

Abstract
Post-transplantation cyclophosphamide (PTCy) reduces the incidences of severe acute and chronic graft-versus-host disease (GVHD) after allogeneic hematopoietic cell transplantation (HCT). Yet, the standard clinical dose and timing of PTCy were partly extrapolated from MHC-matched skin allografting models and partly empirical. Herein, we investigated the impact of differential dosing and timing of PTCy on its efficacy in preventing GVHD in a murine MHC-haploidentical HCT model. Administration of PTCy on days +3/+4 was superior to days +1/+2,+5/+6, or +7/+8, while low-dose (10 mg/kg/day) PTCy on days +1/+2 actually led to accelerated death. Although the optimal timing of PTCy was day +2 or +3 in the skin allografting models, in our MHC-haploidentical HCT model, PTCy on days +2/+3 was inferior to days +3/+4 at lower doses. PTCy administered on days +3/+4, +4/+5, or +3/+5 all were similarly efficacious. Single-day versus two-day dosing schedules demonstrated that PTCy is maximally effective when given on day +4. Flow cytometric analysis showed that optimal PTCy dosing schedules both decreased alloreactive CD4+ T-cell proliferation at day +7 and allowed preferential CD4+CD25+Foxp3+ T-cell reconstitution at day +21, suggesting that this combination may be a potential predictive biomarker of successful GVHD prevention by PTCy. These results reveal that the dose, timing, and cumulative exposure of PTCy all are critical for its efficacy in preventing GVHD. We now are investigating the clinical relevance of these findings in a protocol seeking to optimize PTCy dose and timing and test these T-cell endpoints as candidate biomarkers of successful GVHD prevention by PTCy.
Graphical abstract
Introduction:
The use of post-transplantation cyclophosphamide (PTCy) has overcome historical barriers to hematopoietic cell transplantation (HCT) by safely facilitating human leukocyte antigen (HLA)-haploidentical HCT and by reducing rates of severe acute and particularly chronic graft-versus-host disease (GVHD).1 The clinical implementation of PTCy was based on studies in major histocompatibility complex (MHC)-matched skin allografting models, which suggested that a high 200 mg/kg dose of PTCy on day +2 or +3 after donor splenocyte infusion was effective in prolonging survival of a subsequently placed skin allograft; lower doses or administration at earlier or later times were less effective or ineffective.2 When PTCy was adapted to HCT pre-clinically, 200 mg/kg PTCy on day +2 or +3 promoted engraftment of MHC-mismatched murine allografts and could decrease GVHD.3,4
PTCy then was translated to the clinic to try to overcome the historical barriers to HLA-haploidentical HCT, namely the high rates of graft rejection and severe GVHD associated with HLA-mismatched allografting.5,6 For the first clinical study, PTCy was given at 50 mg/kg on day +3.7 This dosing was chosen as 1) a “high dose” was required in the MHC-matched skin allografting models and 50 mg/kg is nearly the maximal tolerable dose in humans and was effective as high-dose therapy for aplastic anemia,8 and 2) if days +2 and +3 were equivalent in the skin allografting models,2 this timing would maximize the delay between conditioning and PTCy in order to minimize toxicity. Based on promising results from the phase I study,7 a phase II study was initiated at two institutions. One institution administered the same PTCy dosing (50 mg/kg on day +3), whereas the second institution also administered a second PTCy dose of 50 mg/kg on day +4.9 This empiric change was made in an attempt to further improve GVHD and engraftment outcomes. Cross-cohort, cross-institutional comparison suggested that the days +3/+4 dosing schema may be associated with a lower risk of extensive chronic GVHD.9 Based on these limited data, nearly all subequent studies have used PTCy at 50 mg/kg/day on days +3/+4.
This PTCy dosing schema for HLA-haploidentical HCT has shown very encouraging outcomes with survival rates similar to those observed with HLA-matched HCT using standard GVHD prophylaxis while simultaneously significantly reducing rates of chronic GVHD.10–12 Yet, there has never been subsequent pre-clinical or clinical assessment performed to determine if this dosing schema represents the optimized administration of PTCy in HCT. To investigate this question, we assessed the impact of differential dosing and timing of PTCy on its efficacy in reducing GVHD severity in our recently developed MHC-haploidentical murine HCT model.13
Materials and Methods:
Mice:
B6C3F1/Crl (donor) and B6D2F1/Crl (recipient) female mice were obtained from the Charles River Laboratories and were 10–12-weeks-old at HCT. All mice were housed in specific-pathogen-free conditions at the NCI and were provided food and water ad libitum.
Hematopoietic Cell Transplantation:
Recipient B6D2F1 mice were irradiated to 10.5 Gy in a single dose 6–8 hours prior to transplantation via tail vein injection of 10×106 TCD B6C3F1 BM +/− 40×106 red-blood-cell-depleted B6C3F1 splenocytes. Recipient mice received levofloxacin-treated water from days 0 to +14. Evaluations of clinical scores and weights were performed in a blinded manner every three days. Clinical scoring was performed using a standardized rubric.13
Graft Preparation:
Graft preparation was performed as previously described.13 Briefly, B6C3F1 spleens were mechanically dissociated and red-blood-cell lysed. B6C3F1 bone marrow was flushed out of tibias and femurs and then T-cell-depleted using anti-Thy1.2 antibody (BioXCell) followed by treatment with guinea pig complement (CedarLane).
Cyclophosphamide Preparation and Treatment:
Cyclophosphamide (Baxter Oncology) was reconstituted in sterile PBS at 5 or 10 mg/mL, aliquoted, and stored at −80°C. Immediately prior to treatment, aliquots were thawed and diluted with sterile PBS to 0.2 mg/ml (for 5 mg/kg dosing), 0.5 mg/ml (for 10 mg/kg dosing), or 1 mg/ml (for 25 or 50 mg/kg dosing). The dose was determined based on the weight on the day of each treatment. Cyclophosphamide was administered intraperitoneally. Mice not receiving cyclophosphamide on a given day always received similar volumes of sterile PBS vehicle intraperitoneally.
Flow Cytometry:
Blood and spleen collection and processing were as previously described.13 Up to 3×106 viable cells from each sample sequentially were stained with the LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Thermo Fisher), stained with extracellular antibodies, fixed with the eBioscience Foxp3/Transcription Factor Staining Buffer Kit (ThermoFisher), and stained with intracellular antibodies. Data were collected using a BD Fortessa. Fluorescence-minus-one controls were prepared for Ki-67 and CD25. Data analysis was performed using BD FACSDiva 8.0.1. All data, including outliers, were included with the pre-determined exception that cell subsets of <100 were not considered evaluable for subgating to determine percentages (the denominator had to be ≥100).
Fluorochrome-conjugated monoclonal antibodies used for flow cytometry included: APCefluor780 anti-CD4 (clone GK1.5), efluor450 anti-Foxp3 (clone FJK-16S), and PerCP-efluor710 anti-Vβ6 (clone RR4–7) from eBioscience; BUV395 anti-CD3 (clone 145–2611), PE-CF594 antiCD25 (clone PC61), and PE H2kk (clone 36–7-5) from BD Biosciences; and PE-Cy5 anti-CD8 (clone 53–6.7), PE-Cy7 anti-H2kd (clone SF1–1.1), and BV605 anti-Ki-67 (clone 1GA8) from Biolegend.
Histopathologic Evaluation:
Specimens for histopathology were acquired and processed as previously described.13 Coded histopathologic slides were scored according to a standardized scoring rubric13 by a veterinary pathologist (M.A.E.). In brief, each of six organs (skin, liver, stomach, small intestine, cecum, and large intestine) were scored from 0 to 4, and the summated total histopathologic GVHD score from the six organs (possible range 0–24) was determined for each mouse.13
Statistical analysis:
The exact logrank test was used to compare survival distributions. The Wilcoxon rank sum test was used to compare weight and clinical score area-under-the-curve (AUC) values. For AUC analyses, dead mice had their last observation carried forward to subsequent time points. In order to minimize any bias introduced by using this last-observation-carried-forward approach to impute weight and clinical score values for deceased mice, AUC comparisons were restricted to intervals in which there were ≥7 mice per experimental group. The one exception was the PTCy dosing on days +1/+2 vs. +3/+4 vs. +5/+6 vs. +7/+8 experiments in which all mice treated with PTCy 10 mg/kg/day on days +1/+2 died rapidly; therefore, results were compared over the interval in which at least 7 of the vehicle-treated mice survived (days 0 through +21). Weight and clinical score data are shown as the mean +/− the standard error of the mean. Cell-subset percentages were transformed using an Arcsin transformation prior to one-way ANOVA. Cell-subset absolute numbers or median or mean fluorescence intensity data were transformed using a natural logarithmic transformation prior to one-way ANOVA. ANOVAs, when significant, were followed with the Holm-Sidak post hoc test. Although transformed data were used for statistical testing, the non-transformed data are displayed for clarity of understanding and are shown as box-and-whisker plots. SAS/STAT™ version 12 (SAS Institute, Inc.) was used for analyses of survival, weight, and clinical score data. GraphPad Prism (GraphPad Software) version 7.01 was used for statistical analysis of the flow cytometric data and for data presentation. All analyses were two-tailed. P-values <0.05 were considered statistically significant.
Results:
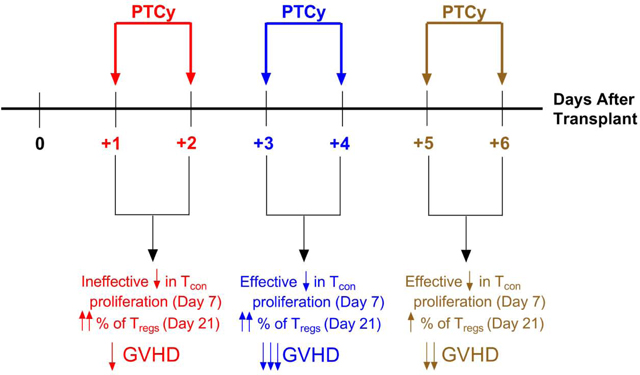
PTCy given on days +3/+4 is superior to administration on days +1/+2, +5/+6, or +7/+8
To explore the effectiveness of various PTCy dosing strategies, we utilized our recently described MHC-haploidentical murine HCT model (B6C3F1→B6D2F1).13 In this model, PTCy doses of 10–50 mg/kg/day on days +3/+4 can effectively prevent fatal GVHD with 25 mg/kg/day PTCy being the optimal dose, associated with less clinical and histopathologic evidence of GVHD than lower or higher doses. 10 mg/kg/day PTCy on days +3/+4 prevents fatal GVHD in a subset of mice, while 5 mg/kg/day on days +3/+4 is ineffective.13
We began by testing the relative efficacy of both the optimal dose (25 mg/kg/day) and the threshold dose (10 mg/kg/day) when given on days +1/+2 vs. +3/+4 vs. +5/+6 vs. +7/+8. Interestingly, mice treated with 10 mg/kg/day PTCy on days +1/+2 died significantly faster than vehicle-treated mice (hazard ratio (HR)=6.3, p=0.0019). By contrast, 10 mg/kg/day PTCy on days +3/+4 or days +5/+6 significantly prolonged survival (HR=0.034, p<0.0001; HR=0.083, p=0.0005; respectively) compared with vehicle-treated mice. 10 mg/kg/day PTCy on days +7/+8 did prolong survival compared with vehicle-treated mice but this difference was not significant (HR=0.44, p=0.16) [Figure 1A, Table 1]. Among the 10 mg/kg/day dosing schedules, PTCy on days +3/+4 was most effective, with 80% of the mice surviving to day +120; this was not significantly different than the days +5/+6 group (HR=0.41, p=0.19), but was significantly longer than the days +7/+8 group (HR=0.077, p=0.0002). These survival differences were mirrored in the weights and clinical scores in which 10 mg/kg/day PTCy given on days +3/+4 was superior not just to vehicle-treated mice but also to mice given PTCy over the other dosing schedules [Figure 1A, Table 1].
Figure 1. PTCy given on days +3/+4 is superior to days +1/+2, +5/+6, or +7/+8 in preventing GVHD.

B6D2F1 female recipient mice were irradiated to 10.5 Gy in a single fraction and 6–8 hours later received intravenous transplantation of 10 × 106 B6C3F1 T-cell-depleted bone marrow (TCD BM) cells +/− 40 × 106 B6C3F1 splenocytes (Splen). Mice were given PTCy at 10 mg/kg/day or 25 mg/kg/day intraperitoneally on designated days; mice not receiving PTCy received PBS vehicle intraperitoneally on the same days. Mice were followed daily for survival and every 3 days for clinical scores and weights. Statistical comparisons were performed between PTCy treatment groups and the TCD BM, Splen, Vehicle control group and between different PTCy treatment groups. Clinical score and weight comparisons were performed using area under the curve (AUC) comparisons of values from days 0 through +21, after which there were less than 7 mice in the vehicle-treated group. A group of mice receiving TCD BM, Vehicle was included in the experiments as a control, but this group was not included in the statistical analyses. The same TCD BM, Vehicle and TCD BM, Splen, Vehicle control groups are shown in all parts. (A) PTCy given at 10 mg/kg/day on days +3/+4 (HR=0.034, p<0.0001) or days +5/+6 (HR=0.083, p=0.0005) significantly prolonged survival compared with vehicle-treated mice, while 10 mg/kg/day PTCy on days +7/+8 did not significantly prolong survival (HR=0.44, p=0.16) and 10 mg/kg/day PTCy on days +1/+2 actually led to accelerated death (HR=6.3, p=0.0019). Mice receiving 10 mg/kg/day PTCy on days +3/+4 had significantly better weights (p=0.023) and clinical scores (p<0.0001) than vehicle-treated mice and were superior to mice treated with other 10 mg/kg/day PTCy dosing schedules including days +5/+6 (weights, p=0.043; clinical scores, p=0.0003, respectively). (B) 25 mg/kg/day PTCy given over any of the four dosing schedules significantly prolonged survival compared with vehicle (days +1/+2, HR=0.13, p=0.0039; days +3/+4, HR=0.042, p=0.0001; days +5/+6, HR=0.19, p=0.0084; days +7/+8, HR=0.31, p=0.0021). 25 mg/kg/day PTCy on days +3/+4 resulted in significantly better weights and clinical scores than vehicle treatment (p=0.0007 and p<0.0001, respectively). 25 mg/kg/day PTCy on days +1/+2 or +5/+6 did not result in significantly better weights than vehicle-treated mice, but both resulted in significantly better clinical scores (p=0.0018 and p=0.0011, respectively). 25 mg/kg/day PTCy on days +3/+4 had significantly superior clinical scores than all other dosing schedules (p<0.0001 for all three comparisons) and significantly superior weights compared with the days +1/+2 and days +7/+8 groups (p=0.052 against days +5/+6). Combined results are shown for two independent experiments of 5 mice/group/experiment
Table 1.
Summary of survival, clinical scores, weights, and histopathologic assessments by PTCy dose.
| PTCy Dose | Survival | Clinical Scores | Weights | Histopathology |
|---|---|---|---|---|
| 5 mg/kg | • Dosing on days +3/+4 resulted in
improved survival compared with days +2/+3 (not statistically
significant, p=0.061) • No significant differences between dosing on days +3/+4, +4/+5, or +3/+5; none significantly prolonged survival compared with vehicle |
• No significant difference between
dosing on days +3/+4 or +2/+3 • No significant differences between dosing on days +3/+4, +4/+5, or +3/+5 |
• No significant difference between
dosing on days +3/+4 or +2/+3 • No significant differences between dosing on days +3/+4, +4/+5, or +3/+5 |
• Not performed |
| 10 mg/kg | • Dosing on days +1/+2 led to
accelerated death compared with vehicle (p=0.0019) • Dosing on days +3/+4 (p<0.0001) or +5/+6 (p=0.0005) significantly prolonged survival compared with vehicle; dosing on days +7/+8 did not prolong survival • Dosing on days +3/+4 significantly prolonged survival compared with days +1/+2 (p<0.0001) or +7/+8 (p=0.0002), but not days +5/+6 (p=0.19) • Dosing on days +3/+4 significantly prolonged survival compared with days +2/+3 (p=0.0075) • Dosing on days +3/+4 (p=0.0054) or +4/+5 (p=0.0001) significantly prolonged survival compared with vehicle, but the effect of dosing on days +3/+5 was less (p=0.06); no significant differences between these dosing schedules • Dosing on day +4 only (p=0.14) or 5 mg/kg/dose BID on days +3/+4 (p=0.058) were only partially effective in prolonging survival compared with vehicle; dosing on day +3 only did not prolong survival |
• Dosing on days +3/+4 resulted in
significantly better scores compared with vehicle-treated mice
(p<0.001) and was superior to dosing on days +1/+2, +5/+6, or
+7/+8 • Dosing on days +3/+4 resulted in significantly better scores than days +2/+3 (p=0.011) • No significant differences between dosing on days +3/+4, +4/+5, or +3/+5; all three dosing schedules were significantly better than vehicle (p=0.0002, p=0.0001, and p=0.0052, respectively) • Dosing on days +3/+4, day +3 only, day +4 only, or 5 mg/kg/dose BID on days +3/+4 resulted in significantly better scores than vehicle (p<0.0001 for all except day +3 only (p=0.035)); dosing on day +3 only resulted in significantly worse scores than other dosing schedules (p=0.0005 vs. days +3/+4, p=0.024 vs. day +4 only, p=0.017 vs. 5 mg/kg/dose BID on days +3/+4), but there were no significant differences between the other dosing groups |
• Dosing on days +3/+4 resulted in
significantly better weights compared with vehicle (p=0.023) and was
superior to dosing on days +1/+2, +5/+6, or
+7/+8 • No significant difference between dosing on days +3/+4 or +2/+3 • No significant differences between dosing on days +3/+4, +4/+5, or +3/+5; all three dosing schedules were significantly better than vehicle (p=0.04, p=0.0002, and p=0.004, respectively) • No significant differences between dosing on days +3/+4, day +3 only, day +4 only, or 5 mg/kg/dose BID on days +3/+4 |
• Day 7: No significant differences
compared with vehicle for dosing on days +1/+2 or
+3/+4 • Day 21: Dosing on days +3/+4 resulted in significantly less GVHD than vehicle (p=0.026); no significant differences compared with vehicle for dosing on days +2/+3, +5/+6, or +7/+8 |
| 25 mg/kg | • Dosing on days +1/+2, +3/+4, +5/+6,
or +7/+8 all significantly prolonged survival compared with vehicle
(p=0.0039, p=0.0001, p=0.0084, and p=0.0021, respectively); dosing on
days +3/+4 significantly prolonged survival compared with days +7/+8
(p=0.0045) but not days +1/+2 (p=0.24) or +5/+6
(p=0.11) • No significant difference between dosing on days +3/+4 or +2/+3 • No significant differences between dosing on days +3/+4, +4/+5, or +3/+5; all three dosing schedules significantly prolonged survival compared with vehicle (p=0.0002, p=0.0001, and p=0.0002, respectively) • Dosing of 50 mg/kg on day +3 only resulted in significantly worse survival compared with 25 mg/kg on day +4 only (p=0.029) or 12.5 mg/kg/dose BID on days +3/+4 (p=0.019); no significant differences between 50 mg/kg on day +3 only and 25 mg/kg/day on days +3/+4 or day +3 only; no significant differences between 25 mg/kg on day +3 only, 25 mg/kg on day +4 only, 25 mg/kg/day on days +3/4, or 12.5 mg/kg/dose BID on days +3/+4 |
• Dosing on days +3/+4 resulted in
significantly better scores compared with vehicle (p<0.0001) or
dosing on days +1/+2, +5/+6, or +7/+8 (p<0.0001 for all three
comparisons) • No significant difference between dosing on days +3/+4 or +2/+3 • No significant differences between dosing on days +3/+4, +4/+5, or +3/+5 • No significant differences between dosing of 12.5 mg/kg/dose BID on days +3/+4 or 25 mg/kg/day on day +3 only, day +4 only, or days +3/+4; dosing of 50 mg/kg on day +3 only resulted in significantly worse scores compared with 12.5 mg/kg/dose BID on days +3/+4 (p=0.014) |
• Dosing on days +3/+4 resulted in
superior weights compared with vehicle (p=0.0007) or dosing on days
+1/+2, +5/+6, or +7/+8 (p=0.0011, p=0.052, and p=0.015,
respectively) • No significant difference between dosing on days +3/+4 or +2/+3 • No significant differences between dosing on days +3/+4, +4/+5, or +3/+5 • No significant differences between dosing of 50 mg on day +3 only, 12.5 mg/kg/dose BID on days +3/+4, or 25 mg/kg/day on either days +3/+4, +3 only, or +4 only |
• Day 7: Dosing on days +3/+4 resulted
in significantly less GVHD compared with vehicle (p<0.001) but
dosing on days +1/+2 did not result in less histopathologic GVHD
compared with vehicle • Day 21: Dosing on days +1/+2, +3/+4, +5/+6, or +7/+8 resulted in significantly less GVHD compared with vehicle (p<0.0001 for each comparison except p=0.0019 for days +7/+8); dosing on days +3/+4 resulted in the least histopathologic GVHD |
Note: The data reported for the clinical scores and weights refer to the comparisons of area under the curve (AUC) analyses over the time points in which there were ≥7 mice per group as per the figure legends for each set of experiments; the only exception was the for the 10 mg/kg groups in Figure 1 in which all mice treated with 10 mg/kg/day PTCy on days +1/+2 died rapidly and so the AUC comparisons were over days 0–21, after which there were <7 mice in the vehicle-treated group.
At the optimal PTCy dose (25mg/kg/day), administration on days +1/+2, +3/+4, +5/+6, or +7/+8 all significantly prolonged survival compared with vehicle treatment (days +1/+2: HR=0.13, p=0.0039; days +3/+4: HR=0.042, p=0.0001; days +5/+6: HR=0.19, p=0.0084; days +7/+8: HR=0.31, p=0.0021), with the day +3/+4 group achieving the highest survival [Figure 1B, Table 1]. 25 mg/kg/day PTCy on days +3/+4 had superior weights and clinical scores compared with vehicle treatment or any of the other three 25 mg/kg/day PTCy dosing schedules [Figure 1B, Table 1].
Histopathologic assessment at day +7 [Figure 2A, Table 1] showed that mice receiving 25 mg/kg/day PTCy on days +3/+4 had only minimal to mild GVHD, which was significantly less severe than mice who received 10 mg/kg/day on days +1/+2 or +3/+4 or 25 mg/kg on days +1/+2 (p<0.0001 for all comparisons). Histopathologic assessment at day +21 mirrored the clinical results with the lowest GVHD histopathologic severity scores at either dose level corresponding with PTCy administration on days +3/+4 [Figure 2B, Table 1]. Overall, these data suggest that PTCy administered on day +3/+4 is superior to days +1/+2, +5/+6, and +7/+8, highlighting the important relationship between the timing of PTCy and prevention of GVHD. Additionally, the worse outcomes in mice treated with 10 mg/kg/day on days +1/+2 compared with vehicle-treated mice suggests that there may be a biologically significant interaction between the dosing and timing of PTCy.
Figure 2. 25 mg/kg/day PTCy on days +3/+4 minimizes histopathologic evidence of GVHD at days +7 and +21.

Mice were transplanted as in Figure 1 with 10 × 106 TCD BM and 40 × 106 splenocytes on day 0 and were given PTCy at various dosing schedules. Mice not receiving PTCy on a given day received PBS vehicle. At day (A) +7 or (B) +21, histopathologic assessment of GVHD was performed. Combined results from two independent experiments are shown. N=6 for all groups in A except for the 10 mg/kg/day PTCy on days +1/+2 and 25 mg/kg/day PTCy on days +1/+2 groups (n=4 each) due to some deaths occurring prior to day +7. N=8 for all groups in B except for the 10 mg/kg PTCy days +2/+3 group (n=4). *p≤0.05, **p≤0.01, ****p≤0.0001 on one-way ANOVA followed by the Holm-Sidak post hoc test using the vehicle-treated group as the control. Only significant results are shown; all other comparisons between treatment groups and the vehicle treatment group are non-significant.
PTCy given on days +3/+4 is superior to days +2/+3 at suboptimal doses
The optimal day for administration of a single dose of PTCy in the MHC-matched skin allografting models was day +2 or +3 with earlier or later dosing being associated with inferior outcomes.2 Therefore, we hypothesized that administration of PTCy on days +2/+3 might be superior to the standard clinical practice of PTCy on days +3/+4. We compared these two dosing schedules using the optimal dose (25mg/kg/day), the threshold dose (10 mg/kg/day), and also the marginal dose of 5 mg/kg/day, which is an ineffective dose in preventing fatal GVHD in this model when given on days +3/+4.13 Contrary to our hypothesis, at both the 5 mg/kg/day and 10 mg/kg/day doses, PTCy on days +3/+4 resulted in superior survival [Figure 3A-B, Table 1]. This finding was most dramatically seen at the threshold dose, where 7 of 10 mice receiving 10 mg/kg/day PTCy on days +3/+4 survived to day +150, while all mice receiving 10 mg/kg/day PTCy on day +2/+3 mice died (HR=5.6, p=0.0075) [Figure 3B, Table 1]. Although there were no significant differences in weights of these two groups (p=0.63), the 10 mg/kg/day PTCy on days +3/+4 resulted in significantly better clinical scores than 10 mg/kg/day on days +2/+3 (p=0.011). By contrast, at the optimal dose (25mg/kg/day), there were no significant differences seen in survival, weights, or clinical scores between days +2/+3 and +3/+4 [Figure 3C, Table 1]. Overall, these data support that PTCy given on days +3/+4 is superior to days +2/+3 although use of the optimal dose may broaden the therapeutic window.
Figure 3. PTCy given on days +3/+4 is superior to days +2/+3 for suboptimal doses.

Mice were transplanted as in Figure 1 and were given PTCy on days +2/+3 or +3/+4 at (A) 5 mg/kg/day, (B) 10 mg/kg/day, or (C) 25 mg/kg/day. Mice were given PBS vehicle on days not receiving PTCy. The same TCD BM, Vehicle and TCD BM, Splen, Vehicle control groups are shown in all parts for comparison purposes. Statistical comparisons are between PTCy on days +2/+3 and on days +3/+4. (A) Survival was better for the 5 mg/kg/day PTCy dosing on days +3/+4, although the difference was not statistically significant (HR=2.7, p=0.061). Weight and clinical score AUCs were compared through day +21 and were not significantly different. (B) For the 10 mg/kg/day dosing, the survival of mice treated with PTCy on days +3/+4 was significantly superior compared with days +2/+3 (HR=5.6, p=0.0075). Although the weight AUCs through day +27 were not significantly different, the clinical score AUCs over that time interval were significantly better for 10 mg/kg/day PTCy on days +3/+4 (p=0.011). (C) There were no differences in survival or AUCs of weights or clinical scores between dosing schedules for mice receiving 25 mg/kg/day PTCy. For all parts, combined results are shown for two independent experiments of 5 mice/group/experiment.
PTCy given on days +3/+4, +4/+5, or +3/+5 has similar efficacy in preventing GVHD
Given that PTCy was more effective on days +3/+4 than days +2/+3 [Figure 3] and dosing on days +5/+6 was partially effective [Figure 1], we next compared PTCy dosing on days +3/+4 versus +4/+5. Since PTCy also has shown efficacy clinically when given on days +3/+5,14,15 we also included this dosing schema as a third comparator group. At 5, 10, or 25 mg/kg/day, PTCy on days +3/+4, +4/+5, or +3/+5 were similarly effective [Figure 4, Table 1].
Figure 4. PTCy has similar efficacy when given on days +3/+4, +4/+5, or +3/+5.

Mice were transplanted as in Figure 1 and received PTCy at (A) 5 mg/kg/day, (B) 10 mg/kg/day, or (C) 25 mg/kg/day on designated days. Mice not receiving PTCy on a given day received PBS vehicle. The same TCD BM, vehicle and TCD BM, Splen, Vehicle control groups are shown in all parts for comparison purposes. Statistical comparisons were performed between PTCy treatment groups and the TCD BM, Splen, Vehicle control group and between different PTCy treatment groups. (A) There were no significant differences in survival, weight AUCs (days 0–15), or clinical score AUCs (days 0–15) between the different 5 mg/kg/day PTCy dosing schedules, and none significantly prolonged survival compared with vehicle-treated mice. (B) 10 mg/kg/day PTCy on either days +3/+4 (HR=0.2, p=0.0054) or days +4/+5 (HR=0.1, p=0.0001) significantly prolonged survival compared with vehicle-treated mice, whereas the impact on survival of PTCy on days +3/+5 was less (HR=0.3, p=0.06). Although the survival after 10 mg/kg/day PTCy on days +4/+5 was the highest, there were no significant differences between treatment groups (days +4/+5 compared with days +3/+5, p=0.07). Weight AUCs (days 0–18) were similar between treatment groups, and all treatment groups were significantly better than vehicle-treated mice. On pointwise comparison, days +4/+5 had higher weights compared with days +3/+4 on day +18 (p=0.02) and compared with days +3/+5 on days +15 and +18 (p<0.003 for both days). Clinical score AUCs were similar between 10 mg/kg/day PTCy treatment groups, and all three treatment groups were significantly better than vehicle-treated mice (days +3/+4, p=0.0002; days +4/+5, p=0.0001; days +3/+5, p=0.0052). (C) At the 25 mg/kg/day dosing, there were no differences in survival between treatment groups, but all three treatment groups significantly prolonged survival compared with the vehicle-treated group (days +3/+4, HR=0.06, p=0.0002; day +4/+5, HR=0.03, p=0.0001; days +3/+5, HR=0.06, p=0.0002). Weight and clinical score AUCs over the entire period were similar between the three 25 mg/kg/day PTCy dosing schedules. Combined results are shown for two independent experiments of 5 mice/group/experiment.
The efficacy of PTCy peaks at day +4, and both the dose and cumulative exposure are critical for PTCy’s efficacy
As the maximal effectiveness of PTCy seemed to center around day +4, we next examined single-day dosing schedules compared with the standard two-day dosing. In these experiments, we also included experimental groups that would receive a half-dose every 12 hours (BID) over two days to simultaneously examine the relative importance of PTCy cumulative exposure. At lower PTCy doses, only 10mg/kg/day PTCy on days +3/+4 significantly prolonged survival compared with vehicle treatment (HR=0.035, p<0.0001), while 10 mg/kg PTCy on day +4 only or 5 mg/kg/dose PTCy BID on days +3/+4 each were partially effective in a subset of mice (HR=0.45, p=0.14; HR=0.38, p=0.058; respectively) [Figure 5A, Table 1]. By contrast, 10 mg/kg PTCy on day +3 only was completely ineffective at preventing lethal GVHD (p=0.64). Consistent with those results, clinical scores were significantly worse in mice treated with 10 mg/kg PTCy on day +3 only compared with the other three PTCy treatment groups (10 mg/kg on day +4 only, p=0.024; 10 mg/kg/day on days +3/+4, p=0.0005; 5 mg/kg/dose BID on days +3/+4, p=0.017).
Figure 5. The efficacy of PTCy peaks at day +4, and both the dose and cumulative exposure are critical.

Mice were transplanted as in Figure 1 and received PTCy at various dosing schedules. Mice not receiving PTCy on a given day received PBS vehicle. The same TCD BM, Vehicle and TCD BM, Splen, Vehicle control groups are shown in all parts for comparison purposes. Statistical comparisons were performed between PTCy treatment groups and the TCD BM, Splen, Vehicle control group and between different PTCy treatment groups. (A) Administration of 10mg/kg/day PTCy on days +3/+4 significantly prolonged survival compared with vehicle treatment (HR 0.035, p<0.0001), while 10 mg/kg PTCy on day +4 only and 5 mg/kg/dose PTCy BID on days +3/+4 each were only partially effective in a subset of mice (HR 0.45, p=0.14; HR=0.38, p=0.058; respectively). 10 mg/kg/day PTCy on day +3 only was completely ineffective at preventing lethal GVHD (p=0.64 compared with vehicle-treated mice). Weight AUCs (days 0–21) were not significantly different between treatment groups. Clinical score AUCs were significantly better for all PTCy groups compared with the vehicle-treated group (10 mg/kg on day +3, p=0.035; all others, p<0.0001). Furthermore, clinical score AUCs were significantly worse in mice treated with PTCy on day +3 only compared with the other three PTCy treatment groups (10 mg/kg on day +4 only, p=0.024; 10 mg/kg/day on days +3/+4, p=0.0005; 5 mg/kg/dose BID on days +3/+4, p=0.017). (B) Although all PTCy treatment groups significantly prolonged survival compared with the vehicle-treated group, mice treated with 50 mg/kg PTCy on day +3 only had significantly worse survival than mice treated with 25 mg/kg PTCy on day +4 only (HR=0, p=0.029) or 12.5 mg/kg/dose PTCy BID on days +3/+4 (HR=0.12, p=0.019), but not significantly worse than 25 mg/kg/day PTCy on days +3/+4 (HR=0.15, p=0.081) or 25 mg/kg PTCy on day +3 only (HR=0.49, p=0.38). None of the other PTCy treatment groups had significant survival differences from each other (all p values ≥0.21). Weight and clinical score AUCs (days 0–21) were significantly better in all PTCy dosing schedules compared with vehicle-treated mice. There were no differences in weight AUCs between PTCy dosing schedules, and no differences in clinical score AUCs between the PTCy groups other than 50 mg/kg PTCy on day +3 only. The 50 mg/kg PTCy group had worse clinical score AUCs than the 12.5 mg/kg/dose PTCy BID on days +3/+4 group (p=0.014). AUC comparisons between PTCy treatment groups were over the intervals of days 0–54 for comparisons including 50 mg/kg PTCy and days 0–141 for all other comparisons between PTCy treatment groups. Combined results are shown for two independent experiments of 5 mice/group/experiment except for the TCD BM group which had n=3/group/experiment (total n=6) and the 25 mg/kg PTCy on day +3 only, 25 mg/kg PTCy on day +4 only, and 50 mg/kg PTCy on day +3 only groups, which had n=4 in one of the two experiments (total n=9/group).
At higher PTCy doses, all dosing schedules significantly prolonged survival compared with vehicle treatment [Figure 5B, Table 1]. Mice receiving 25 mg/kg PTCy on day +4 only had the highest survival rate, followed by 12.5 mg/kg/dose PTCy BID on days +3/+4, 25 mg/kg/day PTCy on days +3/+4, and 25 mg/kg PTCy on day +3 only, respectively; yet, none of these were significantly different from each other (all p-values ≥0.21). However, mice treated with 50 mg/kg PTCy on day +3 only had significantly worse survival than 25 mg/kg PTCy on day +4 only (HR=0, p=0.029) or 12.5 mg/kg/dose PTCy BID on days +3/+4 (HR=0.12, p=0.019), but not significantly worse than 25 mg/kg/day PTCy on days +3/+4 (HR=0.15, p=0.081) or 25 mg/kg PTCy on day +3 only (HR=0.49, p=0.38). There were no differences in weights between PTCy dosing schedules. The 50 mg/kg PTCy on day +3 only group had worse clinical scores than the 12.5 mg/kg/dose PTCy BID on days +3/+4 group (p=0.014), but otherwise there were no significant differences between groups. Overall, these data suggest that PTCy reaches its maximal efficacy when given on day +4 and that PTCy given on day +4 only may be as effective as days +3/+4 at the optimal dose. These results also highlight the importance of both the timing as well as the cumulative exposure of PTCy for its effectiveness.
Effective PTCy dosing schema reduce alloreactive CD4+ conventional T-cell proliferation at day +7
We recently demonstrated that PTCy does not selectively eliminate alloreactive T cells.13 Over the course of that work, we found that 25 mg/kg/day PTCy on days +3/+4 both reduced CD4+ T-cell proliferation at day +7 and resulted in preferential CD4+CD25+Foxp3+ regulatory T-cell (Treg) recovery at day +21.13 100 mg/kg/day PTCy on days +3/+4, which was less effective at GVHD prevention than 25 mg/kg/day, did decrease CD4+ T-cell proliferation at day +7 but did not result in preferential Treg recovery at day +21, while the ineffectively low dose of 5 mg/kg/day PTCy on days +3/+4 did not achieve either.13 Therefore, to gain biological insight into why certain dosing schema are more effective than others, we performed flow cytometric analysis at days +7 and +21 after various PTCy dosing schedules. We hypothesized that these two parameters may be potential biomarkers of effective GVHD prevention by PTCy.
In our prior work, we looked at five different markers of alloreactive T cells.13 Of these, the only one that is applicable to wild-type mice used in the B6C3F1→B6D2F1 HCT model is Vβ6.16 We found that Vβ6+ CD4+CD25−Foxp3− conventional T cells (Tcons) and Vβ6+ CD8+ T cells persisted at day +7 at similar to higher percentages in all PTCy-treated mice compared with vehicle-treated mice although total numbers were reduced for some PTCy-treated groups [Figures 6A and S1]; these percentages also were similar at day +21 [Figure 6B]. However, the proliferation of Vβ6+ CD4+CD25−Foxp3− Tcons at day +7 was affected by PTCy. PTCy given at either 10 mg/kg/day or 25 mg/kg/day on days +3/+4 or +5/+6 significantly reduced Vβ6+ CD4+CD25−Foxp3− Tcon proliferation at day +7 in the spleen, while only 25 mg/kg/day PTCy on days +3/+4 reduced Vβ6+ CD4+CD25−Foxp3− Tcon proliferation in the blood [Figures 6C and S2]. Interestingly, Vβ6+ CD4+CD25−Foxp3− Tcon proliferation was significantly increased in mice treated with 10 mg/kg/day PTCy on days +1/+2 [Figures 6C and S2], a dose associated with more rapid lethality from GVHD, compared with in vehicle-treated mice [Figure 1A].
Figure 6. Effective dosing schedules of PTCy do not eliminate alloreactive (Vβ6+) T cells but do result in decreased proliferation (Ki-67+) of Vβ6+ CD4+ conventional T cells at day +7.

Mice were transplanted as in Figure 1 and received PTCy at various dosing schedules. Mice not receiving PTCy on a given day received PBS vehicle. At designated time points, mice were euthanized, and their spleens and blood were assessed by flow cytometry. (A-B) Percentages of CD4+CD25−Foxp3− or CD8+ T cells that were Vβ6+ were not reduced at day (A) +7 or (B) +21 after any of the PTCy dosing schedules; in fact, some percentages actually were increased at day +7 after PTCy. (C) Vβ6+ CD4+CD25−Foxp3− T-cell proliferation (Ki-67+) was significantly reduced in the spleen after several of the more effective PTCy dosing schedules, but was only significantly reduced in the blood by the optimal dosing (25 mg/kg/day PTCy on days +3/+4). Combined results from two independent experiments are shown. N=6/group for A and C except for the 10 mg/kg/day or 25 mg/kg/day PTCy on days +1/+2 group (n=4 each) and the 25 mg/kg/day PTCy on days +5/+6 group (n=5) due to some deaths occurring prior to day +7. N=8/group for B except 10 mg/kg/day PTCy on days +2/+3 (n=4). *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001 on one-way ANOVA followed by the Holm-Sidak post hoc test using the vehicle-treated group as the control. Only significant results are shown; all other comparisons between treatment groups and the vehicle treatment group are non-significant.
Effective PTCy dosing schema facilitate preferential Treg reconstitution by day +21
Tregs have been shown to play a necessary role in GVHD prevention by PTCy in xenogeneic,17 MHC-matched,18 and MHC-haploidentical13 murine HCT models. In our MHC-haploidentical model, at day +7 there were similar percentages of CD4+CD25+Foxp3+ Tregs in blood and lymph nodes and slightly lower percentages in spleen and liver in PTCy-treated compared with vehicle-treated mice, but at day +21 after 25 mg/kg/day PTCy on days +3/+4, the percentages of Tregs had become significantly increased in all four organs.13
Consistent with our prior results,13 at day +7 we found no differences in Treg percentages in blood regardless of PTCy dosing schedule, whereas the Treg percentages in spleen were lower after most PTCy dosing schedules [Figure 7A]. Moreover, at day +7 we found no significant differences in the percentages of alloantigen-specific (Vβ6+) Tregs [Figure 7B].
Figure 7. Effective dosing schedules of PTCy result in preferential recovery of CD4+CD25+Foxp3+ Tregs at day +21.

Mice were transplanted as in Figure 1 and received PTCy at various dosing schedules. Mice not receiving PTCy on a given day received PBS vehicle. At designated time points, mice were euthanized, and their spleens and blood were assessed by flow cytometry. (A) At day +7, the percentages of CD4+ T cells that were CD25+Foxp3+ were similar or reduced by various PTCy dosing schedules compared with vehicle treatment. (B) The percentages of CD4+CD25+Foxp3+ Tregs that were Vβ6+ were not significantly different between groups at day +7. (C) At day +21, the most effective doses of PTCy (10 mg/kg/day on days +3/+4 or 25 mg/kg/day on days +1/+2, +3/+4, or +5/+6) all were associated with significantly increased percentages of CD4+CD25+Foxp3+ Tregs in both blood and spleen. (D) The percentages of Vβ6+ Tregs were not significantly different at day +21 between treatment groups, although the more effective dosing schedules tended to have higher percentages. (E-F) Pooling across all treatment groups, the percentages of CD4+CD25+Foxp3+ Tregs in either (E) blood or (F) spleen were significantly negatively associated with the histopathologic severity score in the same mice, suggesting that the percentage of Tregs at day +21 in this model may be a biomarker of effective GVHD prevention. Combined results from two independent experiments are shown. N=6/group for A-B except for the 10 mg/kg/day or 25 mg/kg/day PTCy on days +1/+2 group (n=4 each) and the 25 mg/kg/day PTCy on days +5/+6 group (n=5) due to some deaths occurring prior to day +7. N=8/group for C-F except 10 mg/kg/day PTCy on days +2/+3 (n=4). *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001 on one-way ANOVA followed by the Holm-Sidak post hoc test using the vehicle-treated group as the control. Only significant results are shown; all other comparisons between treatment groups and the vehicle treatment group are non-significant.
Supportive of our hypothesis, we found that the most effective PTCy dosing schedules (10 mg/kg/day on days +3/+4 and 25 mg/kg/day on days +1/+2, +3/+4, or +5/+6) all resulted in significantly increased percentages of CD4+CD25+Foxp3+ Tregs at day +21 in both blood and spleens, with maximal increases in these percentages after 25 mg/kg/day PTCy on days +1/+2 or +3/+4 [Figure 7C]. Given the limited power, there were no significant differences in the percentage of Vβ6+ Tregs, although the 25 mg/kg/day PTCy on days +3/+4 group had the highest percentages [Figure 7D]. Absolute numbers of total CD4+CD25+Foxp3+ Tregs and Vβ6+ Tregs at day +21 were similar or significantly increased in PTCy-treated mice compared with vehicle-treated mice [Figure S3]. The percentages of all Tregs in the blood or spleen at day +21 correlated negatively with the GVHD histopathologic severity score within the same mice [Figure 7E-F]. These data suggest that both initial reduction of alloreactive CD4+ Tcon proliferation at day +7 and subsequent Treg reconstitution at day +21 may be potential biomarkers of effective GVHD prevention by PTCy.
Discussion:
Herein, we utilized a murine MHC-haploidentical HCT model to evaluate the impact of the timing of PTCy on GVHD prevention. We found that the efficacy of PTCy peaks at day +4, although the window for effective GVHD prophylaxis was broader at the optimal dose. Moreover, our findings highlighted the importance of the dose and timing of PTCy, but also the cumulative exposure of the drug for effective prevention of GVHD, as demonstrated by the BID and the single-day vs. two-day dosing schedules.
In this work, we also found that optimal PTCy dosing schema both reduced alloreactive CD4+ Tcon proliferation at day +7 and facilitated preferential Treg recovery at day +21, adding potential biologic insight into why certain dosing schema are optimal. These results are consistent with our prior findings that too high a PTCy dose may be ineffective because it blocks the preferential recovery of Tregs compared with Tcons, despite much more effective reduction of alloreactive CD4+ Tcon proliferation at day +7.13 In the current study, PTCy on days +1/+2 allowed Treg recovery at day +21 similar to that observed after PTCy on days +3/+4, but the days +1/+2 dose schedule did not adequately abate alloreactive CD4+ Tcon proliferation at day +7. By contrast, PTCy on days +5/+6 substantially reduced alloreactive CD4+ Tcon proliferation at day +7, but yielded much less robust Treg recovery at day +21. Moreover, 10 mg/kg/day PTCy on days +1/+2 actually increased alloreactive CD4+ Tcon proliferation at day +7, leading to rapid death in these mice. While it is unclear if these immunologic parameters are mechanistic or simply epiphenomena, Tregs are necessary for GVHD prevention by PTCy,13,17,18 and, at the very least, this combination appears to be a potential predictive biomarker for effective GVHD prevention by PTCy.
Further study is needed to understand to what extent these findings apply to human HCT. An important caveat is that we do not know how cyclophosphamide doses correspond between mice and humans, particularly as there are pharmacokinetic differences with mice having quicker clearance of the drug.19 Our prior study suggested that an intermediate dose (25 mg/kg/day) of PTCy was optimal in two different murine HCT models with both lower and higher doses being associated with increased GVHD and mortality.13 In humans, the current clinical dose of 50 mg/kg/day is nearly as high a cyclophosphamide dose as a human can tolerate. 25 mg/kg/day PTCy on days +3/+4 has shown efficacy in small clinical studies20,21 as has 14.5 mg/kg/day in combination with antithymocyte globulin.22 Even PTCy at 7.5 mg/kg/day on serial dosing between days +1 and +100 demonstrated efficacy in reducing chronic GVHD;23 this treatment had minimal impact on acute GVHD, consistent with the results of our mice treated with 10 mg/kg/day who have considerable histopathologic and clinical GVHD early and improve slowly over 4 months. Thus, the clinical observations in mice and humans suggest that doses may have relatively similar effects between the two species.
Our work has several other limitations. Firstly, our murine studies were underpowered to detect small differences between dosing schemas. Even so, we identified that the optimal window centered around day +4. Prior clinical studies have suggested that dosing on days +3/+4 may have lower rates of GVHD than day +3 alone9,21 and that PTCy on days +3/+5 is quite effective.15 The latter approach results in very low rates of acute GVHD as well, but that strategy also begins the adjunct immunosuppression at HCT instead of on day +5. This difference highlights another limitation of the translatability of results from our murine HCT model in which PTCy is used alone for GVHD prevention. The adjunct prophylactic immunosuppression generally given in addition to PTCy in patients impacts clinically on rates of acute GVHD and biologically on alloreactive T-cell proliferation and also may impact Treg recovery.24 Thus, these other agents may obscure the maximal efficacy of PTCy itself on clinical and immunologic parameters and potentially could impact which dosing schema are optimal in specific clinical contexts. Additionally, there may be differences between mouse and human T cells in the kinetics of proliferation and expression of cyclophosphamide resistance pathways such as aldehyde dehydrogenase.17,18 It is also unknown whether the effects on Vβ6+ T cells found here reflect changes in other alloreactive T cells in mice or humans; however, our recent work did show similar effects of PTCy on four other types of alloreactive T cells as PTCy did on Vβ6+ T cells,13 but human data will be required to see whether these results apply to clinical HCT. Furthermore, Ki-67 is a marker of T cells in active cell cycle and consequently is only an indirect marker of cell division. Even so, this marker is easily testable in human HCT, and it is unclear that cell division is more important than proliferation (cell cycle) in this context. Lastly, our B6C3F1→B6D2F1 MHC-haploidentical HCT model is primarily one of acute rather than chronic GVHD, and optimal dosing schemas of PTCy possibly could differ for each.
Our findings contrast with the early skin allografting studies that determined that PTCy most effectively promoted engraftment when given on days +2 or +3 and when using a very high dose (200 mg/kg).2 However, PTCy’s efficacy in those studies was contextual, requiring specific type and dose of donor cells and MHC-matched recipients of a specific age.2,25,26 Moreover, our recent work13 has highlighted important mechanistic differences between PTCy’s activity in preventing GVHD after HCT compared with what has been previously described for preventing MHC-matched skin allograft rejection, possibly contributing to the differing optimal dosing schedules. Furthermore, a high-dose of PTCy at an earlier time point potentially may overcome the less robust impact on alloreactive CD4+ T-cell proliferation of lower doses of PTCy given on days +1/+2 or +2/+3, while taking advantage of the similar preferential Treg recovery occurring at that time point, thereby rebalancing the net impact of different PTCy dosing schema; such a theorized effect may be more active in a mixed chimeric state when trying to induce host tolerance to a subsequently placed skin allograft, rather than controlling active graft-versus-host alloreactivity.
In conclusion, our findings highlight the importance of the dose, timing, and cumulative exposure of PTCy on its clinical effectiveness in preventing GVHD. Our findings support that the current timing of PTCy administration may be among the best options, but suggest that, when given at the optimal dose, PTCy may have equal efficacy when given on day +4 alone. These data support a clinical study initiated at our institution to explore dose de-escalating PTCy first to 25 mg/kg/day on days +3/+4 and then to 25 mg/kg on day +4 only. If successful, this strategy may reduce toxicity and improve hematopoietic and immune reconstitution, while preserving severe acute and chronic GVHD prevention. In this clinical study, we simultaneously are evaluating candidate predictive immunologic biomarkers for effective GVHD prevention by PTCy of CD4+ T-cell proliferation early after PTCy and CD4+CD25+Foxp3+ recovery at day +21 in a quantitative systems pharmacology model. We hope these studies will improve clinical HCT outcomes for patients and provide further insight into the biology of acute and chronic GVHD and their prevention.
Supplementary Material
Highlights:
The dose, timing, and cumulative exposure of post-transplantation cyclophosphamide all are critical for its efficacy.
The maximal efficacy of post-transplantation cyclophosphamide peaks at day +4.
Effective dosing schema both reduce alloreactive conventional CD4+ T-cell proliferation and allow preferential regulatory T-cell recovery.
Acknowledgements:
This work was supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health. We would like to thank Ehydel Castro and Devorah Gallardo for technical assistance.
Footnotes
Conflict of interest disclosures: The authors have declared that no conflict of interest exists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Kanakry CG, Fuchs EJ, Luznik L. Modern approaches to HLA-haploidentical blood or marrow transplantation. Nat Rev Clin Oncol. 2016;13(1):10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mayumi H, Himeno K, Tokuda N, Nomoto K. Drug-induced tolerance to allografts in mice. VII. Optimal protocol and mechanism of cyclophosphamide-induced tolerance in an H-2 haplotype-identical strain combination. Transplant Proc. 1986;18(2):363–369. [PubMed] [Google Scholar]
- 3.Luznik L, Jalla S, Engstrom LW, Iannone R, Fuchs EJ. Durable engraftment of major histocompatibility complex-incompatible cells after nonmyeloablative conditioning with fludarabine, low-dose total body irradiation, and posttransplantation cyclophosphamide. Blood. 2001;98(12):3456–3464. [DOI] [PubMed] [Google Scholar]
- 4.Colson YL, Wren SM, Schuchert MJ, et al. A nonlethal conditioning approach to achieve durable multilineage mixed chimerism and tolerance across major, minor, and hematopoietic histocompatibility barriers. J Immunol. 1995;155(9):4179–4188. [PubMed] [Google Scholar]
- 5.Anasetti C, Amos D, Beatty PG, et al. Effect of HLA compatibility on engraftment of bone marrow transplants in patients with leukemia or lymphoma. N Engl J Med. 1989;320(4):197–204. [DOI] [PubMed] [Google Scholar]
- 6.Szydlo R, Goldman JM, Klein JP, et al. Results of allogeneic bone marrow transplants for leukemia using donors other than HLA-identical siblings. J Clin Oncol. 1997;15(5):1767–1777. [DOI] [PubMed] [Google Scholar]
- 7.O’Donnell PV, Luznik L, Jones RJ, et al. Nonmyeloablative bone marrow transplantation from partially HLA-mismatched related donors using posttransplantation cyclophosphamide. Biol Blood Marrow Transplant. 2002;8(7):377–386. [DOI] [PubMed] [Google Scholar]
- 8.Brodsky RA, Sensenbrenner LL, Smith BD, et al. Durable treatment-free remission after high-dose cyclophosphamide therapy for previously untreated severe aplastic anemia. Ann Intern Med. 2001;135(7):477–483. [DOI] [PubMed] [Google Scholar]
- 9.Luznik L, O’Donnell PV, Symons HJ, et al. HLA-haploidentical bone marrow transplantation for hematologic malignancies using nonmyeloablative conditioning and high-dose, post-transplantation cyclophosphamide. Biol Blood Marrow Transplant. 2008;14(6):641650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ciurea SO, Zhang MJ, Bacigalupo AA, et al. Haploidentical transplant with posttransplant cyclophosphamide vs matched unrelated donor transplant for acute myeloid leukemia. Blood. 2015;126(8):1033–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanate AS, Mussetti A, Kharfan-Dabaja MA, et al. Reduced-intensity transplantation for lymphomas using haploidentical related donors vs HLA-matched unrelated donors. Blood. 2016;127(7):938–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosh N, Karmali R, Rocha V, et al. Reduced-Intensity Transplantation for Lymphomas Using Haploidentical Related Donors Versus HLA-Matched Sibling Donors: A Center for International Blood and Marrow Transplant Research Analysis. J Clin Oncol. 2016;34(26):31413149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wachsmuth LP, Patterson MT, Eckhaus MA, Venzon DJ, Gress RE, Kanakry CG. Post-transplantation cyclophosphamide prevents graft-versus-host disease by inducing alloreactive T cell dysfunction and suppression. J Clin Invest. 2019;130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raiola AM, Dominietto A, Ghiso A, et al. Unmanipulated haploidentical bone marrow transplantation and posttransplantation cyclophosphamide for hematologic malignancies after myeloablative conditioning. Biol Blood Marrow Transplant. 2013;19(1):117–122. [DOI] [PubMed] [Google Scholar]
- 15.Chiusolo P, Bug G, Olivieri A, et al. A Modified Post-Transplant Cyclophosphamide Regimen, for Unmanipulated Haploidentical Marrow Transplantation, in Acute Myeloid Leukemia: A Multicenter Study. Biol Blood Marrow Transplant. 2018;24(6):1243–1249. [DOI] [PubMed] [Google Scholar]
- 16.Hodes RJ, Abe R. Mouse endogenous superantigens: Ms and Mls-like determinants encoded by mouse retroviruses. Curr Protoc Immunol. 2001;Appendix 1:Appendix 1F. [DOI] [PubMed] [Google Scholar]
- 17.Kanakry CG, Ganguly S, Zahurak M, et al. Aldehyde dehydrogenase expression drives human regulatory T cell resistance to posttransplantation cyclophosphamide. Sci Transl Med. 2013;5(211):211ra157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ganguly S, Ross DB, Panoskaltsis-Mortari A, et al. Donor CD4+ Foxp3+ regulatory T cells are necessary for posttransplantation cyclophosphamide-mediated protection against GVHD in mice. Blood. 2014;124(13):2131–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Said R, Abdel-Rehim M, Sadeghia B, Al-Hashemi S, Hassan M. Cyclophosphamide Pharmacokinetics in Mice: A Comparison Between Retro Orbital Sampling Versus Serial Tail Vein Bleeding. The Open Pharmacology Journal. 2007;1:30–35. [Google Scholar]
- 20.Thakar MS, Bonfim C, Sandmaier BM, et al. Cyclophosphamide-based in vivo T-cell depletion for HLA-haploidentical transplantation in Fanconi anemia. Pediatr Hematol Oncol. 2012;29(6):568–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakamae H, Koh H, Katayama T, et al. HLA haploidentical peripheral blood stem cell transplantation using reduced dose of posttransplantation cyclophosphamide for poor-prognosis or refractory leukemia and myelodysplastic syndrome. Exp Hematol. 2015;43(11):921–929 e921. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Chang YJ, Chen L, et al. Low-dose post-transplant cyclophosphamide can mitigate GVHD and enhance the G-CSF/ATG induced GVHD protective activity and improve haploidentical transplant outcomes. Oncoimmunology. 2017;6(11):e1356152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santos GW, Tutschka PJ, Brookmeyer R, et al. Cyclosporine plus methylprednisolone versus cyclophosphamide plus methylprednisolone as prophylaxis for graft-versus-host disease: A randmized double-blind study in patients undergoing allogeneic marrow transplantation. Clin Transplant. 1987;1:21–28. [Google Scholar]
- 24.Kanakry CG, Luznik L. Teaching a Young Dog New Tricks: Modifications to the Post-Transplantation Cyclophosphamide Haploidentical Transplantation Platform. Biol Blood Marrow Transplant. 2018;24(6):1108–1110. [DOI] [PubMed] [Google Scholar]
- 25.Mayumi H, Good RA. Dependency of cyclophosphamide-induced skin allograft tolerance on age of adult recipient mice. Transplantation. 1988;46(3):451–453. [DOI] [PubMed] [Google Scholar]
- 26.Mayumi H, Good RA. The necessity of both allogeneic antigens and stem cells for cyclophosphamide-induced skin allograft tolerance in mice. Immunobiology. 1989;178(4–5):287–304. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.