

Abstract
Integrin receptors enable cells to sense and respond to their chemical and physical environment. As a class of membrane receptors, they provide a dynamic, tightly regulated link between the extracellular matrix or cellular counter-receptors and intracellular cytoskeletal and signaling networks. They enable transmission of mechanical force across the plasma membrane, and particularly for cardiomyocytes, may sense the mechanical load placed on cells. Talins and Kindlins are two families of FERM—domain proteins which bind the cytoplasmic tail of integrins, recruit cytoskeletal and signaling proteins involved in mechano-transduction, and those which synergize to activate integrins, allowing the integrins to physically change and bind to extracellular ligands. In this review, we will discuss the roles of talin and kindlin, particularly as integrin activators, with a focus on cardiac myocytes.
Keywords: Talin, Kindlin, Integrin, Integrin activation, Heart
Introduction to Integrin Biology with a Focus on Heart
Integrins are heterodimeric, transmembrane glycoprotein receptors capable of bidirectionally transmitting signals between the intracellular and extracellular environments. Integrins are expressed on almost all cells and are composed of α and β subunits. Mammals express greater than 18 α and 8 β integrin subunits, which heterodimerize to form 24 receptors. The different integrin heterodimers bind to varied ligands such as laminin, collagen, fibrinogen, tenascin-C, and cadherin. In the cardiomyocyte (CM), the integrin heterodimers most highly expressed are α1β1, α5β1, and α7β1 which are predominantly collagen (Col)-, fibronectin (FN)-, and laminin (LN)-binding receptors, respectively. In addition, α6, α9, and α10 are detected in myocytes. β1 is the dominant β integrin subunit, but β3 and β5 subunits have also been studied in heart [1–3]. In addition, differentially spliced isoforms of the β1 integrin subunit are expressed in embryonic (β1A) versus adult cardiomyocytes (β1D). The β1A versus β1D isoforms are characterized by specific amino acid sequences in their cytoplasmic domains, which provide them with distinct, isoform-specific interactive properties with cytoskeletal and signaling molecules [4].
Integrin subunits range in size from 80 to 180 KDa molecular weight. They generally consist of a large extracellular domain, a single transmembrane spanning domain, and a short cytoplasmic tail. The extracellular domains of the integrins specifically bind in a cation-dependent manner to extracellular matrix (ECM) proteins (e.g., collagens, fibronectin, and laminins) or to counter-receptors, such as Vascular Cell Adhesion Molecule (VCAM), Intercellular Adhesion Molecule (ICAM), or cadherins, on adjacent cells. Their short cytoplasmic tails link to cytoskeletal elements, adaptors, and signaling proteins inside the cell via bridging proteins, such as α-actinin, talin, paxillin, and vinculin–metavinculin [5].
In brief, the extracellular domain of the α subunit consists of a β-propeller, thigh, calf-1 and calf-2 domains [6]. In addition, a subset of α subunits such as α1, α2, α10, and α11 also has I/A domains within the β-propeller [6]. The I/A domain contains a metal ion-dependent adhesion site (MIDAS) that acts as a ligand-binding site, where Mn2+, Ca2+, and Mg2+ can be found [7]. In contrast, the extracellular domain of the β subunit consists of an I/A domain, an immunoglobulin-like hybrid domain, a plexin–semaphorin–integrin (PSI) domain, four EGF-like repeats (EGF1–4) and a β-tail domain. For those α subunits that do not have an I/A domain, the I/A domain located on the β subunit serves as the ligand-binding site instead, after forming a complex with the β-propeller on the α subunit. The I/A domain found in the inactivated integrin is protected, but with activation of the integrin subunit, physical changes occur that expose the I/A domain and enable it to bind to different ligands.
Integrin-adhesive interactions permit transmission of mechanical signals into and out of the cell, and enable conversion of the mechanical information into chemical signals. This information is crucial for a great many processes including embryonic development, tissue maintenance and repair, host defense, and hemostasis [2, 8, 9]. To directly evaluate integrin function in the intact organism, work in our lab along with others has used a floxed β1 integrin mouse. The β1 integrin gene was specifically excised in CMs at different developmental stages, by crossing these floxed mice with a variety of cardiomyocyte-specific Cre mice. In early cardiogenesis, Nkx2.5-Cre-mediated excision led to perturbation of ventricular compaction, along with reduction of CM proliferation, a smaller ventricle with severe fibrosis, and 50% lethality of mutants by weaning. When β1 integrin protein was reduced in CMs soon after birth by means of a constitutively active Cre-recombinase driven by the endogenous myosin light chain (MLC)-2 ventricular (v) promoter, progressive myocardial fibrosis and development of a dilated cardiomyopathy (DCM) were found. These mice were also intolerant of hemodynamic loading and had increased damage after challenge by ischemia/reperfusion [10, 11]. Conversely, when a dominant cardiomyocyte integrin, the laminin-binding α7β1D, was overexpressed in myocytes, the heart was protected from ischemia/reperfusion injury [10].
Integrin Activation
Signals transmitted by integrins from outside to inside the cell regulate cell survival, proliferation, and cell fate. Conversely, intracellular cues can modulate the affinity of integrins for their extracellular ligands a process operationally defined as integrin activation or ‘inside–out’ signaling [10, 11].
Integrins exist in low and high affinity states. On the basis of structural studies, it has been shown that integrins are in a low-affinity state when their extracellular domains are in a bent conformation, and in a high-affinity state when those domains are extended, i.e., allowing the integrin to be in the “activated” state. These conformational changes which allow for integrin activation have been the discussed in great detail in several excellent recent reviews [12–18] and thus a discussion of this topic will not be included here (Fig. 1). In brief, as mentioned above, upon initiation of integrin activation, the ligand-binding site on the I/A domain which is normally hidden in the inactivated state, is exposed to the surrounding environment for ligand binding [19]. The two most significant intracellular activators of integrin are talin [20–22] and kindlin proteins [23–25], which both regulate integrin activation by binding the tails of integrin β subunits. In this review we will mainly focus on the function of talin and kindlin, and specifically their role in heart.
Fig. 1.
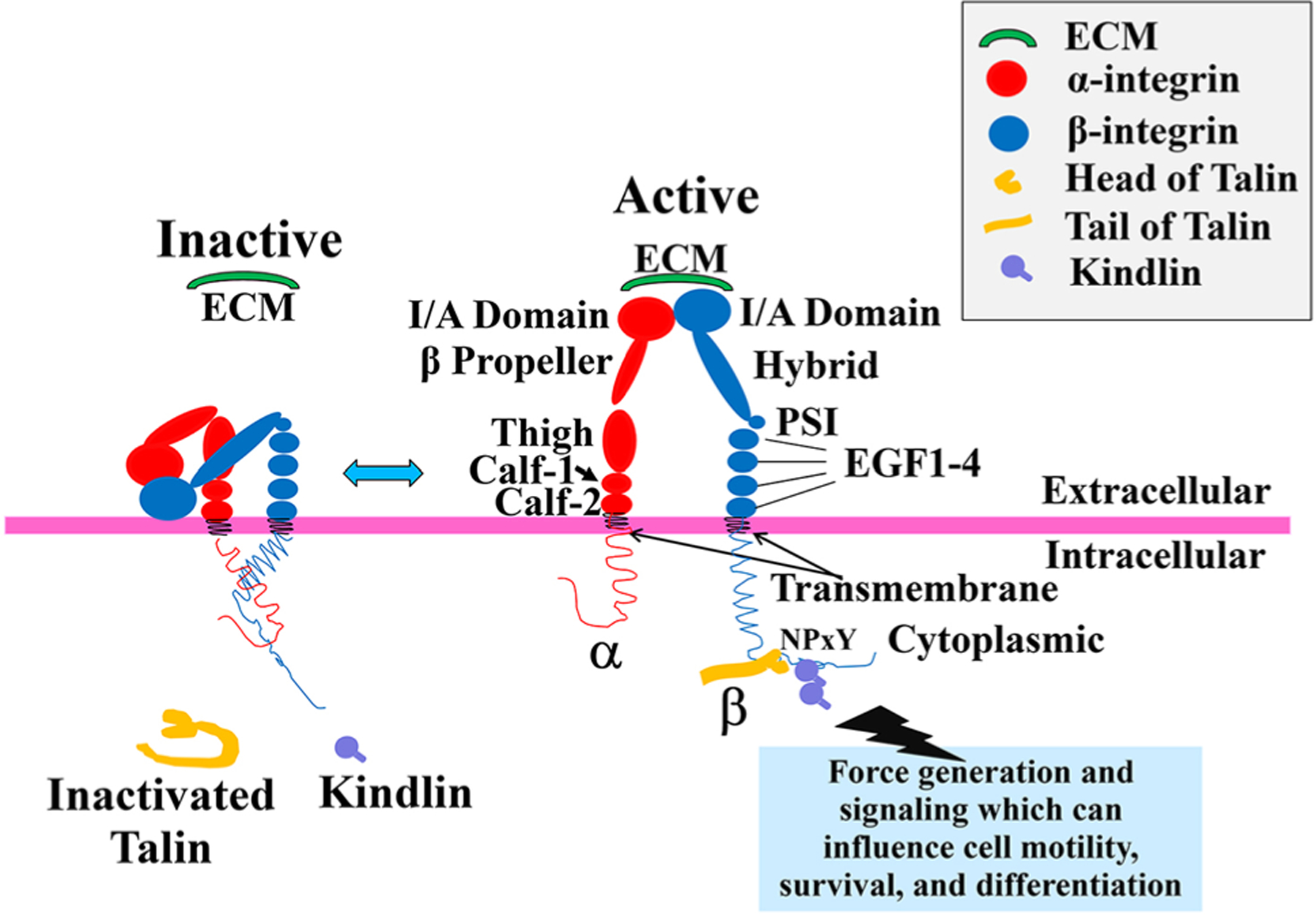
Integrin’s structure and its activation. Activation of integrin heterodimers leads to intracellular signaling (not shown) and results in processes such as cell motility, survival, and differentiation. Schematic shows integrin conformation at the membrane in the inactive and active conformations. In the inactive state, the extracellular domains of integrin heterodimers are present in a closed/bent conformation stabilized by a cytoplasmic salt bridge. This conformation has a very low ligand-binding affinity, e.g., for extracellular matrix (ECM). With initiation of “Inside–Out” signaling, a series of intracellular events occurs. This includes a structural change of talin from its autoinhibitory state, to an activated one as it unfolds. Then, the intracellular integrin activators, talin and kindlin, bind the β integrin cytoplasmic NPxY or Nxxy motif, respectively, leading to a conformational change of the integrin extracellular domain into an open and extended, i.e., active, state. In this state, the integrin I/A domain is exposed, and the integrin receptor has an increase in ligand-binding affinity for extracellular ligands such as the ECM proteins laminin, fibronectin, or collagen. As the extracellular ligands are bound, this initiates “Outside–In” signaling (not shown in this diagram) through the activated integrin, which initiates clustering of a series of intracellular proteins around the integrin cytoplasmic domains, and subsequent intracellular signaling events. In turn, this produces clustering of multiple integrin receptors, enabling a higher affinity and avidity for extracellular ligand, and thus teleologically, a firmer binding of integrins to ligands. Individual names of integrin extracellular domain components have been shown in the main text
Talin
Talin regulates integrin affinity and provides a mechanical link between integrins and the actin cytoskeleton. In vertebrates, there are two forms of talin: talin1 and talin2. Talin1 is expressed in virtually all cell types, while talin2 is found primarily in the brain, as well as skeletal and cardiac muscles [26, 27].
Talin protein is composed of an amino terminal 50-kDa head domain followed by a 220-kDa rod domain [28, 29]. The talin head contains a three-lobed, FERM (4.1, ezrin, radixin, and moesin) domain. Uniquely, the FERM domain is preceded by an F0 domain which is required for activation of β1 integrin [30], and followed by a 33-amino acid stretch. A phosphotyrosine-binding (PTB) domain in the third lobe of the talin FERM region directly binds to the membraneproximal NPxY motif (the canonical PTB domain-binding site) on the β integrin cytoplasmic tail [22, 31]. The talin rod/ tail is composed of a series of helical bundles and contains the major binding sites for actin and vinculin, which are key components of the cytoskeleton at focal adhesions. The talin tail domain can also self-regulate its integrin-binding site on the head domain. Specifically, the C-terminal rod domain of talin structurally interacts with talin head domain, and allosterically restrains talin in a closed conformation. We will discuss additional information about talin regulation of integrin activation in the next section.
Talins activate integrins by binding to the cytoplasmic tail of β integrin, via its FERM domain. With this binding, the cytoplasmic tails of the α and β subunits are separated, leading to a conformational change of the integrin receptor, enabling integrin activation [32]. In addition, talin has been suggested to also function as a scaffolding protein, as its tail binds to multiple integrin-associated proteins and the actin cytoskeleton, while its head binds to integrins. With these activation events, integrins extend and bind to the ECM, triggering a series of intracellular events, such as, cell motility [32, 33]. Hence, talin is widely considered to be a potent integrin activator.
The large size of the entire talin molecule (≈ 2500 amino acids) makes experimental procedures to investigate its function, such as cell transfection of full-length talin, quite difficult. This is particularly noteworthy since the fulllength talin protein is required for activating integrin molecules. Thus, this highlights that studies using genetically engineered mice are one of the best experimental tools to examine talin function. Global talin1 knockout (KO) mice display an embryonic lethal phenotype by embryonic day (E) 8.5 to E9.0 due to gastrulation defects [34], indicating that although both talin genes encode for relatively similar proteins (74% identical), talin2 cannot replace talin1 function in the entire embryo. In contrast, global talin2 KO mice only develop a mild skeletal myopathy later in life attributable to defects in myotendinous junction integrity, indicating that the two talin isoforms have unique properties [1, 35].
In our lab, we used talin2 KO- and talin1-floxed mice to investigate the role of talin in heart. Our laboratory showed that during embryogenesis, talin1 and talin2 are both highly expressed in mouse CMs. In adult mouse CMs, talin1 protein expression becomes reduced, and talin2 becomes the main talin form. With pressure overload (POL)-induced cardiac hypertrophy in mice, talin1 protein expression increases in CMs where it specifically becomes localized to costameres, suggesting that it is involved in the adaptive mechanisms triggered in the stressed heart. The role of talin1 in the cardiac stress response was confirmed by the generation and characterization of talin1 CM-specific KO mice. The talin1 CM-specific KO mice showed normal basal cardiac structure and function, but after chronic pressure overload had blunted hypertrophy, decreased fibrosis, and preserved cardiac function, versus littermate controls. In addition, talin1 CM-specific KO mice showed attenuated Akt, ERK1/2, and p38 activation after acute POL. These data show that normal expression of talin1 is not necessary to maintain basal cardiac structure and function in adult heart. They also demonstrate that talin1 is important in the response of cardiac muscle to mechanical stress, perhaps suggesting that with less talin1, the CM is more compliant (our unpublished work), and is therefore able to accommodate to mechanical stress better than control CMs and hearts, which have a normal expression of talin1 protein.
Interestingly, analysis of human heart samples showed an increase in talin1 expression and localization to the costameres in dilated cardiomyopathy (DCM) specimens, compared with nonfailing heart samples. These human expression data agree with that found in mouse and suggest that the role of talin1 in the mouse cardiac stress response may also be applicable to man. In support of this is a recent study that linked mutation of the mechanosensory protein cardiac ankyrin repeat protein to human familial cardiomyopathy [36]. Interestingly, these cardiac ankyrin repeat protein mutants lost binding to talin1. The fact that talin2 is the most abundant talin isoform in adult CMs suggests that it may have a more important structural role than talin1, supporting the connection between integrins and the sarcomeres at the cardiac costameres.
We also investigated the role of talin2 in the heart by evaluating talin2 KO mice. For this purpose, we first studied cardiac structure and function in these talin mice. In the absence of talin2, CM talin1 was upregulated, and remarkably, normal cardiac structure and function were maintained despite a 50% reduction in levels of β1D integrin, which connects to talin protein.
Next, to fully clarify the role of talin in CM, we analyzed the effects of deleting both talin1 and talin2 proteins from CM, by means of a talin1flox/flox × α-myosin heavy chain (MHC)-Cre/talin2KO mouse model [37]. Talin1cKO/talin2KO mice develop significant cardiac chamber dilation beginning at 4 weeks of age that rapidly progresses to heart failure and premature death beginning at 8 weeks, with 100% mortality by 6 months of age. Concomitant with this, β1 integrin, as well as other proteins (e.g., vinculin), are lost from costameres, and cell damage occurred likely due to loss of membrane integrity. CM-specific overexpression of integrin transgenes could not rescue this phenotype in talin1/talin2-null cells or in the intact mouse heart. Thus, talin protein is essential for proper β1D integrin expression at the CM membrane for the preservation of costamere and cellular integrity, and ultimately for normal myocardial function [38, 39].
To further investigate talin in adult CM, we also used another mouse model where we produced inducible loss of all talin proteins from CMs, but only in the adult heart after myocytes had fully matured with a normal amount of talin protein expression. For this purpose, we used a talin1flox/flox × Mer-Cre-Mer MHC /talin2 KO mouse model [40, 41], and found that severe heart dilation and fibrosis occurred along with reduction of β1D integrin expression 8 weeks after tamoxifen injection (unpublished data from our lab). These results further prove the essential role of talin protein in regulating the proper balance of β1D integrin expression and degradation in CM. This data also suggested talin has vital roles to maintain basal cardiac structure and function in adult heart. It is interesting to note that recent work has demonstrated a role for talin as a molecular mechanosensor and confirmed the importance of talin in providing an initial linkage from integrins to actin that is important for stabilizing cell–ECM binding that is essential for preservation of tissue architecture [42]. Ongoing studies are being performed in our laboratory to elucidate talin function in cardiac development and other postnatal pathologies. One example is as shown in Fig. 2, where our preliminary data show that embryonic deletion of both talin protein forms in cardiac myocytes using the troponin-T Cre mouse that causes deletion beginning at embryonic day (E) 7.5 [43] leads to embryonic lethality with talin knockout embryos at embryonic day 11.5 displaying a thin-walled myocardium and myocardial rupture.
Fig. 2.
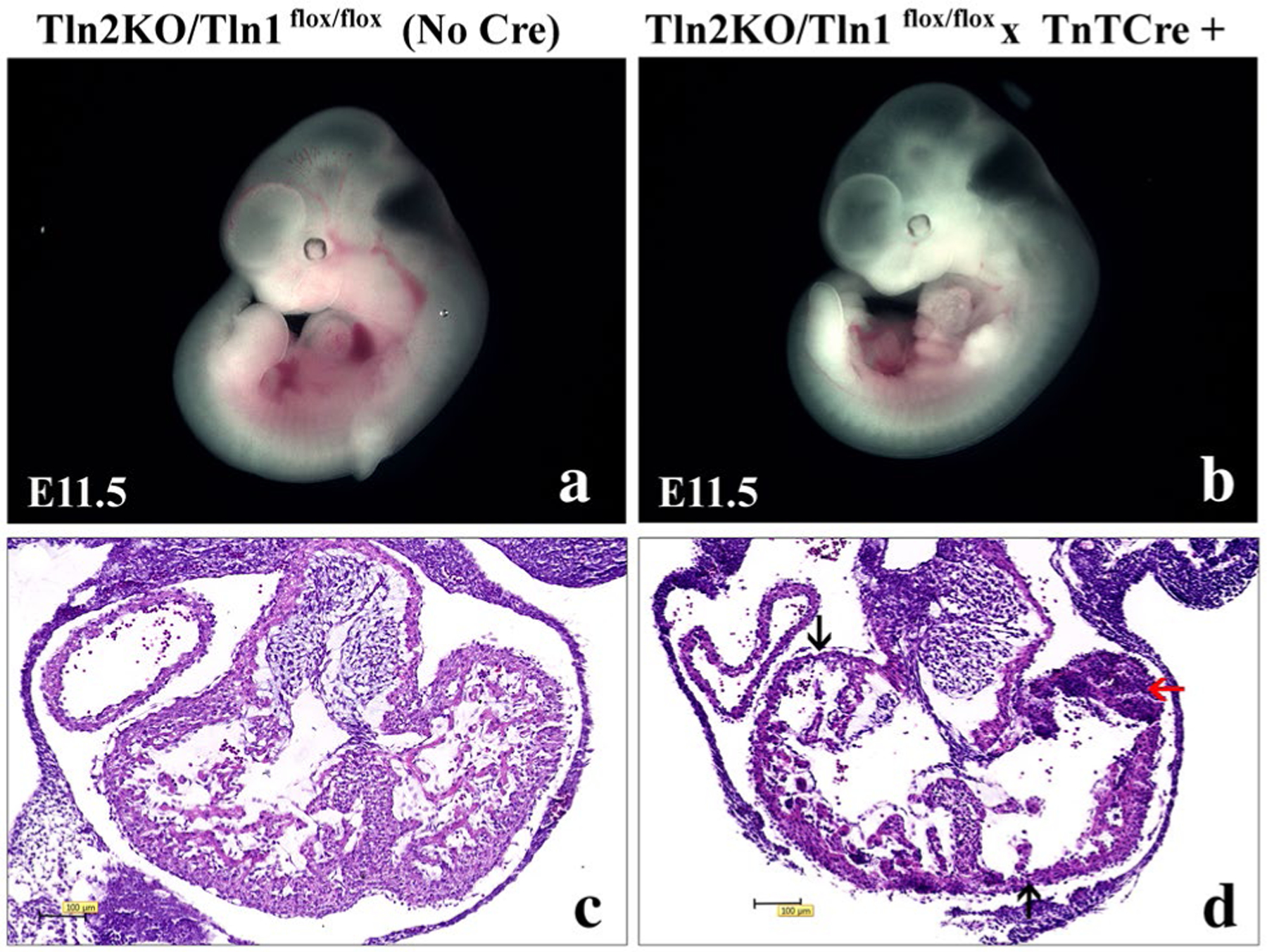
Cardiomyocyte-specific knockout of all talin forms in early embryogenesis leading to embryonic lethality, a thinned wall myocardium, myocardial rupture, and subsequent myocardial hemorrhage. a and b Whole-mount microscopic assessment of a control littermate (Tln2KO/Tln1flox/flox (No Cre) and b Talin 1/2 double knockout—(Tln2KO/Tln1flox/flox × Troponin-T Cre+) embryos (left lateral view) at embryonic day (E) 11.5. c and d Histological evaluation with hematoxylin and eosin staining of a transverse section from the hearts in the upper panels: c Control—(Tln2KO/Tln1flox/flox (No Cre) and d Talin 1/2 double knockout—(Tln2KO/Tln1flox/flox × Troponin-T Cre+) embryos. Arrow in d shows thrombus likely due to rupture of the thin-walled myocardium in the talin 1/2 double knockout embryo
Kindlin
Kindlins are a family of adaptor proteins that are also essential components of the integrin adhesion complex. Kindlin proteins play crucial roles in the integrin-signaling pathway by directly interacting with and activating integrins, which mediate cell–extracellular matrix adhesion and signaling. Mutations of kindlins lead to diseases, such as Kindler syndrome, associated with skin blistering and atrophy; leukocyte adhesion deficiency; and cancers [23]. Our group along with our colleague Dr. Ju Chen has recently begun to study kindlin function in the heart.
There are three members of the kindlin family: Kindlin1, Kindlin2, and Kindlin3, which show different subcellular localization and expression patterns. For instance, kindlin1 is expressed predominantly in epithelial cells, kindlin2 is ubiquitously expressed, and kindlin3 is expressed almost exclusively in hematopoietic cells such as leukocytes and macrophages. Kindlin2, also known as Mig-2, is the only form expressed in cardiac myocytes. In this review, we will focus on kindlin2, but first provide some general background on kindlin proteins [25, 44].
The structural hallmark of kindlins is the evolutionary conserved FERM (4.1, ezrin, radixin, moesin) domain, which is located at the C-terminus of kindlin and is made up of F1, F2, and F3 subdomains [45], like that discussed for talin, described above. All kindlin isoforms bind to the membrane-distal NxxY motif on the β subunit cytoplasmic tail of integrin via the F3 subdomain at the PTB site, resulting in a conformational change and activation of the integrin receptor. Importantly, in contrast to talins, kindlins cannot directly alter the conformation of the integrin transmembrane helix and fail to activate integrin alone. Kindlin needs to have spatial coordination with the talin head domain, in order to support the integrin activation mechanism [46]. In the absence of kindlins, the conformational shift of integrins from the low-to high-affinity state does not occur, providing compelling evidence that kindlins are essential regulators of integrin function [47]. Recently, Huadong et al. determined crystal structures of kindlin2 in the unbound (apo)-form and the β1 and β3-integrin bound forms. The apo-structure shows an overall architecture distinct from talins. The complex-bound structures revealed a unique mode by which kindlins recognize integrins, by combining two binding motifs to provide specificity essential for integrin activation and signaling. Strikingly, this work uncovered an unexpected dimer formation of kindlins. Interrupting dimer formation impaired kindlin-mediated integrin activation [23]. These findings provide data that illustrates a new mechanism for kindlin-mediated integrin activation. Kindlin2 has also been shown to function as cytoskeletal linker molecule involved with “outside–in” integrin signaling, like a number of integrin-binding proteins such as ILK, Paxillin, and Kank2, as shown by methodologies such as proximity-dependent biotinylation [48].
Most studies on kindlin have focused on its role in epithelial cells, leukocytes, and the nervous system; however, the role of kindlin is also of great importance in heart. Genetic deficiency of kindlin2 in mouse results in embryonic lethality [49]. Although ubiquitously expressed, kindlin2 is the most abundantly expressed in cardiac and skeletal muscles. We investigated the role of kindlin2 in heart development by generating a CM-specific KO mouse model. Kindlin2 expression was deleted in developing CMs at approximately E8.0 by crossing kindlin2-floxed mice (f/f) with the αMHC-Cre [37] or cardiac troponin-T (cTnT) Cre [43] mouse models. For simplicity, we term these mice kindlin2 cKO. No viable kindlin2 cKO mice were noted at birth, and embryonic lethality was noted by E11.5. In a related model using kindlin 2-floxed mice, crossed with β-MHC Cre, DNA recombination began at E12.5 in CMs, with low efficiency, with later high efficiency excision by E17.5 [50]. In this model, no morphological, histological, or morphometric (heart weight/body weight) differences were noted between these cKO and control littermates, prior to 7 months of age. By 7 months, decreases in cardiac function as shown by echocardiography (%fractional shortening) were noted, which suggested that loss of kindlin2 leads to progressive heart failure [49, 51]. These results are very similar to the talin cKO mice. The mechanisms leading to the kindlin2 cKO phenotype are still being explored.
Potential Mechanisms to Stimulate Talin/Kindlin‑Mediated Integrin Activation
From the structure of talin, we know the talin is autoinhibited basally, and that it structurally forms a closed conformation by having the large C-terminal rod domain of talin (talin-R) interacting with its own talin head domain. So, a central question in integrin biology is how the talin–integrin interaction is regulated to control integrin activation. Thus, in the next section of the manuscript we will summarize the potential mechanisms regulating talin/kindlin-mediated integrin activation (Fig. 3a).
Fig. 3.
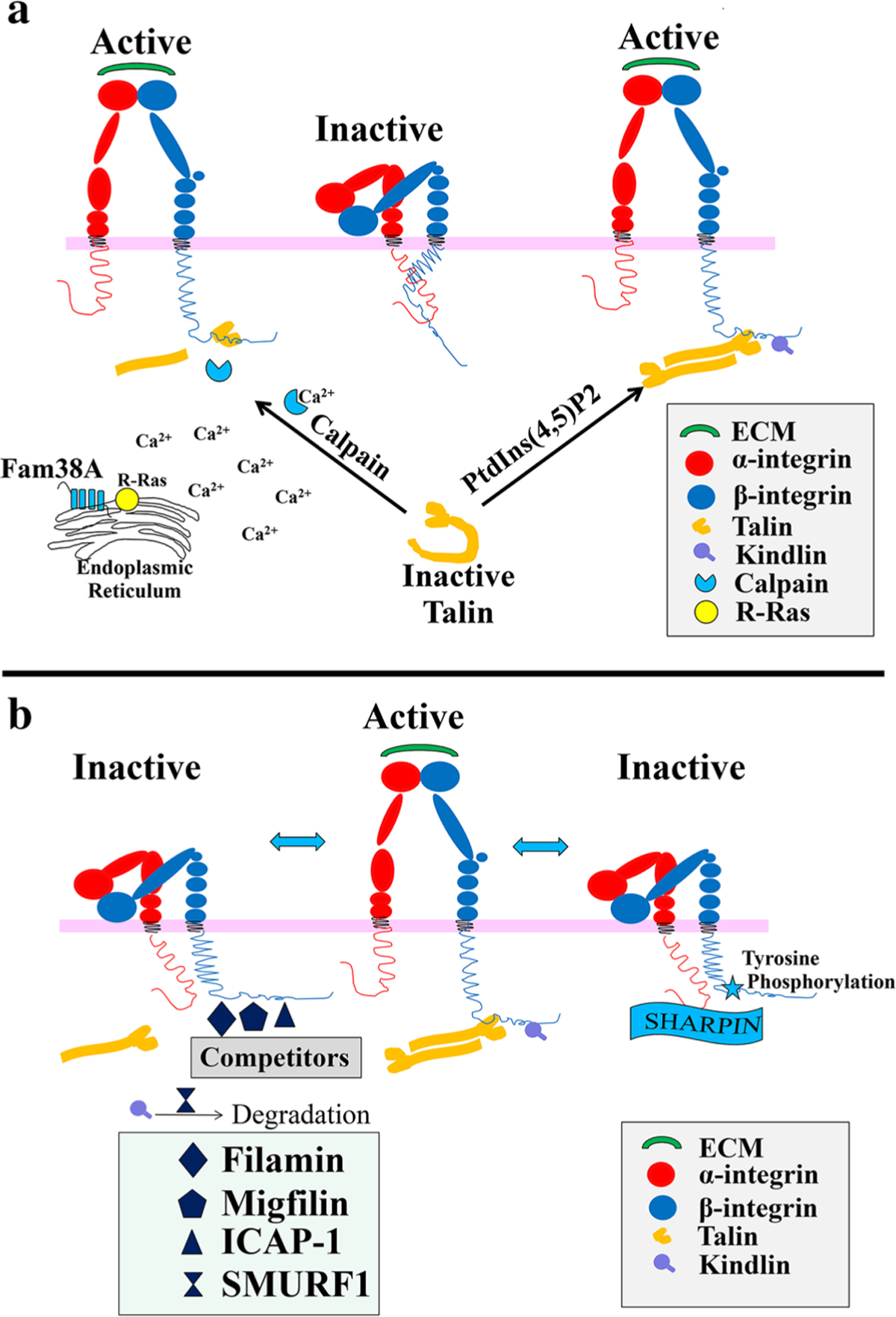
Potential mechanisms regulating talin-mediated integrin activation. a Mechanisms of stimulation of Talin-mediated integrin activation. Talin binding to β integrin tails induces conformational changes in the extracellular domain, increasing their affinity for ligands. (N.B.—The nature of the conformational changes remains controversial, and the model shown represents only one of several possibilities.) Two hypothetical models are shown. In the first, Calpain can cleave talin allowing the talin head to bind integrin cytoplasmic tails and cause activation. In the second model, PtdIns(4,5)P2 binding increased the affinity of talin (specifically talin1) for phospholipid bilayers, leading to activation of talin from its autoinhibited (inactive) form, unmasking the talin-binding site for integrins and then leading to integrin activation. Fam38A can also increase Calpain activation to affect talin-mediated integrin activation. In detail, Fam38A expression causes localization of R-Ras to the endoplasmic reticulum, stimulating Ca2+ release and calpain activation. b Mechanisms of inhibition of Talin/Kindlin-mediated integrin activation. To date, much less is known about inhibition of integrin activation. Two ways that have been published to inhibit talin binding to integrin are (1) Inhibition of talin recruitment, by tyrosine phosphorylation of integrin NPxY motifs or by binding of SHARPIN to conserved cytoplasmic regions of integrin subunits, and (2) competition of other β integrin tail-binding proteins (e.g., Filamin, Migfilin, and ICAP1) for the talin-binding site on integrin. In addition, it has been proposed that SMURF1 can cause Kindlin ubiquitination, leading to its degradation and subsequent inhibition of integrin activation
Recent work has implicated membrane microenvironment, Rap1 (Ras-related protein 1) and Integrin-Activating Transmembrane Protein Fam38A (now named “Piezo1)” as key regulators of talin recruitment to integrin, likely causing stimulation of talin/kindlin activation of integrin [52, 53]. In one study of integrin activation, it was shown that the talin-1 FERM domain binding to the β3 integrin cytoplasmic tail caused activation of integrin αIIbβ3 [54]. Researchers found that the FERM domain also binds to negatively charged acidic phospholipids—phosphatidylinositol 4,5-bisphosphate(PtdIns(4,5)P2), which increased the affinity of talin1 for phospholipid bilayers and activated its autoinhibited form [55]. This action greatly increased the affinity of talin for integrins, so that once talin had been delivered to the plasma membrane, this phospholipid could shift the binding equilibrium in favor of talin–integrin complex formation [54].
In another study, Greensburg and co-workers defined that Rap1 small GTPases interact with the Rap1-GTP-interacting adaptor molecule (RIAM), a member of the MRL (Mig-10/ RIAM/ Lamellipodin) protein family, which directly bound to talin via a short, N-terminal sequence that was predicted to form amphipathic helices. RAP1A–RIAM functions as a scaffold that connects the membrane-targeting sequences in Ras GTPases to talin. In doing so, the authors suggest they recruit talin to the plasma membrane and activate integrins, as evaluated using a Chinese hamster ovary (CHO) cell model [56–58].A few of these works also found that calpain cleavage, which cleaves the talin protein between the head and rod domains, promotes talin binding to the integrin cytoplasmic domain. Importantly, the talin head domain (talin-H) has a sixfold higher binding affinity than intact talin for the β integrin tail.
The above data describe how the activation of integrin occurs by adjusting the affinity of talin for integrins. However, recent work shows that there are also additional integrin activators, like the multi-transmembrane domain protein Fam38A, which can enhance integrin activation by the increment of the integrin–ligand affinity. Fam38A is located in the endoplasmic reticulum (ER) and can mediate integri activation by recruiting the small GTPase, R-Ras, to the ER, which in turn activates the calcium-activated protease calpain, by increasing Ca2+ release from cytoplasmic stores. Fam38A-induced integrin activation is blocked by inhibition of either R-Ras or calpain activity, or by siRNA knockdown of talin expression, perhaps since talin is, as noted above, a well-described calpain substrate. Fam38A is a novel spatial regulator of R-Ras, thereby it can act as an alternative suppressor of integrin activation. By direct re-localization of R-Ras to the ER, combined with a mechanism linking R-Ras and calpain ER signaling, it can perhaps modulate integrin–ligand affinity.
Inhibition of Talin/Kindlin Binding to Integrin: A Means to “Inactivate” Integrins
Binding of talins and kindlins to the cytoplasmic tail of β integrins is widely accepted as being the crucial step in integrin activation. By contrast, much less is known with regard to the counteracting mechanism involved in switching integrins into an inactive conformation. Essentially none of this regulatory data has been evaluated in the context of CM or heart. Still, from a variety of investigations, there have been several ways proposed to inhibit integrin activation (Fig. 3b). These have been recently reviewed [59].
In brief, the most acceptable view includes acknowledgement that there are endogenous inhibitors or inhibitory pathways of β1-integrin activation. One such inhibitor is SHARPIN (SHANK-associated RH domain interacting protein) which is a component of the E3 ubiquitin ligase complex [60], while another suggests that tyrosine phosphorylation of β integrins may also serve as an inhibitory mechanism [61, 62]. SHARPIN inhibits the critical switching of β1 integrins from inactive to active conformations by directly binding to conserved cytoplasmic regions of integrin α subunits. In doing so, it inhibits recruitment of talin and kindlin to the integrin heterodimer. Integrin tyrosine phosphorylation leads to decrease integrin activation both by decreasing talin affinity for integrin, and also by increasing the affinity of competing proteins for integrin, which are themselves incapable of activating the integrin. For example, consider as we have been discussing above that integrin adhesiveness for the ECM is activated by talin via direct binding of its phosphotyrosine-binding-like F3 domain to the cytoplasmic tail of the β integrin subunit. Then, further realize that Docking protein 1 (Dok1) is a signaling protein with a PTB domain capable of binding to integrins, and that it does so in a similar binding region as does talin. Therefore, when Dok1 binding occurs, integrin tyrosine phosphorylation is enhanced, and talin affinity to integrin is decreased. Thus, Dok1 can act as a talin–integrin-binding competitor [61, 62]. Further, it has also been recognized that there are other talin-binding competitors, such as filamin [63], migfilin [64, 65], Integrin Cytoplasmic domain-Associated Protein-1 (ICAP-1), with some acting as inhibitors, and others even acting to “de-inhibit” the inhibitors. For instance, filamins may act as inhibitors of talin binding since the filamin-binding site on the β integrin tails is closely located to the talin-binding site. Thus, filamin and talin compete with each other for integrin binding [66]. In contrast, migfilin, a filamin-binding protein, and enriched at cell–cell and cell–ECM contact sites, can displace filamin from β1 and β3 integrins, thereby promoting integrin activation [64, 65]. Therefore, the balance between filamin and migfilin expressions in cells/tissues can influence the extent to which filamin inhibits integrin activity, thus, the filamin–migfilin interaction provides an additional regulatory layer for filamin-induced inhibition of integrin activation [67]. ICAP1 may also play a role in regulating integrin activation, and it has been well characterized both in vitro and in vivo [68, 69]. ICAP1 has been found to be a negative regulator of β1 integrin activity, although it does not bind β3 integrins. ICAP1 also competes for talin binding, at least as shown in in vitro studies, where it negatively regulates β integrin [70].
Little knowledge is available about the regulation of kindlin which could inhibit their ability to activate integrins. One known example is SMURF1, an E3 ubiquitin ligase that mediates protein degradation, which in turn inhibits integrin activation by controlling kindlin2 ubiquitination and degradation [71].
Summary and Outlook
In this review, we have summarized the roles of talin and kindlin, discussed their function as integrin-activating proteins, and provided a background on integrins as needed for this discussion. To the extent as possible, we emphasized work specifically in heart. As one example, in our studies where we ablated talin or kindlin expression specifically in cardiomyocytes, we found that the mice showed generally more severe cardiac abnormalities than integrin subunit knockout models, suggesting perhaps that talin and kindlin have both integrin-dependent and -independent functions, or that the half-lives of the integrin versus talin/kindlin proteins are variant. Further, in these models, integrin expression levels decreased dramatically as either talin or kindlin expression was reduced, indicating that these two integrinactivating proteins (talin and kindlin) are important for integrin synthesis or degradation in the cardiomyocyte. Further work is needed to understand this result.
Critically, it is important to realize that the majority of work performed in this field has been carried out using mostly cultured cells, but even considering a large number of works exploring integrin, talin, and kindlin functions in vivo, few studies have explored their functions in cardiac myocytes and heart. We posit that understanding the functions of these complex proteins has cell-type-specific effects that are quite distinct in a cell similar to cardiac myocyte that is generally agreed to be terminally differentiated, and of course, has the particularly unique property of having a continuously active contractile state. Although not emphasized extensively earlier in this paper, it has been accepted by most that regulation of integrin activation may also occur through mechanical event. A recent review has emphasized this concept [72]. Yet, as outlined above, the majority of this work is studied and conceptualized in cells that cycle, migrate, and might even frequently attach and disengage from matrix—for instance, leukocytes. In this context, these are the processes that one can more easily relate to integrin activation and inactivation. How these same activation and inactivation events play essential roles in the cardiac myocyte that is not cycling, not migrating, and has less functional need to disengage from binding to its extracellular matrix, are concepts necessary and ripe for future studies on integrin, talin, and kindlin functions in the cardiac myocyte.
Acknowledgements
This work was supported by grants from the National Institutes of Health and The Veterans Administration (Merit Award) to RSR.
Funding Some of the authors and components of this review were funded by grants from NIH/NHLBI (R01HL127806) and the Department of Veterans Affairs Merit Grant (BX003260).
Footnotes
Conflict of interest All authors declare that he/she has no conflict of interest.
Ethical Approval All applicable international, national, and/or institutional guidelines for the care and use of animals were followed for studies performed directly by the authors discussed in this study. This article does not contain any studies with human participants performed by any of the authors.
References
- 1.Israeli-Rosenberg S, Manso AM, Okada H, Ross RS (2014) Integrins and integrin-associated proteins in the cardiac myocyte. Circ Res 114:572–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ross RS (2002) The extracellular connections: the role of integrins in myocardial remodeling. J Card Fail 8:S326–331 [DOI] [PubMed] [Google Scholar]
- 3.Ross RS, Borg TK (2001) Integrins and the myocardium. Circ Res 88:1112–1119 [DOI] [PubMed] [Google Scholar]
- 4.Belkin AM, Retta SF, Pletjushkina OY, Balzac F, Silengo L, Fassler R, Koteliansky VE, Burridge K, Tarone G (1997) Muscle beta1D integrin reinforces the cytoskeleton-matrix link: modulation of integrin adhesive function by alternative splicing. J Cell Biol 139:1583–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pfaff M, Liu S, Erle DJ, Ginsberg MH (1998) Integrin beta cytoplasmic domains differentially bind to cytoskeletal proteins. J Biol Chem 273:6104–6109 [DOI] [PubMed] [Google Scholar]
- 6.Xiong JP, Stehle T, Diefenbach B, Zhang R, Dunker R, Scott DL, Joachimiak A, Goodman SL, Arnaout MA (2001) Crystal structure of the extracellular segment of integrin alpha Vbeta3. Science 294:339–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pesho MM, Bledzka K, Michalec L, Cierniewski CS, Plow EF (2006) The specificity and function of the metal-binding sites in the integrin beta3 A-domain. J Biol Chem 281:23034–23041 [DOI] [PubMed] [Google Scholar]
- 8.Giancotti FG, Ruoslahti E (1999) Integrin signaling. Science 285:1028–1032 [DOI] [PubMed] [Google Scholar]
- 9.Ginsberg MH (2014) Integrin activation. BMB Rep 47:655–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okada H, Lai NC, Kawaraguchi Y, Liao P, Copps J, Sugano Y, Okada-Maeda S, Banerjee I, Schilling JM, Gingras AR, Asfaw EK, Suarez J, Kang SM, Perkins GA, Au CG, Israeli-Rosenberg S, Manso AM, Liu Z, Milner DJ, Kaufman SJ, Patel HH, Roth DM, Hammond HK, Taylor SS, Dillmann WH, Gold-haber JI, Ross RS (2013) Integrins protect cardiomyocytes from ischemia/reperfusion injury. J Clin Invest 123:4294–4308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shai SY, Harpf AE, Babbitt CJ, Jordan MC, Fishbein MC, Chen J, Omura M, Leil TA, Becker KD, Jiang M, Smith DJ, Cherry SR, Loftus JC, Ross RS (2002) Cardiac myocyte-specific excision of the beta1 integrin gene results in myocardial fibrosis and cardiac failure. Circ Res 90:458–464 [DOI] [PubMed] [Google Scholar]
- 12.Kim C, Ye F, Ginsberg MH (2011) Regulation of integrin activation. Annu Rev Cell Dev Biol 27:321–345 [DOI] [PubMed] [Google Scholar]
- 13.Moser M, Legate KR, Zent R, Fassler R (2009) The tail of integrins, talin, and kindlins. Science 324:895–899 [DOI] [PubMed] [Google Scholar]
- 14.Arnaout MA, Goodman SL, Xiong JP (2007) Structure and mechanics of integrin-based cell adhesion. Curr Opin Cell Biol 19:495–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo BH, Springer TA (2006) Integrin structures and conformational signaling. Curr Opin Cell Biol 18:579–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Springer TA, Wang JH (2004) The three-dimensional structure of integrins and their ligands, and conformational regulation of cell adhesion. Adv Protein Chem 68:29–63 [DOI] [PubMed] [Google Scholar]
- 17.Hynes RO (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110:673–687 [DOI] [PubMed] [Google Scholar]
- 18.Shimaoka M, Takagi J, Springer TA (2002) Conformational regulation of integrin structure and function. Annu Rev Biophys Biomol Struct 31:485–516 [DOI] [PubMed] [Google Scholar]
- 19.Vinogradova O, Haas T, Plow EF, Qin J (2000) A structural basis for integrin activation by the cytoplasmic tail of the alpha IIb-subunit. Proc Natl Acad Sci U S A 97:1450–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klapholz B, Brown NH (2017) Talin—the master of integrin adhesions. J Cell Sci 130:2435–2446 [DOI] [PubMed] [Google Scholar]
- 21.Tanentzapf G, Brown NH (2006) An interaction between integrin and the talin FERM domain mediates integrin activation but not linkage to the cytoskeleton. Nat Cell Biol 8:601–606 [DOI] [PubMed] [Google Scholar]
- 22.Calderwood DA, Yan B, de Pereda JM, Alvarez BG, Fujioka Y, Liddington RC, Ginsberg MH (2002) The phosphotyrosine binding-like domain of talin activates integrins. J Biol Chem 277:21749–21758 [DOI] [PubMed] [Google Scholar]
- 23.Li H, Deng Y, Sun K, Yang H, Liu J, Wang M, Zhang Z, Lin J, Wu C, Wei Z, Yu C (2017) Structural basis of kindlin-mediated integrin recognition and activation. Proc Natl Acad Sci USA 114:9349–9354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ye F, Petrich BG, Anekal P, Lefort CT, Kasirer-Friede A, Shattil SJ, Ruppert R, Moser M, Fassler R, Ginsberg MH (2013) The mechanism of kindlin-mediated activation of integrin alphaIIbbeta3. Curr Biol 23:2288–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ussar S, Wang HV, Linder S, Fassler R, Moser M (2006) The kindlins: subcellular localization and expression during murine development. Exp Cell Res 312:3142–3151 [DOI] [PubMed] [Google Scholar]
- 26.Senetar MA, Moncman CL, McCann RO (2007) Talin2 is induced during striated muscle differentiation and is targeted to stable adhesion complexes in mature muscle. Cell Motil Cytoskelet 64:157–173 [DOI] [PubMed] [Google Scholar]
- 27.Monkley SJ, Pritchard CA, Critchley DR (2001) Analysis of the mammalian talin2 gene TLN2. Biochem Biophys Res Commun 286:880–885 [DOI] [PubMed] [Google Scholar]
- 28.Roberts GC, Critchley DR (2009) Structural and biophysical properties of the integrin-associated cytoskeletal protein talin. Biophys Rev 1:61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calderwood DA, Ginsberg MH (2003) Talin forges the links between integrins and actin. Nat Cell Biol 5:694–697 [DOI] [PubMed] [Google Scholar]
- 30.Goult BT, Bouaouina M, Elliott PR, Bate N, Patel B, Gingras AR, Grossmann JG, Roberts GC, Calderwood DA, Critchley DR, Barsukov IL (2010) Structure of a double ubiquitin-like domain in the talin head: a role in integrin activation. EMBO J 29:1069–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X, Jiang G, Cai Y, Monkley SJ, Critchley DR, Sheetz MP (2008) Talin depletion reveals independence of initial cell spreading from integrin activation and traction. Nat Cell Biol 10:1062–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, de Pereda JM, Ginsberg MH, Calderwood DA (2003) Talin binding to integrin beta tails: a final common step in integrin activation. Science 302:103–106 [DOI] [PubMed] [Google Scholar]
- 33.Horwitz A, Duggan K, Buck C, Beckerle MC, Burridge K (1986) Interaction of plasma membrane fibronectin receptor with talin–a transmembrane linkage. Nature 320:531–533 [DOI] [PubMed] [Google Scholar]
- 34.Monkley SJ, Zhou XH, Kinston SJ, Giblett SM, Hemmings L, Priddle H, Brown JE, Pritchard CA, Critchley DR, Fassler R (2000) Disruption of the talin gene arrests mouse development at the gastrulation stage. Dev Dyn 219:560–574 [DOI] [PubMed] [Google Scholar]
- 35.Conti FJ, Monkley SJ, Wood MR, Critchley DR, Muller U (2009) Talin 1 and 2 are required for myoblast fusion, sarcomere assembly and the maintenance of myotendinous junctions. Development 136:3597–3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arimura T, Bos JM, Sato A, Kubo T, Okamoto H, Nishi H, Harada H, Koga Y, Moulik M, Doi YL, Towbin JA, Ackerman MJ, Kimura A (2009) Cardiac ankyrin repeat protein gene (ANKRD1) mutations in hypertrophic cardiomyopathy. J Am Coll Cardiol 54: 334–342 [DOI] [PubMed] [Google Scholar]
- 37.Abel ED, Kaulbach HC, Tian R, Hopkins JC, Duffy J, Doetschman T, Minnemann T, Boers ME, Hadro E, Oberste-Berghaus C, Quist W, Lowell BB, Ingwall JS, Kahn BB (1999) Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J Clin Invest 104:1703–1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manso AM, Okada H, Sakamoto FM, Moreno E, Monkley SJ, Li R, Critchley DR, Ross RS (2017) Loss of mouse cardiomyocyte talin-1 and talin-2 leads to beta-1 integrin reduction, costameric instability, and dilated cardiomyopathy. Proc Natl Acad Sci USA 114:E6250–E6259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manso AM, Li R, Monkley SJ, Cruz NM, Ong S, Lao DH, Koshman YE, Gu Y, Peterson KL, Chen J, Abel ED, Samarel AM, Critchley DR, Ross RS (2013) Talin1 has unique expression versus talin 2 in the heart and modifies the hypertrophic response to pressure overload. J Biol Chem 288:4252–4264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD (2001) Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res 89:20–25 [DOI] [PubMed] [Google Scholar]
- 41.Li R, Wu Y, Manso AM, Gu Y, Liao P, Israeli S, Yajima T, Nguyen U, Huang MS, Dalton ND, Peterson KL, Ross RS (2012) beta1 integrin gene excision in the adult murine cardiac myocyte causes defective mechanical and signaling responses. Am J Pathol 180:952–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hakonardottir GK, Lopez-Ceballos P, Herrera-Reyes AD, Das R, Coombs D, Tanentzapf G (2015) In vivo quantitative analysis of Talin turnover in response to force. Mol Biol Cell 26:4149–4162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiao K, Kulessa H, Tompkins K, Zhou Y, Batts L, Baldwin HS, Hogan BL (2003) An essential role of Bmp4 in the atrioventricular septation of the mouse heart. Genes Dev 17:2362–2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheah M, Andrews MR (2018) Integrin activation: implications for axon regeneration. Cells 7:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Larjava H, Plow EF, Wu C (2008) Kindlins: essential regulators of integrin signalling and cell-matrix adhesion. EMBO Rep 9:1203–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bledzka K, Liu J, Xu Z, Perera HD, Yadav SP, Bialkowska K, Qin J, Ma YQ, Plow EF (2012) Spatial coordination of kindlin-2 with talin head domain in interaction with integrin beta cytoplasmic tails. J Biol Chem 287:24585–24594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Campbell ID, Humphries MJ (2011) Integrin structure, activation, and interactions. Cold Spring Harb Perspect Biol 3:a004994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dong JM, Tay FP, Swa HL, Gunaratne J, Leung T, Burke B, Manser E (2016) Proximity biotinylation provides insight into the molecular composition of focal adhesions at the nanometer scale. Sci Signal 9:rs4 [DOI] [PubMed] [Google Scholar]
- 49.Dowling JJ, Gibbs E, Russell M, Goldman D, Minarcik J, Golden JA, Feldman EL (2008) Kindlin-2 is an essential component of intercalated discs and is required for vertebrate cardiac structure and function. Circ Res 102:423–431 [DOI] [PubMed] [Google Scholar]
- 50.Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD (2006) Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res 98:837–845 [DOI] [PubMed] [Google Scholar]
- 51.Zhang Z, Mu Y, Veevers J, Peter AK, Manso AM, Bradford WH, Dalton ND, Peterson KL, Knowlton KU, Ross RS, Zhou X, Chen J (2016) Postnatal loss of kindlin-2 leads to progressive heart failure. Circ Heart Fail 9:e003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McHugh BJ, Buttery R, Lad Y, Banks S, Haslett C, Sethi T (2010) Integrin activation by Fam38A uses a novel mechanism of R-Ras targeting to the endoplasmic reticulum. J Cell Sci 123:51–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McHugh BJ, Murdoch A, Haslett C, Sethi T (2012) Loss of the integrin-activating transmembrane protein Fam38A (Piezo1) promotes a switch to a reduced integrin-dependent mode of cell migration. PLoS ONE 7:e40346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moore DT, Nygren P, Jo H, Boesze-Battaglia K, Bennett JS, DeGrado WF (2012) Affinity of talin-1 for the beta3-integrin cytosolic domain is modulated by its phospholipid bilayer environment. Proc Natl Acad Sci USA 109:793–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ye X, McLean MA, Sligar SG (2016) Phosphatidylinositol 4,5-bisphosphate modulates the affinity of Talin-1 for phospholipid bilayers and activates its autoinhibited form. Biochemistry 55:5038–5048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lagarrigue F, Kim C, Ginsberg MH (2016) The Rap1-RIAM-talin axis of integrin activation and blood cell function. Blood 128:479–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee HS, Lim CJ, Puzon-McLaughlin W, Shattil SJ, Ginsberg MH (2009) RIAM activates integrins by linking talin to ras GTPase membrane-targeting sequences. J Biol Chem 284:5119–5127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han J, Lim CJ, Watanabe N, Soriani A, Ratnikov B, Calderwood DA, Puzon-McLaughlin W, Lafuente EM, Boussiotis VA, Shattil SJ, Ginsberg MH (2006) Reconstructing and deconstructing agonist-induced activation of integrin alphaIIbbeta3. Curr Biol 16:1796–1806 [DOI] [PubMed] [Google Scholar]
- 59.Nolz JC, Medeiros RB, Mitchell JS, Zhu P, Freedman BD, Shimizu Y, Billadeau DD (2007) WAVE2 regulates high-affinity integrin binding by recruiting vinculin and talin to the immunological synapse. Mol Cell Biol 27:5986–6000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rantala JK, Pouwels J, Pellinen T, Veltel S, Laasola P, Mattila E, Potter CS, Duffy T, Sundberg JP, Kallioniemi O, Askari JA, Humphries MJ, Parsons M, Salmi M, Ivaska J (2011) SHARPIN is an endogenous inhibitor of beta1-integrin activation. Nat Cell Biol 13:1315–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anthis NJ, Haling JR, Oxley CL, Memo M, Wegener KL, Lim CJ, Ginsberg MH, Campbell ID (2009) Beta integrin tyrosine phosphorylation is a conserved mechanism for regulating talin-induced integrin activation. J Biol Chem 284:36700–36710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oxley CL, Anthis NJ, Lowe ED, Vakonakis I, Campbell ID, Wegener KL (2008) An integrin phosphorylation switch: the effect of beta3 integrin tail phosphorylation on Dok1 and talin binding. J Biol Chem 283:5420–5426 [DOI] [PubMed] [Google Scholar]
- 63.Calderwood DA, Huttenlocher A, Kiosses WB, Rose DM, Woodside DG, Schwartz MA, Ginsberg MH (2001) Increased filamin binding to beta-integrin cytoplasmic domains inhibits cell migration. Nat Cell Biol 3:1060–1068 [DOI] [PubMed] [Google Scholar]
- 64.Ithychanda SS, Das M, Ma YQ, Ding K, Wang X, Gupta S, Wu C, Plow EF, Qin J (2009) Migfilin, a molecular switch in regulation of integrin activation. J Biol Chem 284:4713–4722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lad Y, Jiang P, Ruskamo S, Harburger DS, Ylanne J, Campbell ID, Calderwood DA (2008) Structural basis of the migfilin-filamin interaction and competition with integrin beta tails. J Biol Chem 283:35154–35163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kiema T, Lad Y, Jiang P, Oxley CL, Baldassarre M, Wegener KL, Campbell ID, Ylanne J, Calderwood DA (2006) The molecular basis of filamin binding to integrins and competition with talin. Mol Cell 21:337–347 [DOI] [PubMed] [Google Scholar]
- 67.Pouwels J, Nevo J, Pellinen T, Ylanne J, Ivaska J (2012) Negative regulators of integrin activity. J Cell Sci 125:3271–3280 [DOI] [PubMed] [Google Scholar]
- 68.Draheim KM, Huet-Calderwood C, Simon B, Calderwood DA (2017) Nuclear localization of Integrin Cytoplasmic Domain-Associated Protein-1 (ICAP1) influences beta1 integrin activation and recruits Krev/Interaction Trapped-1 (KRIT1) to the nucleus. J Biol Chem 292:1884–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bouvard D, Aszodi A, Kostka G, Block MR, Albiges-Rizo C, Fassler R (2007) Defective osteoblast function in ICAP-1-deficient mice. Development 134:2615–2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fournier HN, Dupe-Manet S, Bouvard D, Luton F, Degani S, Block MR, Retta SF, Albiges-Rizo C (2005) Nuclear translocation of integrin cytoplasmic domain-associated protein 1 stimulates cellular proliferation. Mol Biol Cell 16:1859–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wei X, Wang X, Zhan J, Chen Y, Fang W, Zhang L, Zhang H (2017) Smurf1 inhibits integrin activation by controlling Kindlin-2 ubiquitination and degradation. J Cell Biol 216:1455–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sun Z, Costell M, Fassler R (2019) Integrin activation by talin, kindlin and mechanical forces. Nat Cell Biol 21:25–31 [DOI] [PubMed] [Google Scholar]