicated. (D) Left ventricular FS for mice of genotypes indicated. (B), (C) and (D) show value for each individual mouse with means (longer horizontal lines) and standard errors (shorter horizontal lines). n.s.: not significant, **P < 0.01.
When mice reached 3 weeks of age and all genotypes were of a suitable size, we performed electrocardiography on Lmna+/+;Emd+, Lmna+/−;Emd+, Lmna−/−;Emd+, Lmna−/−;Emd−, Lmna+/−;Emd− and Lmna+/+;Emd− mice (Fig. 5A). Compared to the other genotypes, Lmna−/−;Emd− mice had a significantly prolonged RR interval, which corresponds to a slower heart rate (Fig. 5B). They also had a significantly prolonged PR interval compared to the other genotypes (Fig. 5C). Lmna−/−;Emd+ and Lmna−/−;Emd− mice both had significantly prolonged QRS intervals compared to wild-type mice but they were not statistically significantly different between each other (Fig. 5D). Lmna−/−;Emd− mice also had a prolonged QT interval compared to the other genotypes (Fig. 5E). However, when corrected for the slower heart rate, the QTc of Lmna−/−;Emd− mice was actually significantly shorter than the wild-type mice and not significantly different than that of Lmna−/−;Emd+ mice (Fig. 5F).
Figure 5.
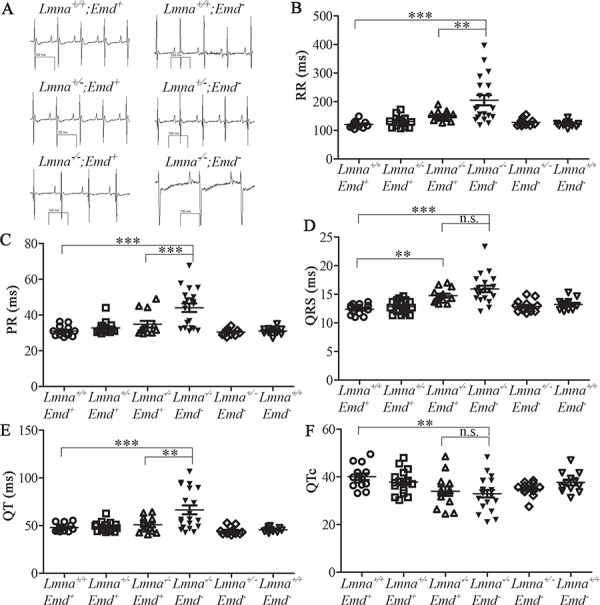
Electrocardiographic analysis of 3-week-old mice with deletions of Lmna and Emd. (A) Representative electrocardiograms of mice with genotypes indicated. (B) RR interval for mice of genotypes indicated. (C) PR interval for mice of genotypes indicated. (D) QRS interval for mice of genotypes indicated. (E) QT interval of mice of genotypes indicated. (F) QTc for mice of genotypes indicated. (B), (C), (D), (E) and (F) show value for each individual mouse with means (longer horizontal lines) and standard errors (shorter horizontal lines). n.s.: not significant, **P < 0.01, ***P < 0.001.
Lmna −/−;Emd+ and Lmna−/−;Emd− mice were the only genotypes with any abnormality on echocardiography and electrocardiography at 2–3 weeks of age. To further assess cardiac function in these mice, we examined expression of Nppa and Nppb. These genes encode natriuretic peptide hormones that are expressed in the myocardium during embryonic and fetal development, downregulated after birth and strongly induced in the ventricles by stress and during heart failure (46). Compared to wild-type, hearts of Lmna−/−;Emd+ and Lmna−/−;Emd− mice both had significantly increased expression of Nppa and Nppb mRNAs; however, there were no significant differences between these two mutant genotypes (Fig. 6).
Figure 6.
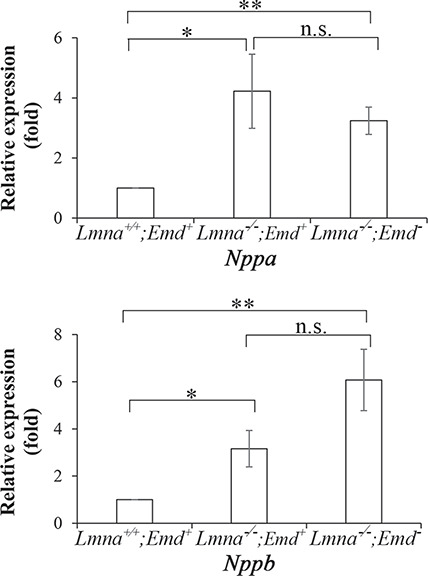
Natriuretic peptide gene expression in 2-week-old mice with deletions of Lmna and Emd determined by RT-PCR. Relative expression of Nppa (top) and Nppb (bottom) mRNAs in hearts of Lmna−/−;Emd+ and Lmna−/−;Emd− mice compared to wild type Lmna+/+;Emd+ mice (mean value arbitrarily set to 1). Values are means ± standard errors, n = 5. n.s.: not significant, *P < 0.05, **P < 0.01.
We next performed electron microscopy to determine if ultrastructural defects are present in Lmna−/−;Emd+ or Lmna−/−;Emd− mice at 3 weeks of age. This analysis revealed normal sarcomere structure; however, disruption of intercalated disks was observed in both mutant genotypes (Fig. 7A). Semi-quantitative assessment of intercalated disk morphology scored by a cardiologist blinded to genotypes showed that both Lmna−/−;Emd+ and Lmna−/−;Emd− mice had significantly abnormal intercalated disks compared to wild-type mice but there was no significant difference between these two genotypes (Fig. 7B).
Figure 7.
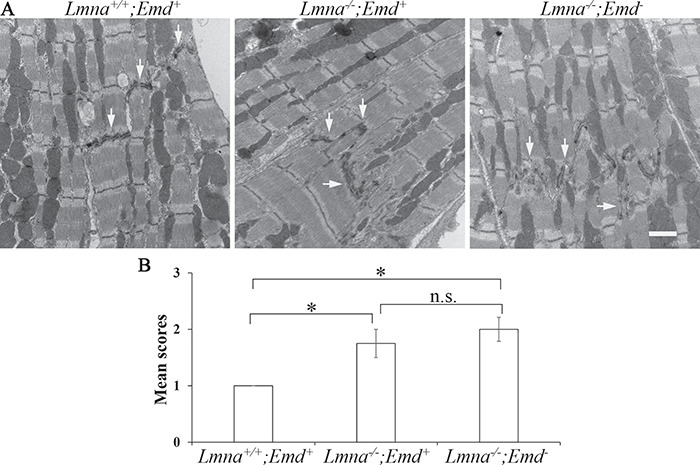
Electron microscopic analysis of heart sections from 3-week-old mice with deletions of Lmna and Emd. (A) Electron micrographs of heart sections from mice of genotypes indicated. Arrows: intercalated disks. Bar = 1 μm. (B) Semi-quantitative analysis of intercalated disk morphology, n = 4–12 sections per genotypes indicated. n.s.: not significant, *P < 0.05.
Lmna +/− ;Emd − mice lived for at least 1 year and their body mass was indistinguishable from that of Lmna+/+;Emd+, Lmna+/−;Emd+ and Lmna+/+;Emd− mice. We therefore examined these mice at 1 year of age. Echocardiography showed no significant differences in left ventricular diameters and FS between these genotypes (Supplementary Material, Fig. S4). One previous study reported increased left ventricular diameters and decreased FS in Lmna+/− mice greater than 50 weeks of age (39). On electrocardiography at 1 year of age Lmna+/−;Emd− mice had slightly but significantly shorter RR and QT intervals than Lmna+/−;Emd+ mice but no significant differences in PR, QRS and QTc intervals (Supplementary Material, Fig. S5). Overall, absence of emerin did not significantly alter cardiac function in Lmna+/− mice at 1 year of age.
Skeletal muscle in mice with deletions of Lmna and Emd
In humans, cardiomyopathy caused by LMNA and EMD mutations is usually accompanied by muscular dystrophy, most frequently in an EDMD distribution (11,26,29). Lmna+/− and Emd− mice do not develop significant myopathy (36,41,42). Lmna−/− mice however display an abnormal gait and have histopathological changes of muscular dystrophy at 3–4 weeks of age. We therefore determined if concurrent deletion of Emd exacerbated the skeletal muscular dystrophy in Lmna−/− mice at 3 weeks of age. Microscopic examination of hematoxylin and eosin-stained cross sections of quadriceps muscle showed that fibers of Lmna−/−;Emd+ and Lmna−/−;Emd− mice had occasional internal nuclei and were smaller than those in wild-type mice (Fig. 8A). The frequency of myofiber cross-sectional areas (CSA) in both of the mutant genotypes was clearly skewed toward smaller sizes compared to wild-type (Fig. 8B). While the mean CSA of myofibers of both Lmna−/−;Emd+ and Lmna−/−;Emd− mice were significantly smaller than wild-type mice, they were not significantly different from each other (Fig. 8C). Hence, loss of emerin did not appear to significantly worsen skeletal muscle pathology in Lmna−/− mice.
Figure 8.
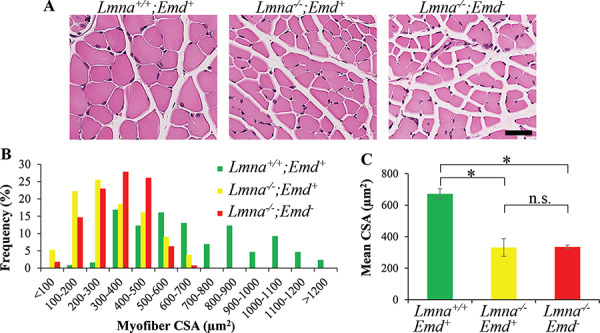
Histological analyses of skeletal muscle from 3-week-old mice with germline deletions of Lmna and Emd. (A) Photomicrographs of representative cross sections of quadriceps muscles stained with hematoxylin and eosin from mice of genotypes indicated. Bar = 50 μm. (B) Frequency distribution of myofiber CSA in quadriceps muscles from three mice per genotype. (C) Mean myofiber CSA of quadriceps. In (C), values are means ± standard errors, n = 3. n.s.: not significant, *P < 0.05.
We analyzed skeletal muscle from Lmna+/−;Emd− mice to determine if they develop evidence of myopathy at 1 year of age. Microscopic examination of hematoxylin and eosin-stained cross sections showed that quadriceps muscle from 1-year-old Lmna+/−;Emd− mice was not different than wild-type (Supplementary Material, Fig. S6A). The frequency of myofiber CSA in Lmna+/−;Emd− mice was also not different than wild-type (Supplementary Material, Fig. S6B). Their mean myofiber CSA also were not significantly different (Supplementary Material, Fig. S6C). Hence, loss of emerin did not cause myopathy in Lmna+/− mice even at 1 year of age.
Mice with deletions of Lmna and Emd and heterozygous germline deletion of Lap1 are born at expected Mendelian ratios
Homozygous disruption of Lap1 with a gene trap insertion mice in 129S6/SvEvTac background is perinatal lethal, with most animals dying at E18 or the first postnatal day (17). We generated another Lap1 deletion strain on the C57BL/6J background by crossing mice with a floxed Lap1 allele to those with an actin-Cre transgene expression. The resulting Lap1+/− mice had normal lifespans without gross abnormalities whereas the resulting Lap1−/− mice all died prenatally. The Lap1+/− mice expressed approximately half the LAP1 in heart as Lap1+/+ mice (Supplementary Material, Fig. S7).
We then performed a series of crosses to generate mice, all on the C57BL/6J genetic background, with various germline deletions of Lmna and Emd combined with heterozygous deletion of Lap1. We first crossed male mice (Lmna+/+;Emd+/y;Lap1+/−) to Lmna+/−;Emd−/−;Lap1+/+ female mice. We then intercrossed the Lmna+/−;Emd+/−;Lap1+/− female to the Lmna+/−;Emd−/y;Lap1+/− male offspring. Next, we intercrossed the Lmna+/−;Emd−/−;Lap1+/− female and Lmna+/−;Emd−/y;Lap1+/− male offspring of the second cross, which led to the generation of 6 different male and 6 different female genotypes (Supplementary Material, Fig. S8).
Both male and female offspring of crosses between Lmna+/−;Emd−/−;Lap1+/− and Lmna+/−;Emd−/y;Lap1+/− mice were born at the expected Mendelian ratios, with no Lap1−/− mice being born (Fig. 9). The live offspring included Lmna−/−;Emd−/−;Lap1+/− and Lmna−/−;Emd−/y;Lap1+/− with complete loss of emerin and lamin A/C combined with approximately half depletion of LAP1. Offspring of all the genotypes that were born appeared grossly normal and were similar in body size for the first few days of life.
Figure 9.

Mice with germline deletions of Lmna and Emd with heterozygous deletion of Lap1 are born at expected Mendelian ratios. Offspring of crosses between Lmna+/−;Emd−/−;Lap1+/− and Lmna+/−;Emd−/y;Lap1+/− (see Fig. S8). Top: numbers of mice born, actual ratios and expected ratios for male mice that were Lmna+/+, Lmna+/− or Lmna−/− combined with hemizygous deletion of Emd (Emd−/y) and full complement of Lap1 (Lap1+/+), heterozygous deletion of Lap1 (Lap1+/−) or homozygous deletion of Lap1 (Lap1−/−). Bottom: numbers of mice born, actual rations and expected ratios for female mice that were Lmna+/+, Lmna+/− or Lmna−/− combined with homozygous deletion of Emd (Emd−/−) with wild-type Lap1 (Lap1+/+), heterozygous deletion of Lap1 (Lap1+/−) or homozygous deletion of Lap1 (Lap1−/−). Homozygous germline deletion of Lap1 in this strain is prenatal lethal so we used an expected birth ration of 0 for mice that were Lap−/−. χ2 and P-value for goodness of fit determination are shown in a rectangular box at the bottom of the results for male and female offspring. Standard χ2 value at α = 0.05 with 5 degrees of freedom is 11.07, which is higher than the calculated χ2 values.
Effect of heterozygous deletion of Lap1 on postnatal growth and survival of mice with deletions of Lmna and Emd
We selected Lmna+/−;Emd−/−;Lap1+/+, Lmna−/−;Emd−/−;Lap1+/+, Lmna−/−;Emd−/−;Lap1+/− and Lmna+/−;Emd−/−;Lap1+/− female mice and their Lmna+/−;Emd−/y;Lap1+/+, Lmna−/−;Emd−/y;Lap1+/+, Lmna−/−;Emd−/y;Lap1+/− and Lmna+/−;Emd−/y;Lap1+/− male littermates for further analysis. This allowed us to assess the heterozygous deletion of Lap1 in mice lacking emerin and either homozygous or heterozygous for loss of lamin A/C. We analyzed these male and female mice in a disaggregated manner, as we did not know if these phenotypes would be influenced by sex in mice of these genotypes. Female Lmna+/−;Emd−/−;Lap1+/+ mice and male Lmna+/−;Emd−/y;Lap1+/+ served as ‘controls,’ as our previous experiments presented above showed that they had normal growth and survival to at least 1 year of age.
Male mice with homozygous Lmna and hemizygous Emd deletion plus heterozygous Lap1 deletion (Lmna−/−;Emd−/y;Lap1+/−) demonstrated a failure to thrive similar to the same mice wild-type for Lap1 (Lmna−/−;Emd−/y;Lap1+/+) whereas Lmna+/−;Emd−/y;Lap1+/− showed no difference in growth compared to Lmna+/−;Emd−/y;Lap1+/+ ‘controls’ (Fig. 10A). Female mice with the corresponding alterations in these genes had similar growth patterns (Fig. 10B). However, male Lmna−/−;Emd−/y;Lap1+/− mice had significantly shorter survival than Lmna−/−;Emd−/y;Lap1+/+ mice (Fig. 10C). Female Lmna−/−;Emd−/−;Lap1+/− mice also had a significantly shorter survival than Lmna−/−;Emd−/y;Lap1+/+ mice (Fig. 10D). There was no significant difference in the medial survival between male Lmna−/−;Emd−/y;Lap1+/− and female Lmna−/−;Emd−/−;Lap1+/− mice. Hence, although they were born as expected, heterozygous deletion of Lap1 in both male and female mice lacking both lamin A/C and emerin significantly shortened their lifespans.
Figure 10.
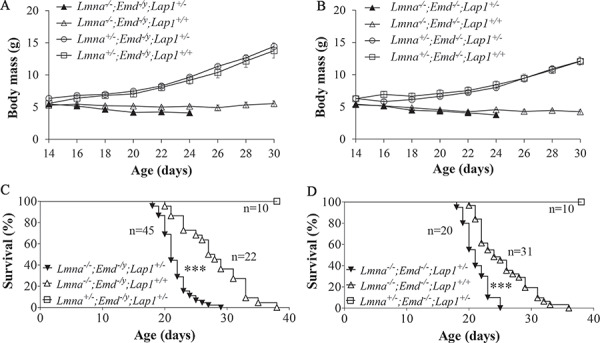
Body mass and survival curves of mice with germline deletions of Lmna and Emd with heterozygous deletion of Lap1. (A) Body mass (means ± standard errors) of male Lmna−/−;Emd−/y;Lap+/− (n = 40), Lmna−/−;Emd−/y;Lap+/+ (n = 19), Lmna+/−;Emd−/y;Lap+/− (n = 10) and Lmna+/−;Emd−/y;Lap+/+ (n = 10) mice versus age. (B) Body mass (means ± standard errors) of female Lmna−/−;Emd−/−;Lap+/− (n = 20), Lmna−/−;Emd−/−;Lap+/+ (n = 26), Lmna+/−;Emd−/−;Lap+/− (n = 15) and Lmna+/−;Emd−/−;Lap+/+ (n = 10) mice versus age. (C) Kaplan–Meier survival plots for male Lmna−/−;Emd−/y;Lap+/−, Lmna−/−;Emd−/y;Lap+/+ and Lmna+/−;Emd−/y;Lap+/− mice, ***P < 0.001 for difference between Lmna−/−;Emd−/y;Lap+/− and Lmna−/−;Emd−/y;Lap+/+. (D) Kaplan–Meier survival plots for female Lmna−/−;Emd−/−;Lap+/−, Lmna−/−;Emd−/−;Lap+/+ and Lmna+/−;Emd−/−;Lap+/− mice, ***P < 0.001 for difference between Lmna−/−;Emd−/−;Lap+/− and Lmna−/−;Emd−/−;Lap+/+ mice. In (C) and (D), numbers of mice (n) in each group are indicated in the figure.
Protein kinase B and extracellular signal-regulated kinase 1/2 signaling in hearts of mice with Lmna, Emd and Lap1 alterations
We have previously demonstrated increased activities in the protein kinase B (AKT) and extracellular signal-regulated kinase 1/2 (ERK1/2) signaling pathways in hearts from humans with cardiomyopathy caused by LMNA mutations and the LmnaH222P/H222P mouse model of the disease (47,48). Increased ERK1/2 activity is also detectable in hearts of Lmna−/− mice at 5–7 weeks of age (49). At 10 weeks of age, hearts from Emd−/y mice also have increased ERK1/2 activity (50). We therefore examined AKT and ERK1/2 signaling in hearts of mice with various Lmna, Emd and Lap1 alterations during the first 3 weeks of their lives. Immunoblotting with antibodies against total and phosphorylated (activated) AKT and ERK1/2 showed no significant increases in AKT or ERK1/2 activity in hearts of any mutant mouse strain examined at 2 weeks of age (Supplementary Material, Fig. S9A). Rather, during the first 2 weeks of life, there was decreased AKT activity in hearts of Lmna−/−;Emd+, Lmna−/−;Emd− and Lmna−/−;Emd−;Lap1+/− mice compared to wild-type mice but no significant differences between surviving genotypes (an insufficient number of Lmna−/−;Emd−;Lap1+/− mice survived to make reproducible measurements) in the third week of life (Supplementary Material, Fig. S9B). Hearts of Lmna−/−;Emd− and Lmna−/−;Emd−;Lap1+/− mice also had decreased ERK1/2 activity compared to wild-type mice during the second week of life but there were no differences among the genotypes in the first week of life or among surviving genotypes in the third week (Supplementary Material, Fig. S9C).
Discussion
Our results are consistent with the fact that, compared to humans, mice are relatively resilient to germline deletions of genes encoding A-type lamins and emerin. In humans, heterozygous mutations in LMNA cause cardiomyopathy and muscular dystrophy (26–32). Heterozygous Lmna+/− mice, however, have essentially normal lifespans and develop only a relatively mild cardiomyopathy at older ages (36,39). Homozygous LMNA loss-of-function mutation is likely prenatal lethal in humans, as only one infant who died at childbirth with a truncating LMNA mutation leading to lack of lamin A/C expression has been described in the literature (51). This infant, who was born preterm and died at birth from respiratory failure, had dysmorphic faces, severe joint contractures and generalized muscular dystrophy (52). In contrast, Lmna−/− mice appear normal at birth but develop cardiomyopathy and muscular dystrophy during the first few postnatal weeks (36,37). Loss of emerin in humans cause cardiomyopathy and muscular dystrophy (11,21–23). However, mice with germline deletion of Emd expressing no emerin demonstrate minimal pathology (41,42). Our results confirm one previous report (45) that mice with total germline deletion for both Lmna and Emd are born at the expected Mendelian ratio and appear grossly normal at birth. However, mice with complete germline deletion of Lmna and Emd have a significantly shorter lifespan than mice with only deletion of Lmna (~21 days versus 41 days). However, we could not detect robust differences in cardiac or skeletal muscle pathology in these two genotypes between the ages of 14 and 21 days.
Mice may be protected from emerin loss because of significantly higher expression of its interacting protein LAP1 relative to humans (18). We have now followed 5 Lmna+/−;Emd−/y;Lap1+/− and 15 Lmna+/−;Emd−/−;Lap1+/− mice for 14 months with no deaths. Hence, one Lap1 allele appears to compensate for loss of emerin in mice. The Lap1−/− mice we generated all died during the prenatal period whereas in another strain, with a different germline deletion on a different genetic background, some are born but die during the first postnatal day (17). In contrast, humans with TOR1AIP1 mutations that lead to lack of expression of all LAP1 isoforms are born and live to between 5 and 10 years of age, albeit with severe developmental abnormalities (53). These findings suggest that emerin and LAP1 may have partially overlapping functions. Emerin, however, appears to play a more important role in human striated muscle as its loss leads to muscular dystrophy and cardiomyopathy, whereas mice lacking emerin do not have significant striated muscle abnormalities.
Tissue-selective depletion of LAP1 from skeletal and cardiac muscle causes muscular dystrophy and cardiomyopathy (18,44). Combined deletion of both Lap1 and Emd from skeletal muscle leads to significantly more severe pathology than deletion of Lap1 alone (18). Skeletal muscle depletion of LAP1 combined with germline Emd deletion is the only mouse model so far in which loss of emerin detectably contributes to muscle pathology. This makes them a useful small animal model that can be used to study interventions such as emerin replacement gene therapy. This model of X-linked EDMD is reminiscent of the utrophin-dystrophin-deficient mouse model of Duchenne muscular dystrophy (54,55), which has also been used to study the effects of gene replacement therapy (56). Our current study also addressed if Lmna+/− mice with combined germline deletion of Emd could similarly be used as such a model. However, we detected no significant pathology at 1 year of age, making them an unsuitable model to study how emerin loss contributes to striated muscle pathology.
In our experiments, we utilized Lmna−/− mice that were generated by Sullivan et al. (36). We confirmed that these mice express low levels of a truncated lamin A/C polypeptide, as previously reported (38). Nonetheless, they develop early onset dilated cardiomyopathy and muscular dystrophy and have been a very widely used animal model. Another Lmna null strain generated using gene trap technology has a somewhat different phenotype, with severe growth retardation, impaired cardiomyocyte hypertrophy, skeletal muscle hypotrophy and death at 2–3 weeks postpartum but without dilated cardiomyopathy (57). Mice with loxP sites flanking the second exon of Lmna crossed to mice with germline Cre expression also die 2–3 weeks postpartum and also have skeletal muscle hypotrophy (58). Mice with Lmna alleles containing loxP sites near the 3′ end of the gene also die at 2–3 weeks of age when globally deleted (59,60). Hence, low-level expression of the truncated lamin A/C polypeptide in the Lmna−/− mice we used may influence phenotype and survival.
In our experiments, all mice were backcrossed onto a pure C57BL/6J background. Mice used for the initial publications of Lmna, Emd and Lap1 null strains were all on different mixed genetic backgrounds (17,36,41). Given the variable clinical presentations of human subjects with even the same LMNA mutations (29,31), studies on how genetic backgrounds influence phenotypes in mice with Lmna deletions could be informative. Studies of the influence of genetic backgrounds on the phenotypes of mice with mutations in other gene encoding nuclear envelope proteins, including Emd and Lap1, may provide information on the variable clinical phenotypes observed in human subjects (11,21–23,33–35,53).
Increased AKT and ERK1/2 activities contribute to cardiomyopathy in LmnaH222P/H222P mice and are also observed in hearts of humans with LMNA mutations and cardiomyopathy (47,48). The decreased left ventricular ejection fraction in 2-week-old Lmna−/−;Emd− mice was not associated with increased AKT or ERK1/2 activity. This was not unexpected. LmnaH222P/H222P mice are a robust model of human cardiomyopathy caused by LMNA mutations, developing late-onset dilated left ventricles, decreased ejection fraction, myocardial fibrosis and a fetal-like gene expression program including upregulation of MHY7 (48,61,62). In contrast, Lmna−/− mice do not develop all of these pathological features, including fibrosis and the fetal-like gene expression pattern (37). Hearts of Lmna−/−;Emd− and Lmna−/−;Emd+ mice rather had decreased AKT activity during the first 2 weeks of life and decreased ERK1/2 activity during the second week of life, all of which increased by the third week. AKT is a positive regulator of physiological postnatal cardiac growth and hypertrophy but long-term activation can lead to pathological hypertrophy with subsequent dilatation and heart failure (63,64). ERK1/2 also functions in cardiac hypertrophy and has both protective and pathologic effects on the heart depending upon the duration, frequency and amplitude of activation as well as the nuclear or cytoplasmic localization of the active kinases (65). Given the positive effects of AKT on postnatal heart growth and possible complementary role of ERK1/2, the decreased activities of these kinases may have contributed to the cardiac defects in 2-week-old Lmna−/− mice with or without Emd deletion. By 4–6 weeks of age, Lmna−/− mice do however have some increase in cardiac ERK1/2 activity (49), which may partially contribute to later-stage pathology.
Lamin A/C, emerin and LAP1 interact with each other and may exist as a complex at the inner nuclear membrane but the stoichiometry is not known. Loss of lamin A/C leads to a partial redistribution of emerin from the inner nuclear membrane to the endoplasmic reticulum and a significant increase in its diffusional mobility in the nuclear envelope (36,66). In fibroblasts lacking LAP1, emerin is sometimes mislocalized to foci along the nuclear envelope, colocalized with lamin A/C, and its diffusional mobility is increased (18). With loss of emerin, however, LAP1 is normally localized to the inner nuclear membrane and its diffusional mobility is unchanged (18). Depletion of emerin also does not significantly affect lamin A/C localization (67). Hence, lamin A/C and LAP1 more profoundly influence emerin localization than emerin influences theirs. This correlates with our genetic results showing that early postnatal defects in mice are primarily driven by loss of lamin A/C or LAP1 rather than by loss of emerin.
Materials and Methods
Mice
Mice with germline deletions of Lmna and Emd have been described previously (36,41). These mice were backcrossed for more than 6 generations onto the C57BL/6J genetic background. To generate mice with germline deletion of Lap1, we intercrossed mice with a floxed Lap1 allele (18), to a transgenic strain expressing Cre recombinase under control of the human β-actin gene promoter, which drives Cre expression in all cells including germline (Jackson Laboratory stock no: 019099). F1 generation mice were genotyped to obtain those heterozygous for the Lap1 allele (Lap flox/+;Cre+/−). To remove the actin-Cre allele present in the F1 generation, Lap1+/−;Cre+/− mice were outcrossed with C57BL/6J wild-type mice. The F2 Lap1+/− mice lacking the Cre transgene were used to maintain the colony and were backcrossed to C57BL/6J mice for more than 6 generations. All mice were fed a chow diet and housed in a barrier facility with 12/12 h light/dark cycles. PCR genotyping was performed on all mice using genomic DNA isolated from tail clippings. The Institutional Animal Care and Use Committee at Columbia University Irving Medical Center approved the protocol.
Growth and survival analyses
To monitor growth, mice were weighed every 2–3 days from birth up to 2 months. For survival analyses, a combined endpoint of death or distress severe enough that a staff veterinarian blinded to genotype determined that euthanasia was necessary. Euthanasia was performed according to the approved protocol of the Institute of Comparative Medicine at Columbia University Irving Medical Center.
Protein isolation and immunoblotting
Proteins were extracted from whole or half mouse hearts in 8.55M urea, 10 mm Tris–HCl (pH 8.0), 2% β-mercaptoethanol, 1 mm phenylmethane sulfonyl fluoride, 1 mm NaF, 10 μM ethylenediaminetetraacetic acid and 1% Protease Inhibitor Cocktail (Sigma-Aldrich). They were separated by electrophoresis in SDS-polyacrylamide slab gels, transferred to nitrocellulose membranes and analyzed by immunoblotting using methods described previously (68). Primary antibodies for immunoblotting were rabbit anti-lamin A/C (Santa Cruz) at 1:5000 dilution, mouse anti-emerin (Vector Laboratories) at 1:50 dilution, rabbit anti-LAP1 (18) at 1:3000 dilution, rabbit anti-lamin B1 (69) at 1:1000 dilution, rat anti-Nup98 (Abcam) at 1:3000 dilution, rabbit anti-SUN2 (Abcam) at 1:1000 dilution, mouse anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Ambion) at 1:3000 dilution, rabbit anti-phosphorylated AKT (Cell Signaling) at 1:1000 dilution, rabbit anti-total AKT(Cell Signaling) at 1:1000 dilution, rabbit anti-phosphorylated ERK1/2 (Cell Signaling) at 1:1000 dilution and rabbit anti-total ERK1/2 (Santa Cruz) at 1:2000 dilution. Secondary antibodies were ECL-horseradish peroxidase-conjugated anti-mouse, anti-rabbit and anti-rat antibodies (GE Healthcare) used at a dilution of 1:5000. Signals were detected using SuperSignal West Pico Chemiluminescent Substrate Kit (Pierce) and X-ray film (Phenix Research Products).
Transthoracic echocardiography
Transthoracic 2D and M-mode echocardiography was performed using a Vevo 770 imaging system (Visualsonics) equipped with a 30 MHz linear transducer as previously described (70). The echocardiographer was blinded to mouse genotype. The transducer was applied to the chest of mice anesthetized with isoflurane (1.0–1.5%)/oxygen at 37°C. LVEDD and LVESD were measured and averaged over 3 cardiac cycles and left ventricular FS was calculated from these parameters.
RT-PCR
Total RNA was extracted from homogenates of whole/half mouse hearts using RNeasy mini kit (Qiagen) according to the manufacturer’s instructions. Total RNA (1 μg) from sample was used to synthesize cDNA using RevertAid RT traverse transcription kit (Thermo Fisher Scientific). Primer sequences of Gapdh, Nppa and Nppb have been reported previously (70). Equally diluted cDNA was used for RT-PCR analysis on an ABI 7300 Real-Time PCR system (Applied Biosystems) using HotStart-IT SYBR Green Supermix (Affymetrix). Relative expression levels were calculated using the ΔΔCt method (71). Expression of Gapdh in individual samples was used as an internal control.
Electrocardiography
Electrocardiography was performed on mice anesthetized with isoflurane (1.0%)/oxygen at room temperature using slightly modified previously described methods (72). Heart rhythm was recorded with electrodes placed under skin of the 4 limbs using an amplifier and software from emka Technologies. An investigator blinded to genotype measured electrocardiogram intervals manually with ecgAUTO software (emka Technologies). Intervals were averaged from 4 consecutive cardiac cycles. QTc was calculated as QTc = QT/(RR/100).
Electron microscopy
Diced mouse hearts were fixed with 2.5% glutaraldehyde in 0.1 M Sorenson's buffer (pH 7.2) for at least 1 h, post-fixed with 1% OsO4 in Sorenson's buffer for 1 h and further processed and examined using a JEOL JEM-1200 EXII transmission electron microscope as reported previously (73). A cardiologist blind to genotype scored the morphology of intercalated disks of 4–12 sections from mice of each genotype examined as: 1 = normal, 2 = mild/moderate disruption and 3 = severe disruption.
Histopathological analysis of skeletal muscle
For light microscopy, dissected mouse quadriceps muscles were placed in 10% neutral-buffered formalin for 48 h, embedded in paraffin and sectioned at 5 μm. Sections were stained with hematoxylin and eosin for histological analysis. The myofiber quantification was performed by a scientist without knowing genotype information of the sections as previously described (43). Briefly, representative images of stained cross sections of quadriceps from mice at 3 weeks of age were photographed, then three different areas of each section were used to assess CSA. Individual myofibers from each image were processed using Adobe Photoshop CS (Adobe Systems). CSA of individual myofibers were measured with Image J software (http://rsb.info.nih.gov/ij/) and graphically represented with Excel 2016 (Microsoft).
Statistics
Chi-squared as χ2 = Σ(C-E)2/E and P-value for goodness of fit test were calculated using Excel and compared with a standard χ2(df) at α = 0.05. The Kaplan–Meier estimator (74) in statistical software GraphPad Prism 5 (Prism Software) was used to generate the survival curves. For comparisons among more than two groups, ANOVA and Tukey’s post hoc multiple comparisons were carried also out using GraphPad Prism 5. Student’s t-tests of comparisons between two means were performed using Excel 2016 (Microsoft); bar graphs and body mass growth curves were generated using the same software.
Supplementary Material
Acknowledgements
We thank Kristy Brown (Columbia University) for assistance with electron microscopy and Christopher B. Damoci (Columbia University) for assistance with echocardiographic analysis. Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under award number R01AR04897. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest statement. None declared.
References
- 1. Gerace L., Blum A. and Blobel G. (1978) Immunocytochemical localization of the major polypeptides of the nuclear pore complex-lamina fraction. Interphase and mitotic distribution. J. Cell Biol., 79, 546–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fisher D.Z., Chaudhary N. and Blobel G. (1986) cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc. Natl. Acad. Sci. U. S. A., 83, 6450–6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goldman A.E., Maul G., Steinert P.M., Yang H.Y. and Goldman R.D. (1986) Keratin-like proteins that coisolate with intermediate filaments of BHK-21 cells are nuclear lamins. Proc. Natl. Acad. Sci. U. S. A., 83, 3839–3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McKeon F.D., Kirschner M.W. and Caput D. (1986) Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature, 319, 463–468. [DOI] [PubMed] [Google Scholar]
- 5. Aebi U., Cohn J., Buhle L. and Gerace L. (1986) The nuclear lamina is a meshwork of intermediate-type filaments. Nature, 323, 560–564. [DOI] [PubMed] [Google Scholar]
- 6. Turgay Y., Eibauer M., Goldman A.E., Shimi T., Khayat M., Ben-Harush K., Dubrovsky-Gaupp A., Sapra K.T., Goldman R.D. and Medalia O. (2017) The molecular architecture of lamins in somatic cells. Nature, 543, 261–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lin F. and Worman H.J. (1993) Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem., 268, 16321–16326. [PubMed] [Google Scholar]
- 8. Wydner K.L., McNeil J.A., Lin F., Worman H.J. and Lawrence J.B. (1996) Chromosomal assignment of human nuclear envelope protein genes LMNA, LMNB1, and LBR by fluorescence in situ hybridization. Genomics, 32, 474–478. [DOI] [PubMed] [Google Scholar]
- 9. Schirmer E.C., Florens L., Guan T., Yates J.R. III. and Gerace L. (2003) Nuclear membrane proteins with potential disease links found by subtractive proteomics. Science, 301, 1380–1382. [DOI] [PubMed] [Google Scholar]
- 10. Senior A. and Gerace L. (1988) Integral membrane proteins specific to the inner nuclear membrane and associated with the nuclear lamina. J. Cell Biol., 107, 2029–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bione S., Maestrini E., Rivella S., Mancini M., Regis S., Romeo G. and Toniolo D. (1994) Identification of a novel X-linked gene responsible for Emery–Dreifuss muscular dystrophy. Nat. Genet., 8, 323–327. [DOI] [PubMed] [Google Scholar]
- 12. Martin L., Crimaudo C. and Gerace L. (1995) cDNA cloning and characterization of lamina-associated polypeptide 1C (LAP1C), an integral protein of the inner nuclear membrane. J. Biol. Chem., 270, 8822–8828. [DOI] [PubMed] [Google Scholar]
- 13. Nagano A., Koga R., Ogawa M., Kurano Y., Kawada J., Okada R., Hayashi Y.K., Tsukahara T. and Arahata K. (1996) Emerin deficiency at the nuclear membrane in patients with Emery–Dreifuss muscular dystrophy. Nat. Genet., 12, 254–259. [DOI] [PubMed] [Google Scholar]
- 14. Manilal S., Nguyen T.M., Sewry C.A. and Morris G.E. (1996) The Emery–Dreifuss muscular dystrophy protein, emerin, is a nuclear membrane protein. Hum. Mol. Genet., 5, 801–808. [DOI] [PubMed] [Google Scholar]
- 15. Fairley E.A., Kendrick-Jones J. and Ellis J.A. (1999) The Emery–Dreifuss muscular dystrophy phenotype arises from aberrant targeting and binding of emerin at the inner nuclear membrane. J. Cell Sci., 112, 2571–2582. [DOI] [PubMed] [Google Scholar]
- 16. Clements L., Manilal S., Love D.R. and Morris G.E. (2000) Direct interaction between emerin and lamin A. Biochem. Biophys. Res. Commun., 267, 709–714. [DOI] [PubMed] [Google Scholar]
- 17. Kim C.E., Perez A., Perkins G., Ellisman M.H. and Dauer W.T. (2010) A molecular mechanism underlying the neural-specific defect in torsinA mutant mice. Proc. Natl. Acad. Sci. U. S. A., 107, 9861–9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shin J.Y., Méndez-López I., Wang Y., Hays A.P., Tanji K., Lefkowitch J.H., Schulze P.C., Worman H.J. and Dauer W.T. (2013) Lamina-associated polypeptide-1 interacts with the muscular dystrophy protein emerin and is essential for skeletal muscle maintenance. Dev. Cell, 26, 591–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Emery A.E. and Dreifuss F.E. (1966) Unusual type of benign x-linked muscular dystrophy. J. Neurol. Neurosurg. Psychiatry, 29, 338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Emery A.E. (2000) Emery–Dreifuss muscular dystrophy—a 40 year retrospective. Neuromuscul. Disord., 10, 228–232. [DOI] [PubMed] [Google Scholar]
- 21. Muntoni F., Lichtarowicz-Krynska E.J., Sewry C.A., Manilal S., Recan D., Llense S., Taylor J., Morris G.E. and Dubowitz V. (1998) Early presentation of X-linked Emery–Dreifuss muscular dystrophy resembling limb-girdle muscular dystrophy. Neuromuscul. Disord., 8, 72–76. [DOI] [PubMed] [Google Scholar]
- 22. Astejada M.N., Goto K., Nagano A., Ura S., Noguchi S., Nonaka I., Nishino I. and Hayashi Y.K. (2007) Emerinopathy and laminopathy clinical, pathological and molecular features of muscular dystrophy with nuclear envelopathy in Japan. Acta Myol., 26, 159–164. [PMC free article] [PubMed] [Google Scholar]
- 23. Ura S., Hayashi Y.K., Goto K., Astejada M.N., Murakami T., Nagato M., Ohta S., Daimon Y., Takekawa H., Hirata K. et al. (2007) Limb-girdle muscular dystrophy due to emerin gene mutations. Arch. Neurol., 64, 1038–1041. [DOI] [PubMed] [Google Scholar]
- 24. Manilal S., Recan D., Sewry C.A., Hoeltzenbein M., Llense S., Leturcq F., Deburgrave N., Barbot J., Man N., Muntoni F. et al. (1998) Mutations in Emery-Dreifuss muscular dystrophy and their effects on emerin protein expression. Hum. Mol. Genet., 7, 855–864. [DOI] [PubMed] [Google Scholar]
- 25. Yates J.R., Bagshaw J., Aksmanovic V.M., Coomber E., Mcmahon R., Whittaker J.L., Morrison P.J., Kendrick-Jones J. and Ellis J.A. (1999) Genotype-phenotype analysis in X-linked Emery–Dreifuss muscular dystrophy and identification of a missense mutation associated with a milder phenotype. Neuromuscul. Disord., 9, 159–165. [PubMed] [Google Scholar]
- 26. Bonne G., Di Barletta M.R., Varnous S., Bécane H.M., Hammouda E.H., Merlini L., Muntoni F., Greenberg C.R., Gary F., Urtizberea J.A. et al. (1999) Mutations in the gene encoding lamin A/C cause autosomal dominant Emery–Dreifuss muscular dystrophy. Nat. Genet., 21, 285–288. [DOI] [PubMed] [Google Scholar]
- 27. Fatkin D., MacRae C., Sasaki T., Wolff M.R., Porcu M., Frenneaux M., Atherton J., Vidaillet H.J. Jr., Spudich S., De Girolami U. et al. (1999) Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med., 341, 1715–1724. [DOI] [PubMed] [Google Scholar]
- 28. Raffaele Di Barletta M., Ricci E., Galluzzi G., Tonali P., Mora M., Morandi L., Romorini A., Voit T., Orstavik K.H., Merlini L. et al. (2000) Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery–Dreifuss muscular dystrophy. Am. J. Hum. Genet., 66, 1407–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bonne G., Mercuri E., Muchir A., Urtizberea A., Bécane H.M., Recan D., Merlini L., Wehnert M., Boor R., Reuner U. et al. (2000) Clinical and molecular genetic spectrum of autosomal dominant Emery–Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann. Neurol., 48, 170–180. [PubMed] [Google Scholar]
- 30. Muchir A., Bonne G., van der Kooi A.J., van Meegen M., Baas F., Bolhuis P.A., de Visser M. and Schwartz K. (2000) Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum. Mol. Genet., 9, 1453–1459. [DOI] [PubMed] [Google Scholar]
- 31. Brodsky G.L., Muntoni F., Miocic S., Sinagra G., Sewry C. and Mestroni L. (2000) Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement. Circulation, 101, 473–476. [DOI] [PubMed] [Google Scholar]
- 32. Quijano-Roy S., Mbieleu B., Bönnemann C.G., Jeannet P.Y., Colomer J., Clarke N.F., Cuisset J.M., Roper H., De Meirleir L., D'Amico A. et al. (2008) De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann. Neurol., 64, 177–186. [DOI] [PubMed] [Google Scholar]
- 33. Kayman-Kurekci G., Talim B., Korkusuz P., Sayar N., Sarioglu T., Oncel I., Sharafi P., Gundesli H., Balci-Hayta B., Purali N. et al. (2014) Mutation in TOR1AIP1 encoding LAP1B in a form of muscular dystrophy: a novel gene related to nuclear envelopathies. Neuromuscul Disord., 24, 624–633. [DOI] [PubMed] [Google Scholar]
- 34. Dorboz I., Coutelier M., Bertrand A.T., Caberg J.H., Elmaleh-Bergès M., Lainé J., Stevanin G., Bonne G., Boespflug-Tanguy O. and Servais L. (2014) Severe dystonia, cerebellar atrophy, and cardiomyopathy likely caused by a missense mutation in TOR1AIP1. Orphanet J. Rare Dis., 9, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ghaoui R., Benavides T., Lek M., Waddell L.B., Kaur S., North K.N., MacArthur D.G., Clarke N.F. and Cooper S.T. (2016) TOR1AIP1 as a cause of cardiac failure and recessive limb-girdle muscular dystrophy. Neuromuscul. Disord., 26, 500–503. [DOI] [PubMed] [Google Scholar]
- 36. Sullivan T., Escalant-Alcalde D., Bhatt H., Anver M., Bhat N., Nagashima K., Stewart C.L. and Burke B. (1999) Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol., 147, 913–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nikolova V., Leimena C., McMahon A.C., Tan J.C., Chandar S., Jogia D., Kesteven S.H., Michalicek J., Otway R., Verheyen F. et al. (2004) Defect in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J. Clin. Invest., 113, 357–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jahn D., Schramm S., Schnölzer M., Hellmann C.J., Koster C.G., Schütz W., Benavente R. and Alsheimer M. (2012) A truncated lamin A in the Lmna−/− mouse line implications for the understanding of laminopathies. Nucleus, 3, 463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wolf C.M., Wang L., Alcalai R., Pizard A., Burgon P.G., Ahmad F., Sherwood M., Branco D.M., Wakimoto H., Fishman G.I. et al. (2008) Lamin A/C haploinsurfficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. J. Mol. Cell. Cardiol., 44, 293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fong L.G., Ng J.K., Lammerding J., Vickers T.A., Meta M., Coté N., Gavino B., Qiao X., Chang S.Y., Young S.R. et al. (2006) Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J. Clin. Invest., 116, 743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Melcon G., Kozlov S., Cutler D.A., Sullivan T., Hernandez L., Zhao P., Mitchell S., Nader G., Bakay M., Rottman J.N. et al. (2006) Loss of emerin at the nuclear envelope disrupts the Rb1/E2F and MyoD pathways during muscle regeneration. Hum. Mol. Genet., 15, 637–651. [DOI] [PubMed] [Google Scholar]
- 42. Ozawa R., Hayashi Y.K., Ogawa M., Kurokawa R., Matsumoto H., Noguchi S., Nonaka I. and Nishino I. (2006) Emerin-lacking mice show minimal motor and cardiac dysfunctions with nuclear-associated vacuoles. Am. J. Pathol., 168, 907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shin J.Y., Méndez-López I., Hong M., Wang Y., Tanji K., Wu W., Shugol L., Krauss R.S., Dauer W.T. and Worman H.J. (2017) Lamina-associated polypeptide 1 is dispensable for embryonic myogenesis but required for postnatal skeletal muscle growth. Hum. Mol. Genet., 26, 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shin J.Y., Le Dour C., Sera F., Iwata S., Homma S., Joseph L.C., Morrow J.P., Dauer W.T. and Worman H.J. (2014) Depletion of lamina-associated polypeptide 1 from cardiomyocytes causes cardiac dysfunction in mice. Nucleus, 5, 260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Smith E.R., Meng Y., Moore R., Tse J.D., Xu A.G. and Xu X.X. (2017) Nuclear envelope structural proteins facilitate nuclear shape changes accompanying embryonic differentiation and fidelity of gene expression. BMC Cell Biol., 18, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sergeeva I.A., Hooijkaas I.B., Ruijter J.M., van der Made I., de Groot N.E., van de Werken H.J., Creemers E.E. and Christoffels V.M. (2016) Identification of a regulatory domain controlling the Nppa-Nppb gene cluster during heart development and stress. Development, 143, 2135–2146. [DOI] [PubMed] [Google Scholar]
- 47. Choi J.C., Muchir A., Wu W., Iwata S., Homma S., Morrow J.P. and Worman H.J. (2012) Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci. Transl. Med., 4, 144ra102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Muchir A., Reilly S.A., Wu W., Iwata S., Homma S., Bonne G. and Worman H.J. (2012) Treatment with selumetinib preserves cardiac function and improves survival in cardiomyopathy caused by mutation in the lamin A/C gene. Cardiovasc. Res., 93, 311–319. [DOI] [PubMed] [Google Scholar]
- 49. Frock R.L., Chen S.C., Da D.F., Frett E., Lau C., Brown C., Pak D.N., Wang Y., Muchir A., Worman H.J. et al. (2012) Cardiomyocyte-specific expression of lamin A improves cardiac function in Lmna−/− mice. PLoS One, 7, e42918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Muchir A., Pavlidis P., Bonne G., Hayashi Y.K. and Worman H.J. (2007) Activation of MAPK in hearts of EMD null mice: similarities between mouse models of X-linked and autosomal dominant Emery–Dreifuss muscular dystrophy. Hum. Mol. Genet., 16, 1884–1895. [DOI] [PubMed] [Google Scholar]
- 51. Muchir A., van Engelen B.G., Lammens M., Mislow J.M., McNally E., Schwartz K. and Bonne G. (2003) Nuclear envelope alterations in fibroblasts from LGMD1B patients carrying nonsense Y259X heterozygous or homozygous mutation in lamin A/C gene. Exp. Cell Res., 291, 352–362. [DOI] [PubMed] [Google Scholar]
- 52. van Engelen B.G., Muchir A., Hutchison C.J., van der Kooi A.J., Bonne G. and Lammens M. (2005) The lethal phenotype of a homozygous nonsense mutation in the lamin A/C gene. Neurology, 64, 374–376. [DOI] [PubMed] [Google Scholar]
- 53. Fichtman B., Zagairy F., Biran N., Barsheshet Y., Chervinsky E., Ben Neriah Z., Shaag A., Assa M., Elpeleg O., Harel A. et al. (2019) Combined loss of LAP1B and LAP1C results in an early onset multisystemic nuclear envelopathy. Nat. Commun., 10, 605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Deconinck A.E., Rafael J.A., Skinner J.A., Brown S.C., Potter A.C., Metzinger L., Watt D.J., Dickson J.G., Tinsley J.M. and Davies K.E. (1997) Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell, 90, 717–727. [DOI] [PubMed] [Google Scholar]
- 55. Grady R.M., Teng H., Nichol M.C., Cunningham J.C., Wilkinson R.S. and Sanes J.R. (1997) Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell, 90, 729–738. [DOI] [PubMed] [Google Scholar]
- 56. Rafael J.A., Tinsley J.M., Potter A.C., Deconinck A.E. and Davies K.E. (1998) Skeletal muscle-specific expression of a utrophin transgene rescues utrophin-dystrophin deficient mice. Nat. Genet., 19, 79–82. [DOI] [PubMed] [Google Scholar]
- 57. Kubben N., Voncken J.W., Konings G., van Weeghel M., van den Hoogenhof M.M., Gijbels M., van Erk A., Schoonderwoerd K., van den Bosch B., Dahlmans V. et al. (2011) Post-natal myogenic and adipogenic developmental: defects and metabolic impairment upon loss of A-type lamins. Nucleus, 2, 195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kim Y. and Zheng Y. (2013) Generation and characterization of a conditional deletion allele for Lmna in mice. Biochem. Biophys. Res. Commun., 440, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Solovei I., Wang A.S., Thanisch K., Schmidt C.S., Krebs S., Zwerger M., Cohen T.V., Devys D., Foisner R., Peichl L. et al. (2013) LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell, 152, 584–598. [DOI] [PubMed] [Google Scholar]
- 60. Wang A.S., Kozlov S.V., Stewart C.L. and Horn H.F. (2015) Tissue specific loss of A-type lamins in the gastrointestinal epithelium can enhance polyp size. Differentiation, 89, 11–21. [DOI] [PubMed] [Google Scholar]
- 61. Arimura T., Helbling-Leclerc A., Massart C., Varnous S., Niel F., Lacène E., Fromes Y., Toussaint M., Mura A.M., Keller D.I. et al. (2005) Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum. Mol. Genet., 14, 155–169. [DOI] [PubMed] [Google Scholar]
- 62. Muchir A., Pavlidis P., Decostre V., Herron A.J., Arimura T., Bonne G. and Worman H.J. (2007) Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J. Clin. Invest., 117, 1282–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shiojima I. and Walsh K. (2006) Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev., 20, 3347–3365. [DOI] [PubMed] [Google Scholar]
- 64. Chaanine A.H. and Hajjar R.J. (2011) AKT signalling in the failing heart. Eur. J. Heart Fail., 13, 825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mutlak M. and Kehat I. (2015) Extracellular signal-regulated kinases 1/2 as regulators of cardiac hypertrophy. Front. Pharmacol., 6, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Östlund C., Sullivan T., Stewart C.L. and Worman H.J. (2006) Dependence of diffusional mobility of integral inner nuclear membrane proteins on A-type lamins. Biochemistry, 45, 1374–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Harborth J., Elbashir S.M., Bechert K., Tuschl T. and Weber K. (2001) Identification of essential genes in cultured mammalian cells using small interfering RNAs. J. Cell Sci., 114, 4557–4565. [DOI] [PubMed] [Google Scholar]
- 68. Wang Y., Östlund C., Choi J.C., Swayne T.C., Gundersen G.G. and Worman H.J. (2012) Blocking farnesylation of the prelamin A variant in Hutchinson-Gilford progeria syndrome alters the distribution of A type lamins. Nucleus, 3, 452–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cance W.G., Chaudhary N., Worman H.J., Blobel G. and Cordon-Cardo C. (1992) Expression of the nuclear lamins in normal and neoplastic human tissues. J. Exp. Clin. Cancer Res., 11, 233–246. [Google Scholar]
- 70. Wu W., Iwata S., Homma S., Worman H.J. and Muchir A. (2014) Depletion of extracellular signal regulated kinase 1 in mice with cardiomyopathy caused by lamin A/C gene mutation partially prevents pathology before isoenzyme activation. Hum. Mol. Genet., 23, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ponchel F., Toomes C., Bransfield K., Leong F.T., Douglas S.H., Field S.L., Bell S.M., Combaret V., Puisieux A., Mighell A.J. et al. (2003) Real-time PCR based on SYBR-Green I fluorescence: an alternative to the TaqMan assay for a relative quantification of gene rearrangements, gene amplifications and micro gene deletions. BMC Biotechnol., 3, 1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Huang H., Amin V., Gurin M., Wan E., Thorp E., Homma S. and Morrow J.P. (2013) Diet-induced obesity causes long QT and reduces transcription of voltage-gated potassium channels. J. Mol. Cell. Cardiol., 59, 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang Y., Herron A.J. and Worman H.J. (2006) Pathology and nuclear abnormalities in hearts of transgenic mice expressing M371K lamin A encoded by an LMNA mutation causing Emery–Dreifuss muscular dystrophy. Hum. Mol. Genet., 15, 2479–2489. [DOI] [PubMed] [Google Scholar]
- 74. Kaplan E.L. and Meier P. (1958) Nonparametric estimation from incomplete observations. J. Amer. Statist. Assn., 53, 457–481. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.