

Abstract
Interstrand crosslinks (ICLs) are adducts of covalently linked nucleotides in opposing DNA strands that obstruct replication and prime cells for malignant transformation or premature cell death. ICLs may be caused by alkylating agents or ultraviolet (UV) irradiation. These toxic lesions are removed by diverse repair mechanisms such as Fanconi anemia (FA) pathway, nucleotide excision repair (NER), translesion synthesis (TLS), and homologous recombination (HR). In mammals, xeroderma pigmentosum group F (XP-F) protein participates in both FA pathway and NER, while DNA polymerase ζ (POLZ-1) and REV-1 mediate TLS. Nevertheless, little is known regarding the genetic determinants of these pathways in ICL repair and damage tolerance in germ cells. In this study, we examined the sensitivity of C. elegans germ cells to ICLs generated by trimethylpsoralen/ultraviolet A (TMP/UV-A) combination, and embryonic mortality was employed as a surrogate for DNA damage in germ cells. Our results show that XPA-1, POLZ-1, and REV-1 were more critical than FA pathway mediators in preserving genomic stability in C. elegans germ cells. Notably, mutant worms lacking both XPA-1 and POLZ-1 (or REV-1) were more sensitive to ICLs compared to either single mutant alone. Moreover, knockdown of XPA-1 and REV-1 leads to retarded disappearance of RPA-1 and RAD-51 foci upon ICL damage. Since DNA repair mechanisms are broadly conserved, our findings may have ramifications for prospective therapeutic interventions in humans.
Keywords: Interstrand DNA Crosslinks (ICLs), Nucleotide Excision Repair (NER), Translesion Synthesis (TLS), Fanconi Anemia Pathway, Caenorhabditis elegans
Graphical Abstract
Introduction
Interstrand DNA crosslinks (ICLs) covalently link nucleotides in antiparallel DNA strands, impeding DNA replication and cell survival. ICLs are formed by ultraviolet (UV) radiation (induces DNA strand breaks) and DNA crosslinking agents including cisplatin, mitomycin C, and trimethylpsoralen (TMP) 1, 2. No or very few DNA ICLs were observed without UV treatment. In particular, TMP induces severe ICLs upon UV-A activation, which facilitates studying molecular mechanisms specifically related to ICL repair in vivo and in vitro 3. Although the Fanconi anemia (FA) pathway plays a major role in ICL repair in mammals 2, 4, 5, other pathways also respond to ICL at different phases of the cell cycle.
In replication-independent (G0/G1) phase (Fig. 1A), proteins of nucleotide excision repair (NER) recognize ICLs and recruit incision factors, XPA, RPA, and TFIIH, to the damaged lesion as to allow XPF-ERCC1 to make an incision at one side of the lesion, while XPG cleaves the other side 6. Translesion synthesis (TLS) recruits polymerases, including POL κ, POL ζ, and REV-1, to the incision site by ubiquitinated proliferating cell nuclear antigen (PCNA) to commence strand extension 1. POL ζ and REV-1 are also activated independently of PCNA ubiquitination upon UV radiation in POL δ mutants 7.
Figure 1. ICL repair pathway at different cell cycle phases.

(A) Replication-independent and (B) replication-dependent ICL repair mechanism. See text for details. Adapted with permission from ref. 1. Copyright 2016 BioMed Central (BMC).
In replication-dependent (late S) phase (Fig. 1B), when replication forks are stalled by ICLs, the FANCM-FAAP24-MHF complex binds the damaged site, and recruits the FA complex FANCA/B/C/E/F/G, FAAP20, and FANCL (a ubiquitin ligase). The FANCI/FANCD2 complex is then mono-ubiquitinated by FANCL, which activates structure-specific endonucleases XPF/FANCQ-ERCC1, SLX/FANCP-SLX1, MUS81-EME1, and FAN1 1. The incision made produces double-strand breaks (DSBs) repaired by homologous recombination (HR). In particular, TLS by REV-1 and POL ζ proceeds during ICL repair, at the step between DSB formation and HR. Mono-ubiquitinated FANCI/FANCD2 complex promotes both incision and TLS before HR 8. Ubiquitinated FANCI/FANCD2 complex is finally deubiquitinated by ubiquitin specific peptidase 1-USP1-associated factor 1 (USP1-UAF1). Collectively, the NER+TLS pathway functions in G0/G1 phase, whereas the FA+HR pathway predominates in late S/G2 phase.
In ICL repair, TLS polymerases insert nucleotides after incisions are made 2. Mammalian cells deficient in POL κ, POL ζ, REV-1, or POL ν are hypersensitive to ICLs, but those deficient in Pol η are not. Also, when unhooking of ICLs generates DSBs at late S/G2 phase, TLS is activated to bypass the unhooked lesion. In replication-independent ICL repair, TLS polymerases are essential to bypass the unhooked ICL and fully extend DNA strands 9. POL ζ and REV-1 insert nucleotides opposite to the unhooked ICL and bypass the lesion in replication-dependent and –independent ICL repair. REV-1 is involved in bypass by inserting a nucleotide to the opposite DNA strand, and POL κ performs strand extension 10.
In mammals, the FA pathway is the main pathway of ICL repair. There are 21 FA genes in mammals 11, some of which are conserved in C. elegans 12, while NER and TLS genes are conserved from C. elegans to mammals 13-15. In this report, we investigate the function of TLS polymerases (POLZ-1 and REV-1) and their genetic relationship with XPA-1 in the germline of the nematode C. elegans. This worm has a short life cycle and its germline has also been widely used as a model system to study cellular processes in vivo. The C. elegans germline is organized in a simple linear fashion that progresses from germline stem cells at one end to maturing gametes at the other (Fig. 2A). The C. elegans germ cells are sensitive to diverse DNA damage that is often associated with infertility or embryonic mortality. Thus, it has been recognized as a suitable model to investigate DNA repair pathways in vivo 3, 4, 16-19. Our genetic and cellular studies demonstrate that XPA-1 and TLS are critical for C. elegans ICL repair, which drive HR progression beyond RPA-1 and RAD-51 recruitment. Since these regulators are broadly conserved, similar molecular mechanism may exist in humans.
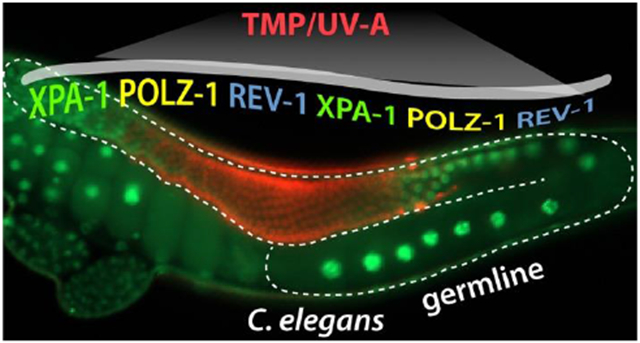
Figure 2. XPA-1, POLZ-1, and REV-1 are required for ICL repair.

(A) Schematic of ICL experiment. TMP/UV-A exposed to only L4 staged whole worms. DNA damages in the germ cells were determined by scoring the survival rate of fertilized embryos. See “Materials and Methods” for details. (B and C) xpa-1(ok698), polz-1(tm8927), or rev-1(tm8701) single mutants are more sensitive to ICLs than fcd-2(tm1298) mutants. L4 staged worms were treated with TMP/UV-A, and their F1 embryos were collected between 24 and 48 h post treatment. Latched embryos were counted 24 h later. (D) Double deficiencies of polz-1(tm8927) or rev-1(tm8701) together with xpa-1(ok698) significantly increase sensitivity to ICLs. Wild type (N2), polz-1(tm8927), and rev-1(tm8701) mutants were fed E. coli expressing double-stranded RNA for xpa-1 from the L1 stage. L4 staged worms were treated with TMP/UV-A and their F1 embryos’ survival was scored. P values: *, P<0.05; **, P<0.01; ***, P<0.001; ns, no statistical significance. See Table S2 for One-way ANOVA analysis.
Materials and Methods
Strains and maintenance
All C. elegans strains were maintained at 20°C as previously described unless otherwise noted 20. Wild type (N2) and xpa-1(ok698) mutants were obtained from the Caenorhabditis Genetics Center (Minneapolis, MN, USA). The fcd-2(tm1298), xpc-1(tm3886), xpf-1(tm2842), polz-1(tm8927), and rev-1(tm8701) mutants were obtained from National BioResource Project (NBRP, Japan). See Tables S1 and S2 for strain information and motif analysis of POLZ-1(tm8927) and REV-1(tm8701). The fcd-2(tm1298), xpf-1(tm2842), xpa-1(ok698), xpc-1(tm3886), polz-1(tm8927), and rev-1(tm8701) mutants were outcrossed with N2 at least three times to eliminate background mutations. All mutations were confirmed by worm PCR (polymerase chain reaction). polz-1(tm8927); rev-1(tm8701) double mutant was generated from polz-1(tm8927) single and rev-1(tm8701) single mutants using a standard genetic method. Genotypes were validated by PCR.
RNA interference (RNAi)
RNAi was conducted by feeding worms with E. coli HT115 (DE3) expressing the double-stranded RNAs of xpa-1 from the C. elegans RNAi V1.1 feeding library (Open Biosystems). RNAi bacteria were cultured in Lysogeny Broth (LB) containing ampicillin (50 μg/ml) for 16 h at 37°C and were seeded onto nematode growth medium (NGM) plates containing ampicillin (50 μg/ml) and IPTG (1 mM). RNAi was started in synchronized L1 larvae at 20°C.
ICL induction
Animals were synchronized as per 21 and grown to L4 stage (48 h past L1 stage).
To induce ICLs, L4 stage worms were treated with 0.8, 1.6, 4, and 20 μM 4,5’,8-Trimethylpsoralen (TMP) (Acros, NJ, USA) for 40 min, and exposed to UV-A light (100 J/m2) by Sylvania UV lamp. Generally, worms are exposed to TMP for 1 hour as per 3. However, to examine the effect of ICLs on C. elegans germ cells, TMP exposure time was slightly reduced to 40 min as per 22.
Embryonic survival
To examine the sensitivity of germ cells to ICLs, embryonic survival was measured. Briefly, L4-larval worms (P0) exposed to ICL-inducing agents were allowed to lay embryos on NGM for 24 h at 20°C. P0 worms were removed, their embryos were maintained at 20°C, and survival rates were determined 24 h later.
Immunostaining
Germline immunostaining was performed as previously described 19. Briefly, after ICL induction, gonads were incised on a glass bath with 1x PBST (0.1% Tween20 in 1x PBS). Gonads were fixed in 10% formalin for 15 min at 25°C, post-fixed in cold 100% methanol for 12 h at −20°C, washed in PBST, treated with blocking solution (goat serum: 1x PBST = 1:1) for 30 min at 25°C, and incubated with primary antibodies overnight at 4°C. Gonads were washed three times in 1x PBST, treated with a FITC-conjugated goat anti-rat secondary antibody (Molecular probes, 1:1000 dilution) for 1 h at 25°C, washed again in 1x PBST, stained with 1 μg/ml DAPI (4,6-diamidino-2-phenylindole) for 15 min, and washed for the last time in 1x PBST. The foci of RPA-1 or RAD-51 were counted in 10 mitotic germ cells close to the distal end of each gonad under a fluorescence microscope (DMR HC, Leica). This was repeated for 10 gonad arms in each strain. Biological experiments were performed at least three times.
Statistics
Data are presented as means ± standard error of the mean (SEM) of at least three independent experiments. One-way ANOVA was used to analyze differences among the experimental groups while comparisons between two groups were accomplished by Student’s t-test. A cutoff for statistical significance was set at a P value of <0.05.
Results
XPA-1 and XPF-1 are required for ICL repair in C. elegans germline
To examine whether NER is required for ICL repair in C. elegans germline, the survival of progeny embryos after L4 stage worms treated with TMP and UV-A (TMP/UV-A) was measured (Fig. 2A). Optimal treatment conditions for TMP/UV-A treatment were determined as per 3, 22 (Fig. S1 and S2). To examine the effect of ICLs on C. elegans germ cells, TMP exposure time was slightly reduced to 40 min instead of 60 min as per 3. Two null mutant alleles fcd-2(tm1298) 23 and xpf-1(tm2842) 24 were used as positive controls for ICL hypersensitivity. The fcd-2 gene, an ortholog of human FANCD2 (FA complementation group D2), participates in FA pathway. The xpf-1 gene, an ortholog of human ERCC4 (ERCC excision repair 4), functions at the initial stage of FA pathway and in NER. We found that xpa-1(ok698) null mutants were more sensitive than fcd-2(tm1298) mutants at 20 μM TMP/UV-A (Fig. 2B; Table S2 for ANOVA analysis). This indicates XPA-1 and XPF-1 are required for ICL repair in C. elegans germline. Similarly, Wilson et al. previously reported that XPA-1 and XPF-1 are also required for ICL repair during C. elegans development and aging 3.
TLS polymerases, POLZ-1 and REV-1, are also required for ICL repair in C. elegans germline
XPA recognizes ICLs, and RPA, TFIIH, XPG, and XPF-ERCC1 make incisions at both sides of the lesion. PCNA and TLS polymerases (e.g., POL κ, POL ζ, and REV-1) are then recruited to the damaged sequence 6 (Fig. 1A and 1B). To test whether TLS polymerases are required for ICL repair in C. elegans germline, we scored embryonic survival of TLS polymerase-deficient mutants, polz-1(tm8927) and rev-1(tm8701), after ICL induction. Notably, both polz-1(tm8927) and rev-1(tm8701) mutants were more sensitive to ICL than fcd-2(tm1298) mutants at low-dose 1.6 μM TMP/UV-A (Fig. 2C). Particularly, the survival of polz-1(tm8927) mutants was non-significantly different to that of xpa-1(ok98) mutants at the same dose (Fig. 2C). The rev-1(tm8701) mutants, were more sensitive to ICL than fcd-2(tm1298) mutants, but were not as sensitive as polz-1(tm8927) mutants (Fig. 2C, Table S2 for ANOVA analysis). Collectively, these results indicate that an ICL repair pathway involving XPA-1 and POLZ-1 may play a more prominent role than the FA pathway and REV-1 in C. elegans germline.
Double POLZ-1 (or REV-1) and XPA-1 deficiency synergistically increase sensitivity to ICLs compared with single deficiencies
To determine the genetic relationship between XPA-1 and TLS polymerases (POLZ-1 and REV-1), wild type (N2), polz-1(tm8927), and rev-1(tm8701) mutants were fed E. coli expressing double-stranded RNA (dsRNA) for xpa-1. The xpa-1(RNAi) worms were not as sensitive to ICLs as xpa-1(ok698) mutants, which indicates that the knockdown of xpa-1 was not complete. Nevertheless, polz-1(tm8927); xpa-1(RNAi) mutants were more sensitive to ICLs than single mutants at 1.6 μM but not at 0.8 μM TMP/UV-A (Fig. 2D, Table S2 for ANOVA analysis). Also, rev-1(tm8701); xpa-1(RNAi) mutants were more sensitive to ICLs at 0.8 and 1.6 μM TMP/UV-A than those with either mutation (Fig. 2D). Altogether, these data suggest that POLZ-1 and REV-1 may work in parallel (Fig. 1A and 1B) and that double deficiency of POLZ-1 (or REV-1) and XPA-1 confers hypersensitivity to ICLs compared to either deficiency.
Loss of REV-1 delays the disappearance of RPA-1 and RAD-51 foci
We next investigated the number of RPA-1 and RAD-51 foci in the mitotic germ cells of C. elegans upon treatment with TMP/UV-A 25. RPA-1 and RAD-51 bind single-stranded DNA (ssDNA) 26, and are thus used as surrogates to measure the efficiency of DNA repair. We measured the formation of RPA-1 and RAD-51 foci in response to ICLs in wild type (N2), xpa-1(RNAi), rev-1(tm8701), and rev-1(tm8701); xpa-1(RNAi) mutants (Fig. 3A-3C). The formation of RPA-1 foci was increased in rev-1(tm8701) and rev-1(tm8701); xpa-1(RNAi) mutants, but not in xpa-1(RNAi) worms. In wild type (N2) and xpa-1(RNAi) mutants, the number of RPA-1 foci reached a peak at 6 h after ICL induction and disappeared almost completely at 18 h (Fig. 3A and 3C). In rev-1(tm8701) mutants, the number of RPA-1 foci reached a peak at 6 h and almost disappeared at 24 h (Fig. 3B and 3C). Remarkably, in rev-1(tm8701); xpa-1(RNAi) mutants, RPA-1 foci remained at 24 h (Fig. 3B and 3C, Table S2 for ANOVA analysis). Furthermore, there were no significant differences among strains in the number of RAD-51 foci at 12 h after ICL induction (Fig. 4A-4C). However, at 24 h, RAD-51 foci disappeared in wild type (N2) and xpa-1(RNAi) mutants (Fig. 4A and 4C). In contrast, in rev-1(tm8701) and rev-1(tm8701); xpa-1(RNAi) mutants, the numbers of RAD-51 foci at 24 h were non-significantly different from those observed at 12 h (Fig. 4B and 4C, Table S2 for ANOVA analysis). These results indicate, in germlines lacking REV-1, HR did not progress further after loading RPA-1 and RAD-51 on ssDNA.
Figure 3. Formation and disappearance of RPA-1 foci after ICL induction are influenced by rev-1 mutation.

(A and B) L4 stage worms of wild type (N2), xpa-1(RNAi), rev-1(tm8701), and rev-1(tm8701); xpa-1(RNAi) were treated with TMP (200 μM) for 40 min and exposed to UV-A light (100 J/m2). Gonads were dissected, fixed, and immunostained using RPA-1 antibody at 0, 6, 12, 18 and 24 h after ICL induction. Scale bar, 10 μm. (C) The number of RPA-1 foci per nuclear focal plane was counted for over 100 mitotic germ cells in each stain. P values: *, P<0.05; **, P<0.01; ***, P<0.001; ns, no statistical significance. See Table S2 for One-way ANOVA analysis.
Figure 4. Disappearance but not formation of RAD-51 foci is delayed by rev-1 mutation after ICL induction in mitotically proliferating germ cells.

(A and B) L4 stage worms of wild type (N2) and xpa-1(RNAi), rev-1(tm8701) and rev-1(tm8701); xpa-1(RNAi) were treated with TMP (200 μM) for 40 min and exposed to UV-A light (100 J/m2). Gonads were dissected, fixed, and immunostained at 12 and 24 h using RAD-51 antibody. Scale bar, 10 μm. (C) The number of RAD-51 foci per nuclear focal plane was scored for over 100 mitotic germ cells in each stain. P values: *, P<0.05; **, P<0.01; ***, P<0.001; ns, no statistical significance. See Table S2 for One-way ANOVA analysis.
POLZ-1 and REV-1 influence another ICL-responsive pathway
POLZ-1 works in tandem with REV-1 during ICL repair at both replication-independent (G0/G1) and replication-dependent (late S/G2) phases 9, 27, 28. Figures 2-4 show that POLZ-1 and REV-1 act in parallel, not serially, in the NER+TLS pathway. To test this, ICL sensitivity of polz-1(tm8927); rev-1(tm8701) double mutants was determined. Significantly enhanced sensitivity to ICLs was exhibited by polz-1(tm8927); rev-1(tm8701) double mutants compared to xpa-1(ok698), polz-1(tm8927), or rev-1(tm8701) single mutants at 0.8 and 1.6 μM TMP/UV-A (Fig. 5, Table S2 for ANOVA analysis). This synergistic increase of sensitivity to ICLs implies that POLZ-1 and REV-1 may function redundantly in the same pathway or work in the undefined parallel pathways to repair ICLs in C. elegans germlines (see Discussion for details).
Figure 5. polz-1 and rev-1 double mutants are more hypersensitive to ICLs than either single mutants.

L4 stage worms were treated with TMP (0.8 and 1.6 μM) and exposed to UV-A (100 J/m2) to score embryonic mortality. P values: *, P<0.05; **, P<0.01; ***, P<0.001; ns, no statistical significance. See Table S2 for One-way ANOVA analysis.
Discussion
In mammals, FA pathway is more prominent during ICL repair than NER 29. However, Wilson et al. recently reported that NER components (e.g., XPA-1 and XPF-1) are more critical for ICL repair than FA pathway during development and aging in C. elegans 3. Notably, increasing evidence indicates that NER plays an important role in ICL repair in mammalian cells 30-36. Our studies also demonstrate that germlines lacking in XPA-1, XPC-1, and XPG-1 display increased sensitivity to ICLs compared to those deficient in fcd-2 (Fig. 2A and Fig. S3). XPA-1 is a damage recognition protein in NER whose mutation induces arrested development and reduced lifespan following TMP/UV-A exposure 3. Similarly, dysfunctional growth and reproduction, DNA lesions, and germ cell apoptosis were more pronounced in XPA1-deficient mutants compared to wild type (N2) worms treated with aflatoxin B1 37. In human keratinocyte HaCaT cells exposed to arsenic, downregulation of XPA exacerbated DNA damage, an effect reversed by inhibition of histone deacetylase 38.
We have also demonstrated that POLZ-1 and REV-1 protect against embryonic mortality caused by germline ICL damage (Fig. 2B-2D and 5). The REV-1-POLZ complex, called the mutasome, inserts nucleotides and initiates replication opposite damaged lesions 39. We have demonstrated that polz-1(tm8927) mutation confers hypersensitivity to ICLs more significantly than rev-1(tm8701) mutation (Fig. 2C). Moreover, polz-1(tm8927); xpa-1(RNAi) and rev-1(tm8701); xpa-1(RNAi) double deficiencies synergistically sensitize worms to ICLs compared to either deficiency (Fig. 2D). In Xenopus eggs, the mutasome extends the leading strand beyond the damage 40; a step significantly obstructed by polz depletion 41. Notably, blocking the catalytic activity of REV-1 does not influence survival following DNA damage in mammalian cells 42. Collectively, these observations suggest POLZ-1 is indispensable to TLS, likely by acting as a strand extender beyond damaged termini.
It has been reported that rev1 knockout mice display stunted growth and reduced lifespan 43. Furthermore, proliferation of fibroblasts and hematopoietic stem cells obtained from these mice was blunted 44, 45. In particular, combined deletion of rev1 and xpc, leads to anemia and death in those mice 45. Accordingly, cells with either deficiency display hypersensitivity to ICL-causing compounds 46. This is in congruence with earlier studies describing the participation of REV1 in repair of alcohol-induced ICLs 47.
RPA-1 and RAD-51 coat ssDNA to form a complex involved in DNA repair 48, 49. Our kinetic studies demonstrate that recruitment of RPA-1 is significantly amplified in rev-1(tm8701) and rev-1(tm8701); xpa-1(RNAi) mutants compared to wild type (N2) and xpa-1(RNAi) mutants (Fig. 3). Likewise, RAD-51 persisted for a significantly longer time in rev-1(tm870) and rev-1(tm870); xpa-1(RNAi) mutants unlike wild type (N2) or xpa-1(RNAi) worms (Fig. 4). This implies that TLS, through REV-1, is indispensable to ICL repair presumably by driving HR beyond RPA-1 and RAD-51 recruitment.
It has been demonstrated that binding of RPA-1 to damaged sites may be a prerequisite for subsequent recruitment of RAD-51 49. Unlike this sequential action of RPA-1 and RAD-51, our results in Figure 5 indicate that REV-1 and POLZ-1 function in tandem as polz-1(tm8927); rev-1(RNAi) mutants were more sensitive to ICLs than wild type (N2) or xpa-1(RNAi) worms. It has been suggested that HR is the main ICL repair pathway in which RAD-51 plays a central role. Congruently, cells with mutated Rad51C are sensitive to mitomycin C 50, and display aberrant centrosome numbers and mitotic spindles 51. Also, in human and yeast cells, RPA-1 appears at the site of damage within 5-10 min of microirradiation 52, and persists for 6 h 49. Similarly, RAD-51 is recruited within the same timeframe in irradiated fibroblasts but exhibits faster kinetics than RPA-1 53.
C. elegans germlines have germ cell population with different cell cycle phases. Distal mitotic region possesses germline stem cells (GSCs) and mitotic germ cells. Once mitotic germ cells enter meiotic cell cycle, they differentiate into either sperm or oocytes in the proximal region. Previous studies by Kimble’s and Schedl’s groups demonstrated that, at any given time, 50-60% of mitotic germ cells exist at the S phase 54, 55 (Fig. 6A). This finding speculates that 50-60% of ICL-induced cells are repaired by replication-dependent mechanism (i.e., FA) while the remaining cells are not (e.g., NER and TLS) (Fig. 6B). As depicted in Figure 1, damaged cells with ICLs employ different repair mediators depending on cell cycle phase. Thus, germ cells lacking either FA repair pathway (e.g., fcd-2(tm1298)) or NER pathway (e.g., xpa-1(ok698)) are partially repaired in a cell cycle-dependent manner. However, both pathways require the activities of TLS polymerase complex including POLZ-1 and REV-1 to remove ICLs, which is reflected in our findings that germ cells lacking polz-1 and/or rev-1 are more sensitive to ICLs than those lacking either fcd-2 or xpa-1 (Fig. 5). It is important to mention that fcd-2 is dispatched to replication foci under stress 56, following incisions to unhook the ICL by endonucleases, which then progresses to HR in order to repair DSBs 57. Accordingly, it is reasonable to assume that the lack of fcd-2 involvement in the genomic maintenance following ICLs suggests that TMP/UV-A-induced ICLs occur outside the S phase. Alternatively, it may take place during replication but that would necessitate for repair proteins to be recruited by upstream, damage-sensing proteins unrelated to FA.
Figure 6. A proposed model for ICL repair in C. elegans germline.

(A) EdU stained adult C. elegans germline. Scale bar: 20 μm (B) Cell cycle-dependent ICL repair mechanism in the C. elegans germline.
In mammals, REV1 and DNA Polymerase ζ (REV3 and REV7) play important roles in TLS and repair of DSBs 28. TLS polymerase complex including REV1 and POLZ-1 may function in the same step. However, double mutation is more sensitive to ICLs than single mutation could be explained by three possibilities. First, polz-1(tm8927) and rev-1(tm8701) alleles may be loss-of-function, but not null, mutants. Second, their function may be redundant with each other. Third, both proteins may have shared and discrete functions in ICL repair pathways. Our recent studies also showed that subunits of the DNA polymerase α-primase complex promote Notch-mediated proliferation with shared and discrete functions in C. elegans germline 58. Distinguishing between these possibilities is beyond the scope of this work but will be an important challenge for the future.
In conclusion, this study presents in vivo evidence in support of the major role played by NER, as opposed to FA pathway, in ICL repair in C. elegans, which may be reciprocated in higher animals. We also report that TLS polymerases, POLZ-1 and REV-1, are essential to ICL repair, and that lack of either protein in addition to XPA-1, synergistically exacerbates ICL-induced mortality and impedes HR repair. Unlike normal cells or post-mitotic cells, cancer cells are continuously dividing without differentiation. Therefore, our findings may provide insights into cell cycle-targeted chemotherapy.
Supplementary Material
Acknowledgments
We thank Dr. Hyeon-Sook Koo (Yonsei University, Seoul, South Korea) and Shohei Mitani (Tokyo Women's Medical University School of Medicine) for sharing unpublished results and providing C. elegans mutants, respectively. The authors are grateful to the Deanship of Scientific Research, King Saud University for funding this research project through Vice Deanship of Scientific Research Chairs (DSRVCH).
Funding
This work was supported by NIA (AG060373-01) to MHL. C. elegans strains were acquired from the Caenorhabditis Genetics Center, funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Footnotes
Declaration of Competing Interest
The authors declare no competing interests.
References
- [1].Hashimoto S, Anai H, and Hanada K (2016) Mechanisms of interstrand DNA crosslink repair and human disorders, Genes and environment : the official journal of the Japanese Environmental Mutagen Society 38, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Roy U, and Scharer OD (2016) Involvement of translesion synthesis DNA polymerases in DNA interstrand crosslink repair, DNA repair 44, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wilson DM 3rd, Rieckher M, Williams AB, and Schumacher B (2017) Systematic analysis of DNA crosslink repair pathways during development and aging in Caenorhabditis elegans, Nucleic acids research 45, 9467–9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lans H, and Vermeulen W (2011) Nucleotide Excision Repair in Caenorhabditis elegans, Molecular biology international 2011, 542795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hoeijmakers JH (2009) DNA damage, aging, and cancer, The New England journal of medicine 361, 1475–1485. [DOI] [PubMed] [Google Scholar]
- [6].Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NG, Beverloo HB, Hoeijmakers JH, and Kanaar R (2004) The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks, Molecular and cellular biology 24, 5776–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tellier-Lebegue C, Dizet E, Ma E, Veaute X, Coic E, Charbonnier JB, and Maloisel L (2017) The translesion DNA polymerases Pol zeta and Rev1 are activated independently of PCNA ubiquitination upon UV radiation in mutants of DNA polymerase delta, PLoS genetics 13, e1007119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ghosal G, and Chen J (2013) DNA damage tolerance: a double-edged sword guarding the genome, Translational cancer research 2, 107–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ho TV, and Scharer OD (2010) Translesion DNA synthesis polymerases in DNA interstrand crosslink repair, Environmental and molecular mutagenesis 51, 552–566. [DOI] [PubMed] [Google Scholar]
- [10].Klug AR, Harbut MB, Lloyd RS, and Minko IG (2012) Replication bypass of N2-deoxyguanosine interstrand cross-links by human DNA polymerases eta and iota, Chemical research in toxicology 25, 755–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sumpter R, and Levine B (2016) Novel functions of Fanconi anemia proteins in selective autophagy and inflammation, Oncotarget 7, 50820–50821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lee KY, Chung KY, and Koo HS (2010) The involvement of FANCM, FANCI, and checkpoint proteins in the interstrand DNA crosslink repair pathway is conserved in C. elegans, DNA repair 9, 374–382. [DOI] [PubMed] [Google Scholar]
- [13].Jones M, and Rose A (2012) A DOG’s View of Fanconi Anemia: Insights from C. elegans, Anemia 2012, 323721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Roerink SF, Koole W, Stapel LC, Romeijn RJ, and Tijsterman M (2012) A broad requirement for TLS polymerases eta and kappa, and interacting sumoylation and nuclear pore proteins, in lesion bypass during C. elegans embryogenesis, PLoS genetics 8, e1002800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Babu V, Hofmann K, and Schumacher B (2014) A C. elegans homolog of the Cockayne syndrome complementation group A gene, DNA repair 24, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Germoglio M, Valenti A, Gallo I, Forenza C, Santonicola P, Silva N, and Adamo A (2020) In vivo analysis of FANCD2 recruitment at meiotic DNA breaks in Caenorhabditis elegans, Scientific reports 10, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rieckher M, Bujarrabal A, Doll MA, Soltanmohammadi N, and Schumacher B (2018) A simple answer to complex questions: Caenorhabditis elegans as an experimental model for examining the DNA damage response and disease genes, Journal of cellular physiology 233, 2781–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bae W, Park JH, Lee MH, Park HW, and Koo HS (2019) Hypersensitivity to DNA double-strand breaks associated with PARG deficiency is suppressed by exo-1 and polq-1 mutations in Caenorhabditis elegans, The FEBS journal 287, 1101–1115. [DOI] [PubMed] [Google Scholar]
- [19].Bae W, Hong S, Park MS, Jeong HK, Lee MH, and Koo HS (2019) Single-strand annealing mediates the conservative repair of double-strand DNA breaks in homologous recombination-defective germ cells of Caenorhabditis elegans, DNA repair 75, 18–28. [DOI] [PubMed] [Google Scholar]
- [20].Brenner S (1974) The genetics of Caenorhabditis elegans, Genetics 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yoon DS, Pendergrass DL, and Lee MH (2016) A simple and rapid method for combining fluorescent in situ RNA hybridization (FISH) and immunofluorescence in the C. elegans germline, MethodsX 3, 378–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lee C, Hong S, Lee MH, and Koo HS (2015) A PHF8 homolog in C. elegans promotes DNA repair via homologous recombination, PloS one 10, e0123865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Adamo A, Collis SJ, Adelman CA, Silva N, Horejsi Z, Ward JD, Martinez-Perez E, Boulton SJ, and La Volpe A (2010) Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia, Molecular cell 39, 25–35. [DOI] [PubMed] [Google Scholar]
- [24].Agostinho A, Meier B, Sonneville R, Jagut M, Woglar A, Blow J, Jantsch V, and Gartner A (2013) Combinatorial regulation of meiotic holliday junction resolution in C. elegans by HIM-6 (BLM) helicase, SLX-4, and the SLX-1, MUS-81 and XPF-1 nucleases, PLoS genetics 9, e1003591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Raderschall E, Golub EI, and Haaf T (1999) Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage, Proceedings of the National Academy of Sciences of the United States of America 96, 1921–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].West SC (2003) Molecular views of recombination proteins and their control, Nature reviews. Molecular cell biology 4, 435–445. [DOI] [PubMed] [Google Scholar]
- [27].Kolas NK, and Durocher D (2006) DNA repair: DNA polymerase zeta and Rev1 break in, Current biology : CB 16, R296–299. [DOI] [PubMed] [Google Scholar]
- [28].Sharma S, Hicks JK, Chute CL, Brennan JR, Ahn JY, Glover TW, and Canman CE (2012) REV1 and polymerase zeta facilitate homologous recombination repair, Nucleic acids research 40, 682–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].McCabe KM, Olson SB, and Moses RE (2009) DNA interstrand crosslink repair in mammalian cells, Journal of cellular physiology 220, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sarkar S, Davies AA, Ulrich HD, and McHugh PJ (2006) DNA interstrand crosslink repair during G1 involves nucleotide excision repair and DNA polymerase zeta, The EMBO journal 25, 1285–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhao J, Jain A, Iyer RR, Modrich PL, and Vasquez KM (2009) Mismatch repair and nucleotide excision repair proteins cooperate in the recognition of DNA interstrand crosslinks, Nucleic acids research 37, 4420–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mukherjee A, and Vasquez KM (2016) HMGB1 interacts with XPA to facilitate the processing of DNA interstrand crosslinks in human cells, Nucleic acids research 44, 1151–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wood RD (2010) Mammalian nucleotide excision repair proteins and interstrand crosslink repair, Environmental and molecular mutagenesis 51, 520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Shen X, Jun S, O’Neal LE, Sonoda E, Bemark M, Sale JE, and Li L (2006) REV3 and REV1 play major roles in recombination-independent repair of DNA interstrand cross-links mediated by monoubiquitinated proliferating cell nuclear antigen (PCNA), The Journal of biological chemistry 281, 13869–13872. [DOI] [PubMed] [Google Scholar]
- [35].Shen X, and Li L (2010) Mutagenic repair of DNA interstrand crosslinks, Environmental and molecular mutagenesis 51, 493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zheng H, Wang X, Legerski RJ, Glazer PM, and Li L (2006) Repair of DNA interstrand cross-links: interactions between homology-dependent and homology-independent pathways, DNA repair 5, 566–574. [DOI] [PubMed] [Google Scholar]
- [37].Feng WH, Xue KS, Tang L, Williams PL, and Wang JS (2016) Aflatoxin B(1)-Induced Developmental and DNA Damage in Caenorhabditis elegans, Toxins 9, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhang AL, Chen L, Ma L, Ding XJ, Tang SF, Zhang AH, and Li J (2020) Role of H3K18ac-regulated nucleotide excision repair-related genes in arsenic-induced DNA damage and repair of HaCaT cells, Human & experimental toxicology 39, 1168–1177. [DOI] [PubMed] [Google Scholar]
- [39].Rizzo AA, and Korzhnev DM (2019) The Rev1-Polzeta translesion synthesis mutasome: Structure, interactions and inhibition, The Enzymes 45, 139–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Budzowska M, Graham TG, Sobeck A, Waga S, and Walter JC (2015) Regulation of the Rev1-pol zeta complex during bypass of a DNA interstrand cross-link, The EMBO journal 34, 1971–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Raschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Scharer OD, and Walter JC (2008) Mechanism of replication-coupled DNA interstrand crosslink repair, Cell 134, 969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ross AL, Simpson LJ, and Sale JE (2005) Vertebrate DNA damage tolerance requires the C-terminus but not BRCT or transferase domains of REV1, Nucleic acids research 33, 1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Jansen JG, Langerak P, Tsaalbi-Shtylik A, van den Berk P, Jacobs H, and de Wind N (2006) Strand-biased defect in C/G transversions in hypermutating immunoglobulin genes in Rev1-deficient mice, The Journal of experimental medicine 203, 319–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jansen JG, Tsaalbi-Shtylik A, and de Wind N (2015) Roles of mutagenic translesion synthesis in mammalian genome stability, health and disease, DNA repair 29, 56–64. [DOI] [PubMed] [Google Scholar]
- [45].Martin-Pardillos A, Tsaalbi-Shtylik A, Chen S, Lazare S, van Os RP, Dethmers-Ausema A, Fakouri NB, Bosshard M, Aprigliano R, van Loon B, Salvatori DCF, Hashimoto K, Dingemanse-van der Spek C, Moriya M, Rasmussen LJ, de Haan G, Raaijmakers M, and de Wind N (2017) Genomic and functional integrity of the hematopoietic system requires tolerance of oxidative DNA lesions, Blood 130, 1523–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kim H, and D’Andrea AD (2012) Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway, Genes & development 26, 1393–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hodskinson MR, Bolner A, Sato K, Kamimae-Lanning AN, Rooijers K, Witte M, Mahesh M, Silhan J, Petek M, Williams DM, Kind J, Chin JW, Patel KJ, and Knipscheer P (2020) Alcohol-derived DNA crosslinks are repaired by two distinct mechanisms, Nature 579, 603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chen R, and Wold MS (2014) Replication protein A: single-stranded DNA’s first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair, BioEssays : news and reviews in molecular, cellular and developmental biology 36, 1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Koury E, Harrell K, and Smolikove S (2018) Differential RPA-1 and RAD-51 recruitment in vivo throughout the C. elegans germline, as revealed by laser microirradiation, Nucleic acids research 46, 748–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wojcik A, Stoilov L, Szumiel I, Legerski R, and Obe G (2005) Rad51C-deficient CL-V4B cells exhibit normal levels of mitomycin C-induced SCEs but reduced levels of UVC-induced SCEs, Biochemical and biophysical research communications 326, 805–810. [DOI] [PubMed] [Google Scholar]
- [51].Renglin Lindh A, Schultz N, Saleh-Gohari N, and Helleday T (2007) RAD51C (RAD51L2) is involved in maintaining centrosome number in mitosis, Cytogenetic and genome research 116, 38–45. [DOI] [PubMed] [Google Scholar]
- [52].Zhang F, Shi J, Chen SH, Bian C, and Yu X (2015) The PIN domain of EXO1 recognizes poly(ADP-ribose) in DNA damage response, Nucleic acids research 43, 10782–10794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Tashiro S, Walter J, Shinohara A, Kamada N, and Cremer T (2000) Rad51 accumulation at sites of DNA damage and in postreplicative chromatin, The Journal of cell biology 150, 283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Crittenden SL, Leonhard KA, Byrd DT, and Kimble J (2006) Cellular analyses of the mitotic region in the Caenorhabditis elegans adult germ line, Molecular biology of the cell 17, 3051–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Fox PM, Vought VE, Hanazawa M, Lee MH, Maine EM, and Schedl T (2011) Cyclin E and CDK-2 regulate proliferative cell fate and cell cycle progression in the C. elegans germline, Development 138, 2223–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Youds JL, Barber LJ, and Boulton SJ (2009) C. elegans: a model of Fanconi anemia and ICL repair, Mutation research 668, 103–116. [DOI] [PubMed] [Google Scholar]
- [57].Dronkert ML, and Kanaar R (2001) Repair of DNA interstrand cross-links, Mutation research 486, 217–247. [DOI] [PubMed] [Google Scholar]
- [58].Yoon DS, Cha DS, Alfhili MA, Keiper BD, and Lee MH (2018) Subunits of the DNA polymerase alpha-primase complex promote Notch-mediated proliferation with discrete and shared functions in C. elegans germline, The FEBS journal 285, 2590–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.