

Abstract
Aim
Chronic intermittent hypoxia (CIH) induces systemic (hypertension) and central (mitochondrial dysfunction underlying cognitive deficits). We hypothesized that agonists of oestradiol receptors (ER) α and β prevent CIH-induced hypertension and brain mitochondrial dysfunction.
Methods
Ovariectomized female rats were implanted with osmotic pumps delivering vehicle (Veh), the ERα agonist propylpyraoletriol (PPT - 30μg/kg/day) or the ERβ agonist diarylpropionitril (DPN - 100μg/kg/day). Animals were exposed to CIH (21–10% FIO2 – 10 cycles/hour – 8 hours/day – 7 days) or normoxia. Arterial blood pressure was measured after CIH or normoxia exposures. Mitochondrial respiration and H2O2 production were measured in brain cortex with high-resolution respirometry, as well as activity of complex I and IV of the electron transport chain, citrate synthase, pyruvate and lactate dehydrogenase (PDH and LDH).
Results
PPT but not DPN prevented the rise of arterial pressure induced by CIH. CIH exposures decreased O2 consumption, complex I activity, and increased H2O2 production. CIH had no effect on citrate synthase activity, but decreased PDH activity and increased LDH activity indicating higher anaerobic glycolysis. PPT and DPN treatments prevented all these alterations.
Conclusions
We conclude that in OVX female rats, the ERα agonist prevents from CIH-induced hypertension while both ERα and ERβ agonists prevent the brain mitochondrial dysfunction and metabolic switch induced by CIH. These findings may have implications for menopausal women suffering of sleep apnoea regarding hormonal therapy.
Keywords: sleep apnoea, hypertension, mitochondrial dysfunction, metabolic switch, selective oestradiol receptor modulators, hormonal therapy
Introduction
Chronic intermittent hypoxia (CIH) is regularly used in animals to reproduce the recurring drops in arterial oxygen saturation that characterize respiratory patterns of sleep apnoea (SA) patients.1–3 CIH induces an elevation of arterial blood pressure that depends on exaggerated activity of peripheral chemoreceptors, leading to high activity of the sympathetic nerves and vascular dysfunction.4,5 In the central nervous system, CIH leads to increased levels of oxidative stress and neuronal apoptosis likely underlying cognitive impairments,6 and support for such findings can also be inferred by recent studies in humans showing reduced cortical thickness in both adult7 and pediatric8 SA patients. It has been postulated that excessive production of reactive oxygen species (ROS) either by cytosolic enzymes (NADPH and xanthine oxidases) or at the mitochondrial level is one of the major mechanisms underlying the systemic and central morbidities observed in SA patients and in rodents exposed to CIH.3,9–11
The prevalence and severity of sleep apnoea (SA) are lower in women than in men but increase after menopause in women.12 The circulating levels of oestradiol (E2) are negatively correlated with the frequency of SA in women,13 and hormone replacement therapy reduces the frequency of SA after adjustment for other known risk factors (age, body mass index and neck circumference).14 Furthermore, the cardiovascular consequences of SA are reduced in women compared to men,15,16 suggesting that ovarian hormones may play a protective role. In line with this conceptual framework, we have previously reported that E2 treatment in ovariectomized female rats exposed to CIH for 7 days prevents the elevation of arterial blood pressure and activation of the arterial peripheral chemoreflex. E2 treatment also reduced the activity of NADPH oxidase, and improved cytosolic and mitochondrial antioxidant enzymatic activities under CIH exposure in the brain cortex.2 These results are consistent with the antioxidant effects of E2.17,18
E2 receptors α and β (ERα and ERβ) are ligand-activated transcription factors able to reduce mitochondrial ROS production and increase oxidative phosphorylation,19–21 leading to substantial interest in the potential clinical use of selective ERα or ERβ agonists in post-menopausal women22 or in the context of neurodegenerative disorders.23,24 ERα and ERβ are expressed in the central and peripheral nervous system. In the brain cortex, ERα mediates neuroprotection during hypoxic/ischemic insults.25 ERα and ERβ reduce arterial blood pressure through the sympathetic nuclei in the brainstem and hypothalamus.26 In the model of angiotensin II-induced hypertension, ERα in the suprafornical organ controls arterial blood pressure.27 We have previously reported that mRNA encoding ERα and ERβ are present in peripheral chemoreceptors28 while it is largely acknowledged that peripheral chemoreceptors are necessary for the elevation of arterial blood pressure during CIH exposure.29 However, it remains unclear if agonists of ERα or ERβ could abrogate the increase in arterial blood pressure induced by CIH exposure. On the other hand, both ERα and ERβ are able to increase mitochondrial O2 consumption21,30,31 and estradiol prevents the reduction of mitochondrial antioxidant enzymes activity in the brain cortex of female rats exposed to CIH.2
In the present study, we used ovariectomized female rats exposed to CIH to assess the roles of ERα and Erβ on arterial blood pressure, mitochondrial O2 consumption and ROS production on brain cortical samples.
Under the hypothetical construct that CIH exposures reduce mitochondrial respiration, this effect could be associated with a metabolic “switch” in the brain, that favours glycolytic activity and facilitates ATP production.32,33 Accordingly, we also measured the activities of pyruvate and lactate dehydrogenase, the enzymes that control the rate of the aerobic and anaerobic glycolysis, respectively.
Results
PPT, but not DPN, prevents the elevation of arterial blood pressure induced by CIH.
Exposure to CIH increased the mean, systolic, and diastolic blood pressures (Table 1). This effect was prevented by treatment with the ERα agonist (PPT), but not with the ERβ agonist (DPN). In all groups, body weight increased during exposure to CIH or room air, without significant effects of PPT or DPN treatments.
Table 1:
Body weight before and after exposure to CIH, and mean, systolic, and diastolic arterial pressured after exposure to CIH in ovariectomized female rats treated with vehicle (Veh), the ERα (PPT), or the ERβ (DPN) agonist and exposed to room air (AIR) or chronic intermittent hypoxia (CIH). All data are mean ± SD.
| Group |
Veh-AIR | Veh-CIH | PPT-CIH | DPN-CIH |
|---|---|---|---|---|
|
Body weight (g) Before CIH After CIH |
304 ± 16 323 ± 15 |
302 ± 19 314 ± 17 |
307 ± 25 321 ± 26 |
317 ± 32 330 ± 31 |
| MAP (mmHg) | 87.6 ± 2.1 | 98.8 ± 3.6*** | 89.6 ± 1.9†† | 98.2 ± 5.8** |
| SAP (mmHg) | 110.4 ± 1.7 | 122 ± 2.9*** | 109.1 ± 2.3††† | 123.7 ± 7.2*** |
| DAP (mmHg) | 76.0 ± 2.5 | 84.7 ± 2.8** | 77.8 ± 4.6† | 86.5 ± 5.7** |
p<0.01
p<0.001 vs Veh-Air.
p<0.05
p<0.01
p<0.001 vs Veh-CIH.
PPT and DPN prevent the decrease of mitochondrial O2 consumption induced by CIH.
Typical recordings of O2 concentration within the Oroboros chamber are presented in Figure 1. Compared to animals exposed to room air (Veh AIR), CIH exposures reduced O2 consumption when mitochondrial respiration was induced by addition of the substrates of complex I (pyruvate + malate) and ADP (state 3). This effect was abrogated by PPT (Figure 1) and DPN (not shown).
Figure 1:
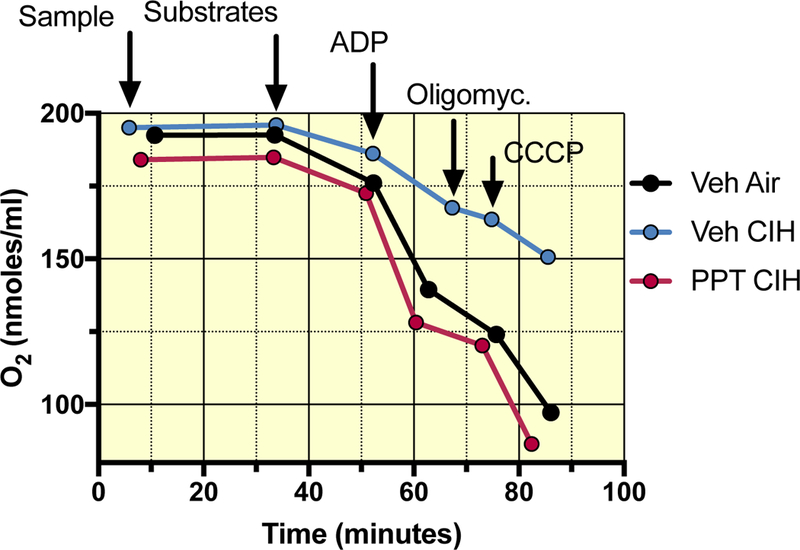
Typical recordings of mitochondrial respiration measured in permeabilized brain cortex samples from ovariectomized female rats treated with vehicle, and exposed to either room air (Veh AIR) or CIH (Veh CIH), and one ovariectomized female rat treated with the ERα agonist and exposed to CIH (PPT CIH). X-axis reflects the time from the start of the experiment, Y-axis depicts the O2 concentration in the recording chamber. O2 consumption corresponds to the slope of each step of the experiment. The different steps are indicated by the arrows showing the sequential addition of the sample, the substrates (respiratory state 2), ADP (state 3), oligomycin (state 4) and CCCP. Note the effect of CIH exposure on O2 consumption and the protective effect of the ERα agonist (PPT). The ERβ agonist (DPN – not shown) had a similar protective effect.
Figure 2A shows that CIH exposures reduced NADH-linked mitochondrial respiration by about 35%, and that the ERα (PPT) and the ERβ (DPN) agonists prevented this effect. In contrast, FADH2-linked respiration was unaffected by CIH exposures or by PPT or DPN treatment (Figure 2B). When mitochondrial respiration was induced simultaneously by the substrates of complexes I and II (NADH+FADH2-linked respiration), O2 consumption was reduced almost by 50% after CIH exposures, and this effect was prevented by PPT and DPN (Figure 2C). However, in female rats treated with PPT and exposed to CIH, NADH and NADH+FADH2 linked respiration rates were higher than in corresponding controls (by about 20% and 45 % respectively), while such effects were not apparent with DPN, suggesting that agonists of ERα and ERβ act through distinct mechanisms under CIH exposures.
Figure 2:
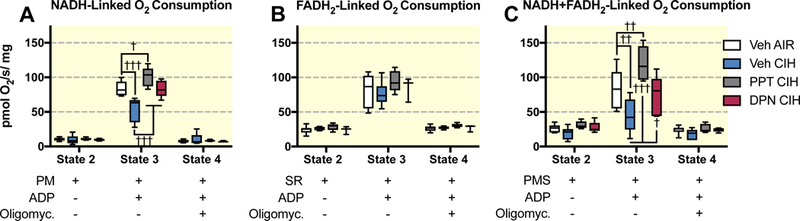
Mitochondrial oxygen consumption during respiratory states 2, 3 and 4 measured on permeabilized brain cortex samples in ovariectomized female rats treated with vehicle (Veh), the ERα (PPT), or the ERβ (DPN) agonist and exposed to room air (AIR) or chronic intermittent hypoxia (CIH). A: NADH-linked mitochondrial respiration (complex I – activated by pyruvate and malate - PM). B: FADH2-linked mitochondrial respiration (complex II – activated by succinate and complex I blocked by rotenone - SR). D: NADH+FADH2-linked mitochondrial respiration (complexes I + II – activated by pyruvate + malate + succinate - PMS). All data are box and whiskers (median, 25th and 75th percentiles, min and max values). †, ††, ††† : p<0.01, p<0.001, and p 0.0001 vs Veh AIR.
The respiratory control ratio (RCR - obtained by dividing O2 consumption rates in state 3 by state 4) indicates the capacity of mitochondria to respond to increasing metabolic demands, whereby a high RCR would indicate a high respiratory and ATP turnover capacities 34. CIH exposures reduced the RCR for NADH (from 7.4 ± 1.8 to 4.6 ± 2.0 – P=0.0001 – Figure 3) and NADH+FADH2-linked respiration (from 3.3 ± 0.8 to 2.0 ± 0.6 – P=0.048). In female rats treated with PPT and exposed to CIH, the RCR for the NADH-linked respiration (8.8 ± 1.0) was higher than in control female rats (7.4 ± 1.8 – P=0.04), but this effect was not observed for DPN.
Figure 3:
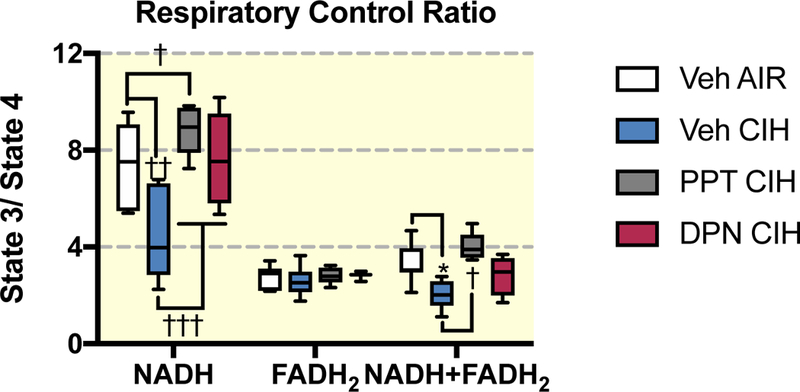
Respiratory control ratio (RCR) calculated as the ratio of oxygen consumption in state 3 to state 4 with mitochondrial respiration activated by pyruvate + malate (PM), succinate + rotenone (SR) and pyruvate + malate + succinate (PMS) in ovariectomized female rats treated with vehicle (Veh) or the agonist of E2 receptor α (PPT) and E2 receptor β (DPN) and exposed to room air (AIR) or chronic intermittent hypoxia (CIH). All data are box and whiskers (median, 25th and 75th percentiles, min and max values). *, ††: p<0.05, and p<0.001 vs Veh AIR. vs Veh AIR.
Citrate synthase activity, the key enzyme of the citric acid cycle which could also be used as a marker of mitochondrial content 35,36, and the maximum activity of complex IV were similar across groups (Figure 4 A-B). The specific activity of complex I was reduced 3-fold by CIH exposures (P=0.003), and the effect was prevented by PPT and DPN (Figure 4C).
Figure 4:
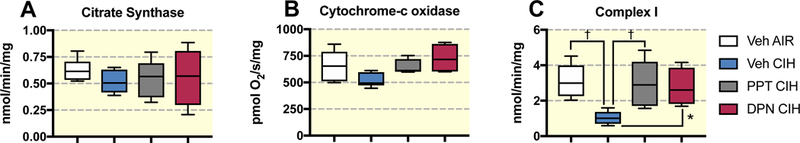
Activity of citrate synthase (A), cytochrome-c oxidase (complex IV - B), and complex I (C) measured on brain cortex samples in ovariectomized female rats treated with vehicle (Veh), the ERα (PPT), or the ERβ (DPN) agonist and exposed to room air (AIR) or chronic intermittent hypoxia (CIH). All data are box and whiskers (median, 25th and 75th percentiles, min and max values) † : p<0.01, vs Veh AIR.
PPT and DPN prevent the elevation of mitochondrial H2O2 production induced by CIH.
When mitochondrial O2 consumption is low (respiratory states 2 and 4) it is more likely that electrons will react with molecular oxygen and form superoxide molecules (∙O2-) within the complexes I and III of the electron transport chain 37. ∙O2- is almost immediately transformed to H2O2 by the mitochondrial superoxide dismutase. We measured the production of mitochondrial H2O2 release during the states 2, 3 and 4 of the NADH+FADH2-linked respiration. In animals exposed to CIH, H2O2 production in respiratory state 3 was 2-fold higher than in controls, and this CIH effect was prevented by PPT or DPN treatments (Figure 5A). An index of the electrons leaking from the electron transport chain to react with O2 can be obtained by the ratio of H2O2 produced to O2 consumed for each respiratory state. This ratio is reported in Figure 5B, showing that for all respiratory states, it was higher in animals exposed to CIH compared to all other exposure or treatment groups.
Figure 5:
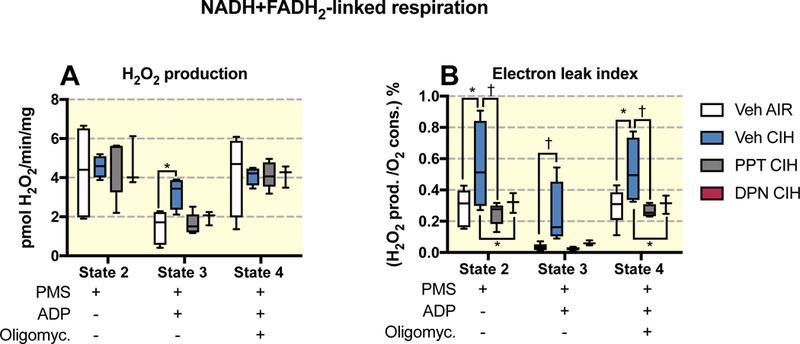
H2O2 production and electron leak index during respiratory states 2, 3 and 4 measured using fluorometric detection on brain cortex samples and NADH+FADH2-linked mitochondrial respiration (with pyruvate + malate + succinate as substrates - PMS) in ovariectomized female rats treated with vehicle (Veh) or the agonist of E2 receptor α (PPT) and E2 receptor β (DPN) and exposed to room air (AIR) or chronic intermittent hypoxia (CIH). All data are box and whiskers (median, 25th and 75th percentiles, min and max values). *, † : p<0.05, and p<0.01 vs Veh AIR.
PPT and DPN prevent the metabolic switch induced by CIH
Reduced mitochondrial respiration could compromise neuronal integrity when energy requirements are elevated. Such limited respiration could however be compensated by increased glycolytic capacity. The maximum activity of lactate dehydrogenase in brain cortex samples was 2-fold higher in rats exposed to CIH (P=0.008 - Figure 6A), this effect was prevented by treatment with PPT, but not by DPN. In parallel, the maximum activity of pyruvate dehydrogenase, which controls the entry of pyruvate into the citric acid cycle, was reduced by CIH exposures (by ~40%). PPT and DPN treatments prevented this effect, and induced an elevation of PDH activity above the normoxic control animals (Figure 6B). Because both lactate and pyruvate dehydrogenase activities use pyruvate as a substrate, we calculated the ratio of LDH/PDH as an index of anaerobic to aerobic metabolism. This ratio was increased by CIH exposures (P=0.0001), and this change was prevented by PPT and DPN treatments (Figure 6C).
Figure 6:
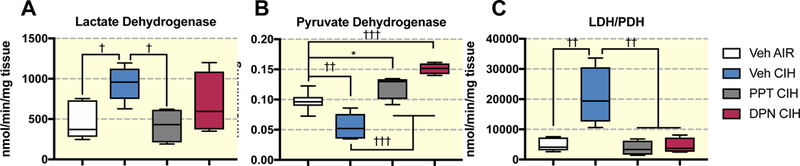
Activity of pyruvate dehydrogenase (A), lactate dehydrogenase (B), and activity ratio of lactate dehydrogenase to pyruvate dehydrogenase (LDH/PDH - C) on brain cortex samples in ovariectomized female rats treated with vehicle (Veh), the ERα (PPT), or the ERβ (DPN) agonist and exposed to room air (AIR) or chronic intermittent hypoxia (CIH). All data are box and whiskers (median, 25th and 75th percentiles, min and max values) *, † , ††, ††† : p<0.05, p<0.01, p<0.001, and p<0.0001 vs Veh AIR.
Discussion
This study shows that exposures of ovariectomized female rats to CIH reduces NADH-linked mitochondrial respiration in brain cortex tissues, alters the activity of the complex I of the electron transport chain, and increases mitochondrial ROS production. The reduced mitochondrial respiration was accompanied by a lower activity of pyruvate dehydrogenase and higher activity of lactate dehydrogenase, likely indicating that the brain cortex adapts to the reduced mitochondrial respiration by favouring anaerobic glycolysis. Treatment with specific agonists of ERα (PPT) or ERβ (DPN) during exposure to CIH prevented these changes, indicating that these two receptors can be targeted to attenuate the effects of CIH on mitochondrial functions in the brain. However, only the ERα agonist prevented the increase of arterial blood pressure during exposure to CIH. These results are in line with our recent study showing that E2 treatment prevents oxidative stress in the brain cortex, adrenal medulla,2 and in the thoracic aorta38 of ovariectomized female rats exposed to a similar pattern of CIH.
PPT, but not DPN prevent the elevation of arterial blood pressure induced by CIH
Our results indicate that only the ERα agonist prevents the elevation of arterial blood pressure in female rats exposed to CIH. Our experiments were not designed to elucidate at which anatomical level this effect occurs, but it is worth mentioning that ERα is expressed in the level of carotid bodies,28 within the central structures controlling sympathetic nerve activities,26 and in blood vessels.39 Thus, the effects of PPT within these different substrates may underlie the observed anti-hypertensive effects. Of note, we showed previously that E2 prevents the elevation of arterial chemoreflex during exposures to CIH in female rats,2 such that an effect of PPT on peripheral chemoreceptors might be expected. It is also relevant to mention that high blood pressure can compromise the cerebral microcirculation, increasing arterial walls and reducing arterial lumen, and high blood pressure also alters metabolism in the brain cortex.40,41 In hypertensive patients, a reduced cortical thickness has been reported,40 a clear sign of cortical damage probably linked to reduced blood perfusion. In that regard, because our results indicate that only PPT reduces arterial blood pressure, while both PPT and DPN prevent the mitochondrial dysfunction and metabolic switch in the brain cortex, we are confident that the central effect of CIH and ERs agonists is not the result of the induction of (or protection against) high blood pressure.
PPT and DPN prevent the mitochondrial dysfunction induced by CIH
We have previously shown in female rats that CIH exposures enhance oxidative stress damage and reduce mitochondrial antioxidant defences in brain cortex.2 The present results expand on these observations, and show that CIH increases mitochondrial ROS production, the latter being associated with reduced mitochondrial respiration and activity of complex I of the electron transport chain. It has been previously demonstrated that mitochondria are the major source of ROS production during CIH exposures in cultured mouse brain cortical neurons.10 Additional studies have reported that CIH exposures reduce the activity of the complex I of the electron transport chain in peripheral chemoreceptors in adult male rats, and also reduce aconitase activity, a marker of mitochondrial ROS production.42 Similar findings have been described in PC12 cells exposed to CIH.43 Taken together, these results indicate that CIH exposures impose a profound effect on mitochondrial function in both the central and peripheral nervous systems, likely contributing to the enhanced oxidative stress reported among SA patients.
It is now well established that E2 acts directly on isolated mitochondria in vitro, and at normal intracellular concentrations (in the nM range) E2 activation prevents the formation of ROS by mitochondria, and leads to increased mitochondrial membrane potentials.30 In brain endothelial cells, ERα increases the expression of cytochrome c, a protein that transfers electron between complexes III and IV of the electron transport chain.31 On the other hand, in ovariectomized female rats, DPN (but not PPT) increases the expression of cytochrome oxidase subunit I, encoded by mitochondrial DNA,21 and both PPT and DPN increase the activity of complex IV of the electron transport chain on isolated whole brain mitochondria. Furthermore, PPT and DPN enhance the transcription of several other mitochondrial proteins coded by the nuclear DNA, including the cytochrome oxidase subunit IV, ATP synthase F1 subunit α, and mitochondrial superoxide dismutase.21
In contradistinction with these previous findings, current results show that under exposure to CIH, PPT and DPN do not increase the activity of complex IV, but rather prevent the reduction of complex I activity. We have recently reported that in the thoracic aorta of ovariectomized female rats the effects of E2 on pro- and antioxidant enzyme activities are different under normoxic and CIH conditions.38 Therefore, it is possible that the discrepancy observed between the present results and those reported by Irwin et al.21 may be accounted for the CIH exposures in our study.
PPT and DPN prevent the metabolic switch induced by CIH
In control ovariectomized female rats, exposure to CIH induces 4-fold increases in the LDH/PDH activity ratio, indicating enhancements in anaerobic glycolysis. It is noteworthy that glycolysis is substantially less efficient than oxidative phosphorylation to generate ATP, but since glycolysis is much faster, it remains a viable option to sustain cellular survival when mitochondrial respiration is compromised.44 CIH increases the expression of HIF-1α in the brain cortex of adult mice45 and the expression levels of LDH and PDH are increased and decreased by HIF-1α, respectively.46 Under CIH exposure, HIF-1α expression is increased by the enhanced ROS production generated by xanthine and NADPH oxidases.47,48 We previously reported that E2 supplementation reduces the activity of these pro-oxidant enzymes in the brain cortex during CIH exposures.2 Because both PPT and DPN prevent the elevation of the LDH/PDH ratio induced by CIH, it is likely that ERα and ERβ contribute to reduce the up-regulation of HIF-1α by preventing ROS generation. In line with this assumption, ERα and ERβ have been shown to reduce HIF-1α expression in cell cultures.49,50
An alternative hypothesis linking mitochondrial dysfunction to glycolysis proposes that reduced oxidative phosphorylation will increase the mitochondrial NADH/NAD+ ratio, which then reduces PDH activity. On the other hand, anaerobic glycolysis is activated by a high NADH/NAD+ ratio, with LDH consuming the NADH to produce NAD+, contributing to restore the cellular redox state.51 This process could therefore contribute to the high LDH/PDH ratio observed after CIH exposures. In this conceptual framework, ER agonists could restore metabolic activity in the brain cortex simply by preventing the mitochondrial dysfunction induced by CIH. Other mechanisms linking estradiol receptors, CIH and mitochondrial functions in the brain might depend on molecular metabolic sensors such as the AMPK-mTOR pathway (which is responsive to ATP/ADP ratio), the sirtuin family pathway (responsive to NAD+/NADH ratio) both of which are modulated by CIH, estradiol, or brain oxygen levels.52–55 Finally, it is worth mentioning that complex I of the electron transport chain can switch between an activated form and a deactivated form, and that hypoxia or hypoxic/ischemia “deactivates” complex I within minutes, but the effects of intermittent hypoxia, or estradiol receptors on this process are so far unknown.56,57
Conclusion & Perspectives
We conclude that the ERα agonist prevents the hypertension induced by CIH exposures in ovariectomized female rats. Constrastingly the ERα and ERβ are both efficient to avoid the reduced rates of mitochondrial oxygen consumption, lower activity of complex I, enhanced production of ROS and electron leaks, and enhanced glycolysis induced by CIH exposures. These findings are congruent with data showing estrogenic effects on mitochondrial respiration and ROS production. Several mechanisms might underlie these protective effects of the ERs agonists against the mitochondrial dysfunction induced by CIH, and future mechanistic experiments might help discriminating which processes are involved. Finally, it is worth mentioning that we used ovariectomized female rats as a model of hormonal depletion induced by menopause. Given the close associations between menopause, circulating E2 levels, and the occurrence of sleep apnoea in women,12–14 we propose that the present study may provide the rationale for designing specific therapeutic interventions based on selective ER agonists to protect cardiovascular and brain mitochondrial functions among menopausal women suffering from sleep apnoea.
Material and methods
Animals
The protocol was approved by the committee on the protection of animal of the CHUQ Research Center (#: 2014–156) in accordance with the Canadian Council on Animal Care. We used 24 female Sprague-Dawley rats (body weight 220–250g when purchased) from Charles-Rivers Laboratories (Saint-Constant, QC, Canada). All animals had access to food and water ad-libitum and were maintained on a 12:12h light-dark cycle.
General experimental design and drugs
Two weeks after arrival to the vivarial facility, animals were anesthetized under isoflurane (4% induction then 2%) for ovariectomy through bilateral flank incisions. All animals received pre- and post-operative analgesics for 48 hours (bupevacaïne, lidocaine, subcutaneous injection, respectively 3.5 and 7 mg/kg in 2.5 ml/kg) according to our normalized protocols. The level of anaesthesia was verified before and during the surgery by the lack of reflex responses to frequent tail pinching. During the surgery, the animals were implanted subcutaneously in the upper mid-dorsal region with an osmotic pump (Alzet®; model 2ML4 – flow of 60μl/day during 28 days) for continuous delivery of either vehicle (2-hydroxypropyl-β-cyclodextrin, Cayman Chemicals, Ann Arbor, MI, USA), the ERα agonist (propylpyraoletriol-PPT - 30μg/kg/day) or the ERβ agonist (diarylpropionitril-DPN, 100μg/kg/day - both from Tocris, Bio-Techne Canada, Oakville, ON). These doses have been demonstrated to modulate brain mitochondrial activity without inducing uterine growth 21. Two weeks following the surgery, the animals were exposed to either intermittent hypoxia or left under room air conditions for one week. We used a total of 4 groups of rats, vehicle exposed to room air, vehicle exposed to CIH, PPT exposed to CIH, and DPN exposed to CIH. After completion of the 7-day exposures, all rats were sacrificed in the morning following the last day of exposures.
Chronic intermittent hypoxia exposure
After 2 weeks of recovery, the rats were weighed and housed in a Plexiglas chamber (internal volume 0.05m3), connected to an oxycycler (Biospherix, Redfield, NY, USA). As previously described 2, oxygen dropped from 21% to 10% in 90 seconds, holding O2 at 10% for 30 seconds, then returned to 21% in 70 seconds and holding at 21% for 120 seconds, at the rate of 10 cycles/hour for 8 consecutive hours between 8:30am - 4:30pm during 7 days. Rats exposed to room air and treated with vehicle were placed in the same room that those exposed to CIH.
Measurements of arterial blood pressure
Arterial blood pressure was measured by the tail cuff method by volume pressure recording (CODA system – Kent Scientific, Torrington, CT, USA) in conscious rats between noon and 1:00pm as described previously.2 The animal was placed in a restrainer tube over a warmed blanket. After 30 minutes of habituation, several recordings were performed, separated at least by 5 minutes. We have reported the mean of the 3 lowest values for systolic, diastolic, and mean arterial pressures.
Measurements of mitochondrial respiration and H2O2 production on permeabilized brain cortex
Rats were weighed and sacrificed with an overdose of anaesthetic (ketamine/xylazine), the brain cortex was rapidly dissected and separated through the midline. A first part was immediately frozen and kept at −80°C. From the second part, we immediately used fresh samples for measurements of oxygen consumption and H2O2 production rates using a high‐resolution fluorespirometry system (Oroboros 2k, Oroboros Instruments, Innsbruck, Austria). After calibration of the Oroboros chambers, cortex samples were weighted (2–3 mg), and recordings of O2 consumption performed at 37°C in a respiration buffer (0.5 mM EGTA, 3mM MgCl2, 60 mM potassium lactobionate, 10 mM KH2PO4, 20 mM Hepes, 110 mM sucrose, 1 g/l BSA). Based on a previous study establishing the optimal conditions 58, the samples were incubated in the recording chamber for 20 min with saponin (50 μg/ml). For each sample, we have differentiated mitochondrial oxygen consumption linked to NADH oxidation through the mitochondrial respiratory complex I (pyruvate 5mM, malate 2mM), mitochondrial oxygen consumption linked to FADH2 oxidation through the mitochondrial respiratory complex II (succinate 5mM and rotenone 0.5μM to block complex I activity) and mitochondrial oxygen consumption linked to NADH+FADH2 oxidation through the mitochondrial respiratory complex I and II (pyruvate 5mM, malate 2mM, succinate 10mM).
After equilibration with the substrates (mitochondrial respiratory state 2), ADP (2.5mM) is added to the chambers to measure O2 consumption under normal phosphorylating state (ATP synthesis - state 3). We then add cytochrome-c (10 μM) to assess the integrity of mitochondrial membranes, and then oligomycin (2.5 μM) was added to block ATP synthesis and measure O2 consumption due to leakage of protons in non-phosphorylating state (no ATP synthesis - state 4). Uncoupling is further exaggerated by adding graded bolus of carbonyl cyanide m-chlorophenylhydrazone (CCCP – 0.5 μM in 1 μl/bolus; 3–5 boluses in total) until reaching stable O2 consumption. We then blocked the activity of the complex 3 with antimycin A (2.5 μM) to measure non-mitochondrial O2 consumption (Residual oxygen flux - ROX) due to cytosolic oxidases. ROX was subtracted from all other measurements to report mitochondrial oxygen consumption.
H2O2 production
The rate of hydrogen peroxide formation was measured using the samples incubated with pyruvate, malate, and succinate (respiration through complexes I and II) by fluorimetric detection 59. We used Horseradish peroxidase (HRP, 1 U/mL) and Amplex Red fluorescent dye (10 μM). The excitation wavelength was 525 nm and fluorescence detection at 587 nm. Calibration was done by using known amounts of hydrogen peroxide (0.1 μM) added to the recording chamber at the end of all experiments.
In vitro measurement of enzymatic activities
Complex I
Complex I activity was measured using a kit from Abcam (#ab109721) following the manufacturer’s instruction. Briefly, 100 mg of brain cortex was homogenized in 500 μl of ice-cold PBS. The concentration of proteins was determined by a standard colorimetric BCA assay kit (Thermo-Fisher Scientific, Ottawa, ON, Canada), and adjusted at 5.5 mg/ml with ice-cold PBS. Then, the samples were centrifuged at 10,000 g for 10 minutes and the supernatant was transferred to a fresh tube. 200 μl of sample was added (in duplicate) to wells of 96-well plate pre-coated with anti-complex I antibody. After an incubation of 3h, the plate was emptied and 200 μl of assay solution (buffer, NADH and dye) was added. The plate was placed in the reader and the absorbance was recorded at 450nm every 30 seconds for 30 minutes at room temperature. The activity of complex I was determined by the oxidation of NADH and the simultaneous reduction of the provided dye. Since the protein concentration was similar across all groups (see table 1), all enzymatic activities are normalized to tissue mass (in mg).
Cytochrome c oxidase (COX – complex IV)
We measured the maximum activity of cytochrome c oxidase (COX – complex IV of the mitochondrial respiratory chain) as the O2 consumption on homogenized cortex samples using the Oroboros 2k. We used 50–150 μl of sample, with 1 μl of Antimycin A, 5 μl of ascorbate and 5 μl of Tetramethyl-paraphenylenediamine (TMPD) in 2 ml of respiration buffer (0.5 mM EGTA, 3mM MgCl2, 60 mM potassium lactobionate, 10 mM KH2PO4, 20 mM Hepes, 110 mM sucrose, 1 g/l BSA). The maximal activity of COX was read when O2 consumption rate was stable (typically a few minutes after starting the recording).
Citrate Synthase (CS)
CS activity was used as a marker of mitochondrial content 35,36. CS was measured using a kit from Sigma aldrich (#MAK193) following the manufacturer’s instruction. Briefly, 10 mg of brain cortex was homogenized in 100 μl of ice-cold CS Assay Buffer. Then, the samples were centrifuged at 10,000 g for 5 minutes and the supernatant was transferred to a fresh tube. 25 μl of sample was added to wells of 96-well plates in duplicate with appropriate reaction mixes (CS assay buffer, developer and substrate mix). A standard-curve was obtained with serial dilutions of GSH solution (0 to 40 nmol/well). The plate was incubated for 3 minutes at 25°C and the absorbance was recorded at 412nm every 5 minutes for 30 minutes. The colorimetric product (GSH) was proportional to the enzymatic activity of CS and normalized to the quantity of tissue.
Pyruvate Dehydrogenase (PDH)
PDH activity was measured using a kit from Sigma Aldrich (#MAK183) following the manufacturer’s instruction. Briefly, 10 mg of brain cortex was homogenized in 100 μl of ice-cold PDH Assay Buffer. Then, the samples were centrifuged at 10,000 g for 5 minutes and the supernatant was transferred to a fresh tube. 40 μl of sample was added to wells of 96-well plate in duplicate with appropriate reaction mixes (PDH assay buffer, developer and substrate). A standard-curve was obtained with serial dilutions of NADH solution (0 to 12.5 nmol/well). The plate was incubated for 3 minutes at 37 °C and the absorbance was recorded at 450nm every 5 minutes for 30 minutes. The colorimetric product (NADH) was proportional to the enzymatic activity of PDH and normalized to the quantity of tissue.
Lactate Dehydrogenase (LDH)
LDH activity was measured using a kit from Sigma Aaldrich (#MAK066) following the manufacturer’s instruction. Briefly, 100 mg of brain cortex was homogenized in 500 μl of ice-cold LDH Assay Buffer. Then, the samples were centrifuged at 10,000 g for 5 minutes and the supernatant was transferred to a fresh tube. 1 μl of sample was added to wells of 96-well plate in duplicate with appropriate reaction mixes (LDH assay buffer, developer and LDH substrate Mix). A standard-curve was obtained with serial dilutions of NADH solution (0 to 12.5 nmol/well). The plate was incubated for 3 minutes at 37 °C and the absorbance was recorded at 450nm every 5 minutes for 30 minutes. The colorimetric product (NADH) was proportional to the enzymatic activity of LDH and normalized to the quantity of tissue.
Statistical analysis
All analyses were done with the GraphPad prism software. For experiments of O2 consumption we used two-way ANOVA with the respiratory states as the repeated variable. When significant effects of groups or interaction between groups and states appeared, a post-hoc test (Fisher’s Least Significance Difference) was applied. For all other variables, one-way ANOVAs followed by a post-hoc test (Fisher’s Least Significance Difference) were used. Statistical significance was set for P values <0.05. All data are presented as means ± SD in the text and as boxes and whiskers in the figures.
Table 2:
Concentration of proteins in brain samples lysates in ovariectomized female rats treated with vehicle (Veh), the ERα (PPT), or the ERβ (DPN) agonist and exposed to room air (AIR) or chronic intermittent hypoxia (CIH). All data are mean ± SD
| Groupe | Veh-AIR | Veh-CIH | PPT-CIH | DPN-CIH |
|---|---|---|---|---|
| Protein (μg/mg tissue) |
43.6 ± 6.2 | 44.4 ± 5.5 | 42.1 ± 4.5 | 42.5 ± 3.9 |
Acknowledgements:
SL supported by Région Rhône Alpe (Programme Explo’ra Doc) and Consulat Général de France à Québec (Programme Frontenac). Study funded by CIHR (VJ: MOP-102715 and AB: MOP-119272). DG is supported by National Institutes of Health grant HL130984. VJ and AB are members of the FRQ-S Respiratory Health Network.
Footnotes
Conflict of interest
None.
References
- 1.Gileles-Hillel A, Almendros I, Khalyfa A, et al. Prolonged Exposures to Intermittent Hypoxia Promote Visceral White Adipose Tissue Inflammation in a Murine Model of Severe Sleep Apnea: Effect of Normoxic Recovery. Sleep. 2017;40(3). [DOI] [PubMed] [Google Scholar]
- 2.Laouafa S, Ribon-Demars A, Marcouiller F, et al. Estradiol Protects Against Cardiorespiratory Dysfunctions and Oxidative Stress in Intermittent Hypoxia. Sleep. 2017;40(8):zsx104. [DOI] [PubMed] [Google Scholar]
- 3.Lavie L Oxidative stress in obstructive sleep apnea and intermittent hypoxia--revisited--the bad ugly and good: implications to the heart and brain. Sleep Med Rev. 2015;20:27–45. [DOI] [PubMed] [Google Scholar]
- 4.Guo QH, Tian YL, Wang Z, et al. Endothelin receptors in augmented vasoconstrictor responses to endothelin-1 in chronic intermittent hypoxia. Clin Exp Pharmacol Physiol. 2013;40(7):449–457. [DOI] [PubMed] [Google Scholar]
- 5.Lesske J, Fletcher EC, Bao G, Unger T. Hypertension caused by chronic intermittent hypoxia--influence of chemoreceptors and sympathetic nervous system. J Hypertens. 1997;15(12 Pt 2):1593–1603. [DOI] [PubMed] [Google Scholar]
- 6.Xu W, Chi L, Row BW, et al. Increased oxidative stress is associated with chronic intermittent hypoxia-mediated brain cortical neuronal cell apoptosis in a mouse model of sleep apnea. Neuroscience. 2004;126(2):313–323. [DOI] [PubMed] [Google Scholar]
- 7.Macey PM, Haris N, Kumar R, Thomas MA, Woo MA, Harper RM. Obstructive sleep apnea and cortical thickness in females and males. PLoS One. 2018;13(3):e0193854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Macey PM, Kheirandish-Gozal L, Prasad JP, et al. Altered Regional Brain Cortical Thickness in Pediatric Obstructive Sleep Apnea. Front Neurol. 2018;9:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nair D, Dayyat EA, Zhang SX, Wang Y, Gozal D. Intermittent hypoxia-induced cognitive deficits are mediated by NADPH oxidase activity in a murine model of sleep apnea. PLoS One. 2011;6(5):e19847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shan X, Chi L, Ke Y, et al. Manganese superoxide dismutase protects mouse cortical neurons from chronic intermittent hypoxia-mediated oxidative damage. Neurobiol Dis. 2007;28(2):206–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y, Zhang SX, Gozal D. Reactive oxygen species and the brain in sleep apnea. Respir Physiol Neurobiol. 2010;174(3):307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Young T, Finn L, Austin D, Peterson A. Menopausal status and sleep-disordered breathing in the Wisconsin Sleep Cohort Study. Am J Respir Crit Care Med. 2003;167(9):1181–1185. [DOI] [PubMed] [Google Scholar]
- 13.Galvan T, Camuso J, Sullivan K, et al. Association of estradiol with sleep apnea in depressed perimenopausal and postmenopausal women: a preliminary study. Menopause. 2017;24(1):112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shahar E, Redline S, Young T, et al. Hormone replacement therapy and sleep-disordered breathing. Am J Respir Crit Care Med. 2003;167(9):1186–1192. [DOI] [PubMed] [Google Scholar]
- 15.Cano-Pumarega I, Barbe F, Esteban A, et al. Sleep Apnea and Hypertension: Are there gender differences? The Vitoria Sleep Cohort. Chest. 2017. [DOI] [PubMed] [Google Scholar]
- 16.Mokhlesi B, Ham SA, Gozal D. The effect of sex and age on the comorbidity burden of OSA: an observational analysis from a large nationwide US health claims database. Eur Respir J. 2016;47(4):1162–1169. [DOI] [PubMed] [Google Scholar]
- 17.Lagranha CJ, Silva TLA, Silva SCA, et al. Protective effects of estrogen against cardiovascular disease mediated via oxidative stress in the brain. Life Sci. 2018;192:190–198. [DOI] [PubMed] [Google Scholar]
- 18.Boukari R, Laouafa S, Ribon-Demars A, Bairam A, Joseph V. Ovarian steroids act as respiratory stimulant and antioxidant against the causes and consequences of sleep-apnea in women. Respir Physiol Neurobiol. 2017;239:46–54. [DOI] [PubMed] [Google Scholar]
- 19.Chen JQ, Cammarata PR, Baines CP, Yager JD. Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochim Biophys Acta. 2009;1793(10):1540–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rettberg JR, Yao J, Brinton RD. Estrogen: a master regulator of bioenergetic systems in the brain and body. Front Neuroendocrinol. 2014;35(1):8–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Irwin RW, Yao J, To J, Hamilton RT, Cadenas E, Brinton RD. Selective oestrogen receptor modulators differentially potentiate brain mitochondrial function. J Neuroendocrinol. 2012;24(1):236–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valera MC, Fontaine C, Dupuis M, et al. Towards optimization of estrogen receptor modulation in medicine. Pharmacol Ther. 2018. [DOI] [PubMed] [Google Scholar]
- 23.Brinton RD. Estrogen regulation of glucose metabolism and mitochondrial function: therapeutic implications for prevention of Alzheimer’s disease. Adv Drug Deliv Rev. 2008;60(13–14):1504–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bourque M, Morissette M, Di Paolo T. Repurposing sex steroids and related drugs as potential treatment for Parkinson’s disease. Neuropharmacology. 2018. [DOI] [PubMed] [Google Scholar]
- 25.Dubal DB, Zhu H, Yu J, et al. Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci U S A. 2001;98(4):1952–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hay M, Xue B, Johnson AK. Yes! Sex matters: sex, the brain and blood pressure. Curr Hypertens Rep. 2014;16(8):458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xue B, Zhang Z, Beltz TG, Guo F, Hay M, Johnson AK. Genetic knockdown of estrogen receptor-alpha in the subfornical organ augments ANG II-induced hypertension in female mice. Am J Physiol Regul Integr Comp Physiol. 2015;308(6):R507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joseph V, Doan VD, Morency CE, Lajeunesse Y, Bairam A. Expression of sex-steroid receptors and steroidogenic enzymes in the carotid body of adult and newborn male rats. Brain Res. 2006;1073–1074: [DOI] [PubMed] [Google Scholar]
- 29.Fletcher EC. Invited review: Physiological consequences of intermittent hypoxia: systemic blood pressure. J Appl Physiol (1985). 2001;90(4):1600–1605. [DOI] [PubMed] [Google Scholar]
- 30.Borras C, Gambini J, Lopez-Grueso R, Pallardo FV, Vina J. Direct antioxidant and protective effect of estradiol on isolated mitochondria. Biochim Biophys Acta. 2010;1802(1):205–211. [DOI] [PubMed] [Google Scholar]
- 31.Razmara A, Sunday L, Stirone C, et al. Mitochondrial effects of estrogen are mediated by estrogen receptor alpha in brain endothelial cells. J Pharmacol Exp Ther. 2008;325(3):782–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malthankar-Phatak GH, Patel AB, Xia Y, et al. Effects of continuous hypoxia on energy metabolism in cultured cerebro-cortical neurons. Brain Res. 2008;1229:147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vega C, R. Sachleben LJ, Gozal D, Gozal E Differential metabolic adaptation to acute and long-term hypoxia in rat primary cortical astrocytes. J Neurochem. 2006;97(3):872–883. [DOI] [PubMed] [Google Scholar]
- 34.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435(2):297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larsen S, Nielsen J, Hansen CN, et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol. 2012;590(14):3349–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kilbaugh TJ, Karlsson M, Byro M, et al. Mitochondrial bioenergetic alterations after focal traumatic brain injury in the immature brain. Exp Neurol. 2015;271:136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ribon-Demars A, Pialoux V, Boreau A, et al. Protective roles of estradiol against vascular oxidative stress in ovariectomized female rats exposed to normoxia or intermittent hypoxia. Acta Physiol (Oxf). 2018:e13159. [DOI] [PubMed] [Google Scholar]
- 39.Darblade B, Pendaries C, Krust A, et al. Estradiol alters nitric oxide production in the mouse aorta through the alpha-, but not beta-, estrogen receptor. Circ Res. 2002;90(4):413–419. [DOI] [PubMed] [Google Scholar]
- 40.Pires PW, Dams Ramos CM, Matin N, Dorrance AM. The effects of hypertension on the cerebral circulation. Am J Physiol Heart Circ Physiol. 2013;304(12):H1598–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seryapina AA, Shevelev OB, Moshkin MP, Markel AL, Akulov AE. Stress-sensitive arterial hypertension, haemodynamic changes and brain metabolites in hypertensive ISIAH rats: MRI investigation. Exp Physiol. 2017;102(5):523–532. [DOI] [PubMed] [Google Scholar]
- 42.Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci U S A. 2003;100(17):10073–10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yuan G, Adhikary G, McCormick AA, Holcroft JJ, Kumar GK, Prabhakar NR. Role of oxidative stress in intermittent hypoxia-induced immediate early gene activation in rat PC12 cells. J Physiol. 2004;557(Pt 3):773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science. 2001;292(5516):504–507. [DOI] [PubMed] [Google Scholar]
- 45.Peng YJ, Yuan G, Ramakrishnan D, et al. Heterozygous HIF-1alpha deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J Physiol. 2006;577(Pt 2):705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3(3):187–197. [DOI] [PubMed] [Google Scholar]
- 47.Yuan G, Nanduri J, Khan S, Semenza GL, Prabhakar NR. Induction of HIF-1alpha expression by intermittent hypoxia: involvement of NADPH oxidase, Ca2+ signaling, prolyl hydroxylases, and mTOR. J Cell Physiol. 2008;217(3):674–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nanduri J, Vaddi DR, Khan SA, et al. HIF-1alpha activation by intermittent hypoxia requires NADPH oxidase stimulation by xanthine oxidase. PLoS One. 2015;10(3):e0119762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsieh DJ, Kuo WW, Lai YP, et al. 17beta-Estradiol and/or Estrogen Receptor beta Attenuate the Autophagic and Apoptotic Effects Induced by Prolonged Hypoxia Through HIF-1alpha-Mediated BNIP3 and IGFBP-3 Signaling Blockage. Cell Physiol Biochem. 2015;36(1):274–284. [DOI] [PubMed] [Google Scholar]
- 50.Li Y, Liu Y, Lu Y, Zhao B. Inhibitory effects of 17beta-estradiol or a resveratrol dimer on hypoxia-inducible factor-1alpha in genioglossus myoblasts: Involvement of ERalpha and its downstream p38 MAPK pathways. Int J Mol Med. 2017;40(5):1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gaude E, Schmidt C, Gammage PA, et al. NADH Shuttling Couples Cytosolic Reductive Carboxylation of Glutamine with Glycolysis in Cells with Mitochondrial Dysfunction. Mol Cell. 2018;69(4):581–593 e587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lejri I, Grimm A, Eckert A. Mitochondria, Estrogen and Female Brain Aging. Front Aging Neurosci. 2018;10:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koronowski KB, Perez-Pinzon MA. Sirt1 in cerebral ischemia. Brain Circ. 2015;1(1):69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wen ZW, Liang DS, Cai XH, Chen J. The role of AMPK/mTOR signal pathway in brain injury following chronic intermittent hypoxia in growing rats. Eur Rev Med Pharmacol Sci. 2018;22(4):1071–1077. [DOI] [PubMed] [Google Scholar]
- 55.Yang S, Wang J. Estrogen Activates AMP-Activated Protein Kinase in Human Endothelial Cells via ERbeta/Ca(2+)/Calmodulin-Dependent Protein Kinase Kinase beta Pathway. Cell Biochem Biophys. 2015;72(3):701–707. [DOI] [PubMed] [Google Scholar]
- 56.Gorenkova N, Robinson E, Grieve DJ, Galkin A. Conformational change of mitochondrial complex I increases ROS sensitivity during ischemia. Antioxid Redox Signal. 2013;19(13):1459–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Galkin A, Abramov AY, Frakich N, Duchen MR, Moncada S. Lack of oxygen deactivates mitochondrial complex I: implications for ischemic injury? J Biol Chem. 2009;284(52):36055–36061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Herbst EA, Holloway GP. Permeabilization of brain tissue in situ enables multiregion analysis of mitochondrial function in a single mouse brain. J Physiol. 2015;593(4):787–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krumschnabel G, Fontana-Ayoub M, Sumbalova Z, et al. Simultaneous high-resolution measurement of mitochondrial respiration and hydrogen peroxide production. Methods Mol Biol. 2015;1264:245–261. [DOI] [PubMed] [Google Scholar]