

Abstract
Application of the original vitrification protocol used for pieces of heart valves to intact heart valves has evolved over time. Ice-free cryopreservation by Protocol 1 using VS55 is limited to small samples where relatively rapid cooling and warming rates are possible. VS55 cryopreservation typically provides extracellular matrix preservation with approximately 80 % cell viability and tissue function compared with fresh untreated tissues. In contrast, ice-free cryopreservation using VS83, Protocols 2 and 3, has several advantages over conventional cryopreservation methods and VS55 preservation, including long-term preservation capability at −80 °C; better matrix preservation than freezing with retention of material properties; very low cell viability, reducing the risks of an immune reaction in vivo; reduced risks of microbial contamination associated with use of liquid nitrogen; improved in vivo functions; no significant recipient allogeneic immune response; simplified manufacturing process; increased operator safety because liquid nitrogen is not used; and reduced manufacturing costs.
Keywords: Heart valves, Vitrification, Tissue banking
1. Introduction
1.1. Overview of Heart Valve Tissue Banking
Three types of heart valves are employed for replacement of defective valves in patients: mechanical, xenogeneic tissue, and allogeneic human valves derived from donors’ post-mortem. Most patients receive either xenogeneic tissue or mechanical valves; however, the use of cryopreserved human valve allografts became established during the 1970s and 1980s for certain patient subsets. Two trileaflet valves, the aortic and pulmonary, with associated arterial conduit and cardiac muscle band, are dissected from cadaver donor hearts for patient use. Following cryopreservation, aortic and pulmonary valve allografts have typically been used to reconstruct the left or right ventricular outflow tract for repair of diverse congenital cardiac anomalies. Cryopreserved allografts have especially benefited children with congenital heart disease since the use of alternative mechanical and xenogeneic tissue valves has historically been limited in this patient population. They are also used in women of childbearing age and older patients with memory problems who may not be relied upon to keep up with the medications required for mechanical valves.
In the last century, cryopreserved, control rate frozen Me2SO-protected, human heart valves were used in approximately 20 % of the tissue heart valve procedures performed annually [1–4]. They are currently less utilized because of new xenogeneic valves, which are impacting aortic valve utilization, and because of failures in young patients [5], particularly infants [6,7]. The pathophysiology of allograft heart valve failure is not fully understood [8–10]. Immunology definitely played a significant role in failure in young patients; this led to the development of decellularization methods to minimize the recipient immune response upon implantation. We, however, hypothesized that the rapid deterioration seen in some allograft heart valve recipients might be due, at least in part, to disruptive interstitial ice damage that occurred during cryopreservation leading subsequently to accelerated valve degeneration upon implantation. About 75 % of the area of conventional Me2SO-cryopreserved heart valve leaflets, the most important functional component of a heart valve, was occupied by ice [11] (see Fig. 1). It was hard to imagine that such extensive ice formation was not damaging the tissue. Both light and electron microscopy demonstrated that the ice was extracellular. Upon thawing and rehydration, the leaflets looked normal histologically because the ice domains within the extracellular matrix closed up as the ice melted.
Fig. 1.

Montage of micrographs from a cryo-substituted porcine heart valve leaflet demonstrating the presence of ice (white spaces) throughout the tissue. This tissue sample was slow frozen in culture medium containing 1 M Me2SO. Cryo-substitution was performed with methanol at −90 °C as previously reported [21]
Development of ice-free cryopreservation by vitrification methods for heart valves was initially stimulated by our demonstration of large ice crystal domains within heart valve tissues [11] (see Fig. 1). Vitrification as we have practiced it for heart valve tissues is a non-equilibrium approach (Protocol 1) in contrast to the equilibrium approach developed by Pegg and his colleagues (reviewed in 12). This method for the avoidance of ice was based on the early work of Fahy and Rall [13–15]. The basic principles of this approach have been discussed and reviewed in great detail many times [12, 13, 15–20]. In addition to heart valve tissues, we have used Protocol 1 for many other types of tissue including blood vessels [21–24], cartilage [25–28], pancreatic islets [29], and engineered tissues [30–32]. The functional survival of these vitrified tissues was approximately 80 % or higher, whereas the frozen counterparts yielded less than 20 % survival (see the “The emerging 80/20 rule” [12]). This marked contrast (~80 % vs. 20–30 %) was consistent irrespective of the nature of the tissue except in the case of cardiovascular tissues where equivalent post-cryopreservation cell viability can be obtained by either freezing or vitrification methods. In the case of tissues that were particularly susceptible to cryoprotectant cytotoxicity, the final steps in addition and first steps in removal of the cryoprotectants could be performed at subzero temperatures.
Our first heart valve vitrification studies utilized pieces of porcine aortic valves and intact rat aortic valves in small volumes of a vitrification solution (VS) called VS55 to reflect its 55 % (w/v) of cryoprotectant solutes that was originally designated VS41A by its originators [14]. VS55 consists of an 8.4 M mixture of 1,2- propanediol, formamide, and dimethyl sulfoxide in Euro-Collins (EC) solution. EC is an extracellular-type preservation solution that was developed in the 1970s by Collins and subsequently modified in Europe, where it was the gold standard for hypothermic storage of human kidneys for several years. The protocol is described in US Patent #6,740,484 [33, 34] and in Protocol 1 below. In the porcine studies, we demonstrated that in contrast to conventionally frozen heart valves, vitrified leaflets had no visible ice by light microscopy. There was some discussion of whether or not very small ice crystals could be observed by electron microscopy. In vitro studies with animal tissues have repeatedly demonstrated that Protocol 1 maintained ~80 % heart valve leaflet cell viability immediately after rewarming, returning to control values over a few days in tissue culture (example, Table 1). The emphasis in our rat studies was assessment of transplant outcomes. Two implant models were employed (1) a subcutaneous implantation calcification model and (2) descending thoracic aorta implants [35]. The calcification rate of frozen valves was significantly greater (p < 0.01) than that of vitrified valves in both syngeneic and allogeneic recipients, supporting prior observations that ice-free cryopreservation reduces allogeneic heart valve calcification. However, cryopreservation by freezing and vitrification resulted in only mild morphological changes in 2- and 4-week syngeneic explants, a slight decrease in leaflet cellularity, and a more rapid onset of intimal hyperplasia than in fresh valve explants. The allograft explant groups exhibited changes, fresh allograft leaflets were relatively acellular, and frozen leaflets were hypercellular due to acute inflammatory reactions compared with the vitrified allografts (see Fig. 2). At that time, we decided that “these observations provided only weak support for the hypothesis that the rapid deterioration seen in some allograft heart valve recipients was due to disruptive interstitial ice damage that occurs during cryopreservation by freezing” [35]. In hindsight it is likely that the decreased inflammation observed in ice-free vitrified heart valve explants was actually a sign of the decreased immune response subsequently observed in sheep.
Table 1.
Viability post-VS55 heart valve leaflet preservation
| Post-recovery time incubation (days) | 0 | 1 | 2 |
| Relative florescent units/mg dry weighta (mean, N = 6) | 2,047 | 2,318 | 2,743 |
| Results in % of fresh untreated controls | 80 | 90 | 107 |
Resazurin metabolic assay [45]
Fig. 2.

Representative heterotopic rat heart valve explant histology. The valves were explanted at 2 weeks (4 week explants were similar in appearance). The syngeneic valves all appear similar; however, both the fresh allograft and frozen valve leaflets appear thickened relative to vitrified valves. Fresh allograft leaflets were relatively acellular, and frozen leaflets were hypercellular due to acute inflammatory reactions. Leaflets indicated by arrows, hematoxylin and eosin stain, 10× magnification
1.2. Development of a Vitrification Protocol
When our rat studies were published in 2004 [35], our vitrified heart valve research had encountered a significant hurdle; a stronger demonstration of superiority to conventional cryopreservation was needed to warrant further development. In 2006 the publication of a paper using advanced morphological methods that demonstrated significant differences in extracellular matrices of fresh and Me2SO-cryopreserved frozen heart valves [36, 37] provided the tools and experience to compare vitrified and frozen heart valves. Autofluorescence and particularly second harmonic generated microscopy images revealed that conventional frozen cryopreservation of cardiac valves, when compared with fresh or vitrified tissues, led to the loss of normal ECM structures in valve leaflets [38]. Similar results were found in associated cardiac muscle and artery suggesting that structural deterioration of the ECM is a common consequence of frozen cryopreservation. These observations led to a renewed enthusiasm for further development and optimization of the vitrification protocol for heart valves. Our first protocol was excellent for small-volume tissue samples where relatively rapid cooling and warming rates that avoided ice formation could be achieved. However, upon scale up to full-sized heart valves, 80–100 mL volume, we encountered two problems: (1) cracking at vapor-phase nitrogen temperatures, particularly in dry nitrogen shippers, and (2) the VS55 solution demonstrated ice formation during rewarming [39]. The solution to both problems was to increase the concentrations of the three cryoprotectants in VS55 from 55 to 83 % to make VS83. Furthermore, modulated differential scanning calorimetry studies indicated that this new formulation was potentially stable at −80 °C, free from ice at −80 °C [39], which would make it easier and cheaper to store and ship the tissue samples.
Three in vivo juvenile sheep implant studies were performed to test the VS83 protocol (Protocol 2). The first two studies are unpublished. Protocol 2 was similar to Protocol 1 except that VS83 was employed and after cooling the tissues were stored at −80 °C and shipped on dry ice to the implant site. There were differences in the post-warming wash in each study. Another issue that should be made clear is that in contrast to early work with VS55, there was no effort to keep heart valve tissue cells alive, viable, with VS83. The reason for this is that at this time there is no convincing evidence in the literature suggesting that maintenance of graft cell viability after cryopreservation is beneficial in allogeneic heart valve recipients. In the early years, 1980s and 1990s, of allograft heart valve processing, cell viability was thought to be very important for long-term maintenance of the heart valve post-transplant in patients. Since then analysis of explanted heart valves has supported the assumption that living cells are not necessary in a functioning transplant. Explant analyses indicated that most of the original cells either vanished, especially in the leaflet cusps, or were replaced by donor fibroblasts [40, 41]. Only a few living cells from the original donor could be detected after long-term transplantation and normal graft leaflet cellularity was not observed in a single explant [42]. Furthermore, many groups are now decellularizing heart valves in order to reduce their concerns regarding immunogenicity and its consequences in patients [43, 44]. Therefore, we did not attempt to preserve cell viability during further development of the VS83 method for heart valves. We had previously demonstrated that the VS83 formulation cryoprotectant components are cytotoxic for cultured porcine myofibroblasts at high concentrations [45].
In the first study, we compared conventional frozen aortic sheep heart valves with VS83 vitrified valves stored at −80 °C or in vapor-phase nitrogen over a period of 6 months in vivo. These valves went through a multistep wash protocol similar to that described in Protocol 1. Explant pathology studies indicated that frozen heart valves had most deterioration at explant and 2/4 explants had changes that may have been due to ice damage (Fig. 3 and Table 2). Even though all heart valves exhibited changes, the VS83 formulation combined with −80 °C storage demonstrated the least structural deterioration. Explant gross pathology and histology revealed little difference, except that two cryopreserved by freezing controls had single holes present at explant that could have been due to ice damage (example, Fig. 3d). There were no significant differences in calcium content in explanted leaflet or artery samples (Fig. 4) and least development of aortic insufficiency (AI) (Table 2).
Fig. 3.

Sheep heart valve explant gross and histopathology. Heart valves were explanted at 6 months and photographed before further processing. (a) VS83 formulation stored at −80 °C in a mechanical storage freezer, (b) H&E-stained section of VS83 −80 °C explant after 6 months in vivo demonstrating cell-free leaflet with recipient cell-derived pannus overgrowth on the artery (bottom and leaflet top), (c) and (d) cryopreserved by freezing controls, (d) also demonstrating a hole in the base of leaflet, (e) and (f) VS83 formulation stored in vapor-phase nitrogen
Table 2.
Pathology overview
| Group | Description | Implant pathologya | AI at 6 monthsb |
|---|---|---|---|
| 1 | Conventional freezing method | Mild diffuse leaflet fibrosis. 3/4 exhibited focal nodular vegetation (organized thrombi) and 1/4 exhibited calcified foci. 1/4 exhibited a basal dehiscence and 1/4 a paravalvular aneurysm. 1/4 conduits mineralized | 1–2+, 1–2+, 2+, 2–3+ |
| 2 | VS83 stored at −80 °C | Implants all exhibited mild diffuse fibrosis; 2 of 4 conduits were mineralized | −2+, none,1+, trace-1+ |
| 3 | VS83 stored in vapor-phase nitrogen | Diffuse and focal fibrosis, 2 of 4 exhibited thrombi and 1 of 4 exhibited focal nodular vegetation on leaflets. 3 of 4 conduits were mineralized | None, 2–3+, 2–3+, 2–3+ |
Leaflets in all groups were devoid of cells except for recipient overgrowth that appears to be a chronic low-grade inflammatory response with a marked tendency for all leaflets to exhibit some thinning with thicker fibrous overgrowth at the base of the cusps resulting in leaflet thickening and stiffening
Aortic Insufficiency(AI) was assessed by echocardiographic evaluation just prior to sacrifice
Fig 4.

Calcium contents in sheep heart valve explants. Leaflet results are on the left and conduit results on the right. VPN = vapor-phase nitrogen. Calcium-specific mineralization occurring in aortic allografts was quantified using atomic absorption spectroscopy [59]. There were no significant differences
These test results suggested that preservation methods based upon modifications of VS83 combined with −80 °C storage in a mechanical freezer may provide an alternative for liquid nitrogen storage of allogeneic heart valves, reducing the costs of both long- term storage and distribution. Due to the small size of each experimental group (n = 4), it was not possible to state that the benefits of the VS83 protocol combined with −80 °C storage were significantly different from cryopreservation by freezing. However, this test group demonstrated less structural deterioration and less aortic insufficiency than traditionally cryopreserved by freezing allograft heart valves after 6 months in vivo in sheep.
The second study was an unpublished Current Good Laboratory Practice (cGLP) study in which decellularized arterial tissue, sheep pulmonary artery, was vitrified (n = 5 sheep) and compared with cryopreserved tissue (n = 2 sheep) by implantation as patches in the pulmonary artery of juvenile female sheep (results on file pending future regulatory submissions). The cryopreserved/decellularized/vitrified samples had an average 99.8 ± 0.05 % reduction in extractable DNA. Two patches were implanted in each sheep, and they were explanted after 140 days. One explant from each sheep was used for calcium analysis (non-GLP inductively coupled plasma optical emission spectroscopy) and the other for histopathology. Histopathology included review of H&E, α-actin smooth muscle, TUNEL for apoptosis, Hsp47 (marker for the production of type I and III collagens) (Fig. 5), and factor VIII endothelial cell marker-stained sections. Panel reactive antibody analysis was performed using serum obtained pre-implant and 10 and 20 weeks post implant. None of the animals demonstrated MHC Class I antibodies. One of the two controls demonstrated a 54.65 % shift to the right over baseline for MHC Class II. One of five experimental sheep experienced an 11.16 % shift over baseline for MHC Class II. There were no differences in blood work, echocardiography, or necropsy results between the experimental and control groups. The cryopreserved patches decreased in cellularity and calcified. The decellularized vitrified patches recellularized and resisted calcification, and the infiltrating cells were producing α-smooth muscle actin, factor VIII, and Hsp47 after 20 weeks in vivo. An increase in TUNEL-positive cells was observed in vitrified tissue compared with cryopreserved controls, 45 vs. 34 % positive. The cellularity was the result of fibroblast and myofibroblast infiltration, and the amount of inflammation seen was little to none. The results were similar to published work on the decellularization method employed using ovine patch grafts in the same animal model [46–48]. These results suggest that VS83-preserved tissues should be safe in humans. However, the increased apoptosis noted in the TUNEL staining led to in vitro studies to determine whether rewarmed, washed vitrified tissues were cytotoxic (discussed later).
Fig. 5.
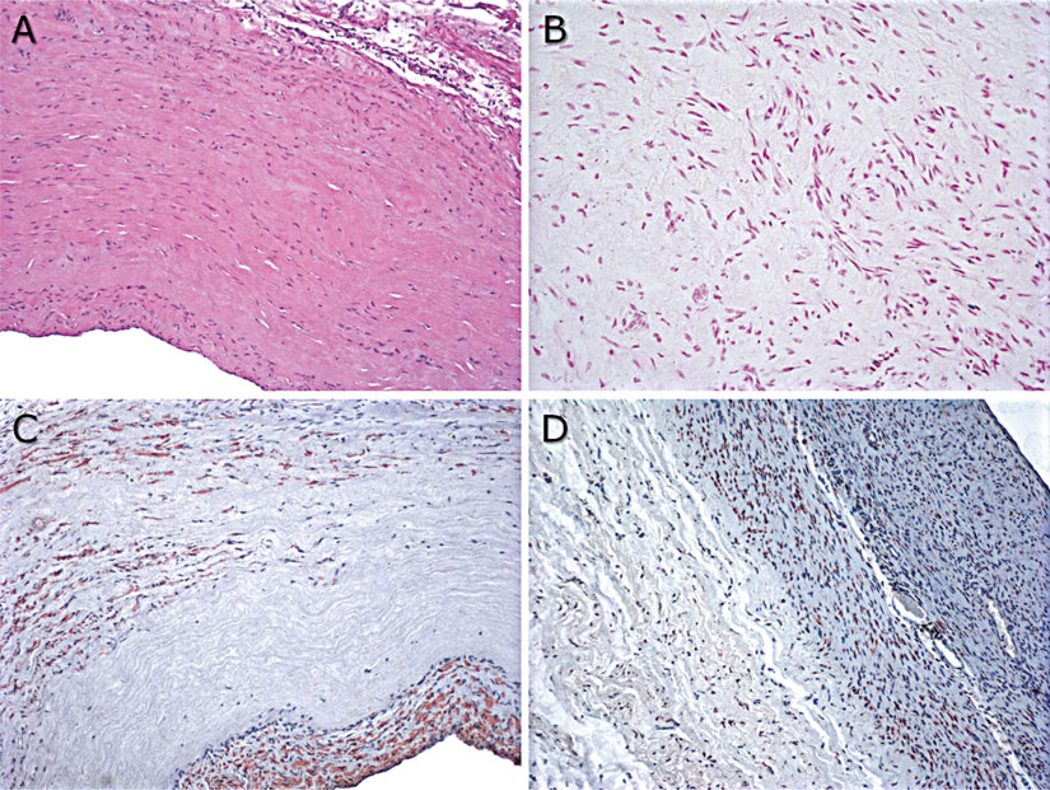
Representative explant histology from 20-week decellularized VS83-preserved artery patch explants: (a) hematoxylin and eosin stain, 100×; (b) TUNEL stain to identify apoptotic cells, 200×; (c) actin stain to identify smooth muscle cells, 100×; and (d) HSP47-stained cells to identify cells actively producing collagen, 100×. Factor VIII-positive endothelial cells were also observed (not shown)
The third study was a research study in which the VS83 protocol was employed with simplified washing steps to process ovine pulmonary heart valves (Protocol 2 [49, 50]). Vitrified and conventionally frozen valves were then stored at −80 °C and in vapor- phase nitrogen, respectively, for approximately 1 year. Vitrified pulmonary valves were shipped from Charleston, South Carolina, to the implantation site in Berlin, Germany, on dry ice and stored at −80 °C until implantation. The control frozen pulmonary valves were shipped in a nitrogen dry shipper and stored at −135 °C in Berlin. Aortic valve tissues were kept in Charleston to evaluate the impact of preservation without implantation. The pulmonary valves were implanted in the pulmonary position. We had concerns that our earlier unpublished sheep studies, presented above, utilized sheep that may have been highly inbred because crossbred Whiteface heart donors and recipients were used. Therefore, two different strains of sheep were chosen, crossbred Whiteface heart valve donors from Minnesota and Heidschnucke, a Nordic short- tailed breed, recipients from Germany to increase the possibility of using a true allogeneic model.
Multiphoton microscopy revealed reduced, but not significantly, damaged extracellular matrix before implantation in frozen valves compared with ice-free tissues. Viability assessment revealed significantly less metabolic activity in the ice-free valve leaflets and artery samples compared with frozen tissues (p < 0.05). After 3 and 6 months, in vivo valve function was determined by two- dimensional echo-Doppler, and at 7 months the valves were explanted. Severe valvular stenosis with right heart failure was observed in recipients of frozen valves; the echo data revealed increased velocity and pressure gradients compared to ice-free valve recipients (p = 0.0403, p = 0.0591). Histopathology showed significantly thickened leaflets in the frozen valves (p < 0.05) and infiltrating CD3+ T-cells (p < 0.05) compared with ice-free vitrified valve leaflets. Multiphoton microscopy at explant revealed reduced inducible autofluorescence and extracellular matrix damage in the frozen explants and well-preserved structures in the ice-free explant leaflets. We concluded that ice-free cryopreservation of heart valve transplants at −80 °C avoids ice formation and tissue-glass cracking and preserves extracellular matrix integrity resulting in minimal inflammation and improved hemodynamics in allogeneic juvenile sheep [49–51].
Biomaterial properties of ice-free preserved heart valves have also been assessed [39]. We observed occasional tissue cracking and fracturing of the vitrified solution in experiments involving storage of heart valves in VS55 below the solution glass transition temperature in vapor-phase nitrogen. Substitution of VS83 for VS55 corrected this for samples stored at −135 °C. Biomechanical testing was performed on fresh untreated control and VS83 cryopreserved leaflets and stored at −80 and −135 °C. There were no significant differences in Young’s modulus and ultimate stress (strain) observed between the −80 and −135 °C stored leaflets for any of the mechanical test comparisons; similarly neither group differed significantly when compared with the fresh untreated control leaflet samples (Fig. 6). Thakrar [52] reported that vitreous cryopreservation, employing 40 % w/v 1,2-propanediol in DMEM culture medium maintains the viscoelastic property of human vascular grafts in contrast to conventional cryopreservation by freezing with Me2SO. We also assessed the impact of VS83 preservation upon ECM integrity at the two storage temperatures. Multiphoton imaging of VS83-preserved heart valves stored at each temperature demonstrated branched elastic fibers and wavy bundles of collagen in almost all vitrified heart valve leaflets, aortic trunk, and cardiac muscle specimens. There were no significant differences in SHG signal intensities (Fig. 7) or in qualitative mean rating scores on tissue quality determined by the operator during imaging. These studies provided further support for the storage of VS83-preserved tissues at −80 °C provided that cell viability was not required. However, these tissues are technically not vitrified since we are storing them above the glass transition temperature. Therefore, in most of our more recent publications, we refer to tissues preserved at −80 °C as being ice-free cryopreserved.
Fig. 6.

Summary of VS83 biomechanical testing. Results comparing VS83-preserved leaflets, stored at −80 or −135 °C, and fresh untreated control leaflets are presented as (a) ultimate force, (b) ultimate stress, (c) ultimate strain, and (d) Young’s modulus. Data presented as the mean ± 1 standard deviation, n = 5–6, for each individual sample mean. There were no significant differences [39]
Fig. 7.

Summary of second harmonic generated signal intensities. Collagenous structures in heart valve tissues were observed by spectral fingerprinting after VS83 preservation at −80 or −135 °C. Data presented as the mean ± 1 standard deviation, n = 4, of individual sample mean maximal intensity emission values detected at peak emission wavelengths of 414–425 nm. There were no significant differences [39]
Further process simplification has since been performed (Protocol 3) with in vitro evaluation studies to assess the impact of VS83 vitrification on cell viability and tissue hemocompatibility. During the development of Protocol 3, we examined the impact of varying the number of addition or removal steps in combination with cooling and found that it made little if any difference for cell survival. Room temperature incubation in VS83 without subsequent cooling and storage had similar effects on cell viability. The vitrified artery and leaflet components were significantly less viable than either fresh or Me2SO-cryopreserved frozen tissues. The muscle component, while less viable, was not significantly different. The changes in tissue viability were a function of cryoprotectant exposure and cytotoxicity, not temperature reduction during storage. Therefore, the simple one-step addition at room temperature was selected for use in further experiments. Multiple changes of washing solution were employed to minimize the residual cryoprotectant concentrations after rewarming. Subsequent studies confirmed the impact of VS83 on heart valve tissue cell viability using Protocol 3 [52] and demonstrated that heart valve tissues were hemocompatible using multiple assays [53, 54]. Protocol 3 will be tested in vivo with either ice-cold or room temperature wash solutions in future studies.
1.3. Current State
In the evolution of our vitrification protocols, we started with Protocol 1 based upon VS55 (reviewed in 12, 55). This protocol resulted in excellent avoidance of ice formation with retention of cell viability and matrix integrity provided that the sample size and geometry permitted rapid cooling and warming. Ice formed during slow rewarming of VS55 but not during rewarming of a more concentrated cryoprotectant solution, VS83 [39]. The use of Protocol 2 with VS83 permitted the use of large sample volumes and heart valve tissue storage at −80 °C at the expense of cell viability.
A variety of reasons for allograft heart valve failure were discussed in the past, and most investigators have emphasized immunological issues [41, 56]. Standard quantitative and qualitative cellular and matrix evaluations such as biochemical, immunohisto-chemical screening, and routine histology did not help to solve the controversial discussion whether remaining allogeneic cells or potentially altered extracellular matrix contributed to the observed degeneration. Preliminary data on patients treated with decellularized allografts has recently demonstrated that decellularization did not significantly improve outcome in terms of pressure gradients and structural deterioration compared to nondecellularized allografts [57]. These early clinical results question the validity of theories suggesting that an immune reaction to the remaining donor cells in allogeneic heart valves is the sole cause of structural deterioration. The impact of vitrification on the immunoreactivity of tissue grafts was first clearly observed in the in vivo sheep model study employing different sheep strains discussed above [49, 50]. Explant leaflets demonstrated well-preserved extracellular matrix structures, with few CD3+ T-cells and significantly improved hemodynamics. In marked contrast, leaflets in the frozen control group demonstrated significant matrix damage and T-cell-mediated inflammation. The observation that explanted vitrified leaflets were nearly devoid of active immune cells suggested that a state equivalent to decellularization was accomplished by the ice-free cryopreservation step. Further support for this observation was obtained in studies combining human cells with rewarmed, washed tissues in vitro [58]. Concerns that the apparent benefits of vitrification were due to cytotoxic cryoprotectant residuals were dismissed by demonstration that cells cultured with these tissues survived [58] (Figs. 8 and 9). Our current working hypothesis to explain these changes in vitrified tissue immunoreactivity is that this vitrification method is modifying or masking tissue signals perceived by responder cells. These tissue signals are most likely damage-associated molecular pattern molecules that can initiate and perpetuate immune responses.
Fig. 8.

Assessment of VS83-preserved cytotoxicity in vitro. Human umbilical vein-derived endothelial cells were cultured with ice-free preserved vessels for 7 days in non-tissue culture grade tubes. The constructs were tested on days 2, 3, 4, and 7 using the resazurin reduction assay. There was a gradual decrease over time in culture for all groups, no significant differences except for comparison of the no cell group with the HUVEC groups (p < 0.05). No cell = ice-free preserved vessels without added endothelial cells, Decell = decellularized vessels plus cells. Rewarmed ice-free preserved vessels were subjected to 2, 4, and 8 wash steps. Note: The no cell and 2–8 step groups were not decellularized. The gradually increasing viability in the no cell group is due to proliferation of a small number of smooth muscle cells, which were used to engineer the vessel, surviving the ice-free preservation process
Fig. 9.

Endothelial cell-vessel co-culture histology after 7 days. Human umbilical vein-derived endothelial cells were cultured with ice-free preserved vessels for 7 days in non-tissue culture grade tubes. The tissues were then fixed, embedded, sectioned, and stained for the CD31 endothelial cell marker and photographed at 40× magnification. (a) No cell = ice-free preserved vessel without added endothelial cells, (b) control decellularized vessel plus endothelial cells, and ice-free preserved vessel with either (c) 2 wash steps, (d) 4 wash steps, or (e) 8 wash steps. No qualitative differences were observed between the decellularized and IFC groups. Note: CD31-positive endothelial cells at the upper edge of the tissue in b–e. The interstitial staining is not associated with endothelial cells since it is also seen in a, the “no cell” added group
The potential advantages of VS83 heart valve storage at −80 °C employing Protocol 3 includes reduced infrastructural needs for preservation, storage, and shipping in comparison with traditional freezing methods while maintaining extracellular matrix integrity and material properties. The loss of cell viability is probably a benefit contributing to the reduction of immunogenicity observed in vivo and in vitro. The typical methods employed for conventional cryopreserved frozen heart valves require a control rate freezer, storage in nitrogen-cooled tanks, and a continuous supply of liquid nitrogen. Furthermore, conventionally cryopreserved valves also require shipment in nitrogen dry shippers to the implantation site where the valves need to be kept in nitrogen-cooled freezers until implantation. Dry shippers are expensive, and the cost of return shipment adds to the cost of the heart valve to the end user. Liquid nitrogen is also a safety hazard for employees, and precautions must be taken during operation of nitrogen-cooled equipment. In contrast, no expensive equipment and liquid nitrogen are needed for the Protocol 3 VS83 storage method that has evolved. There may also be a reduced risk of microbial contamination associated without use of liquid nitrogen. The only equipment required is a mechanical storage freezer, and long-term preservation can be performed at −80 °C. Shipping requires only an insulated box with dry ice. The removal of liquid nitrogen from the process also reduces the training required and safety hazards for employees. In the future, we anticipate removal of the cooling below −100 °C step in the initial cooling procedure in order to further reduce the complexity and cost of this method. In vivo large animal studies of VS83 preservation combined with −80 °C storage employing pulmonary valves in juvenile sheep have already demonstrated better function and explant pathology than traditionally frozen cryopreserved heart valves [49, 50]. VS83 preservation protocols combined with −80 °C storage method should be particularly beneficial in developing countries with limited financial and logistic resources that have a high incidence of aortic heart valve disease.
2. Materials
Prepare all solutions using deionized water (prepared with an activated carbon filter, a mixed bed working deionizer, and a mixed bed polishing deionizer to attain a sensitivity of 10 MΩ cm at room temperature). The solutions are prepared with raw materials that meet or exceed requirements of the American Chemical Society. If no such specifications exist, chemicals of the highest purity available should be used. The solutions are made at room temperature and stored at 4 °C for up to 4 weeks. The solution formulations described are for 1 L batches.
2.1. Euro-Collins Solutions
Put 0.5 L water in a container (graduated cylinder or glass beaker), then add a stir bar followed by addition of each component in the following order, 174.76 g Dextrose, 10.2 g KH2PO4, 36.5 g K2HPO4, 5.6 g KCl, 4.2 g NaHCO 3 make up to 1 L with water to make a 5× stock solution. Stir continuously during formulation; when the solution is clear of particles, it should be filtered using 0.2 μm filters. We do not test the osmolality or pH of 5× Euro-Collins. Dilute 1:5 with water to make 1× Euro- Collins and check the pH (~7.4) and osmolality (365 ± 5). Store at 4 °C.
2.2. Vitrification Solutions
VSs are formulated by placing a stir bar and then each component, in the order that they appear in the list below (Table 3), in a graduated container. Stir continuously; when finished and it is clear of particles, it should be filtered. We use bottle top 0.2 μm filters, and it takes a long time (hours) to filter due to the high viscosity of the solutions. At the conclusion of preparation, check the final pH and make a 1:20 dilution for assessment of osmolality. Store at 4 °C.
Table 3.
VS formulation, quantities
| VS55 | VS83 | |
|---|---|---|
| 5× Euro-Collins (mL) | 200 | 200 |
| HEPES buffer (g) | 2.39 | 2.39 |
| Propylene glycola (g) | 168.38 | 252.57 |
| Formamidea (g) | 139.56 | 209.34 |
| Me2SOa (g) | 242.14 | 363.21 |
| Water (mL) | to 1 L | to 1 L |
| pH (units) | 7.9–8.1 | 7.9–8.1 |
| Osmolalityb (mOsm/kg) | 460 ± 5 | 670 ± 5 |
Liquids measured by weight
20× dilution = 50 μL in 950 μL of distilled water
2.3. Vitrification Solutions
Sequential vitrification solutions, both addition and removal solutions, are used in certain protocols (see Note 1). The VS chemical component concentration can be increased from 1/8, ¼, ½, and ¾ to 100 % of the final full-strength VS formulation being employed. The diluted VS solutions are prepared by the addition of appropriate volumes of 1× EC. The solutions employed for removal are similar to those employed for addition except that 200–400 mM mannitol is added to the formulation as an osmotic buffer. For example, using seven steps, the VS CPAs are decreased from full strength to 0 M plus mannitol to 0 M, plain EC, and finally Dulbecco’s Modified Eagle Medium (DMEM).
3. Methods
3.1. Protocol 1: Suitable for Small Heart Valve Tissue Samples (≤3 mL Total Volume, Sample Plus Solution) with Maximum Retention of Cell Viability and Matrix Integrity
Tissues should be gradually infiltrated with VS55 at 4 °C (see Note 1). Precooled diluted VS solutions are employed in six 15 min steps at 4 °C of increasing CPA concentration as previously described [12] (see Note 2).
The top of the vitrification solution is then covered with 2-methylbutane (isopentane; see Note 3).
The samples should be cooled rapidly to −100 °C followed by slow cooling to ≥ −135 °C (see Note 4).
Finally the samples should be stored at −135 °C in a mechanical storage freezer or at ≥ −135 °C near the top in a vapor-phase nitrogen freezer.
Rewarming of vitrified tissues is a two-stage process including slow warming to −100 °C and then rapid warming to ~4 °C (see Note 5). Wipe the container with 70 % ethanol to minimize the risk of microbial contamination and use sterile pickups to transfer the tissue to the first VS removal solution.
The vitrification solution is then removed in a stepwise manner at 4 °C in six or seven sequential, 15 min steps of increasingly dilute VS solutions containing mannitol as previously described [12] (see Note 1). Finally the tissues are placed in DMEM culture medium.
3.2. Protocol 2: Suitable for Large Heart Valve Tissue Samples (Up to 100 mL Total Volume) with Preservation of Matrix Integrity and Minimal Residual Cell Viability
Tissues are gradually infiltrated with VS83 in six 15 min steps of increasing concentration at 4 °C.
After the final step, the tissues are placed individually in sterile polyester bags containing 70–80 mL of the vitrification solution. Each bag is then evacuated of air by squeezing (see Note 3) and heat-sealed.
The bags containing tissues need to be cooled rapidly to −100 °C in a precooled 2-methylbutane bath and then transferred to storage (see Note 4).
Store at −80 °C in a mechanical storage freezer (see Note 6).
Rewarming needs to be performed by placing each bag in a 37 °C water bath until the solution moves freely.
The bag is then transferred onto ice and the bag externally cleaned with 70 % ethanol to reduce the risk of microbial contamination. Cut the bag open with sterile scissors and use sterile pickups to transfer the tissue to the first VS removal solution.
A multistep VS removal protocol was employed in our first unpublished sheep study, similar to that used in Protocol 1, above. Simpler, washing methods have been employed in a subsequent sheep study [49, 50] and an unpublished study with arterial patches (see Note 7).
3.3. Protocol 3: Simplified Versions of Protocol 2 in Development for Heart Valves
Place the tissues in sterile polyethylene bags containing 80 mL of VS83 at room temperature (see Note 1).
Evacuate the air and heat-seal the bags (see Note 3).
Incubate the tissues with continuous shaking for at least 1 h at room temperature prior to cooling.
The cooling process can then be achieved by placing the bags for 10 min in a precooled bath of 2-methylbutane (< −100 °C, see Note 4).
The tissues can then be placed at −80 °C for storage.
Rewarming of the tissue is then performed by submersion of the tissue in its bag in a 37 °C water bath.
The bag is then dried and externally cleaned with 70 % ethanol to reduce the risk of microbial contamination. Cut the bag open with sterile scissors and use sterile pickups to transfer the tissue to the first wash step.
Washing to remove CPAs has been performed using five wash steps of 5 min duration each with ice Euro-Collins solution or with several washes with Lactated Ringers solution containing 5 % dextrose (see Note 8).
Acknowledgments
This work was supported in part by a US Public Health Grant from the National Institute of Biomedical Imaging and Bioengineering, Grant # R43 EB014614, to KGMB. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Biomedical Imaging and Bioengineering or the National Institutes of Health. Commercial use of protocols disclosed in this work is subject to several issued US Patents (6,194,137; 6,596,531; 6,740,484; 7,157,222; 8,440,390) and International Patents (available upon request).
Footnotes
Notes
Stepwise VS addition and removal procedures are employed in Protocol 1 in order to minimize damage to cells as they respond to osmotic changes in their environment as a consequence of being placed in VS solutions with high concentrations of CPAs (reviewed in 12). This is not an issue in Protocols 2 and 3 because we are not trying to maximize cell viability. Loss of cell viability is considered advantageous because it appears to reduce immunogenicity in vivo [49, 50].
In the last step, the tissues are placed in vials; we usually use glass scintillation vials (25 × 60 mm), containing 1–2 mL of precooled full-strength VS solution. In circumstances where unexpected cytotoxicity is observed, the last one or two steps can be performed at subzero temperatures in a cold bath to minimize cytotoxicity.
We use 0.7–1.0 mL of 2-methylbutane for the glass vials described in Note 2. The 2-methylbutane has a freezing point of −160 °C and density of 0.62 g/cm3. The 2-methylbutane layer prevents direct air contact minimizing the risk of ice nucleation. Alternatively, the vial can be purged with a dry gas, such as nitrogen, or a container without excess air space can be used.
Rapid cooling to < −100 °C is done by placing the vials in a precooled (−135 °C) 2-methylbutane bath; slow cooling is performed by placing the vials in air in the top of storage freezer, either a −135 °C mechanical freezer or near the top of a vapor- phase nitrogen-cooled freezer. More rapid cooling can be obtained, if needed, using liquid ethane at ~ −183 °C. However, this strategy may result in the development of thermal stresses within the sample because the edges will cool rapidly and the inside more slowly. Such stresses may lead to cracking during storage or upon rewarming.
A dummy sample prepared under the same conditions as the preserved samples with a thermocouple is used to monitor cooling and warming. Make sure that the thermocouple has been calibrated using a NIST standard. The dummy sample is placed in close proximity to the samples to be rewarmed in the storage freezer. Plug the dummy sample thermocouple into a reader. Place the samples and dummy sample at the top of the storage freezer and hold just inside the lid. When the temperature reaches −100 °C (~1 min), initiate the rapid warming step. Place both vials in a room temperature 30 % Me2SO bath and swirl the vials manually until the glass begins to melt (the VS starts to move). Open the vial and remove the VS with a transfer pipette and add the first dilution solution. Start timer for 15 min.
The samples can be stored in vapor-phase nitrogen but VS solution fractures may be observed. Although the visual impact of the fracturing is disturbing, we have rarely seen tissue fractures, and on the rare occasions when tissue cracking has occurred, it has been during the use of nitrogen dry shippers for transport. If samples are stored in vapor-phase nitrogen, the two-step rewarming should be performed as described in Protocol 1.
Ice-free artery patches were rewarmed by placing the bag containing the tissue in a 37 °C water bath for 1 min and then washed by transferring the contents of the bag to an AlloFlow® continuous gradient washout chamber with 1 L of room temperature isotonic saline over 20 min. Ice-free cryopreserved valves were rinsed briefly in ice-cold EC solution containing 200 mM mannitol. Then three sequential rinses were performed for 15 min each with continuous careful agitation in ice-cold EC solution with 200 mM mannitol, followed by EC solution alone and finally DMEM. The valves were stored briefly on ice in DMEM culture medium until implantation or evaluation.
We are presently evaluating the use of Lactated Ringers solution containing 5 % dextrose (LRD5) for washing because it is commonly available in surgery suites where ice-free cryopreserved tissues may be used. Human vessels generated in the laboratory by culturing human smooth muscle cells on a polyglycolic acid scaffold [31] were employed. The tissues were subjected to a total of 2, 4, 8, or 15 min room temperature wash steps in Lactated Ringers solution containing 5 % dextrose to remove residual cryoprotectants. In control decellularized tissues, the cellular material was removed using enzymes and detergents leaving an extracellular matrix tube as previously described [31], which was refrigerated in phosphate-buffered saline until tested. Trypan blue excluding human umbilical vein endothelial cells was added to each tissue sample under physiological cell culture conditions in non-tissue culture tubes (to minimize cell migration from the tissues). There were no statistically significant viability differences comparing endothelial cell seeded decellularized vessels and ice-free preserved tissue-engineered blood vessels that were not decellularized after 2, 4, or 8 washes in cell culture (Fig. 8). The endothelial cells formed confluent monolayers on all surfaces of the washed tissues, regardless of the number of post-rewarming washes (Fig. 9), demonstrating that any residual cryoprotectants present were not cytotoxic. In parallel xenograft tissue studies, we have also demonstrated that viability of human peripheral blood mononuclear cells is not impacted by culture with rewarmed tissues washed 5× with EC.
References
- 1.Angell WW, DeLanerolle P, Shumway NE (1973) Valve replacement: present status of homograft valves. Prog Cardiovasc Dis 15: 589–622 [DOI] [PubMed] [Google Scholar]
- 2.Stelzer P, Jones DJ, Elkins RC (1989) Aortic root replacement with pulmonary autograft. Circulation 80:209–213 [PubMed] [Google Scholar]
- 3.Angell WW, Oury JH, Lamberti JJ, Koziol J (1989) Durability of the viable aortic allograft. J Thorac Cardiovasc Surg 98:48–56 [PubMed] [Google Scholar]
- 4.O’Brien MF, McGiffin DC, Stafford EG, Gardner MA, Pohlner PF, McLachlan GJ, Gall K, Smith S, Murphy E (1991) Allograft aortic valve replacement: long-term comparative clinical analysis of the viable cryopreserved and antibiotic 4C stored valves. J Card Surg 6: 534–543 [DOI] [PubMed] [Google Scholar]
- 5.O’Brien MF, Stafford EG, Gardner MAH, Pohlner PF, Tesar PJ, Cochrane AD, Mau TK, Gall KL, Smith SE (1995) Allograft aortic valve replacement: long-term follow-up. Ann Thorac Surg 60:565–570 [DOI] [PubMed] [Google Scholar]
- 6.Clarke DR, Campbell DN, Hayward AR, Bishop DA (1993) Degeneration of aortic valve allografts in young recipients. J Thorac Cardiovasc Surg 105:934–942 [PubMed] [Google Scholar]
- 7.Yankah AC, Alexi-Meskhishvili V, Weng Y, Schorn K, Lange RE, Hetzer R (1995) Accelerated degeneration of allografts in the first two years of life. Ann Thorac Surg 60:71–77 [DOI] [PubMed] [Google Scholar]
- 8.Wolfinbarger L Jr, Hopkins RA (1989) Biology of heart valve cryopreservation In: Hopkins PA (ed) Cardiac reconstructions with allograft valves. Springer, New York, pp 21–36 [Google Scholar]
- 9.Mitchell RN, Jonas RA, Schoen FJ (1995) Structure-function correlations in cryopreserved allograft cardiac valves. Ann Thorac Surg 60:8108–8113 [DOI] [PubMed] [Google Scholar]
- 10.Mitchell RN, Jonas RA, Schoen FJ (1998) Pathology of explanted cryopreserved allograft heart valves: comparison with aortic valves from orthotopic heart transplants. J Thorac Cardiovasc Surg 115:118–127 [DOI] [PubMed] [Google Scholar]
- 11.Brockbank KGM, Lightfoot FG, Song YC, Taylor MJ (2000) Interstitial ice formation in cryopreserved homografts: a possible cause of tissue deterioration and calcification in vivo. J Heart Valve Dis 9:200–206 [PubMed] [Google Scholar]
- 12.Taylor MJ, Song YC, Brockbank KGM (2004) Vitrification in tissue preservation: new developments In: Benson E, Fuller B, Lane N (eds) Life in the frozen state. Taylor and Francis Books, London, pp 603–641 [Google Scholar]
- 13.Fahy GM, MacFarlane DR, Angell CA, Meryman HT (1984) Vitrification as an approach to cryopreservation. Cryobiology 21: 407–426 [DOI] [PubMed] [Google Scholar]
- 14.Rall WF, Fahy GM (1985) Ice-free cryopreservation of mouse embryos at –196 °C by vitrification. Nature 313:573–575 [DOI] [PubMed] [Google Scholar]
- 15.Rall WF (1987) Factors affecting the survival of mouse embryos cryopreserved by vitrification. Cryobiology 24:387–402 [DOI] [PubMed] [Google Scholar]
- 16.Fahy GM (1988) Vitrification In: McGrath JJ, Diller KR (eds) Low temperature biotechnology: emerging applications and engineering contributions. The American Society of Mechanical Engineers, New York, pp 113–146 [Google Scholar]
- 17.Fahy GM (1989) Vitrification as an approach to organ cryopreservation: past, present, and future In: Smit Sibinga CT, Das PC, Meryman HT (eds) Cryopreservation and low temperature biology in blood transfusion. Kluwer Academic Publishers, Dordrecht, pp 255–268 [Google Scholar]
- 18.Armitage WJ, Rich SJ (1990) Vitrification of organized tissues. Cryobiology 27:483–491 [DOI] [PubMed] [Google Scholar]
- 19.Pegg DE, Diaper MP (1990) Freezing versus vitrification: basic principles In: Smit Sibinga CT, Das PC, Meryman HT (eds) Cryopreservation and low temperature biology in blood transfusion, vol 24 Kluwer Academic Publishers, Dordrecht, pp 55–69 [Google Scholar]
- 20.MacFarlane DR, Forsyth M, Barton CA (1992) Vitrification and devitrification in cryopreservation In: Steponkus PL (ed) Advances in low temperature biology, vol 1 JAI Press, Greenwich, CT, pp 221–278 [Google Scholar]
- 21.Song YC, Khirabadi BS, Lightfoot F, Brockbank KGM, Taylor MJ (2000) Vitreous cryopreservation maintains the function of vascular grafts. Nat Biotechnol 18:296–299 [DOI] [PubMed] [Google Scholar]
- 22.Rabin Y, Taylor MJ, Walsh JR, Baicu S, Steif PS (2005) Cryomacroscopy of vitrification, Part I: a prototype and experimental observations on the cocktails VS55 and DP6. Cell Preserv Technol 3:169–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baicu S, Taylor MJ, Chen Z, Rabin Y (2006) Vitrification of carotid artery segments: an integrated study of thermophysical events and functional recovery towards scale-up for clinical applications. Cell Preserv Technol 4: 236–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brockbank KGM, Taylor MJ (2007) Tissue preservation In: Baust JG (ed) Advances in biopreservation. CRC Press, Boca Raton, pp 157–196 [Google Scholar]
- 25.Song YC, An YH, Kang QK, Li C, Boggs JM, Chen Z, Taylor MJ, Brockbank KGM (2004) Vitreous preservation of articular cartilage grafts. J Invest Surg 17:65–70 [DOI] [PubMed] [Google Scholar]
- 26.Song YC, Lightfoot FG, Chen Z, Taylor MJ, Brockbank KGM (2004) Vitreous preservation of rabbit articular cartilage. Cell Preserv Technol 2:67–74 [Google Scholar]
- 27.Brockbank KGM, MacLellan WR, Xie J, Hamm-Alvarez SF, Chen ZZ, Schenke-Layland K (2008) Quantitative second harmonic generation imaging of cartilage damage. Cell Tissue Bank 9:299–308 [DOI] [PubMed] [Google Scholar]
- 28.Brockbank KGM, Chen ZZ, Song YC (2010) Vitrification of porcine articular cartilage. Cryobiology 60:217–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song YC, Chen ZZ, Mukherjee N, Lightfoot FG, Taylor MJ, Brockbank KGM, Sambanis A (2005) Vitrification of tissue engineered pancreatic substitute. Transplant Proc 37: 253–255 [DOI] [PubMed] [Google Scholar]
- 30.Elder E, Chen Z, Ensley A, Nerem R, Brockbank K, Song Y (2005) Enhanced tissue strength in cryopreserved, collagen-based blood vessel constructs. Transplant Proc 37: 4625–4629 [DOI] [PubMed] [Google Scholar]
- 31.Dahl S, Chen Z, Solan A, Lightfoot F, Li C, Brockbank KGM, Niklason L, Song YC (2006) Tissue engineered blood vessels. Tissue Eng 12:291–300 [DOI] [PubMed] [Google Scholar]
- 32.Farooque TM, Chen ZZ, Schwartz Z, Wick TM, Boyan BD, Brockbank KGM (2009) Protocol development for vitrification of tissue-engineered cartilage. Bioprocessing (Williamsbg Va) 8:29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khirabadi BS, Song YC, Brockbank KGM (2004) Method of cryopreservation of tissues by vitrification. United States Patent #6,740, 484, 2004
- 34.Khirabadi BS, Song YC, Brockbank KGM (2007) Method of cryopreservation of tissues by vitrification. United States Patent #7, 157,222
- 35.Brockbank KGM, Song YC (2004) Morphological analyses of ice-free and frozen cryopreserved heart valve explants. J Heart Valve Dis 13:297–301 [PubMed] [Google Scholar]
- 36.Schenke-Layland K, Madershahian N, Riemann I, Starcher B, Halbhuber KJ, König K, Stock UA (2006) Impact of cryopreservation on extracellular matrix structures of heart valve leaflets. Ann Thorac Surg 81:918–926 [DOI] [PubMed] [Google Scholar]
- 37.Schenke-Layland K, Riemann I, Damour O, Stock UA, König K (2006) Two-photon microscopes and in vivo multiphoton tomographs – powerful diagnostic tools for tissue engineering and drug delivery. Adv Drug Deliv Rev 58:878–896 [DOI] [PubMed] [Google Scholar]
- 38.Schenke-Layland K, Xie J, Haydarkhan-Hagvall S, Hamm-Alvarez SF, Stock UA, Brockbank KGM, MacLellan WR (2007) Optimized preservation of extracellular matrix in cardiac tissues: implications for long-term graft durability. Ann Thorac Surg 83:1641–1650 [DOI] [PubMed] [Google Scholar]
- 39.Brockbank KGM, Wright GJ, Yao H, Greene ED, Chen ZZ, Schenke-Layland K (2011) Allogeneic heart valve preservation – allogeneic heart valve storage above the glass transition at −80 °C. Ann Thorac Surg 91:1829–1835 [DOI] [PubMed] [Google Scholar]
- 40.Hazekamp MG, Koolbergen DR, Braun J, Sugihara H, Cornelisse CJ, Goffin YA, Huysmans HA (1995) In situ hybridization: a new technique to determine the origin of fibroblasts in cryopreserved aortic homograft valve explants. J Thorac Cardiovasc Surg 110: 248–257 [DOI] [PubMed] [Google Scholar]
- 41.Koolbergen DR, Hazekamp MG, de Heer E, Bruggemans EF, Huysmans HA, Dion RA, Bruijn JA (2002) The pathology of fresh and cryopreserved homograft heart valves: an analysis of forty explanted homograft valves. J Thorac Cardiovasc Surg 124:689–697 [DOI] [PubMed] [Google Scholar]
- 42.Armiger LC (1998) Postimplantation leaflet cellularity of valve allografts: are donor cells beneficial or detrimental? Ann Thorac Surg 66(suppl):S233–S235 [DOI] [PubMed] [Google Scholar]
- 43.Navarro FB, Costa FD, Mulinari LA, Pimentel GK, Roderjan JG, Vieira ED, Noronha L, Miyague NI (2010) Evaluation of the biological behavior of decellularized pulmonary homografts: an experimental sheep model. Rev Bras Cir Cardiovasc 25:377–387 [DOI] [PubMed] [Google Scholar]
- 44.da Costa FD, Costa AC, Prestes R, Domanski AC, Balbi EM, Ferreira AD, Lopes SV (2010) The early and midterm function of decellularized aortic valve allografts. Ann Thorac Surg 90:1854–1860 [DOI] [PubMed] [Google Scholar]
- 45.Campbell LH, Brockbank KGM (2010) Cryopreservation of porcine aortic heart valve leaflet-derived myofibroblasts. Biopreserv Biobank 8:211–217 [DOI] [PubMed] [Google Scholar]
- 46.Ketchedjian A, Jones AL, Krueger P, Robinson E, Crouch K, Wolfinbarger L Jr, Hopkins R (2005) Recellularization of decellularized allograft scaffolds in ovine great vessel reconstructions. Ann Thorac Surg 79:888–896 [DOI] [PubMed] [Google Scholar]
- 47.Ketchedjian A, Krueger P, Lukoff H, Robinson E, Jones A, Crouch K, Wolfinbarger L, Hopkins RA (2005) Ovine panel reactive antibody assay of HLA responsivity to allograft bioengineered vascular scaffolds. J Thorac Surg 129:155–166 [DOI] [PubMed] [Google Scholar]
- 48.Linthurst Jones A, Moore M (2009) MATRACELL TM Decellularized allograft bio- implants – critical applications for cardiovascular surgery a bio-implants brief. LifeNet Health Nosfolk, VA, USA [Google Scholar]
- 49.Lisy M, Pennecke J, Brockbank KGM, Fritze O, Schleicher M, Schenke-Layland K, Kaulitz R, Riemann I, Weber CN, Braun J, Mueller KE, Fend F, Scheunert T, Gruber AD, Albes JM, Ziemer G, Stock UA (2010) The performance of ice-free cryopreserved heart valve allografts in an orthotopic pulmonary sheep model. Biomaterials 31:5306–5311 [DOI] [PubMed] [Google Scholar]
- 50.Brockbank KGM, Schenke-Layland K, Greene ED, Chen Z, Fritze O, Schleicher M, Kaulitz R, Riemann I, Fend F, Albes JM, Stock UA, Lisy M (2012) Ice-free cryopreservation of heart valve allografts: better extracellular matrix preservation in vivo and preclinical results. Cell Tissue Bank 13:663–671 [DOI] [PubMed] [Google Scholar]
- 51.Brockbank KGM (2013) Methods for ice-free preservation of tissues. US Patent #8,440,390
- 52.Thakrar RR, Patel VP, Hamilton G, Fuller BJ, Seifalian AM (2006) Vitreous cryopreservation maintains the viscoelastic property of human vascular grafts. FASEB J 20:874–881 [DOI] [PubMed] [Google Scholar]
- 53.Huber AJ, Brockbank KGM, Aberle T, Schleicher M, Chen Z, Greene ED, Lisy M, Stock UA (2012) Development of a simplified ice-free cryopreservation method for heart valves. Biopreserv Biobank 10:479–484 [DOI] [PubMed] [Google Scholar]
- 54.Huber AJT, Brockbank KGM, Riemann I, Schleicher M, Fritze O, Wendel H, Stock UA (2012) Preclinical evaluation of ice-free cryopreserved arteries: structural integrity and hemocompatibility. Cells Tissues Organs 196: 262–270 [DOI] [PubMed] [Google Scholar]
- 55.Brockbank KGM, Song YC, Greene ED, Taylor MJ (2007) Quantitative analyses of vitrified autologous venous arterial bypass graft explants. Cell Preserv Technol 5:68–76 [Google Scholar]
- 56.Rajani B, Mee RB, Ratliff NB (1998) Evidence for rejection of homograft cardiac valves in infants. J Thorac Cardiovasc Surg 115: 111–117 [DOI] [PubMed] [Google Scholar]
- 57.Bechtel JF, Stierle U, Sievers HH (2008) Fifty-two months mean follow up of decellularized SynerGraft-treated pulmonary valve allografts. J Heart Valve Dis 17:98–104 [PubMed] [Google Scholar]
- 58.Brockbank KGM, Campbell LH, Chen Z, Greene ED, Stock UA, Seifert M (2013) Minimization of allogeneic tissue immunogenicity by cryopreservation. Presented at the 22nd annual congress of the European Association of Tissue Banks, November (abstract) [Google Scholar]
- 59.Valente M, Faggian G, Billingham ME, Talentie E, Calabrese F, Casula R, Shumway NE, Thiene G (1995) The aortic valve after heart transplantation. Ann Thorac Surg 60(suppl):S135–S140 [DOI] [PubMed] [Google Scholar]