

Abstract
β-Coronaviruses are a family of positive-strand enveloped RNA viruses that includes the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Much is known regarding their cellular entry and replication pathways, but their mode of egress remains uncertain. Using imaging methodologies and virus-specific reporters, we demonstrate that β-coronaviruses utilize lysosomal trafficking for egress rather than the biosynthetic secretory pathway more commonly used by other enveloped viruses. This unconventional egress is regulated by the Arf-like small GTPase Arl8b and can be blocked by the Rab7 GTPase competitive inhibitor CID1067700. Such non-lytic release of β-coronaviruses results in lysosome deacidification, inactivation of lysosomal degradation enzymes, and disruption of antigen presentation pathways. β-Coronavirus-induced exploitation of lysosomal organelles for egress provides insights into the cellular and immunological abnormalities observed in patients and suggests new therapeutic modalities.
Keywords: coronavirus, SARS-CoV-2, viral egress, lysosome, pH, viral immunology, antigen presentation, acidification/deacidification ARL8b, Rab7, CD1067700
Graphical Abstract
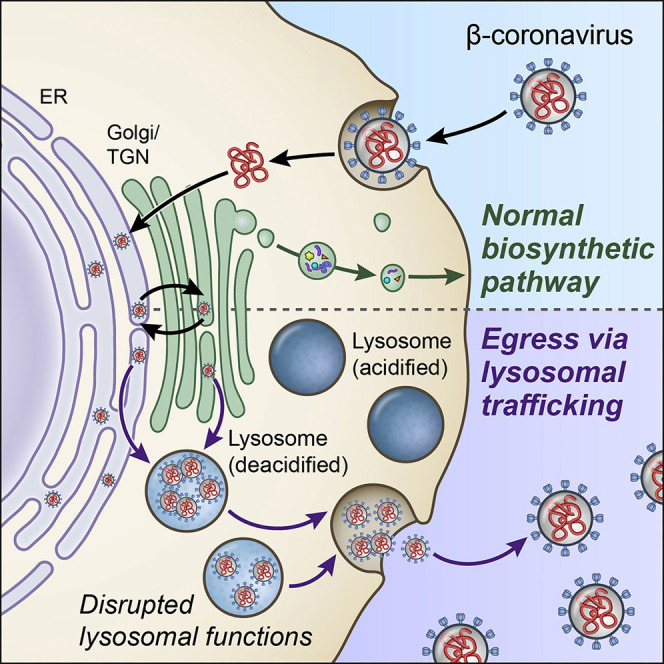
Ghosh et al. provide evidence that β-coronaviruses do not use the biosynthetic secretory pathway typically used by enveloped viruses to leave infected cells. Instead, these viruses traffic to lysosomes for unconventional egress by Arl8b-dependent lysosomal exocytosis. Their non-lytic release results in lysosome deacidification, inactivation of lysosomal degradation enzymes, and disruption of antigen presentation.
Introduction
β-Coronaviruses are positive-strand enveloped RNA viruses that comprise one of the four genera of the Coronaviridae family of viruses. β-Coronaviruses infect humans and other mammals, with infection resulting in a range of diseases with considerable morbidity and mortality. In late 2019, one member, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), originating in bats, spread to humans and caused a worldwide pandemic (Lu et al., 2020).
The ability of these viruses to infect many different cell types, including those of the pulmonary, cardiovascular, hepatic, gastrointestinal, central nervous, and immune systems, results in complex multi-organ disease manifestations that can vary from individual to individual (Puelles et al., 2020; Ziegler et al., 2020). Especially with regard to the immune system, these viruses appear to deregulate the traditional innate and adaptive immune responses to pathogens (Vardhana and Wolchok, 2020). Currently there is no cure, and the antiviral treatment options are few (Williamson et al., 2020), and whether lasting immune responses can be generated to infection by natural means or through vaccine administration remains open to question (Long et al., 2020).
One of the major reasons for the lack of antiviral therapies is the paucity of knowledge regarding the β-coronavirus-host cell interface. When the viral envelope fuses with the plasma membrane and/or endosome membranes, and the viral RNA genome is released into the cytosol, it translates into nonstructural and structural proteins. The nonstructural proteins assemble on endoplasmic reticulum (ER)-derived membranes and replicate the viral RNA (Snijder et al., 2006, 2020). Although a large amount of molecular detail is known regarding coronavirus entry and replication, very little is known regarding how the newly assembled coronaviruses egress from cells, including which cellular pathways they exploit and whether they induce cell lysis (Fung and Liu, 2019; Machamer, 2013; Tooze et al., 1987).
The egress pathway for all β-coronaviruses starts with newly synthesized viral genomic RNA, coated with viral N proteins, budding into the lumen of the ER and the ER-Golgi intermediate compartment (ERGIC) (Cohen et al., 2011; McBride et al., 2007; Perrier et al., 2019; Tooze et al., 1987, 1988). This results in viral particles enveloped with host membranes containing viral M, E, and S transmembrane structural proteins (de Haan et al., 1998; Ruch and Machamer, 2012; Siu et al., 2008). When in the ER/ERGIC, virus particles traffic to the Golgi apparatus and trans-Golgi network (TGN) for glycosylation and other post-translational modifications (Fung and Liu, 2018; McBride et al., 2007; Oostra et al., 2006; Tooze et al., 1987). But after the Golgi apparatus/TGN, it has been assumed that coronaviruses use vesicles of the biosynthetic secretory pathway to track to the plasma membrane and egress (Machamer, 2013; Tooze et al., 1987), similar to other enveloped RNA viruses, such as hepatitis C virus, dengue virus, and West Nile virus (Ravindran et al., 2016; Robinson et al., 2018).
Here we investigated the egress pathway of β-coronaviruses and found that, rather than the biosynthetic secretory pathway, these viruses use a lysosomal, Arl8b-dependent exocytic pathway for release into the extracellular environment. We show that GRP78/BIP, an ER chaperone that facilitates coronavirus infectivity (Chu et al., 2018; Ha et al., 2020), is co-released with β-coronaviruses through this pathway. As a consequence of viral exploitation of lysosomal exocytosis, we demonstrate that late endosomes/lysosomes are deacidified and lysosomal proteases are inactive. Significantly, we show that this perturbation of lysosome physiology has important functional consequences on the host cell, including disruption of antigen presentation pathways.
Results
β-Coronaviruses Egress from Cells Independent of the Biosynthetic Secretory Pathway
We began investigating the mechanism of β-coronavirus egress using mouse hepatitis virus (MHV), as it is the prototype of the family that can be studied under biosafety level 2 (BSL-2) conditions, with intranasal MHV infection in mice inducing pathogenesis similar to SARS, including acute pneumonia, lung injury, and hepatic and neurological disease (Channappanavar et al., 2016; De Albuquerque et al., 2006; Khanolkar et al., 2007).
First we investigated the kinetics of replication and egress in this model system. HeLa-ATCC cells stably expressing murine CEACAM1 (HeLa-mCC1a) were infected with the MHV-A59 strain virus. Cell lysates and extracellular medium were collected at different intervals and processed for quantitative PCR (qPCR) determination of viral genomic RNA. The results were plotted as fold change over uninfected cell lysates and extracellular medium (Figure 1 A). We found that the replication rate was highest until 8 h post infection (pi) and then plateaued. Virus egress was highest from 8–12 h pi before leveling off at 14 h pi (Figure 1A). The extracellular genomic RNA (i.e., egressed virus), released between 8 and 14 h pi, was infectious and could be titered (Figure S1 A). Notably, viral egress took place in the absence of any cell lysis because there was no significant change in the permeability of the plasma membrane to the membrane-impermeant cell viability dyes trypan blue and propidium iodide when the virus accumulated extracellularly (Figure 1B; Figures S1B and S1C).
Figure 1.

β-Coronaviruses Egress Independent of the Biosynthetic Secretory Pathway and Are Enriched in Late Endosomes/Lysosomes
(A) Kinetics of MHV replication and release. Viral genomic RNA was quantified in cell lysates and extracellular medium. Experiments were done in triplicates.
(B) Plasma membrane permeability in MHV-infected cells, measured by trypan blue exclusion along with virus release. Experiments were done in triplicates.
(C) Cargo transport kinetics through the biosynthetic secretory pathway in the absence and presence of MHV infection. Experiments were done in triplicates.
(D) Effect of Brefeldin A (BFA) (5 μg/mL) treatment on MHV egress and Gaussia luciferase secretion. Cells were treated with BFA for 4, 6, or 8 h prior to collection of extracellular medium at 14 h pi. Experiments were done in triplicates.
(E) MHV-infected cells treated with BFA (8–14 h pi) or left untreated and coimmunostained with anti-Golgi apparatus (mannosidase II, green) and anti-MHV (MJ1.3, red) antibodies. Scale bar, 10 μm.
(F) Immunoelectron micrograph of MHV-infected cells coimmunostained with anti-MHV (MJ1.3) primary and 10-nm gold-coupled secondary antibodies. The scale bar is indicated on the micrograph.
(G) MHV-infected cells coimmunostained with anti-E (green) and anti-MHV (MJ1.3) (red) antibodies. Scale bar, 5 μm.
(H) MHV-infected cells coimmunostained with anti-LAMP1 (green) and anti-MHV (MJ1.3) (red) antibodies. Arrows point to LAMP1+/MHV+ puncta. Scale bar, 5 μm.
(I) Quantification of colocalization between LAMP1 and MHV, calculated at 6 h (n = 6 cells) and 12 h pi (n = 20 cells).
(J) SARS-CoV-2-infected cells coimmunostained with anti-LAMP1 (green) and anti-CoV-2 M (red) antibodies. Arrows point to LAMP1 puncta containing the M label. Scale bar, 2 μm.
(K and L) MHV-infected cells fractionated at 12 h pi. MHV genomic RNA associated with LAMP1+ fractions (K) was quantified and plotted (L). Dyngo-4a (30 μM) or vehicle was added from 6–12 h pi (L). Fractionation experiments were done in duplicate; qPCR measurements in each were done in triplicate. Mean data from 2 independent experiments are presented.
Representative blot and images are shown. Data are shown as mean ± SEM. p values were considered significant when p < 0.05 and denoted as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 10−5; ns, not significant. See also Figures S1, S2, and S3.
Figure S1.

Coronavirus Egress and Infectivity, Related to Figure 1
(A) Infected cells were treated with/without BFA at 8 h pi or 10 h pi. Supernatants collected at 14 h pi were reinoculated into new HeLa-mCC1a cells and TCID50/ml was calculated at 72 h .
(B) Propidium iodide labeling to detect changes in plasma membrane permeability in MHV-infected cells. As a positive control, cells were treated with staurosporine which induced apoptosis and disrupted the plasma membrane.
(C) Trypan blue exclusion was used to detect changes in plasma membrane permeability in MHV-infected cells at 14 h pi. Cells were imaged and the number of trypan blue positive cells quantified and plotted. Scale bar 200 μm.
(D) HeLa-mCC1a cells transfected with Gaussia Luciferase and infected with MHV were coimmunostained with anti-Gaussia luciferase (green) and anti-MHV (MJ1.3) (red) antibodies. Scale bar 5 μm.
(E) Trypan blue exclusion at 14 h pi was used to detect changes in plasma membrane permeability of MHV-infected cells treated with/without BFA at 8 h pi and 10 h pi. Extracellular viral genomic RNA was quantified with qPCR and plotted as fold increase over uninfected cells. Experiments done in triplicates.
Representative images are shown. Data shown as mean ± SEM; ns = not significant.
β-Coronaviruses are widely thought to use the biosynthetic secretory pathway for egress. Given this, we next interrogated the status of the secretory pathway in infected cells and whether this pathway was utilized for MHV egress. Cells were transfected with Gaussia luciferase, a reporter for the biosynthetic secretory pathway (Tannous, 2009), and infected with MHV or left uninfected. We confirmed that Gaussia luciferase transfection of cells did not block their subsequent infection by MHV (Figure S1D). Extracellular luciferase levels were measured by luminescence, and released viral genomes were quantified by qPCR (Figure 1C). We found that the kinetics of Gaussia luciferase secretion was not altered significantly throughout the MHV egress period, consistent with previous reports (Machamer, 2013; Tooze et al., 1987).
Given that the secretory pathway remained operational, we next asked whether β-coronaviruses utilized it for egress. We treated Gaussia luciferase transfected cells with Brefeldin A (BFA), a small molecule that rapidly shuts down all anterograde biosynthetic secretory traffic from the ER/ERGIC out to the plasma membrane and leads to resorption of the Golgi apparatus into the ER (Lippincott-Schwartz et al., 1989; Miller et al., 1992). The uninfected/Gaussia luciferase-transfected and MHV-infected/Gaussia luciferase-transfected cells were treated with or without BFA at 6, 8, and 10 h pi, and extracellular media were collected at 14 h pi. This resulted in 8, 6, and 4 h of total BFA treatment time, respectively. From these collected media, Gaussia luciferase and MHV extracellular genomic RNA levels were quantified and plotted simultaneously (Figure 1D). Remarkably, the presence of BFA, from 6 h pi onward, did not affect MHV egress; neither quantification of extracellular viral genomic RNA (Figure 1D) nor viral titers (Figure S1A) of BFA-treated cells showed any significant change relative to untreated infected cells. The secretory pathway was sensitive to BFA in MHV-infected cells because the Golgi apparatus was completely disrupted (Figure 1E), and Gaussia luciferase secretion was blocked (Figure 1D). Furthermore, the viral egress observed was not due to cell lysis, irrespective of BFA treatment (Figure S1E). Thus, MHV could egress even when trafficking through the biosynthetic secretory pathway was blocked.
β-Coronaviruses Are Enriched in Late Endosomes/Lysosomes during Egress
We then investigated the spatiotemporal distribution of MHV during egress to identify which cellular trafficking pathway it exploited. The M protein is the most abundant protein in the envelope of β-coronaviruses and drives virus assembly, membrane curvature, and budding into the ER/ERGIC by oligomerizing with itself and with viral RNA, N, E, and S proteins (de Haan and Rottier, 2005; Ruch and Machamer, 2012). Immunolabeling cells at peak egress (12 h pi) with the J1.3 monoclonal anti-M antibody (Narayanan et al., 2000; Stohlman et al., 1982) and subsequent immunoelectron microscopy revealed antibody labeling to be concentrated 3-fold more on the envelopes of viral particles compared with membranes elsewhere (ER, ERGIC, Golgi apparatus, etc.) (Figure 1F; Figure S2 A). The J1.3 antibody may recognize free M proteins, but our quantitative analysis of electron micrographs indicated that the antibody mostly detected M in the context of assembled particles. In addition, consistent with recognition of assembled virus particles, MHV(MJ1.3) antibody labeling colocalized with E and S envelope proteins throughout infection (Figure 1G; Figures S2B and S2C).
Figure S2.

Spatio-temporal Organization of Subcellular Organelles and Coronavirus during Infection, Related to Figure 1
(A) The density of MHV (MJ1.3) gold particles per 0.5μm2 area (n=10 areas with and n=10 areas without virions) of infected cells is plotted.
(B) HeLa-mCC1a cells, infected with MHV, fixed (6 h and 12 h pi), and coimmunostained with anti-E(green) and anti-S (red) antibodies.
(C) HeLa-mCC1a cells, infected with MHV, fixed (6 h and 12 h pi), and coimmunostained with anti-E(green) and anti-MHV(MJ1.3) (red) antibodies.
(D) HeLa-mCC1a cells infected with MHV, fixed (6 h, 9 h and 12 h pi)) and coimmunostained with anti-TGN46 (green) and anti-MHV(MJ1.3) (red).
(E) HeLa-mCC1a cells infected with MHV, fixed (5 h and 14 h pi) and coimmunostained with anti-Golgin97 (green) and anti-MHV(MJ1.3) (red). Scale bar 10 μm.
(F) HeLa-mCC1a cells infected with MHV, fixed (6 h and 10 h pi) and coimmunostained with anti-mannosidase II (green) and anti-MHV(MJ1.3) (red).
(G) MHV-infected cells, washed, fixed (6 h and 12 h pi), and coimmunostained with anti-cathepsin D (green) and anti-MHV(MJ1.3) (red) antibodies. Arrows point to Cathepsin+/MHV+ puncta.
(H) HeLa-mCC1a cells infected with MHV, fixed at 12 h pi and coimmunostained with anti-LAMP1 (blue), anti-MHV(MJ1.3) (red) and anti-Atlastin 3 (green) antibodies. White arrows point to LAMP1+/MHV+ puncta; turquoise arrows point to Atlastin-3+/MHV+ puncta.
Representative images are shown. Scale bar 5μm unless indicated.
We infected cells with MHV, washed off the virus, and fixed the cells at different times post-inoculation. We co-stained the fixed cells with anti-MHV(MJ1.3), anti-E, and other antibodies against organelle-resident host proteins. At 6 h pi, consistent with previous reports showing newly synthesized viruses trafficking to the Golgi apparatus and TGN at the early stages of infection (Machamer, 2013; Tooze et al., 1987), MHV(M J1.3) labeling was perinuclear (Figure 1G, 6 h pi) and colocalized with TGN46, Golgin 97, and mannosidase II by immunofluorescence (Figures S2D–S2F).
However, at 12 h pi, the bulk of the MHV(MJ1.3) labeling was no longer perinuclear and did not colocalize with these Golgi apparatus/TGN markers (Figures S2D–S2F). Instead, it was concentrated in puncta dispersed across the cytoplasm (Figure 1G, 12 h pi). The late endosomal/lysosomal transmembrane protein LAMP1 (Figure 1H, 12 h pi) and lumenal enzyme cathepsin D (Figure S2G) were colocalized with many of the MHV(MJ1.3)-labeled puncta. We quantified the colocalization of fluorescence signals in the MHV(M J1.3) and LAMP1 channels (Manders et al., 1993), which revealed an ~5-fold increase in LAMP1+/ MHV+ organelles during the egress period (Figure 1I). MHV(MJ1.3) puncta not with LAMP1 were localized to the ER (Figure S2H). In SARS-CoV-2-infected Vero E6 cells, M labeling could also be detected within LAMP1+ organelles (Figure 1J).
We then fractionated MHV-infected cells at 12 h pi using Nycodenz gradients (Graham et al., 1990) and quantified MHV genomic content in each fraction by qPCR. This revealed viral genomic RNA to be enriched in the LAMP1+ fractions (2 and 3), which correspond to late endosomes/lysosomes, and in ERGIC53+ fractions (4 and 5), corresponding to Golgi apparatus/ER/ERGIC membranes, where MHV replication takes place (Figures 1K and 1L; Figures S3 A and S3B). In contrast, a similar fractionation done on poliovirus-infected HeLa cells revealed poliovirus to be enriched in ERGIC53+/LC3+ fractions but not in LAMP1+ fractions (Figure S3C), a result consistent with poliovirus using ER/Golgi apparatus/ERGIC-derived autophagosomes for egress that are blocked from fusing with lysosomes (Chen et al., 2015).
Figure S3.

Coronavirus Distribution in Subcellular Organelles, Related to Figure 1
(A) MHV genomic RNA levels at 12hr pi throughout fractions isolated from Nycodenz gradient. Fractionation experiment was done in duplicate; qPCR measurements in each were done in triplicate. Mean qPCR measurements from the 2 independent experiments plotted.
(B) Fractions from (A) processed for SDS-PAGE/Western blotting; probed with antibodies against LAMP1, CI-MPR and ERGIC53 proteins.
(C) HeLa cells infected with poliovirus, incubated for 6 hr and fractionated using Nycodenz gradient as in (A). Isolated fractions processed for SDS-PAGE/Western blotting; probed with antibodies against LAMP1, CI-MPR, ERGIC53 and LC3.
(D). HeLa-mCC1a cells were infected with MHV, fixed at 12 hr pi and coimmunostained with anti-CI-MPR (green), anti-LAMP1 (blue) and anti-MHV (J1.3) (red) antibodies. White arrows point to LAMP1+/MHV+/CI-MPR- puncta. Scale bar 5 μm.
(E) Quantification of colocalization between LAMP1 and CI-MPR in uninfected and infected cell groups (n = 20 cells/group).
(F) Dextran uptake in HeLa-mCC1a cells is inhibited by Dyngo-4a treatment. Dyngo-4a (30 μM) or DMSO was incubated with the cells for 6 hr. Alexa 555 Dextran (2mg/ml) was incubated with the cells in the last hour of Dyngo-4a or DMSO treatment. Scale bar 10 μm.
Representative blots and images are shown. Data shown as mean ± SEM; where∗∗∗p < 0.0002.
The LAMP1+ fractions are a mixture of lysosomes and late endosomes that overlap in many membrane and lumenal proteins (Huotari and Helenius, 2011). However, the cation-independent mannose 6-phosphate receptor (CI-MPR) mainly cycles between late endosomes and the Golgi apparatus/TGN (Brown et al., 1986). Indeed, in poliovirus-infected cells, CI-MPR was detectable throughout fractions 2–5, reflecting this broad distribution (Figure S3C). In MHV-infected cells, it was mostly detectable in fraction 5 (Figure S3B). Consistent with this, imaging of MHV-infected cells coimmunostained with anti-CI-MPR and anti-LAMP1 antibodies revealed a 3-fold decrease in colocalization between CI-MPR and LAMP1 relative to uninfected cells (Figures S3D and S3E). This suggests that, in infected cells, MHV is largely associated with lysosomes and/or atypical late endosomes.
Significantly, MHV association with the lysosome fractions was not a result of endocytic reuptake of egressed virus. When infected cells were treated with Dyngo-4a, a potent inhibitor of endocytosis (Park et al., 2013; Figure S3F) during egress, the quantity of MHV genomic RNA associated with the LAMP1+ fractions remained significant (Figure 1L).
Ultrastructural Characterization of β-Coronavirus Egress
Using transmission electron microscopy (TEM) and immunoelectron microscopy (immuno-EM) methodologies, which have a resolution of a few nanometers, we further interrogated the association between β-coronaviruses and lysosomes in MHV- and SARS-CoV-2-infected cells (Figure 2 ). By TEM, lysosomes can be easily recognized by specific hallmarks, such as intralumenal electron dense material and intralumenal membrane swirls (Fawcett, 1966). TEM of MHV- or SARS-CoV-2-infected cells during the egress stage revealed many such organelles filled with intact viruses of typical 70- to 90-nm particle size. These organelles were likely lysosomes because they displayed all of the physiological hallmarks (Figures 2A–2C). Immuno-EM against native LAMP1 revealed this protein to be on the membrane of these virus-filled organelles (Figure 2B), further confirming the colocalization of LAMP1 and MHV(MJ1.3) observed by confocal imaging (Figures 1H–1J) and the biochemical association between LAMP1+ fractions and MHV genomes (Figures 1K and 1L).
Figure 2.

Ultrastructural Localization of MHV and SARS-CoV-2 to Lysosomes during Egress
(A) TEM images of MHV-infected cells (14 h pi). Representative images are shown. Cartoon traces of viruses, electron-dense cargo, and membrane swirls are shown in parallel.
(B) MHV-infected cells (12 h pi) were processed for immuno-EM and labeled with anti-LAMP1 and 10-nm gold-coupled protein A.
(C) TEM images of VeroE6 cells infected with SARS-CoV-2 (24 h pi). Cartoon traces of viruses, electron-dense cargo, and membrane swirls are shown in parallel.
Representative images are shown. Scale bars are shown on the micrographs.
β-Coronaviruses and ER Chaperones Are Co-released during Infection
We next investigated whether any host proteins co-trafficked with MHV during egress. The KDEL receptor, an ER/Golgi apparatus-cycling transmembrane protein critical for retrieving escaped ER-resident proteins from the Golgi apparatus (Munro and Pelham, 1987) and its cargo, the KDEL sequence containing the ER lumenal chaperones GRP78/BIP (Figures 3 A–3C) and calreticulin (data not shown), were found to colocalize with LAMP1 and MHV during peak virus egress. Soluble ER chaperones generally do not escape the ER/Golgi apparatus and get secreted (Munro and Pelham, 1987). However, in MHV-infected cells, these chaperones were co-released with the virus, and this was not due to cell lysis because actin was undetectable in the extracellular medium (Figure 3D). Remarkably, chaperone release, much like MHV, was not inhibited by BFA when the drug was added during the peak virus egress period (8–14 h pi) (Figure 3E). Note that since the BFA molecular target GBF1 is required for the generation of coronavirus replication organelles, addition of BFA before 6 h pi suppressed replication (Verheije et al., 2008) and GRP78/BIP, similar to uninfected cells, was not released. These data demonstrate that, during egress, MHV and ER chaperones are co-trafficked to lysosomal organelles and released from cells through a route bypassing the BFA-sensitive biosynthetic secretory pathway.
Figure 3.

β-Coronaviruses and the Chaperone GRP78/BIP Are Co-released during Infection
(A and B) MHV-infected and uninfected cells coimmunostained with anti-KDEL receptor (green) and anti-MHV(MJ1.3) (red) antibodies. Scale bars, 5 μm (A) and 10 μm (B).
(C) MHV-infected cells coimmunostained with anti-GRP78/BIP (green), anti-LAMP1 (blue), and anti-MHV (MJ1.3) (red) antibodies. Scale bar, 2 μm.
(D) GRP78/BIP release from MHV-infected cells. The blot is representative of 2 independent experiments.
(E) GRP78/BIP release from MHV-infected cells with or without BFA(5 μg/mL) at the indicated times. All cell lysates and extracellular media were collected at 14 h pi. The blot is representative of 2 independent experiments.
Representative images are shown.
β-Coronaviruses and GRP78/BIP Use an Arl8b-Dependent Lysosomal Exocytic Pathway for Egress
Lysosome exocytosis is a known BFA-insensitive pathway whereby lysosomes traffic to the cell periphery and fuse with the plasma membrane to release their lumenal contents (Laulagnier et al., 2011). We conjectured that MHV may be exploiting this route for egress. In support of this, we found that plasma membrane LAMP1 levels were ~2.5-fold higher in infected cells (Figures 4A and 4B), implying significant fusion of lysosomes with the plasma membrane.
Figure 4.

β-Coronaviruses, Lysosomal Enzymes, and the Chaperone GRP78/BIP Are Co-released through Arl8b-Dependent Lysosome Exocytosis
(A) Surface LAMP1 (red) levels on uninfected and MHV-infected cells. Scale bar, 10 μm.
(B) Quantification of surface LAMP1 levels (n = 10 cells/group).
(C) Extracellular pro-cathepsin D and cathepsin D in uninfected and MHV-infected cells.
(D) Quantification of extracellular cathepsin D. Mean levels of 2 independent experiments were plotted.
(E) Quantification of extracellular pro-cathepsin D. Mean levels of 2 independent experiments were plotted.
(F) Frequency of lysosome plasma membrane fusion events (n = 7 cells for uninfected; n = 10 cells for MHV infected). Data represented are mean ± SD.
(G) Immunoelectron micrographs of LAMP1+/MHV+ organelles (pink arrows) docked at the plasma membrane. Note LAMP1 at the plasma membrane (white arrows). The scale bar is indicated on the micrograph.
(H) Arl8b-GFP-transfected, MHV-infected cells coimmunostained with anti-GFP (green), anti-LAMP1 (blue), and anti-MHV(MJ1.3) (red) antibodies. Arrows point to LAMP1/MHV/Arl8b-GFP+ lysosomes. Scale bar, 1 μm.
(I) MHV release in Arl8b siRNA-treated cells. Quantified viral genomes were plotted relative to non-target siRNA-treated cells. A representative triplicate dataset of genome levels from 4 independent experiments was plotted.
(J) GRP78/BIP release in Arl8b siRNA-treated and MHV-infected cells. Mean levels of 3 independent experiments were plotted.
Representative blot and images are shown. Data are shown as mean ± SEM unless otherwise indicated. p values were considered significant when p < 0.05 and denoted as ∗p < 0.05, ∗∗∗p < 0.0002. See also Figure S4.
Cathepsin D (~30 kD) is a proteolytic enzyme that is synthesized as pro-cathepsin D (~50 kD) and is cleaved into the mature form by lysosomal proteases (Samarel et al., 1989). Collection of extracellular media from MHV-infected cells at peak egress revealed ~2-fold more cathepsin D and ~3-fold more pro-cathepsin D secreted relative to uninfected cells (Figures 4C–4E).
Cell surface total internal reflection fluorescence (TIRF) imaging of transiently expressed pHluorin-LAMP1-mCherry (Raiborg et al., 2015) also showed ~3-fold more lysosome fusion events in MHV-infected than in uninfected cells (Figure 4F). Immuno-EM of infected cells showed LAMP1+/MHV+ organelles just beneath (Figure 4G, pink arrows) and LAMP1 on (Figure 4G, white arrows) the plasma membrane.
Arl8b is a small Arf-like Ras family GTPase that localizes to late endosomes/lysosomes (Boda et al., 2019; Khatter et al., 2015; Michelet et al., 2015, 2018; Xu et al., 2014) and regulates their movement to the plasma membrane and, ultimately, their exocytosis (Michelet et al., 2015). Cells transiently transfected with Arl8b-GFP and infected and coimmunostained with anti-GFP, anti-MHV (MJ1.3), and anti-LAMP1 antibodies revealed numerous Arl8b+/MHV+/LAMP+ organelles at peak egress stages (Figure 4H, arrows).
To interrogate the potential role of Arl8b in MHV release, we treated cells with non-targeting or targeting Arl8b small interfering RNAs (siRNA) for 48–72 h, infecting the cells with MHV, and collecting extracellular supernatant and cell lysates at peak egress times (12–14 h pi). Cell lysate measurements of protein levels demonstrated an ~50% downregulation of Arl8b in siRNA-treated cells (Figures S4 A and S4B). qPCR measurements of genomic MHV RNA demonstrated that Arl8b-depleted cells had decreased viral genome release by ~30-fold compared with non-target siRNA-treated cells, whereas RNA replication was unaffected (Figure 4I). Moreover, Arl8b depletion also resulted in a more than 50% decrease in extracellular GRP78/BIP levels (Figure 4J), confirming that MHV and GRP78/BIP utilize Arl8b-dependent lysosomal exocytic pathways for egress during infection.
Figure S4.

Modulating Coronavirus Egress, Related to Figure 4
(A) Extent of Arl8b depletion after siRNA treatment. Cell lysates probed with anti-Arl8b and anti-actin antibodies.
(B) Quantification of Arl8b depletion from 4 independent experiments.
(C) Effect of Rab27 depletion on MHV egress. TCID50/ml was calculated from extracellular media of non-target and Rab27a siRNA treated, MHV-infected cells (10hr pi). Mean data of 2 independent sets of experiments plotted.
(D) Effect of GW4869 on MHV egress. Infected cells in serum-free media were treated with GW4869 (10 μM) or DMSO (8-14 hr pi); extracellular media and cell lysates collected at 14hrpi and MHV genomic RNA quantified. Mean data of 2 independent sets of experiments plotted.
(E) Western blot of extracellular media from (D) probed with antibody against CD81, a marker for exosomes.
(F) Effect of BAPTA-AM calcium chelation on MHV egress and replication. Infected cells treated with BAPTA-AM (30 μM) or DMSO (8-12 hr pi) in calcium-free media with EGTA. Mean data of 2 independent sets of experiments plotted.
(G) Effect of Synaptotagmin VII depletion on MHV egress. Cells incubated with Synaptotagmin VII (50nM) or non-target siRNA (50nM) for 72 hr and infected with MHV. Extracellular medium and cell lysates collected at 14 hr pi. Mean data of 2 independent sets of experiments plotted.
(H) Extent of Synaptotagmin VII depletion. Western blot of cell lysates from (G) probed with anti-synaptotagmin VII and anti-actin antibodies.
Representative blots are shown. Data shown are mean ± SEM. p values were considered significant for p < 0.05 and denoted as ∗ where p < 0.05; ∗∗ where p < 0.01; ns = not significant
We then wanted to find out whether, in addition to lysosomes, direct fusion of late endosomes or multivesicular bodies (MVBs) with the plasma membrane contributed to MHV egress. Neither Rab27 GTPase depletion nor GW4869 treatment, both implicated in blocking this fusion pathway (Catalano and O’Driscoll, 2019) had any significant effect on MHV egress (Figures S4C–S4E). However, late endosomes/MVBs are intermediates in the biogenesis of lysosomes (Huotari and Helenius, 2011) and, thus, may have a role in viral trafficking to lysosomes.
CID1067700 Inhibits β-Coronavirus Egress
Rab7 is a small GTPase found on lysosomes, late endosomes, and MVBs (Figure 5 A). It has a critical role in lysosome biogenesis and lysosome maintenance (Bucci et al., 2000; Langemeyer et al., 2018); depletion of Rab7 inhibits maturation of late endosomes/MVBs and leads to reduced lysosome numbers in cells (Vanlandingham and Ceresa, 2009). Dual immuno-EM performed on Rab7-GFP-transfected/MHV-infected cells with anti-GFP and anti-MHV(MJ1.3) revealed MHV-loaded and empty lysosomes with characteristic electron-dense bodies, membrane swirls, and limiting membranes studded with Rab7-GFP (Figure 5A).
Figure 5.

CID1067700 Blocks β-Coronavirus Egress
(A) Immunoelectron micrographs of Rab7+ lysosomes with or without MHV particles. Cells were coimmunostained with anti-GFP, anti-MHV(MJ1.3), and gold-coupled secondary antibodies. Cartoon traces of viruses, electron-dense cargo, and membrane swirls are shown in parallel. Scale bars are indicated on the micrographs.
(B) Intracellular LAMP1 levels in MHV-infected cells treated with CID1067700 (8–14 h pi) or DMSO.
(C) LAMP1 and anti-MHV (MJ1.3) coimmunostaining in DMSO- and CID1067700-treated (40 μM, 8–14 h pi) cells. Scale bar, 5 μm.
(D) Quantification of (C); n = 10 cells/DMSO or CID1067700.
(E) Intracellular and extracellular MHV genomic RNA in DMSO- and CID1067700-treated (4 and 40 μM, 8–14 h pi) MHV-infected cells. MHV genomic RNA was plotted as fold change over uninfected from 3 independent experiments.
Representative blots and images are shown. Data are shown as mean ± SEM. p values were considered significant when p < 0.05 and denoted as ∗p < 0.05, ∗∗p < 0.01.
To investigate the effect of perturbing Rab7 activity on β-coronavirus egress, we treated MHV-infected cells from 8–14 h pi with the Rab7-selective competitive nucleotide binding inhibitor CID1067700 (Agola et al., 2012). After CID1067700 treatment, we measured an ~40% decrease in intracellular LAMP1 protein levels by western blot (Figure 5B) relative to DMSO-treated MHV-infected cells. Furthermore, we observed an ~50% decrease in LAMP1+ punctate organelles (per cell), suggesting a decrease in lysosome numbers (Figures 5C and 5D). These phenotypes are consistent with previous reports of Rab7 depletion (Vanlandingham and Ceresa, 2009), but the full lysosome proteome will need to be characterized to determine how CID1067700 affects lysosomes.
Most significantly, although CID1067700 had no effect on cell viability or replication (Figure 5E, intracellular), this compound potently decreased viral egress in a dose-dependent manner: by 100-fold at 4 μM and by 1,000-fold at 40 μM (Figure 5E, extracellular). These data further support a critical role of lysosomal biogenesis and exocytic pathways in regulating egress of β-coronaviruses and potentially identify a new class of potent therapeutic agents to impede their spread.
Lysosomes Are Deacidified and Lysosomal Enzymes Are Inactive in β-Coronavirus-Infected Cells
We then assessed the functional consequences of the path of egress taken by β-coronaviruses in terms of lysosomal function. We used LysoTracker Red DND-99, a cell-permeable weak base dye that is acidotropic and accumulates in acidified organelles (Sanman et al., 2016). Indeed, labeling cells with the dye prior to fixing with aldehydes and staining with anti-LAMP1 antibodies (without detergents) revealed near-complete localization of the dye fluorescence to LAMP1+ organelles; i.e., lysosomes and late endosomes (Figure 6 A).
Figure 6.

Lysosomes Are Deacidified and Lysosomal Enzymes Are Inactive in β-Coronavirus-Infected Cells
(A) LysoTracker Red DND-99 and LAMP1 co-staining in HeLa-mCC1a and VeroE6 cells. Scale bar, 5 μm.
(B) LysoTracker Red DND-99 staining of MHV-infected HeLa-mCC1a cells (12 h pi), primary mouse macrophages (12 h pi), and SARS-CoV-2-infected Vero E6 cells (24 h pi). Scale bars, 5 μm for HeLa-mCC1a and macrophages and 10 μm for Vero E6 cells.
(C) Mean LysoTracker Red DND-99 fluorescence intensity per punctum (i.e., late endosome/lysosome) in MHV/HeLa-mCC1, MHV/primary mouse macrophage, and CoV-2/VeroE6 infected cell groups; n = 20 cells/group; 10 puncta/cell scored.
(D) Number of LysoTracker Red DND-99 positive puncta (i.e., late endosome/lysosome) per MHV/HeLa-mCC1 or CoV-2/VeroE6 infected cell groups; n = 30 cells/group.
(E) pH of lysosomes in MHV-infected HeLa-mCC1a cells. Mean LysoSensor Green fluorescence intensity in uninfected and MHV-infected cell groups (n = 18 cells/group, 10 lysosome/cell) was converted to a pH value from calibration of the dye. Data represented are mean ± SD.
(F) Lysosome enzyme activity in uninfected and MHV-infected HeLa-mCC1a cell groups; n = 55 cells/group. The mean fluorescence intensity of lysosome substrate colocalizing with endocytosed dextran was measured for each group.
Representative images are shown. Data are shown as mean ± SEM unless otherwise indicated. p values were considered significant when p < 0.05 and denoted as ∗p < 0.05, ∗∗∗p < 0.0002. See also Figure S5.
HeLa-mCC1a cells and primary mouse macrophages infected with MHV for 12 h and Vero E6 cells infected with SARS-CoV-2 for up to 24 h were labeled with LysoTracker Red before imaging. We observed a stark decrease in LysoTracker Red fluorescence intensity per punctum and in the number of LysoTracker Red+ puncta, indicating that the acidity and number of acidified lysosomes in β-coronavirus-infected cells were decreased compared with uninfected cells (Figures 6B–6D). Separate LAMP1 staining confirmed that the decrease in LysoTracker Red+ puncta in β-coronavirus-infected cells was not due to a decrease in late endosome/lysosome quantities (Figures 1H and 1J).
LysoSensor Green DND-189 is a useful dye for quantifying pH in lysosomes because it accumulates in acidified organelles; it has a low pK of 5.2, which renders it non-fluorescent except in highly acidic organelles such as lysosomes, and its fluorescence amplitude changes with pH in a calibratable manner (Brazill et al., 2000). Using LysoSensor Green, we found that the mean pH of lysosomes in uninfected cells was pH 4.7 (with a range of 4.2–5.2), and in MHV-infected cells it was 5.7 (with a range of 5.0–6.4), very significant deacidification, one full pH unit higher than under uninfected conditions (Figures 6E).
Lysosomal enzymes are optimized to function in these organelles’ highly acidic pH, and even small increases in pH can be sufficient to decrease protease activity (Lie and Nixon, 2019; Sanman et al., 2016). Given our observations above, we quantified the in situ lysosomal enzyme activities using self-quenched enzymatic substrates that are taken up by endocytosis and targeted to lysosomes and turn fluorescent upon enzymatic activity (Humphries and Payne, 2012). To account for any potential change in endocytic uptake, we co-incubated cells with a pH-insensitive fluorophore-coupled dextran that was endocytosed along with substrate into lysosomes. The mean fluorescence intensity of the substrate was quantified in lysosomes of uninfected and MHV-infected cells with a similar mean dextran fluorescence intensity. These measurements revealed that, consistent with the observed increased lysosomal pH in MHV-infected cells, lysosome enzyme activities were reduced by ~40% relative to uninfected cells (Figure 6F). Furthermore, the increased secretion of pro-cathepsin D from MHV-infected cells (Figure 4E) likely reflects this decreased lysosomal enzymatic activity.
In that context, it has also been reported that the SARS-CoV-1 open reading frame protein 3A (ORF3a) is a viroporin that localizes to lysosomes, disrupts their acidification (Yue et al., 2018), and contributes to viral egress (Castaño-Rodriguez et al., 2018; Lu et al., 2006; Yue et al., 2018). We found that SARS-CoV-2 ORF3a (Gordon et al., 2020) was also targeted to lysosomes (Figure S5 A) and limited LysoTracker Red accumulation within them (Figure S5B). Although further investigation will be needed, these data suggest that ORF3a may be responsible for deacidifying lysosomes so that SARS-CoV-2 can use them for egress.
Figure S5.

SARS-CoV2-ORF3a and Lysosomes, Related to Figure 6
(A) Cells expressing strep-tagged SARS-CoV2-ORF3a were coimmunostained with anti-strep (red) and anti-LAMP1 (green). Inset shows extensive co-labeling of LAMP1 structures with ORF3a. Scale bar 5 μm.
(B) Cells ectopically expressing SARS-CoV2-ORF3a-GFP labeled with LysoTracker Red DND-99. Scale bar 10 μm.
Representative images shown.
Lysosome-Dependent Antigen Cross-Presentation Pathways Are Disrupted in β-Coronavirus-Infected Cells
Finally, we investigated the functional consequences of the altered lysosomal functions during β-coronavirus infection in terms of antigen processing. Myeloid cells rely on active lysosomal degradation of proteins to produce short peptides that are loaded and presented on major histocompatibility complex (MHC) class I of cells (Trombetta and Mellman, 2005). We tested whether the decrease in protein degradation by cells infected with MHV would limit antigen cross-presentation of antigens derived from large proteins while minimally affecting presentation of short peptides. We exposed bone marrow-derived primary macrophages to extracellular chicken ovalbumin (OVA1–385 protein) or to an OVA-derived MHC class I-restricted oligopeptide (OVA257–264 peptide) with or without MHV infection (Figures 7A and 7B). First we measured endocytosis of fluorescent OVA1–385 protein by macrophages and found that it was not significantly affected by MHV infection (Figure 7C). Then we measured the amount of OVA antigen being presented by macrophages using the H-2Kb/ OVA257–264-responsive OT-1 T cell receptor (TCR) transgenic T cells (Figure 7D). We found that coronavirus infection made macrophages induce stronger T cell activation when presenting OVA257–264 peptide, but weaker T cell activation was measured when cross-presenting OVA1–385 protein (Figure 7D). Such results point out how disrupted lysosomal function in infected cells alters antigen cross-presentation from proteins while possibly boosting presentation from peptides (e.g., by enhancing open conformers of MHC on the surface of cells through increased delivery by lysosome exocytosis).
Figure 7.

Lysosome-Dependent Antigen Cross-Presentation Pathways Are Disrupted in β-Coronavirus-Infected Cells
(A and B) Sketch and (B) timeline of assay to test the effect of MHV infection on antigen uptake and cross-presentation.
(C) Measurement of fluorescent OVA uptake by bone marrow-derived macrophages with or without prior infection with MHV.
(D) Measurement of OVA antigen presentation bone marrow-derived macrophages (with or without viral infection), measured as the percentage of CD69+ activated T cells.
(E and F) Sketch and (F) Timeline of the assay to measure the amount of HLA-F open conformers on the surface of infected HeLa-mCC1a cells, using a Jurkat KIR3DL1 reporter cell.
(G) ERK phosphorylation in KIR3DL1 reporter cells as a measure of the amount of open HLA-F conformers on the surface of HeLa cells (with or without viral infection). Phorbol myristate acetate (PMA) and null activation were used to normalize ERK phosphorylation, respectively.
All experiments were done in triplicate. Data are shown as mean ± SEM. p values are indicated on the plots.
We further tested whether coronavirus infection induces presentation of open conformers of MHC in human cells. Specifically, open conformers of human leukocyte antigen F (HLA-F) could stem from lysosomal dysfunction, serve as activating ligands for natural killer (NK) cells, and determine innate immune responses (Garcia-Beltran et al., 2016; Goodridge et al., 2013). We infected HeLa-mCC1a cells with MHV. At 12 h pi, we added KIR3DL1- immunoreceptor tyrosine based activation motif (ITAM) Jurkat reporter cells (Garcia-Beltran et al., 2016) and measured their ERK phosphorylation after 15 min by flow cytometry (Figures 7E and 7F). In parallel, we used acid stripping to open up MHC conformers. We found that MHV-infected cells triggered open HLA-F-responding Jurkat cells more efficiently, whereas acid stripping of cells induced a similar (enhanced) response with or without infection (Figure 7G). This demonstrated that β-coronavirus infection results in enhanced levels of open conformers of HLA-F, another functional immunologically relevant consequence of altered lysosomal activity and cellular stress.
Discussion
Since the 1970s, intact coronaviruses have been detected in lysosomes at late stages of infection (Ducatelle and Hoorens, 1984), but the significance of these observations remained unexplored. Here we demonstrated that β-coronaviruses egress from infected cells by tracking a path through lysosomal organelles. This is unlike other enveloped RNA viruses, whose egress tracks with the biosynthetic secretory pathway or directly buds out of the plasma membrane (Robinson et al., 2018; Ravindran et al., 2016; Pornillos et al., 2002). Consistent with previous reports (Machamer, 2013; McBride et al., 2007; Tooze et al., 1987), our experiments show that the secretory pathway remains largely operational in β-coronavirus-infected cells, with newly assembled virus particles budding into the lumen of the ER/ERGIC and trafficking to the Golgi apparatus/TGN during the early stages of infection. However, after reaching the Golgi apparatus/TGN, β-coronaviruses traffic to lysosomes and use exocytic lysosomes instead of the biosynthetic secretory pathway to egress (Figures 1, 2, 3, 4, and 5), as demonstrated by our experiments.
Work is in progress to delineate the route β-coronaviruses take to reach lysosomes, but least two BFA-insensitive (i.e., non-biosynthetic secretory) trafficking routes are possible (Strous et al., 1993). First is a direct route from the Golgi apparatus/TGN to lysosomes via late endosomes/MVBs. These organelles are known to mature into lysosomes through a Rab7 GTPase-regulated pathway (Stroupe, 2018). Indeed, when we treated cells with CID1067700, a competitive inhibitor of Rab7 activation, lysosome numbers appeared to be reduced, and β-coronavirus egress was inhibited by ~3 logs. CID1067700 has been used in vivo with little toxicity (Lam et al., 2016). Given this, it will be important to test whether CID1067700 can be an inhibitor of SARS-CoV-2 spread in mice and non-human primate models. A second, more circuitous route to lysosomes would involve retrograde transport back to the ER/ERGIC. From there, viruses would reach lysosomes, again through late endosome/MVB intermediates or through the little-understood process of microphagy, where lysosomes directly engulf the ER (Chino and Mizushima, 2020).
We report here that the KDEL receptor and its cargo, the KDEL sequence-containing ER chaperones GRP78/BIP and calreticulin (data not shown) were selectively co-trafficked with coronaviruses to lysosomes and co-released with them outside of the cell (Figure 3). In contrast, other secretory pathway resident proteins, such as mannosidase II, TGN46, Golgin 97, ERGIC53, and CI-MPR, remained behind and did not co-traffic to lysosomes (Figures S2 and S3). In the ER, GRP78/BIP, in its role as a chaperone, likely binds and helps fold newly synthesized coronavirus proteins. Surprisingly, our findings indicate that this interaction is maintained while coronaviruses egress through exocytic lysosomes. Notably, during entry, GRP78/BIP interaction with SARS/MERS has been reported to facilitate the infectivity of these viruses (Chu et al., 2018), and the S protein of SARS-CoV-2 has been postulated to directly bind GRP78/BIP (Ha et al., 2020). Therefore, one benefit of maintaining this interaction through exocytic lysosomes may be that coronaviruses are ready to infect a nearby cell as soon as they egress and are not limited by extracellular GRP78/BIP availability.
Lysosome exocytosis can be regulated by calcium (Rodríguez et al., 1997). When intracellular calcium stores were chelated with BAPTA-AM, we measured an ~2-log decrease in MHV egress, but replication was also decreased (Figure S4F). On the other hand, depleting synaptotagmin VII, considered a calcium-dependent trigger for lysosomal fusion with the plasma membrane (Martinez et al., 2000), resulted in decreased viral egress without significantly affecting replication (Figures S4G and S4H). The context of β-coronavirus egress and the precise roles of calcium, synaptotagmins, and others remain to be investigated further.
Lysosomal proteolytic enzyme activities are central in many critical cellular processes, including autophagy, cell motility, cholesterol metabolism, release of cell killing enzymes by T cells, pathogen degradation by macrophages, and self/non-self antigen presentation by all cells. Lysosome acidification is required for lysosomal enzyme stability and enzymatic activity, and even a small increase in pH is sufficient to inhibit these enzymes and stop their critical biological functions (Mindell 2012). Here we report significant deacidification of lysosomes in β-coronavirus-infected cells in conjunction with a reduction in lysosomal enzyme activity (Figures 6B–6F). Moreover, infected cells were found to secrete greater amounts of lysosomal enzymes to the extracellular environment than uninfected cells. The mechanism of deacidification is currently under investigation. One possibility is that lysosomes become deacidified indirectly because of being loaded with too much cargo (i.e., viruses) and/or perturbations in proton pump or ion channel trafficking (Ballabio and Bonifacino, 2020). Alternatively, deacidification may be a consequence of the action of specific coronavirus proteins that behave like viroporins. For example, the γ-coronavirus E protein increases Golgi apparatus/TGN pH levels from 6.8 to 7.1 (Ruch and Machamer, 2012; Westerbeck and Machamer, 2019). SARS-CoV-1/CoV-2 and Middle East respiratory syndrome coronavirus (MERS-CoV) express ORF3a, which has been shown previously (Yue et al., 2018) and here (Figures S5A and S5B) to traffic to lysosomes and disrupt their acidification. Importantly, SARS/MERS viruses deficient in ORF3a may be unable to egress (Castaño-Rodriguez et al., 2018; Lu et al., 2006; Yue et al., 2018), a phenotype that is consistent with acidic lysosomes being prohibitive to trafficking viral cargo.
Our findings here indicate that the altered lysosomal function of β-coronavirus-infected cells can result in perturbation of antigen presentation and lead to altered immune responses (Figure 7). The flurry of studies triggered by the coronavirus disease 2019 (COVID-19) pandemic has pointed out how unusual and problematic immune responses against coronaviruses can be (Vardhana and Wolchok, 2020). Indeed, clinicians as well as basic immunologists have difficulties reconciling observations (e.g., the delayed and erratic macrophage-driven cytokine release syndrome and the severe lymphopenia of CD4+ T and NK lymphocytes) with our current knowledge of immune responses against viruses or cancers (Vardhana and Wolchok, 2020). Our findings regarding the cellular biology of coronaviruses and their functional consequence in terms of altered antigen presentation may open new research avenues; e.g., focusing on the NK cell response against coronavirus-infected cells. The relevance of KIR3DS1-expressing NK cells in delaying progression to AIDS in HIV-1-infected patients (Martin et al., 2002) or exacerbating the severity of H1N1 infections (Aranda-Romo et al., 2012) points out how our findings of increased presentation of open conformers of HLA-F (a known ligand for activating KIR3DS1 and inhibiting KIR3DL1 receptors) could have positive and negative effects on immune responses against coronavirus infection (Garcia-Beltran et al., 2016). Furthermore, egress through lysosomal trafficking may also result in disruption of endolysosomal Toll-like receptor signaling, which requires acidification (de Bouteiller et al., 2005). A non-enveloped virus has been shown recently to also use lysosomes for its egress (Fernández de Castro et al., 2020). Given our findings of the functional consequences of harnessing lysosomes on host cell physiology, including antigen presentation and innate immunity, it is likely that more and more cases of viruses exploiting these organelles for egress will come to light.
In summary, our findings reveal that β-coronaviruses use an unexpected lysosome-based egress pathway, and this potentially opens up new therapeutic avenues to mitigate coronavirus infection and slow virus spread by targeting regulators of lysosomal trafficking and biogenesis, such as Arl8b and Rab7, and by reversing deacidification and/or enhancing immune responses against lysosomal defects.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-LAMP1 | Abcam | Cat.# ab24170; RRID: AB_775978 |
| Anti-LAMP1 (extracellular epitope) | R&D Systems | Cat.# AF4800; RRID: AB_1026176 |
| Anti-Cathepsin-D | Abcam | Cat.# ab6313; RRID: AB_305416 |
| Anti-Synaptotagmin VII | Invitrogen | Cat.# PA5-50270; RRID: AB_2635723 |
| Anti−β-Actin | Invitrogen | Cat.# MA5-15739;RRID: AB_10979409 |
| Anti-GRP78/BIP | Abcam | Cat.# ab21685;RRID: AB_2119834 |
| Anti-TGN46 | Abcam | Cat.# Ab50595;RRID: AB_2203289 |
| Anti-Golgin 97 | ThermoFisher | Cat.# A21270;RRID: AB_221447 |
| Anti-CI-MPR | Abcam | Cat.# Ab124767;RRID: AB_10974087 |
| Anti-SARS-CoV2/M | Antibodies-online | Cat.# ABIN6952906 |
| Anti-GFP | Millipore | Cat.# AB3080;RRID: AB_91337 |
| Anti-Arl8b (western) | Abcam | Cat.# Ab207697 |
| Anti-Mannosidase II | Abcam | Cat.# Ab12277;RRID: AB_2139551 |
| APC Anti-H-2Kb | Biolegend | Cat.# 116517;RRID: AB_10568693 |
| BV650 Anti-CD25 | Biolegend | Cat.# 102038;RRID: AB_2563060 |
| BV711 Anti-CD69 | Biolegend | Cat.# 104537;RRID: AB_2566120 |
| FITC Anti-Va2 TCR | Biolegend | Cat.# 127805;RRID: AB_1134186 |
| PE Anti-SIINFEK/H-2Kb | Invitrogen | Cat.# 12-5743-82;RRID: AB_925774 |
| PE-Cy7 Anti-CD11b | Biolegend | Cat.# 557743;RRID: AB_396849 |
| Anti-Gaussia Luciferase | ThermoFisher | Cat.# PA1-181;RRID: AB_2539912 |
| Reagents | ||
| DMEM high glucose | GIBCO | Cat.# 11965118 |
| Pen/Strep | Corning | Cat.# 30-002-CI |
| EMEM | GIBCO | Cat.# 670086 |
| FBS | Atlas Biologicals | Cat.# EF-05-00A |
| PFA | EMS | Cat.# 15710 |
| DPBS | GIBCO | Cat.# 14190144 |
| Fluoromont-G | Invitrogen | Cat.# 17984-24 |
| Gaussia Luciferase Glow Assay Kit | ThermoFisher Scientific | Cat.# 16160 |
| Glutaraldehyde | EMS | Cat.# 111-30-8 |
| Dyngo-4a | Sigma | Cat.# S7163 |
| BAPTA-AM | Sigma | Cat.# A4926 |
| BFA | Sigma | Cat.# 20350-15-6 |
| TNE Buffer | Quality Biological | Cat.# 351-302-101 |
| Nycodenz | Fisher Scientific | Cat.# AN1002423 |
| 0.4% Trypan Blue | Invitrogen | Cat.# T10282 |
| BCA Protein Assay Kit | ThermoFisher Scientific | Cat.# 23225 |
| TCA | Fisher Chemical | Cat.# A322-100 |
| Laemmli Sample Buffer | Bio Rad | Cat.# 1610747 |
| Trans-Blot Turbo 5X Buffer | Bio Rad | Cat.# 10026938 |
| TBS | Bio Rad | Cat.# 1706435 |
| Tween-20 | Affymetrix | Cat.# 90005-04-5 |
| Blotting Grade Blocker (Milk) | Bio Rad | Cat.# 1706404 |
| SuperBlock T20 Blocking Buffer | Thermo Scientific | Cat.# 37536 |
| SuperSignal West Pico Plus | Thermo Scientific | Cat.# 34580 |
| Quick-RNA Microprep Kit | Zymo Research | Cat.# R1051 |
| Maxima FirstStrand cDNA Synthesis | Fisher Scientific | Cat.# FERK1642 |
| SYBR Green Supermix | Bio Rad | Cat.# 1725124 |
| Lysosome-Specific Self-Quenched Substrate |
Abcam | Cat.# Ab234622 |
| Lysotracker Red DND-99 | ThermoFisher Scientific | Cat.# L7528 |
| Lysotracker Green DND-189 | ThermoFisher Scientific | Cat.# L7526 |
| Nigericin | Sigma | Cat.# 28643-80-3 |
| M-CSF | R&D | Cat.# 416-ML-010 |
| UltraComp e-beads | Invitrogen | Cat.# 01-222-42 |
| Trypsin-versene-EDTA mixture | Lonza | Cat.# 17-161E |
| PBS (for FACS Buffer) | Lonza | Cat.# 17-516F |
| Sodium Azide | Sigma | Cat.# S2002 |
| Primer Sequences | ||
| MHV A59 b Forward CTGACTTGCCCGCTTATGT | IDT | N/A |
| MHV A59 b Reverse GCTGATTCCTTCTGCCTCTATT | IDT | N/A |
| β−Actin Forward AGAGCTACGAGCTGCCTGAC | IDT | N/A N/A |
| β−Actin Reverse AGCACTGTGTTGGCGTACAG | IDT | |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57Bl6/J | Jackson labs | Cat.# Jax-00064 |
| Software & Algorithms | ||
| GraphPad Prism v8 | GraphPad | https://www.graphpad.com/ |
| FlowJo v10 | TreeStar | https://www.flowjo.com/ |
| Python 3.7 | Anaconda | https://www.anaconda.com/ |
| Zen2 Blue Edition | Zeiss | https://www.zeiss.com/corporate/int/home.html |
| Gen5 software | Synergy | https://www.biotek.com/ |
| Amersham Imager 600 Software | GE Lifesciences | https://www.cytivalifesciences.co.kr/ |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nihal Altan-Bonnet (nihal.altan-bonnet@nih.gov).
Materials Availability
This study did not generate any new unique resources or reagents. Further information on materials, dataset and protocols should be directed to and will be fulfilled by the Lead Contact, Nihal Altan-Bonnet (nihal.altan-bonnet@nih.gov).
Data and Code Availability
This study did not generate any unique datasets or code.
Experimental Model and Subject Details
Cell lines and cultures
HeLa-mCC1a cells were cultured in complete (with 10% Fetal Bovine Serum (FBS)) or serum-free Dulbecco’s Minimal Essential Medium (DMEM)/high glucose/Penicillin-/Streptomycin (Pen/Strep) and maintained at 37°C. Vero E6 cells were cultured in Eagle’s Minimal Essential Medium (EMEM) supplemented with 10% FBS and Pen/Strep and maintained at 37°C.
In-vitro macrophage culture
Primary mouse macrophages were prepared starting from bone marrow precursors harvested from 6-12-week old female C57Bl16 mice (Jackson Labs, Bar Harbor, ME). Femoral aspirates were collected, washed with complete RPMI once, resuspended at 1 million per ml in complete RPMI medium augmented with 1nM recombinant mouse M-CSF (R&D Systems, Minneapolis MN). The single cell suspension was then placed in Fluoroethyl polymer culture bags (Origen Biomedical, Austin TX) to expand and to differentiate. Medium was replaced with complete RPMI augmented with 1nM M-CSF after 3 days of culture. Macrophages were harvested after 7 days of culture, washed in complete RPMI, seeded in 96-flat well plates (10,000 cells per well, with 100μl complete RPMI) and left to adhere overnight for additional experiment.
Complete RPMI consists of RPMI 1640 supplemented with 10% heat–inactivated fetal bovine serum, 2 mM L–glutamine, 10 mM HEPES (pH 7.4), 0.1 mM non–essential amino acids, 1 mM sodium pyruvate, 100 μg/ml of penicillin, 100 μg/ml of streptomycin and 50 μM β–mercaptoethanol.
Method Details
Virus Infection
HeLa-mCC1a cells were infected with MHV-A59 for 4hrs, washed, and then kept in either complete (with 10%FBS) or serum-free DMEM/high glucose/Pen/Strep for desired period of time. Vero E6 cells were grown in Millicell EZ 8-well glass slides (Millipore) in infection media (EMEM, 4% FBS (Corning)) to a confluency of 90 – 100%. Cells were then infected with the SARS-CoV2 isolate USA-WA1/2020 at a MOI 1 for 24hrs.
TCID50/ml determination
Extracellular medium collected from infected cell cultures was serially diluted and dilutions used to inoculate multiple cell cultures for up to 72hr. Cytopathic changes in the cells were tabulated to calculate TCID50/mL values, as described (Leibowitz et al., 2011). Briefly, supernatant (media) from MHV infected and BFA treated cells were obtained and inoculated to HeLa-mCC1a cells seeded at 4X104 cells/well in 96-well format. 11 serial dilution of the media was prepared in DMEM (10−1 to 10−11) and added in triplicates for statistical significance. Cells were left in 37°C incubator for 72hrs. Thereafter the media was discarded and each well received 100 μL of Crystal Violet solution (25% crystal violet supplemented with 20% ethanol in double distilled water) and incubated for 15min. The solution was discarded, each well was washed twice with water and the image of the 96-well plate was captured. Dilution that showed 50% cell death (i.e., where 50% of the cells in dish were left behind, stained with Crystal Violet) was used for calculating TCID50/ml.
DNA transfections
All DNA transfections were carried out with Fugene 6 according to manufacturer instructions (Promega Corp.).Briefly, Fugene 6 was incubated with serum-free media for 5min and then mixed with plasmid DNA (Gaussia Luciferase, pHluorin-LAMP1-mCherry, Arl8b-GFP) in a separate tube and incubated for another 20min. The plasmid DNA / Fugene 6 mixture was pipetted slowly onto the culture of cells. Cells were kept at 37°C and utilized after 18-24hrs.
Drug Treatments
Brefeldin A (BFA) was prepared as a 5mg/ml stock solution in ethanol and Dyngo-4a was prepared as a 100mM stock in DMSO; both were stored long term at −20°C.
BAPTA-AM was prepared fresh (prior to experiment) as a 10mM stock solution in DMSO and not stored. BFA and Dyngo-4a treatments were done in cell culture media; For BAPTA-AM treatment, cells at 8hr pi were switched to calcium-free media supplemented with 2mM EGTA and 30 μM BAPTA-AM.
Immunofluorescence staining
Unless otherwise indicated, cells were fixed in 3.7% paraformaldehyde (PFA)/phosphate buffer solution (PBS) for 10 min; blocked in PBS/10%FBS. All primary and secondary antibody incubations were carried out in PBS/10%FBS supplemented with saponin at 0.2% for 1 hr at room temperature. Cells were rinsed in PBS and mounted with Fluoromount-G (Invitrogen).
For cell surface LAMP1 staining, cells were pre-chilled at 4°C for 20 min and incubated on ice with anti-LAMP1 (R & D) antibody in PBS for 30 min. After rinses with chilled PBS, cells were kept on ice and incubated with appropriate secondary antibody in PBS for 30min. Cells were rinsed with chilled PBS, fixed in chilled 2% PFA for 5min, rinsed and mounted.
Confocal Light Microscopy
All microscopy and image acquisition were performed on the LSM780 confocal microscope (Carl Zeiss USA) with a 63X/1.4 NA or 40X/1.3 NA oil objectives. Live cells were imaged on a heated stage. Cells were imaged with 12-bit resolution using pinhole settings optimized for either high resolution imaging or for fluorescence quantification where the pinhole was kept open such that fluorescence from entire organelle or cell volume could be acquired. Zen software (Carl Zeiss USA) were used for all image analysis including quantification of LAMP1, CI-MPR, Lysotracker Red DND-99 etc. positive organelles.
Total Internal Reflection Fluorescence (TIRF) Imaging
pHluorin-LAMP1-mCherry (gift of Harald Stanmark, University of Oslo, Norway) transfected cells were plated on coverglass chambers (Nunc Lab-Tek II, ThermoFisher). Cells were left uninfected or infected with MHV. After 4 hr, cells were washed with phenol red free DMEM/10%FBS/25mMHEPES pH 7.3 and kept in this media. After 10hr of infection, cells were placed on the heated ELYRA.PS1 microscope stage. Fluorescent fusion events at the plasma membrane (facing the coverslip) were imaged using the ELYRA.PS1 in its TIRF setting mode (488nm laser excitation and 505-550BP filter emission to image the pHluorin; 565 excitation and 575LP to image the mCherry) with an incident angle providing an evanescent field < 100nm. Time series, with no delay in between frames, was collected for a total of 3min. Appearance of punctate pHluorin fluorescence above background were counted as fusion events, quantified and plotted.
Immunoelectron Microscopy
HeLa-mCC1a cells were infected with MHV and fixed in 4% formaldehyde and 0.1% glutaraldehyde in 1x PHEM buffer for 90 min. Cryo-sectioning and immunolabelling were performed as described elsewhere (Griffiths et al., 1993; Tokuyasu, 1973). In brief, ultrathin sections (55–70 nm) from gelatin-embedded and frozen cell pellets were obtained using an FC7/UC7-ultramicrotome (Leica, Vienna, Austria). Immunogold labeling was carried out on thawed sections with anti-GFP (2.5 mg/ml, rabbit, Rockland, 600-401-215), J1.3 (1:50, mouse), anti-LAMP1 (1:20, rabbit) antibodies. Mouse primary antibodies were detected with polyclonal rabbit anti-mouse immunoglobulin Gs (0.5 mg/ml, Rockland, 610-40120). All samples were incubated with 5 or 10 nm protein A gold (1:50, UMC Utrecht University, Utrecht, Netherlands), as described (Griffiths et al., 1993), and stained/embedded in 4% uranyl acetate / 2% methylcellulose mixture (ratio 1:9) (Slot and Geuze, 2007; Tokuyasu, 1980). Sections were examined with a JEM-1200EX (JEOL USA) transmission electron microscope (accelerating voltage 80 keV) equipped with a bottom-mounted AMT 6-megapixel digital camera (Advanced Microscopy Techniques Corp).
Transmission Electron Microscopy
Vero E6 cells infected with SARS-CoV-2 and HeLa-mCC1a cells infected with MHV were fixed in 4% formaldehyde and 0.1% glutaraldehyde in 1x PHEM buffer for 12 hr. Cryo-sectioning were performed as described elsewhere (Griffiths et al., 1993; Tokuyasu, 1973). In brief, ultrathin sections (55–70 nm) from gelatin-embedded and frozen cell pellets were obtained using an FC7/UC7-ultramicrotome (Leica, Vienna, Austria) and stained/embedded in 4% uranyl acetate / 2% methylcellulose mixture (ratio 1:9) (Slot and Geuze, 2007; Tokuyasu, 1980). Sections were examined with a JEM-1200EX (JEOL USA) transmission electron microscope (accelerating voltage 80 keV) equipped with a bottom-mounted AMT 6-megapixel digital camera (Advanced Microscopy Techniques Corp).
Colocalization coefficient calculations
Rather than a Pearson correlation coefficient, we calculated colocalization coefficients as recommended in Manders et al. (1993) and computed the weighted colocalization coefficients WCC for the LAMP1 (lysosome) and protein M (MHV particle) channels. Using the colocalization toolbox from the ZEN software, we defined four quadrants for high/low fluorescence in each channel. Then we compute
where:
is the fluorescence of pixel high in both LAMP-1 and protein M fluorescence
is the total fluorescence for the LAMP-1 channel.
WCC varies between 0 and 1, with 0 corresponding to none of the lysosome pixel colocalizing with the M protein, and 1 corresponding to a perfect overlap where each lysosome pixel has a high M protein fluorescence. Calculation of colocalization coefficients for LAMP1 and CI-MPR was carried out as described above.
Lysotracker and Lysosensor pH measurements
Uninfected and MHV/SARS-Cov2-infected cells were incubated with Lysotracker Red DND-99 (100nM) or Lysosensor Green DND-189 (1 μM) according to manufacturer (ThermoFisher Scientific) instructions. For Lysosensor Green pH measurements, Step 1: cells were imaged with 458nm Argon laser excitation and 500-550nm bandpass emission filters. Step 2: the cells in step 1 were treated sequentially with potassium buffers of known pH containing 10 μM Nigericin. The images at each buffer condition were collected using the same image acquisition settings as in Step 1 to generate a standard pH curve. This standard curve was then used to convert the fluorescence values collected in step 1 to pH values.
Lysosomal in situ Enzyme Activity
Uninfected and MHV-infected cells were incubated with Alexa-555 10kD dextran (1mg/ml) and Lysosome-Specific Self-Quenched Substrate (Abcam Cat No. ab234622) at manufacturers recommended dosage for 1hr before they were fixed with 4% PFA at room temperature for 15 min. Cells were mounted with Fluoromount G (Invitrogen) containing DAPI and imaged with Zeiss LSM780 Confocal Laser Scanning microscope. Images were analyzed using Zen software. Mean fluorescence intensity of substrate was quantified in lysosomes with similar mean dextran fluorescence intensity in uninfected and MHV-infected cells.
Trypan blue staining for plasma membrane permeability/cell viability
After the incubation period cells were harvested from individual treatment groups and resuspended in 1ml of 1X PBS. A 1:1 mixture of cell suspension and 0.4% Trypan Blue stain (Invitrogen, Eugene, OR, Catalogue No. T10282) was made and incubated for 3 min at room temperature. 10μl of the mixture was added to Countess cell counting chamber slide (Invitrogen, Eugene, OR, Catalogue No. 100078809) and quantified cell viability in Countess Automated Cell Counter (Invitrogen, Eugene, OR, Catalogue No. C10227). In order to image the Trypan blue staining, cells were seeded in 4-well cover-glass bottom chamber slides and incubated overnight before being inoculated with virus. One group received 300nM of staurosporine, an apoptosis inducer, used as a positive control for comparing cell viability for uninfected and MHV-infected cells. At 14 hr pi media was replaced with 100μl of PBS and 0.4% Trypan Blue was added in a 1:1 ratio followed by incubation for ~3 min at room temperature. PBS was removed and cells were imaged by DIC.
Propidium Iodide staining for plasma membrane permeability/cell viability
After the incubation period cells were harvested from individual treatment groups and washed in 200μl of 1X PBS. 50μl of Trypsin Versene was then added to each well, and the plate was incubated for 10 min at 37°C. 150μl of FACS buffer was then added and cells were spun down and washed once in 200μl FACS buffer. Cells were then resuspended in 75μl of propidium iodide (PI) in FACS buffer (final concentration = 10μg/ml), incubated for 5min at room temperature and immediately run on a Fortessa Flow Cytometer: fluorescence of PI was acquired in the PE-CF594 channel, forward scattering (FSC) was also acquired. Live/Dead cells were then counted post-acquisition using FlowJo: Live cells were defined as FSC+PI- and dead cells were defined as FSC-PI+ (see Figure S1B). Each time point was set up in quadruplicate.
Gaussia Luciferase Assay
Gaussia Luciferase Assay was performed using the Pierce Gaussia Luciferase Glow assay kit (Thermo Scientific, USA, Catalog No. 16160). Fugene 6 was used to transfect HeLa-mCC1a cells with Gaussia luciferase plasmid and after mock or MHV infection and with and without BFA treatment, supernatants were collected and analyzed for Gaussia Luciferase activity as dictated by manufacturer instructions. Briefly, 15μl of the media from the treated wells was added to a black opaque 96-well plate and to that added 50μl of the working solution. After a 10 min incubation detected the glow luminescence in a Synergy H1 Hybrid Multi-Mode Reader (BioTek, Winooski, VT, USA).
Cell fractionation
Lysosome isolation was adapted from Graham et al. (1990). Cells were grown in 150cm2 dishes and divided into 3 groups: uninfected, MHV and MHV + Dyngo-4a. Cells were infected with MHV; washed at 4 hr pi with 1X PBS. 30 μM of Dyngo- 4a was added to one group at 6 hr pi. Cells were harvested at 12 hr pi from all 3 groups. Medium was removed, cells were washed with ice-cold PBS, scraped on ice, resuspended in 1mL of ice-cold TNE buffer (Tris NaCl EDTA buffer, Quality Biological, Gaithesburg, MD, Catalog No. 351-302-101; DNase, RNase, and Protease tested) and collected in a 2mL Eppendorf tube. After a quick freeze-thaw, cells were lysed on ice using a 28G syringe (15 strokes) and centrifuged 10 min at 800 g at 4°C to pellet nuclei and residual non-lysed cells. The supernatant was harvested and centrifuged 15 min at 20,000 g at 4°C. The obtained pellet was resuspended in 1mL TNE buffer and centrifuged once more 15 min at 20,000 g at 4°C. After a final resuspension in 1mL TNE buffer, it was loaded on top of a 25%–40% Nycodenz (Fisher Scientific, USA, Catalog No. AN1002423) discontinuous gradient and centrifuged for 2 hr at 100,000xg at 4°C using a SW40 Ti rotor (Beckman Coulter, Indianapolis, IN, Catalog No. 331301). Fractions were harvested from the top; aliquots of each fraction were set aside for western blot analysis with organelle markers; the rest was processed for qPCR analysis of genomic MHV RNA.
SDS-PAGE/Western Blot
Western Blot was performed from both cell lysates and supernatants. For cell lysates protein was quantified by Pierce BCA Protein Assay kit (Thermo Scientific, Rockford, IL, Catalogue No. 23225) while for supernatants protein was precipitated using Trichloroacetic Acid (Fisher Chemical, Fair Lawn, NJ, Catalogue No. A322-100) followed by acetone washes. Samples were boiled in Laemmli Sample Buffer (Bio Rad, USA, Catalogue No. 1610747) for 10min before running on pre-made SDS-PAGE gels (Bio Rad, USA). Gels were transferred on nitrocellulose membrane provided in the kit provide along with Trans-Blot Turbo 5X Buffer (Bio Rad, USA, Catalogue No. 10026938) and run in a Trans-Blot Turbo system as per the user manual. Following transfer blots were blocked using 5% non-fat milk blocking followed by probing with primary antibodies diluted in 5% Bovine Serum Albumin (BSA) and overnight incubation. Blots were thereafter washed with 1X TBS buffer (Bio-Rad, USA, Catalogue No. 1706435) supplemented with Tween-20 (Affymetrix, Maumee, OH, Catalogue No. 90005-04-5). Further re-probed with corresponding HRP conjugated secondary antibodies and incubated for 1hr followed by washing steps. Blots were developed using SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Scientific, Rockford, IL, Catalogue No. 34580) and imaged in Amersham Imager 600 (GE Healthcare Biosciences, Pistcataway, NJ). All densitometric quantification was done in Amersham Imager 600 software.
Extracellular protein detection and quantification
Serum free extracellular media from cells was collected and concentrated in a spin concentrator followed by TCA precipitation, acetone wash and air dry. Samples were resuspended in SDS-PAGE loading buffer with β-ME, boiled for 5 min and ran on 4%–20% gradient acrylamide gels (Bio Rad, USA) before being transferred onto nitrocellulose for western blotting with antibodies against targets of interest (cathepsin D, BIP/GRP78, actin etc.).
Quantitative(q) PCR Analysis
Cell lysates and supernatants were obtained from specific time points of virus and mock infections and lysed using RNA lysis buffer provided in the RNA isolation kit (Quick-RNA Microprep Kit, Zymo Research, Irvine, CA, Catalog No. R1051). RNA isolation was performed as per the manufacturer’s instructions and cDNA was prepared using Thermo Scientific Maxima First Strand cDNA Synthesis Kit for RT-qPCR (Fisher Scientific, Pittsburgh, PA, Catalog No. FERK1642). RT-PCR was performed using iTaq Universal SYBR® Green Supermix (BioRad, Hercules, CA, Catalog No. 1725124) in Roche LightCycler 96 system (Roche, Product No. 05815916001). The thermal cycling conditions were composed of a pre-incubation step of 95°C for 90 s followed by 40 cycles at 95°C for 10 sec, 54°C for 10 sec and 72°C for 110 sec. The samples were run in duplicate for each data point for an experiment. The primers used are mentioned below:
Primers with sequences
| MHV A59 b Forward | CTGACTTGCCCGCTTATGT |
| MHV A59 b Reverse | GCTGATTCCTTCTGCCTCTATT |
| β−Actin Forward | AGAGCTACGAGCTGCCTGAC |
| β−Actin Reverse | AGCACTGTGTTGGCGTACAG |
siRNA treatment
Arl8b, synaptotagmin VII and Non-Target siRNA (Horizon Therapeutics), with 25nM-50nM concentration range, were transfected with Dharmafect 1 (Horizon Therapeutics) and incubated for up to 72 hr pi. They were infected with MHV for 4 hr, before washing off the virus and switching to serum free media. At 12-14 hr pi, intracellular and extracellular virus was quantified by qPCR. For intracellular RNA levels, cells were lysed with RNA lysis buffer (Zymo Research); for intracellular protein level quantifications cells were scraped and lysed in cell lysis buffer (Invitrogen) containing protease inhibitors. For Rab27 depletion, siRNA was purchased from Ambion. In all siRNA treatments, both intracellular and extracellular proteins were TCA precipitated from serum-free media, acetone washed, air-dried and suspended in Laemmli gel loading buffer before SDS-PAGE/Western analysis.
Antigen cross-presentation by infected macrophages
Bone-marrow derived macrophages were prepared from femoral aspirates cultured for 7 days in complete RPMI medium with 1nM M-CSF in Fluoroethyl polymer culture bags (Origen). 2.104 cells were harvested and plated on plastic (96-flat-well plate), let to adhere for 2hrs, then exposed to MHV (or not) for 24 hr. Macrophages were then infected with SIINFEKL peptides or with chicken ovalbumin at varied concentrations for 2 hr, then washed with complete RPMI. C57Bl6 Rag1−/− OT-1 TCR Transgenic mouse splenocytes were then harvested, cleared of their red blood cells by ACK lysis, added and spun onto macrophages (105 cells per well), and incubated for 6 hr. Cell cultures were then harvested using a 15 min trypsin-versene treatment, washed and antibody-stained for flow cytometry (see panel below) with DAPI added just before acquisition. Cells were analyzed using a 5-laser FORTESSA flow cytometer (BD Bioscience) as well as single-stained compensatory UltraComp ebeads (Invitrogen). Data were compensated and processed using FlowJo (TreeStar) and a custom-written Python pipeline (https://www.python.org). The level of antigen presentation was quantified using the percentage of live activated T cells (itself estimated as % CD69+ among FSCintDAPI-Va2+ cells).
Antibody panel for cross-presentation assay
| Epitope | Fluorophore | Clone | Target specie |
|---|---|---|---|
| H-2Kb | APC | AF6-88.5 | mouse |
| CD25 | BV650 | PC61 | mouse |
| CD69 | BV711 | H1.2F3 | mouse |
| Va2 TCR | FITC | B20.1 | mouse |
| SIINFEK/H-2Kb | PE | 25D1.16 | mouse |
| CD11b | PE-Cy7 | M17/4 | human/mouse |
Endocytosis by infected primary macrophages
Chicken ovalbumin was fluorescently labeled with the Alexa647 dye using a bioconjugation kit (ThermoFisher). Bone-marrow derived macrophages were prepared as described in the previous paragraph. 2.104 cells were harvested, and plated on plastic (96-flat-well plate), let to adhere for 2 hr, then exposed to MHV (or not) for 24 hr. Macrophages were then infected with varying concentrations of fluorescently-labeled chicken ovalbumin for 2 hr, harvested with a 15 min exposure to a solution of trypsin-versene-EDTA mixture (Lonza), washed with FACS buffer (PBS/4%FBS/0.1%Sodium Azide) and immediately analyzed by flow cytometry.
Upregulation of HLA-F open conformers upon MHV infection
HeLa-mCC1a cells were plated and infected (or not) with MHV for 24 hr. Cells were lifted up using a trypsin-versene solution, washed with PBS, incubated for 1min at room temperature with a 0.1 M solution of Glycine in PBS (pH adjusted to 2.4) for acid stripping, or with PBS for control, then washed with complete RPMI twice. KIR3DL1-reporter Jurkat cells (Garcia-Beltran et al., 2016) were cultured in complete RPMI and harvested by aspiration. 5.104 HeLa-mCC1a cells were washed with Jurkat cell culture medium then resuspended with 5.104 Jurkat cells, spun at 100 g for 15 sec and incubated at 37°C for 15 min (some wells received only Jurkat cells or Jurkat cells and 1μMol of phorbol 12-myristate 13-acetate for negative and positive controls, respectively). Cells were then immediately resuspended with ice-cold 2% PFA for 115 min, permeabilized with ice-cold 90% Methanol for 15 min, washed with FACS buffer and stained for phospho-ERK (E10 clone, Cell Signaling Technology) and anti-mouse secondary antibody (Jackson Immunochemicals). A more detailed protocol can be found in Vogel et al., (Vogel et al., 2016). Cells were analyzed using a 5-laser FORTESSA flow cytometer (BD Bioscience). Data were processed using FlowJo (TreeStar) and a custom-written Python pipeline (python.org). The levels of open HLA-F conformers were quantified by monitoring Jurkat cell activation (itself estimated geometric mean of phosphor-ERK staining in FSCint Jurkat cells).
Quantification and Statistical Analysis
All graphs were plotted and unpaired two-tailed Student-t Test was performed using the GraphPad Prism 8 software or the SciPy Statistics library in Python. p values were considered significant for p < 0.05 unless otherwise indicated and denoted as ∗ where p < 0.05; ∗∗ where p < 0.01; where ∗∗∗ p < 0.0002; ∗∗∗∗ where p < 0.00001; and ns = not significant.
Acknowledgments
The authors thank Mary Carrington (NCI, Bethesda, MD), Rosa Puertollano (NHLBI, Bethesda MD), Jyoti Jaiswal (Children’s National Medical Center, Washington, DC), Norma Andrews (University of Maryland, College Park, MD), and Graham Brogden (University of Veterinary Medicine, Hannover, Germany) for critical discussions and reagents. P.M.T. was supported by NIH R01 A1091985-05, S.P. by NIH R01 NS36592, A.F. by F32-AI113973, V.H. by NIH R37GM058615, G.W. by NIH R01AI35270, and all other authors by intramural NIH and NCI funds.
Author Contributions
N.A.-B. and G.A.-B. wrote the manuscript. N.A.-B., G.A.-B., S.G., T.A.D.-R., A.K., and C.B. designed all experiments. S.P., C.A.M.d.H., J.K., and V.W.H. developed reagents. S.G., T.A.D.-R., A.K., P.M.T., C.B., S.R.A., G.A.-B., and N.A.-B. performed all experiments.
Declaration of Interests
The authors declare no competing interests.
Published: October 27, 2020
References
- Agola J.O., Hong L., Surviladze Z., Ursu O., Waller A., Strouse J.J., Simpson D.S., Schroeder C.E., Oprea T.I., Golden J.E., et al. A competitive nucleotide binding inhibitor: in vitro characterization of Rab7 GTPase inhibition. ACS Chem. Biol. 2012;7:1095–1108. doi: 10.1021/cb3001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda-Romo S., Garcia-Sepulveda C.A., Comas-García A., Lovato-Salas F., Salgado-Bustamante M., Gómez-Gómez A., Noyola D.E. Killer-cell immunoglobulin-like receptors (KIR) in severe A (H1N1) 2009 influenza infections. Immunogenetics. 2012;64:653–662. doi: 10.1007/s00251-012-0623-3. [DOI] [PubMed] [Google Scholar]
- Ballabio A., Bonifacino J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020;21:101–118. doi: 10.1038/s41580-019-0185-4. [DOI] [PubMed] [Google Scholar]
- Boda A., Lőrincz P., Takáts S., Csizmadia T., Tóth S., Kovács A.L., Juhász G. Drosophila Arl8 is a general positive regulator of lysosomal fusion events. Biochim. Biophys. Acta Mol. Cell Res. 2019;4:533–544. doi: 10.1016/j.bbamcr.2018.12.011. [DOI] [PubMed] [Google Scholar]
- Brazill D.T., Caprette D.R., Myler H.A., Hatton R.D., Ammann R.R., Lindsey D.F., Brock D.A., Gomer R.H. A protein containing a serine-rich domain with vesicle fusing properties mediates cell cycle-dependent cytosolic pH regulation. J. Biol. Chem. 2000;275:19231–19240. doi: 10.1074/jbc.M000900200. [DOI] [PubMed] [Google Scholar]
- Brown W.J., Goodhouse J., Farquhar M.G. Mannose-6-phosphate receptors for lysosomal enzymes cycle between the Golgi complex and endosomes. J. Cell Biol. 1986;103:1235–1247. doi: 10.1083/jcb.103.4.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci C., Thomsen P., Nicoziani P., McCarthy J., van Deurs B. Rab7: a key to lysosome biogenesis. Mol. Biol. Cell. 2000;11:467–480. doi: 10.1091/mbc.11.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaño-Rodriguez C., Honrubia J.M., Gutiérrez-Álvarez J., DeDiego M.L., Nieto-Torres J.L., Jimenez-Guardeño J.M., Regla-Nava J.A., Fernandez-Delgado R., Verdia-Báguena C., Queralt-Martín M., et al. Role of Severe Acute Respiratory Syndrome Coronavirus Viroporins E, 3a, and 8a in Replication and Pathogenesis. mBio. 2018;9:e02325-17. doi: 10.1128/mBio.02325-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalano M., O’Driscoll L. Inhibiting extracellular vesicles formation and release: a review of EV inhibitors. J. Extracell. Vesicles. 2019;9:1703244. doi: 10.1080/20013078.2019.1703244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Channappanavar R., Fehr A.R., Vijay R., Mack M., Zhao J., Meyerholz D.K., Perlman S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe. 2016;19:181–193. doi: 10.1016/j.chom.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.H., Du W., Hagemeijer M.C., Takvorian P.M., Pau C., Cali A., Brantner C.A., Stempinski E.S., Connelly P.S., Ma H.C., et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell. 2015;160:619–630. doi: 10.1016/j.cell.2015.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chino H., Mizushima N. ER-Phagy: Quality Control and Turnover of Endoplasmic Reticulum. Trends Cell Biol. 2020;30:384–398. doi: 10.1016/j.tcb.2020.02.001. [DOI] [PubMed] [Google Scholar]
- Chu H., Chan C.M., Zhang X., Wang Y., Yuan S., Zhou J., Au-Yeung R.K., Sze K.H., Yang D., Shuai H., et al. Middle East respiratory syndrome coronavirus and bat coronavirus HKU9 both can utilize GRP78 for attachment onto host cells. J. Biol. Chem. 2018;293:11709–11726. doi: 10.1074/jbc.RA118.001897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J.R., Lin L.D., Machamer C.E. Identification of a Golgi complex-targeting signal in the cytoplasmic tail of the severe acute respiratory syndrome coronavirus envelope protein. J. Virol. 2011;85:5794–5803. doi: 10.1128/JVI.00060-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Albuquerque N., Baig E., Ma X., Zhang J., He W., Rowe A., Habal M., Liu M., Shalev I., Downey G.P., et al. Murine hepatitis virus strain 1 produces a clinically relevant model of severe acute respiratory syndrome in A/J mice. J. Virol. 2006;80:10382–10394. doi: 10.1128/JVI.00747-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bouteiller O., Merck E., Hasan U.A., Hubac S., Benguigui B., Trinchieri G., Bates E.E., Caux C. Recognition of double-stranded RNA by human toll-like receptor 3 and downstream receptor signaling requires multimerization and an acidic pH. J. Biol. Chem. 2005;280:38133–38145. doi: 10.1074/jbc.M507163200. [DOI] [PubMed] [Google Scholar]
- de Haan C.A.M., Rottier P.J.M. Molecular interactions in the assembly of coronaviruses. Adv. Virus Res. 2005;64:165–230. doi: 10.1016/S0065-3527(05)64006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A., Kuo L., Masters P.S., Vennema H., Rottier P.J. Coronavirus particle assembly: primary structure requirements of the membrane protein. J. Virol. 1998;72:6838–6850. doi: 10.1128/jvi.72.8.6838-6850.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducatelle R., Hoorens J. Significance of lysosomes in the morphogenesis of coronaviruses. Arch. Virol. 1984;79:1–12. doi: 10.1007/BF01314299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett D.W. W.B. Saunders; 1966. The Cell. [Google Scholar]
- Fernández de Castro I., Tenorio R., Ortega-González P., Knowlton J.J., Zamora P.F., Lee C.H., Fernández J.J., Dermody T.S., Risco C. A modified lysosomal organelle mediates nonlytic egress of reovirus. J. Cell Biol. 2020;219:e201910131. doi: 10.1083/jcb.201910131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung T.S., Liu D.X. Post-translational modifications of coronavirus proteins: roles and function. Future Virol. 2018;13:405–430. doi: 10.2217/fvl-2018-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung T.S., Liu D.X. Human Coronavirus: Host-Pathogen Interaction. Annu. Rev. Microbiol. 2019;73:529–557. doi: 10.1146/annurev-micro-020518-115759. [DOI] [PubMed] [Google Scholar]
- Garcia-Beltran W.F., Hölzemer A., Martrus G., Chung A.W., Pacheco Y., Simoneau C.R., Rucevic M., Lamothe-Molina P.A., Pertel T., Kim T.-E., et al. Open conformers of HLA-F are high-affinity ligands of the activating NK-cell receptor KIR3DS1. Nat. Immunol. 2016;17:1067–1074. doi: 10.1038/ni.3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodridge J.P., Lee N., Burian A., Pyo C.W., Tykodi S.S., Warren E.H., Yee C., Riddell S.R., Geraghty D.E. HLA-F and MHC-I open conformers cooperate in a MHC-I antigen cross-presentation pathway. J. Immunol. 2013;191:1567–1577. doi: 10.4049/jimmunol.1300080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O’Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L., et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham J.M., Ford T., Rickwood D. Isolation of the major subcellular organelles from mouse liver using Nycodenz gradients without the use of an ultracentrifuge. Anal. Biochem. 1990;187:318–323. doi: 10.1016/0003-2697(90)90463-j. [DOI] [PubMed] [Google Scholar]
- Griffiths G., Burke B., Lucocq J. Springer; 1993. Fine structure immunocytochemistry. [Google Scholar]
- Ha D.P., Van Krieken R., Carlos A.J., Lee A.S. The stress-inducible molecular chaperone GRP78 as potential therapeutic target for coronavirus infection. J. Infect. 2020;81:452–482. doi: 10.1016/j.jinf.2020.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries W.H., 4th, Payne C.K. Imaging lysosomal enzyme activity in live cells using self-quenched substrates. Anal. Biochem. 2012;424:178–183. doi: 10.1016/j.ab.2012.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huotari J., Helenius A. Endosome maturation. EMBO J. 2011;30:3481–3500. doi: 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanolkar A., Pewe L., Tifrea D., Perlman S., Harty J.T. Developing MHV-1 infection as an animal model for Severe Acute Respiratory Syndrome (SARS) (43.30) J. Immunol. 2007;178:S42. [Google Scholar]
- Khatter D., Sindhwani A., Sharma M. Arf-like GTPase Arl8: Moving from the periphery to the center of lysosomal biology. Cell. Logist. 2015;5:e1086501. doi: 10.1080/21592799.2015.1086501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam T., Kulp D.V., Wang R., Lou Z., Taylor J., Rivera C.E., Yan H., Zhang Q., Wang Z., Zan H., et al. Small Molecule Inhibition of Rab7 Impairs B Cell Class Switching and Plasma Cell Survival To Dampen the Autoantibody Response in Murine Lupus. J. Immunol. 2016;197:3792–3805. doi: 10.4049/jimmunol.1601427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langemeyer L., Fröhlich F., Ungermann C. Rab GTPase Function in Endosome and Lysosome Biogenesis. Trends Cell Biol. 2018;28:957–970. doi: 10.1016/j.tcb.2018.06.007. [DOI] [PubMed] [Google Scholar]
- Laulagnier K., Schieber N.L., Maritzen T., Haucke V., Parton R.G., Gruenberg J. Role of AP1 and Gadkin in the traffic of secretory endo-lysosomes. Mol. Biol. Cell. 2011;22:2068–2082. doi: 10.1091/mbc.E11-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibowitz J., Kaufman G., Liu P. Coronaviruses: propagation, quantification, storage, and construction of recombinant mouse hepatitis virus. Curr. Protoc. Microbiol. 2011 doi: 10.1002/9780471729259.mc15e01s21. Chapter 15, Unit 15E.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie P.P.Y., Nixon R.A. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol. Dis. 2019;122:94–105. doi: 10.1016/j.nbd.2018.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J., Yuan L.C., Bonifacino J.S., Klausner R.D. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell. 1989;56:801–813. doi: 10.1016/0092-8674(89)90685-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Q.X., Tang X.J., Shi Q.L., Li Q., Deng H.J., Yuan J., Hu J.L., Xu W., Zhang Y., Lv F.J., et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat. Med. 2020;26:1200–1204. doi: 10.1038/s41591-020-0965-6. [DOI] [PubMed] [Google Scholar]
- Lu W., Zheng B.J., Xu K., Schwarz W., Du L., Wong C.K., Chen J., Duan S., Deubel V., Sun B. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc. Natl. Acad. Sci. USA. 2006;103:12540–12545. doi: 10.1073/pnas.0605402103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N., et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machamer C.E. Accommodation of large cargo within Golgi cisternae. Histochem. Cell Biol. 2013;140:261–269. doi: 10.1007/s00418-013-1120-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders E.M.M., Verbeek F.J., Aten J.A. Measurement of co localization of objects in duaå colour confocal images. J. Microsc. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- Martin M.P., Gao X., Lee J.H., Nelson G.W., Detels R., Goedert J.J., Buchbinder S., Hoots K., Vlahov D., Trowsdale J., et al. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat. Genet. 2002;31:429–434. doi: 10.1038/ng934. [DOI] [PubMed] [Google Scholar]
- Martinez I., Chakrabarti S., Hellevik T., Morehead J., Fowler K., Andrews N.W. Synaptotagmin VII regulates Ca(2+)-dependent exocytosis of lysosomes in fibroblasts. J. Cell Biol. 2000;148:1141–1149. doi: 10.1083/jcb.148.6.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride C.E., Li J., Machamer C.E. The cytoplasmic tail of the severe acute respiratory syndrome coronavirus spike protein contains a novel endoplasmic reticulum retrieval signal that binds COPI and promotes interaction with membrane protein. J. Virol. 2007;81:2418–2428. doi: 10.1128/JVI.02146-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelet X., Garg S., Wolf B.J., Tuli A., Ricciardi-Castagnoli P., Brenner M.B. MHC class II presentation is controlled by the lysosomal small GTPase, Arl8b. J. Immunol. 2015;194:2079–2088. doi: 10.4049/jimmunol.1401072. [DOI] [PubMed] [Google Scholar]
- Michelet X., Tuli A., Gan H., Geadas C., Sharma M., Remold H.G., Brenner M.B. Lysosome-Mediated Plasma Membrane Repair Is Dependent on the Small GTPase Arl8b and Determines Cell Death Type in Mycobacterium tuberculosis Infection. J. Immunol. 2018;200:3160–3169. doi: 10.4049/jimmunol.1700829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S.G., Carnell L., Moore H.H. Post-Golgi membrane traffic: brefeldin A inhibits export from distal Golgi compartments to the cell surface but not recycling. J. Cell Biol. 1992;118:267–283. doi: 10.1083/jcb.118.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell J.A. Lysosomal acidification mechanisms. Annu. Rev. Physiol. 2012;74:69–86. doi: 10.1146/annurev-physiol-012110-142317. [DOI] [PubMed] [Google Scholar]
- Munro S., Pelham H.R. A C-terminal signal prevents secretion of luminal ER proteins. Cell. 1987;48:899–907. doi: 10.1016/0092-8674(87)90086-9. [DOI] [PubMed] [Google Scholar]
- Narayanan K., Maeda A., Maeda J., Makino S. Characterization of the coronavirus M protein and nucleocapsid interaction in infected cells. J. Virol. 2000;74:8127–8134. doi: 10.1128/jvi.74.17.8127-8134.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostra M., de Haan C.A., de Groot R.J., Rottier P.J. Glycosylation of the severe acute respiratory syndrome coronavirus triple-spanning membrane proteins 3a and M. J. Virol. 2006;80:2326–2336. doi: 10.1128/JVI.80.5.2326-2336.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park R.J., Shen H., Liu L., Liu X., Ferguson S.M., De Camilli P. Dynamin triple knockout cells reveal off target effects of commonly used dynamin inhibitors. J. Cell Sci. 2013;126:5305–5312. doi: 10.1242/jcs.138578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrier A., Bonnin A., Desmarets L., Danneels A., Goffard A., Rouillé Y., Dubuisson J., Belouzard S. The C-terminal domain of the MERS coronavirus M protein contains a trans-Golgi network localization signal. J. Biol. Chem. 2019;294:14406–14421. doi: 10.1074/jbc.RA119.008964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pornillos O., Garrus J.E., Sundquist W.I. Mechanisms of enveloped RNA virus budding. Trends Cell Biol. 2002;12:569–579. doi: 10.1016/s0962-8924(02)02402-9. [DOI] [PubMed] [Google Scholar]
- Puelles V.G., Lütgehetmann M., Lindenmeyer M.T., Sperhake J.P., Wong M.N., Allweiss L., Chilla S., Heinemann A., Wanner N., Liu S., et al. Multiorgan and Renal Tropism of SARS-CoV-2. N. Engl. J. Med. 2020;383:590–592. doi: 10.1056/NEJMc2011400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiborg C., Wenzel E.M., Pedersen N.M., Olsvik H., Schink K.O., Schultz S.W., Vietri M., Nisi V., Bucci C., Brech A., et al. Repeated ER-endosome contacts promote endosome translocation and neurite outgrowth. Nature. 2015;520:234–238. doi: 10.1038/nature14359. [DOI] [PubMed] [Google Scholar]
- Ravindran M.S., Bagchi P., Cunningham C.N., Tsai B. Opportunistic intruders: how viruses orchestrate ER functions to infect cells. Nat. Rev. Microbiol. 2016;14:407–420. doi: 10.1038/nrmicro.2016.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M., Schor S., Barouch-Bentov R., Einav S. Viral journeys on the intracellular highways. Cell. Mol. Life Sci. 2018;75:3693–3714. doi: 10.1007/s00018-018-2882-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez A., Webster P., Ortego J., Andrews N.W. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J. Cell Biol. 1997;137:93–104. doi: 10.1083/jcb.137.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruch T.R., Machamer C.E. The coronavirus E protein: assembly and beyond. Viruses. 2012;4:363–382. doi: 10.3390/v4030363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samarel A.M., Ferguson A.G., Decker R.S., Lesch M. Effects of cysteine protease inhibitors on rabbit cathepsin D maturation. Am. J. Physiol. 1989;257:C1069–C1079. doi: 10.1152/ajpcell.1989.257.6.C1069. [DOI] [PubMed] [Google Scholar]
- Sanman L.E., van der Linden W.A., Verdoes M., Bogyo M. Bifunctional Probes of Cathepsin Protease Activity and pH Reveal Alterations in Endolysosomal pH during Bacterial Infection. Cell Chem. Biol. 2016;23:793–804. doi: 10.1016/j.chembiol.2016.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu Y.L., Teoh K.T., Lo J., Chan C.M., Kien F., Escriou N., Tsao S.W., Nicholls J.M., Altmeyer R., Peiris J.S.M., et al. The M, E, and N structural proteins of the severe acute respiratory syndrome coronavirus are required for efficient assembly, trafficking, and release of virus-like particles. J. Virol. 2008;82:11318–11330. doi: 10.1128/JVI.01052-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slot J.W., Geuze H.J. Cryosectioning and immunolabeling. Nat. Protoc. 2007;2:2480–2491. doi: 10.1038/nprot.2007.365. [DOI] [PubMed] [Google Scholar]
- Snijder E.J., van der Meer Y., Zevenhoven-Dobbe J., Onderwater J.J., van der Meulen J., Koerten H.K., Mommaas A.M. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J. Virol. 2006;80:5927–5940. doi: 10.1128/JVI.02501-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Limpens R.W.A.L., de Wilde A.H., de Jong A.W.M., Zevenhoven-Dobbe J.C., Maier H.J., Faas F.F.G.A., Koster A.J., Bárcena M. A unifying structural and functional model of the coronavirus replication organelle: Tracking down RNA synthesis. PLoS Biol. 2020;18:e3000715. doi: 10.1371/journal.pbio.3000715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohlman S.A., Brayton P.R., Fleming J.O., Weiner L.P., Lai M.M. Murine coronaviruses: isolation and characterization of two plaque morphology variants of the JHM neurotropic strain. J. Gen. Virol. 1982;63:265–275. doi: 10.1099/0022-1317-63-2-265. [DOI] [PubMed] [Google Scholar]
- Stroupe C. This Is the End: Regulation of Rab7 Nucleotide Binding in Endolysosomal Trafficking and Autophagy. Front. Cell Dev. Biol. 2018;6:129. doi: 10.3389/fcell.2018.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strous G.J., van Kerkhof P., van Meer G., Rijnboutt S., Stoorvogel W. Differential effects of brefeldin A on transport of secretory and lysosomal proteins. J. Biol. Chem. 1993;268:2341–2347. [PubMed] [Google Scholar]
- Tannous B.A. Gaussia luciferase reporter assay for monitoring biological processes in culture and in vivo. Nat. Protoc. 2009;4:582–591. doi: 10.1038/nprot.2009.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuyasu K.T. A technique for ultracryotomy of cell suspensions and tissues. J. Cell Biol. 1973;57:551–565. doi: 10.1083/jcb.57.2.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuyasu K.T. Immunochemistry on ultrathin frozen sections. Histochem. J. 1980;12:381–403. doi: 10.1007/BF01011956. [DOI] [PubMed] [Google Scholar]
- Tooze J., Tooze S.A., Fuller S.D. Sorting of progeny coronavirus from condensed secretory proteins at the exit from the trans-Golgi network of AtT20 cells. J. Cell Biol. 1987;105:1215–1226. doi: 10.1083/jcb.105.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze S.A., Tooze J., Warren G. Site of addition of N-acetyl-galactosamine to the E1 glycoprotein of mouse hepatitis virus-A59. J. Cell Biol. 1988;106:1475–1487. doi: 10.1083/jcb.106.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombetta E.S., Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu. Rev. Immunol. 2005;23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538. [DOI] [PubMed] [Google Scholar]
- Vanlandingham P.A., Ceresa B.P. Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J. Biol. Chem. 2009;284:12110–12124. doi: 10.1074/jbc.M809277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardhana S.A., Wolchok J.D. The many faces of the anti-COVID immune response. J. Exp. Med. 2020;217:e20200678. doi: 10.1084/jem.20200678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheije M.H., Raaben M., Mari M., Te Lintelo E.G., Reggiori F., van Kuppeveld F.J., Rottier P.J., de Haan C.A. Mouse hepatitis coronavirus RNA replication depends on GBF1-mediated ARF1 activation. PLoS Pathog. 2008;4:e1000088. doi: 10.1371/journal.ppat.1000088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel R.M., Erez A., Altan-Bonnet G. Dichotomy of cellular inhibition by small-molecule inhibitors revealed by single-cell analysis. Nat. Commun. 2016;7:12428. doi: 10.1038/ncomms12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerbeck J.W., Machamer C.E. The Infectious Bronchitis Coronavirus Envelope Protein Alters Golgi pH To Protect the Spike Protein and Promote the Release of Infectious Virus. J. Virol. 2019;93:e00015–e00019. doi: 10.1128/JVI.00015-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson B.N., Feldmann F., Schwarz B., Meade-White K., Porter D.P., Schulz J., van Doremalen N., Leighton I., Yinda C.K., Pérez-Pérez L., et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS-CoV-2. Nature. 2020;585:273–276. doi: 10.1038/s41586-020-2423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M., Liu K., Swaroop M., Sun W., Dehdashti S.J., McKew J.C., Zheng W. A phenotypic compound screening assay for lysosomal storage diseases. J. Biomol. Screen. 2014;19:168–175. doi: 10.1177/1087057113501197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y., Nabar N.R., Shi C.S., Kamenyeva O., Xiao X., Hwang I.Y., Wang M., Kehrl J.H. SARS-Coronavirus Open Reading Frame-3a drives multimodal necrotic cell death. Cell Death Dis. 2018;9:904. doi: 10.1038/s41419-018-0917-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler C.G.K., Allon S.J., Nyquist S.K., Mbano I.M., Miao V.N., Tzouanas C.N., Cao Y., Yousif A.S., Bals J., Hauser B.M., et al. HCA Lung Biological Network. Electronic address: lung-network@humancellatlas.org. HCA Lung Biological Network SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell. 2020;181:1016–1035.e19. doi: 10.1016/j.cell.2020.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate any unique datasets or code.