

Abstract
We show that accurate exchange–correlation hybrid functionals give very physically optimized effective-correlation potentials, capable of correctly describing the quantum oscillations of atoms and molecules. Based on this analysis and on understanding the error cancellation between semilocal exchange and correlation functionals, we propose a very simple, semilocal correlation potential model compatible with the exact exchange of density functional theory, which performs remarkably well for charge densities and orbital energies.
1. Introduction
The ground-state Kohn–Sham (KS)1 self-consistent, orbital formulation of density functional theory (DFT)2 is the most used method for electronic structure calculations. In KS–DFT, the noninteracting kinetic energy functional, representing usually a dominant/important part of the total energy,3−5 is treated exactly with KS one-particle orbitals, and only the exchange–correlation (XC) energy Exc[ρ] must be approximated as a functional of the electronic density ρ(r). Even if the XC energy is just a small fraction of the total energy, it contains all the many-body effects beyond the Hartree method, having crucial theoretical and computational importance.
Starting from the local density approximation (LDA),1 many useful semilocal density functional approximations (DFA) such as generalized gradient approximations (GGAs)6 and meta-GGAs7 have been developed. Most of the standard semilocal XC functionals work by mutual error cancellation effects between the exchange and correlation parts.8,9 The error cancellation is significantly diminished for the sophisticated meta-GGA functionals (in comparison with the LDA), but this problem is still important especially in the low-density regime, where the correlation energy starts to behave as the exchange energy under the uniform density scaling of the density.10 Noting that in most of the non-covalent molecular bonds, the density is small,11 we may conclude that the error cancellation issue plays important role in semilocal DFT calculations of material science. To exemplify this problem, we show in Figure 1 the relative errors (RE) in % of jellium surface exchange, correlation, and XC energies. Even if the LDA is one of the most accurate functionals for jellium and metal surface energies,12,13 its outstanding performance is based on a huge error cancellation between exchange and correlation. The popular PBE GGA14 improves the exchange and correlation parts, as reported in the figure, but the total XC results are not as accurate as the LDA ones. Another class of DFAs widely applied in quantum chemistry calculation are the hybrid functionals.17 They are mixture of a fraction of the Hartree–Fock (HF) exchange energy with fractions of given semilocal exchange and correlation functionals. Similar in popularity are the functionals based on range-separation18,19 of Coulomb electron–electron interaction wee(r12) = 1/r12 into the short-range and long-range contributions (wee(r12) = weesr(r12) + wee(r12)). The former interaction is described by specially developed short-range semilocal DFAs,20−23 whereas the later one, in general, takes the form of long-range HF exchange energy which captures the right −1/r dependence in the tail of XC potential. From here on, we will refer to these two types of functionals as hybrid functionals.
Figure 1.

RE (in %) of the LDA and PBE jellium surface exchange, correlation, and XC energies, vs the bulk parameter rs. The correlation and exchange-only reference values are the diffusion Monte Carlo15 and the exact exchange (EXX)16 ones, respectively.
Because the HF optimum potential is non-local, the calculations for both types of functionals (and meta-GGAs as well) are typically done within the generalized KS framework.24,25 However, in order to remain in the true KS self-consistent scheme, the HF should be replaced with the KS EXX that gives a local-multiplicative exchange potential through the optimized effective potential (OEP) approach.26−29 The HF- and OEP-based hybrids give practically the same accuracy for ground-state properties30−32 (see also Figure S4 of ref (33)), differing only for quantities involving excited states, electron affinities, and band gaps.34 Moreover, EXX satisfies the uniform scaling relation (Ex[ρλ] = λEx[ρ], with ρλ(r) = λ3ρ(λr) being the uniformly scaled density and λ ≥ 0), in contrast to HF that (slightly) violates this condition.35
In the following, we show that XC hybrid functionals give very physically optimized effective correlation potentials, capable of correctly describing the quantum oscillations of atoms and molecules. Moreover, based on the analysis of error cancellation between semilocal exchange and correlation functionals, we propose a very simple, semilocal correlation potential model compatible with the EXX potential supported by analysis of charge densities and orbital energies.
2. Theory
In general, for a given KS orbital-(ϕpσ) and/or eigenvalue-(εpσ) dependent XC energy functional, the OEP equation for the XC potential reads26,27,29,36−40
| 1 |
which is an integral equation (Fredholm of the first kind) with the inhomogeneity given by
| 2 |
where Xσ–1 is the inverse of the static KS linear response function
| 3 |
The KS orbitals and eigenvalues are determined by solutions of standard KS equations
| 4 |
with vext(r) and vJ(r) being the external (nuclear) and the Coulomb/Hartree potentials, respectively. In all equations, we use the convention that i, j, ... label occupied KS orbitals, a, b, ... label virtual ones; the indexes p, q, ... are used otherwise, while σ, τ denotes the spin degrees of freedom. In particular, the corresponding OEP potential of EXX energy expression
| 5 |
labeled as OEPx is defined by the
| 6 |
with (pσqσ|rσsσ) being a two-electron integral (in the Mulliken notation) computed using KS orbitals. Let us now consider the OEP-versions of several most popular hybrid functionals, namely, the global hybrid B3LYP42 and PBE043 XC functionals, and one example of range-separated XC functional, that is, ωPBE,44,45 where only exchange-part of functional is divided into the short- and long-range part and correlation is kept in the original PBE14 form.
Now, using the OEP formalism, we are able to calculate the correlation potential vc(r) = δEc[ρ]/δρ(r), where the correlation energy functional is by definition
| 7 |
Keeping eq 7 in mind, the corresponding correlation energy functionals (for all aforementioned XC functionals) have the following expressions
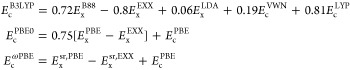 |
8 |
which, as one can note, depend explicitly on EXX energy.
3. Computational Details
All calculation have been carried out with locally modified version of ACES II46 program. As in our previous studies8,31,32,47−49 in order to solve OEP equation (eq 1), we have employed the finite-basis set procedure of refs (50) and (51). To calculate pseudo-inverse of density–density response matrix, we have utilized a truncated singular-value decomposition (TSVD). This step is essential for determining stable and physically meaningful OEP solutions.48,52,53 The cutoff criteria in the TSVD procedure was set to 10–6. For more technical details on this type of calculations, we refer the reader to refs (8) and (48).
In all calculations, we employed uncontracted triple zeta quality basis sets as in refs (9) and (48), namely, an even tempered 20s10p2d basis set8 for He and He2, an uncontracted ROOS-ATZP basis set54 for Ne atom and Ne2, and for Ar atom, we used a modified basis set which combines s and p type basis functions from the uncontracted ROOS-ATZP54 with d and f functions coming from the uncontracted aug-cc-pwCVQZ basis set.55 Remaining systems were treated in uncontracted cc-pVTZ basis set of dunning.56 For all molecular systems, we considered their equilibrium geometries from refs (57−59) also used in our previous studies.8,48 To assess the results, we considered the reference data from the Ab Initio DFT OEP2-sc method60,61 and CCSD(T)62 calculations. The reference KS[CCSD(T)] correlation potentials (depicted on Figures 2 and 4) and CCSD(T) KS orbital energies (used to calculated error statistics in Table 1) have been obtained using inverse KS method from ref (41) taking as a starting point the relaxed densities63−66 constructed using the Lagrangian approach.67−70 For more technical details, we refer the reader to refs (8, 41, 71).
Figure 2.

Correlation potentials of the Ne (upper panel), Ar (middle panel) atoms, and CO molecule (lower panel) for various methods. The reference KS[CCSD(T)] have been obtained using the method from ref (41).
Figure 4.

Correlation potentials for the He and Ne atoms and CO molecule. The reference KS[CCSD(T)] have been obtained using method from ref (41).
Table 1. Error Statistics of Several Properties [∑iMICDDi/(Ne)i))]/Ma.
| OEPx- |
|||||||
|---|---|---|---|---|---|---|---|
| PBE | +ACSC | –ACSC | +PBEc | –PBEc | OEPx | OEP2-sc | |
| ICDD | |||||||
| MAE | 19.31 | 21.37 | 13.95 | 18.64 | 13.45 | - | 3.43 |
| MAE/Ne | 1.58 | 1.71 | 1.07 | 1.53 | 1.03 | - | 0.29 |
| HOMO (IP) | |||||||
| MAE [eV] | 6.17 | 1.78 | 0.51 | 1.81 | 0.50 | 0.90 | 0.66 |
| MARE [%] | 38.03 | 11.54 | 3.26 | 11.68 | 3.17 | 5.67 | 4.43 |
| LUMO | |||||||
| MAE [eV] | 4.04 | 1.66 | 0.91 | 1.65 | 0.85 | 1.30 | 0.64 |
| MARE [%] | 111.49 | 45.01 | 27.94 | 44.57 | 26.07 | 36.70 | 19.77 |
| HOMO–LUMO Energy Gap | |||||||
| MAE [eV] | 1.11 | 1.14 | 0.39 | 1.13 | 0.39 | 0.64 | 0.22 |
| MARE [%] | 8.38 | 10.96 | 4.50 | 10.76 | 4.46 | 6.56 | 1.91 |
For the ICDD of eq 10, we report the mean absolute error (MAE) in units of 10–2, and the MAE weighted with the number of electrons (MAE/Ne = [∑iMICDDi/(Ne)i))]/M, where (Ne)i is the number of electrons of the i-th system and M = 14 is the total number of systems). In case of HOMO, LUMO and HOMO–LUMO gaps results, we report additionally the mean absolute RE (MARE) calculated with respect to the reference data.47,94
4. Results
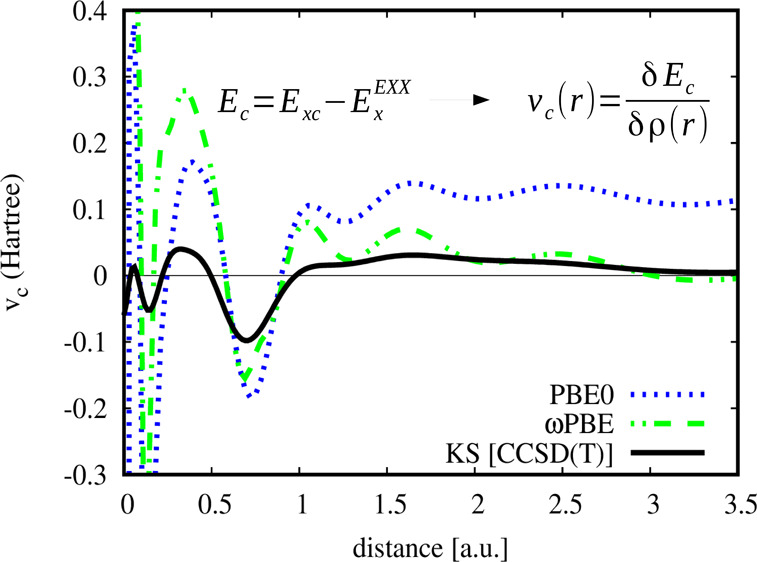
The correlation potentials corresponding to eq 8 are shown on Figure 2 for three representative cases of Ne and Ar atoms and CO molecule. Inspection of Figure 2 reveals that:
-
(i)
The B3LYP, PBE0, and ωPBE correlation potentials are very physical, being in phase with and having the shape of the reference (coupled cluster singles–doubles with perturbative triples62 - CCSD(T)) and Ab Initio DFT OEP2-sc60,61) curves in all the cases. Note that the correlation potentials of, EcLYP, and Ec fail badly to describe such a feature.8,9 Consequently, even if the semilocal exchange is very accurate, as are ExB88(72) and Ex,14 they still contain correlation effects that are crucial for the shape of the correlation potential.
-
(ii)
Figure 2 additionally reveals the physics behind the hybrid functionals and can be considered an elegant proof on the hybrid functional construction. Until now, the widely accepted rationale for mixing EXX with semilocal functionals was based on the adiabatic connection formula with a heuristic model for the hybrid coupling-constant dependence given in eq 6 of ref (73). We also note that the correlation potential vc is well defined, entering the KS scheme, as shown in eq 4, in sharp contrast to the correlation energy per particle ϵc (defined by Ec = ∫dr ρ(r)ϵc(r)) which is not unique, being defined up to a gauge transformation.74 Thus, this finding shows that inspection of OEP correlation potentials should be seen as a powerful criterion in the construction of new hybrid functionals, whose parameters can be found such that to give the optimal correlation potential.
-
(iii)
In the tail of the density, the EXX potential behaves as −1/r, while the semilocal exchange potentials are usually vanishing much faster (e.g., B8872 and xPBE14 behave as −1/r2 and e–r, respectively). This issue gives significant errors of vc in the asymptotic region (see Figure 2) directly transferring on the quality of ionization and excited state energies. On the other hand, this region is energetically evanescent, being not relevant for most of ground state properties. Additionally, Figure 2 shows that in the case of range-separated ωPBE functional, the vc decays much faster in the tail, being similar to the reference OEP2-sc and CCSD(T) potentials, thus explaining the dramatically improvement upon global hybrids XC functionals in the calculation of many properties.75−77
In Figure 3 we report the correlation densities8,9,48 Δρc = ρmethod – ρEXX obtained for same systems. All hybrid densities are reasonably accurate everywhere, including the tail region. Overall, B3LYP slightly outperforms PBE0 and ωPBE for these systems being much more similar to reference CCSD(T) results.
Figure 3.

Correlation densities Δρc = ρmethod – ρEXX, of the Ne and Ar atoms and CO molecule for various methods. The reference CCSD(T) correlation density was calculated with respect to HF density as Δρc = ρCCSD(T) – ρHF.
As shown above for the case of PBE0 and ωPBE hybrids, the exchange potential difference 0.75(vxPBE – vx) and vxsr,PBE – vx, respectively, give the right phase of the correlation potential, almost inverting the vcPBE. Therefore, they can be seen as EXX compatible functionals in some sense.
In several works,11,78−80 it has been observed that vcPBE is out of phase in comparison with the exact correlation potential. On the other hand, the −vc exhibits similar features as shown in Figure 2. Thus, in the following, we combine vxOEPx with vc, where we chose very simple test cases
| 9 |
with vcACSC being the adiabatic connection semilocal correlation (ACSC) GGA potential of ref (78) which gives a correlation potential close to but smoother than the vc one, as shown in Figure S5 of ref (33). We recall that the ACSC GGA correlation functional has been constructed from the modified interaction strength interpolation method.78 Note that the GGA–OEPx correlation potential of eq 9 is a stray potential (is not a functional derivative of any correctly defined correlation energy functional), failing the line-integral test proposed in ref (81). This fact gives severe limitations, such as for electronic excitation calculations.82 However, most of model XC potentials that are very useful in DFT calculations (e.g., Becke–Johnson potential and its modifications83−85) are not functional derivatives.81
In Figure 4, we compare the correlation potentials of vcPBE and −vc with high-level Ab Initio OEP2-sc method and the CCSD(T) reference curve. Indeed, vcGGA–OEPx can remarkably reproduce the features of the exact correlation potential, with the exception of the inherent GGA divergence at the nucleus. Nevertheless, comparing to the hybrid functionals behavior of Figure 2, we observe a less prominent description of the quantum oscillations, especially for the CO molecule, but with a better performance in the density tail region (the decay is similar to the one of ωPBE hybrid potential).
Next, in Figure 5, we show the correlation density Δρc(r) obtained from the self-consistent OEPx method combined with vcGGA–OEPx correlation potential (denoted as OEPx–PBEc/ACSC) and also (for comparison) with standard PBEc/ACSC potentials (denoted as OEPx + PBEc/ACSC). The OEPx–PBEc and OEPx–ACSC methods give the right shapes of the correlation densities (reported for ACSC in ref (33)) with exception of nucleus region, while both OEPx + PBEc and OEPx + ACSC fail badly. The compatibility between semilocal correlation functionals and the EXX is one of the most difficult DFT challenges.50,51,86−88 Even the sophisticated meta-GGA non-empirical correlation functionals, developed to satisfy many constraints of the exact correlation energy, do not perform better than simple GGAs for various molecular properties,89,90 and the reason behind that seems to be intimately related to the shape of their OEP-based correlation potentials which basically is GGA quality.91 (In Figure S1 of ref (33), we show for Ne atom that the vxc(92) of PBE GGA, TPSS meta-GGA, and SCAN meta-GGA agree closely. All these semilocal functionals give accurate densities,93 see also Figure S2 of ref (33), because of a large error cancellation between exchange and correlation parts, as proved in Figure S3 of ref (33).). In this respect, we recall that very complex, non-local, and orbital-dependent hyper-GGA correlation functionals have been developed,87,88 with the aim to be compatible with full EXX, but still they do not achieve good overall accuracy.
Figure 5.

Correlation densities for the He and Ne atoms and CO molecule. The reference CCSD(T) correlation density was calculated with respect to HF density as Δρc = ρCCSD(T) – ρHF.
To quantify the method accuracy, we consider several small atoms and molecules (He, Ne, Ar, H2, He2, HF, CO, Cl2, N2, Ne2, HCl, H2O, NH3, and C2H6) used in our previous studies,9,48 and we calculate
the integrated correlation density differences (ICDDs) defined as
| 10 |
where δρcmethod(r) = ρmethod(r) – ρxref(r), and xref denotes the corresponding exchange method. ICDDs provide a direct test of the quality of the correlation potential.9,48 Note that δρc is a generalization of Δρc;
the energy of the highest occupied molecular orbital (HOMO), and we compare it with the results from ref (47)
the energy of the lowest unoccupied molecular orbital (LUMO), and we use the CCSD(T) results as reference values.94
the HOMO–LUMO gap energies, considering the CCSD(T) results as refs (94) and (95)
The full results are reported in ref (33), while in Table 1, we summarize them showing the error statistics. The vxOEP + vc method gives a significant and systematic improvement over the vxOEP, vx + vcGGA and PBE, for all the properties, but it is not as accurate as the high-level Ab Initio OEP2-sc method, with exception of the HOMO energies where it gives the best performance. The ACSC results are in line with the PBEc ones. Somehow, this was expected because the ACSC construction is based on PBE functional.78 Similar trends can also be observed for HOMO and HOMO–LUMO gap energies (see Tables S5 and S6 of ref (33)) obtained with Becke–Johnson potential83 combined with PBEc and vc correlation potentials. This can be important from the standpoint of further application of the potential model in solid-state calculations.
To assess in detail the performance of these functionals, we show in Figure 6 the correlation plots for HOMO and HOMO–LUMO gap energies, respectively, calculated with respect to reference data. For all the benchmark systems, OEPx–PBEc is remarkably accurate despite its simplicity.
Figure 6.

Correlation plots for HOMO energies (upper panel) and HOMO–LUMO gap energies (lower panel).
We note that even for the exact HOMO–LUMO gap energy, one needs to add the uniform shift Δ because of derivative discontinuity of the KS potential when the particle number changes, in order to obtain the band gap energy Eg.96 The derivative discontinuity can be rigorously taken into account by perturbation theory methods97 but can also be modeled by non-local potentials of the generalized KS, in both hybrid98,99 and meta-GGA calculations,84,85,100 and, to some extent, even by the semilocal GGA potentials.101
5. Conclusions
We have shown that the correlation potential/densities of the OEP-based hybrid functionals can reproduce the quantum shell oscillations of the high-level reference CCSD(T) correlation potential/densities, providing an alternative but very physical explanation on the success of this type of functionals. The presented analysis might shed some light on how to overcome some current DFT limitations and face challenges, as described in ref (102). Thus, for example, using the reverse engineering method, one can build more accurate hybrid XC functionals by fixing the functional parameters (e.g., the mixing fraction of EXX), eventually using the machine learning techniques,103 to give the optimum OEP correlation potential.
We have also explained the error cancellation between the semilocal exchange and correlation functionals. Thus, as in the case of popular PBE GGA,14 even if both exchange (Ex) and correlation (Ec) energies are very accurate for a given system, still only the XC potential is realistic, while the correlation potential is out of phase. Based on these considerations, we have shown that the OEPx–PBEc potential method gives an outstanding performance in spite of its simplicity, showing accurate correlation densities and orbital energies.
Semilocal correlation potential models compatible with OEPx are of utmost theoretical and computational importance, having the ability to become the next generation method in electronic structure calculations.104 The simplicity of the semilocal correlation potential combined with the almost-exact description of the many-electron self-interaction given by OEP EXX can be of high interest for solid-state applications.
In this sense, the here developed vcGGA–OEPx may open such a path, but more accurate potential models can be built considering exact conditions on correlation potentials (including derivative discontinuity problem96,101), and right semilocal ingredients, such as the Laplacian of the density (∇2ρ), which is crucial for describing various quantum oscillations105 and to impose exact properties in the bond and core regions.106,107
Acknowledgments
We thank Dr. Zeng-hui Yang for providing us data for Figure S1 of ref (33). S.S thanks the Polish National Science Center for the partial financial support under grant no. 2016/21/D/ST4/00903.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpca.0c04156.
Data of all calculations and accompanying statistical analysis and additional figures and tables (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Kohn W.; Sham L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. 10.1103/physrev.140.a1133. [DOI] [Google Scholar]
- Hohenberg P.; Kohn W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. 10.1103/physrev.136.b864. [DOI] [Google Scholar]
- Constantin L. A.; Fabiano E.; Śmiga S.; Della Sala F. Jellium-with-gap model applied to semilocal kinetic functionals. Phys. Rev. B 2017, 95, 115153. 10.1103/physrevb.95.115153. [DOI] [Google Scholar]
- Constantin L. A.; Fabiano E.; Della Sala F. Semilocal Pauli-Gaussian Kinetic Functionals for Orbital-Free Density Functional Theory Calculations of Solids. J. Phys. Chem. Lett. 2018, 9, 4385–4390. 10.1021/acs.jpclett.8b01926. [DOI] [PubMed] [Google Scholar]
- Śmiga S.; Siecińska S.; Fabiano E. Methods to generate reference total and Pauli kinetic potentials. Phys. Rev. B 2020, 101, 165144. 10.1103/physrevb.101.165144. [DOI] [Google Scholar]
- Scuseria G. E.; Staroverov V. N.. Progress in the development of exchange-correlation functionals. In Theory and Applications of Computational Chemistry; Elsevier, 2005; pp 669–724. [Google Scholar]
- Della Sala F.; Fabiano E.; Constantin L. A. Kinetic-energy-density dependent semilocal exchange-correlation functionals. Int. J. Quantum Chem. 2016, 116, 1641–1694. 10.1002/qua.25224. [DOI] [Google Scholar]
- Grabowski I.; Teale A. M.; Śmiga S.; Bartlett R. J. Comparing ab initio density-functional and wave function theories: The impact of correlation on the electronic density and the role of the correlation potential. J. Chem. Phys. 2011, 135, 114111. 10.1063/1.3636114. [DOI] [PubMed] [Google Scholar]
- Grabowski I.; Teale A. M.; Fabiano E.; Śmiga S.; Buksztel A.; Sala F. D. A density difference based analysis of orbital-dependent exchange-correlation functionals. Mol. Phys. 2014, 112, 700–710. 10.1080/00268976.2013.854424. [DOI] [Google Scholar]
- Levy M. Mathematical thoughts in DFT. Int. J. Quantum Chem. 2016, 116, 802–804. 10.1002/qua.25133. [DOI] [Google Scholar]
- Buksztel A.; Śmiga S.; Grabowski I.. The Correlation Effects in Density Functional Theory Along the Dissociation Path. Advances in Quantum Chemistry; Elsevier, 2016; Vol.73; pp 263–283. [Google Scholar]
- Chiodo L.; Constantin L. A.; Fabiano E.; Della Sala F. Nonuniform scaling applied to surface energies of transition metals. Phys. Rev. Lett. 2012, 108, 126402. 10.1103/physrevlett.108.126402. [DOI] [PubMed] [Google Scholar]
- Vega L.; Ruvireta J.; Viñes F.; Illas F. Jacob’s Ladder as Sketched by Escher: Assessing the Performance of Broadly Used Density Functionals on Transition Metal Surface Properties. J. Chem. Theory Comput. 2017, 14, 395–403. 10.1021/acs.jctc.7b01047. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/physrevlett.77.3865. [DOI] [PubMed] [Google Scholar]
- Wood B.; Hine N.; Foulkes W.; García-González P. Quantum Monte Carlo calculations of the surface energy of an electron gas. Phys. Rev. B: Condens. Matter Mater. Phys. 2007, 76, 035403. 10.1103/physrevb.76.035403. [DOI] [Google Scholar]
- Pitarke J.; Eguiluz A. Jellium surface energy beyond the local-density approximation: Self-consistent-field calculations. Phys. Rev. B: Condens. Matter Mater. Phys. 2001, 63, 045116. 10.1103/physrevb.63.045116. [DOI] [Google Scholar]
- Schmidt T.; Kümmel S. One-and many-electron self-interaction error in local and global hybrid functionals. Phys. Rev. B 2016, 93, 165120. 10.1103/physrevb.93.165120. [DOI] [Google Scholar]
- Savin A.On Degeneracy, Near Degeneracy and Density Functional Theory. In Recent Developments of Modern Density Functional Theory; Seminario J. M., Ed.; Elsevier: Amsterdam, 1996; pp 327–357. [Google Scholar]
- Toulouse J.; Colonna F.; Savin A. Long-range–short-range separation of the electron-electron interaction in density-functional theory. Phys. Rev. A 2004, 70, 062505. 10.1103/physreva.70.062505. [DOI] [Google Scholar]
- Toulouse J.; Savin A.; Flad H.-J. r. Short-range exchange-correlation energy of a uniform electron gas with modified electron-electron interaction. Int. J. Quantum Chem. 2004, 100, 1047. 10.1002/qua.20259. [DOI] [Google Scholar]
- Toulouse J.; Colonna F.; Savin A. Short-range exchange and correlation energy density functionals: Beyond the local-density approximation. J. Chem. Phys. 2005, 122, 014110. 10.1063/1.1824896. [DOI] [PubMed] [Google Scholar]
- Paziani S.; Moroni S.; Gori-Giorgi P.; Bachelet G. B. Local-spin-density functional for multideterminant density functional theory. Phys. Rev. B: Condens. Matter Mater. Phys. 2006, 73, 155111. 10.1103/physrevb.73.155111. [DOI] [Google Scholar]
- Goll E.; Werner H.-J.; Stoll H.; Leininger T.; Gori-Giorgi P.; Savin A. A short-range gradient-corrected spin density functional in combination with long-range coupled-cluster methods: Application to alkali-metal rare-gas dimers. Chem. Phys. 2006, 329, 276. 10.1016/j.chemphys.2006.05.020. [DOI] [Google Scholar]
- Fritsche L. Generalized Kohn-Sham theory for electronic excitations in realistic systems. Phys. Rev. B: Condens. Matter Mater. Phys. 1986, 33, 3976–3989. 10.1103/physrevb.33.3976. [DOI] [PubMed] [Google Scholar]
- Seidl A.; Görling A.; Vogl P.; Majewski J. A.; Levy M. Generalized Kohn-Sham schemes and the band-gap problem. Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 53, 3764. 10.1103/physrevb.53.3764. [DOI] [PubMed] [Google Scholar]
- Sharp R. T.; Horton G. K. A Variational Approach to the Unipotential Many-Electron Problem. Phys. Rev. 1953, 90, 317. 10.1103/physrev.90.317. [DOI] [Google Scholar]
- Talman J. D.; Shadwick W. F. Optimized effective atomic central potential. Phys. Rev. A 1976, 14, 36–40. 10.1103/physreva.14.36. [DOI] [Google Scholar]
- Norman M. R.; Koelling D. D. Towards a Kohn-Sham potential via the optimized effective-potential method. Phys. Rev. B: Condens. Matter Mater. Phys. 1984, 30, 5530–5540. 10.1103/physrevb.30.5530. [DOI] [Google Scholar]
- Kümmel S.; Kronik L. Orbital-dependent density functionals: Theory and applications. Rev. Mod. Phys. 2008, 80, 3. 10.1103/revmodphys.80.3. [DOI] [Google Scholar]
- Arbuznikov A. V. Hybrid exchange correlation functionals and potentials: Concept elaboration. J. Struct. Chem. 2007, 48, S1–S31. 10.1007/s10947-007-0147-0. [DOI] [Google Scholar]
- Śmiga S.; Franck O.; Mussard B.; Buksztel A.; Grabowski I.; Luppi E.; Toulouse J. Self-consistent double-hybrid density-functional theory using the optimized-effective-potential method. J. Chem. Phys. 2016, 145, 144102. 10.1063/1.4964319. [DOI] [PubMed] [Google Scholar]
- Śmiga S.; Grabowski I.; Witkowski M.; Mussard B.; Toulouse J. Self-Consistent Range-Separated Density-Functional Theory with Second-Order Perturbative Correction via the Optimized-Effective-Potential Method. J. Chem. Theory Comput. 2020, 16, 211–223. 10.1021/acs.jctc.9b00807. [DOI] [PubMed] [Google Scholar]
- Śmiga, S.; Constantin, L. A. Supporting Information.
- Kim J.; Hong K.; Hwang S.-Y.; Ryu S.; Choi S.; Kim W. Y. Effects of the locality of a potential derived from hybrid density functionals on Kohn-Sham orbitals and excited states. Phys. Chem. Chem. Phys. 2017, 19, 10177–10186. 10.1039/c7cp00704c. [DOI] [PubMed] [Google Scholar]
- Levy M.; Perdew J. P. Hellmann-Feynman, virial, and scaling requisites for the exact universal density functionals. Shape of the correlation potential and diamagnetic susceptibility for atoms. Phys. Rev. A 1985, 32, 2010–2021. 10.1103/physreva.32.2010. [DOI] [PubMed] [Google Scholar]
- Krieger J. B.; Li Y. Derivation and application of an accurate Kohn-Sham potential with integer discontinuity. Phys. Lett. A 1990, 146, 256–260. 10.1016/0375-9601(90)90975-t. [DOI] [PubMed] [Google Scholar]
- Görling A.; Levy M. DFT ionization formulas and DFT perturbation theory for exchange and correlation. Through adiabatic connection. Int. J. Quantum Chem. 1995, 56, 93–108. 10.1002/qua.560560810. [DOI] [Google Scholar]
- Grabowski I.; Hirata S.; Ivanov S.; Bartlett R. J. Ab initio density functional theory: OEP-MBPT(2). A new orbital-dependent correlation functional. J. Chem. Phys. 2002, 116, 4415–4425. 10.1063/1.1445117. [DOI] [Google Scholar]
- Della Sala F.Orbital-dependent exact-exchange methods in density functional theory. In Chemical Modelling; Springborg M., Ed.; Royal Society of Chemistry: London, U.K., 2011; Vol. 7, pp 115–161. [Google Scholar]
- Engel E.Orbital-dependent functional for the Exchange-Correlation Energy: A Third Generation of DFT. In A Primer in Density Functional Theory; Fiolhais C., Nogueira F., Marques M. A., Eds.; Springer: Berlin, 2003. [Google Scholar]
- Wu Q.; Yang W. A direct optimization method for calculating density functionals and exchange-correlation potentials from electron densities. J. Chem. Phys. 2003, 118, 2498–2509. 10.1063/1.1535422. [DOI] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Adamo C.; Barone V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. 10.1063/1.478522. [DOI] [Google Scholar]
- Weintraub E.; Henderson T. M.; Scuseria G. E. Long-Range-Corrected Hybrids Based on a New Model Exchange Hole. J. Chem. Theory Comput. 2009, 5, 754–762. 10.1021/ct800530u. [DOI] [PubMed] [Google Scholar]
- Goll E.; Werner H.-J.; Stoll H. A short-range gradient-corrected density functional in long-range coupled-cluster calculations for rare gas dimers. Phys. Chem. Chem. Phys. 2005, 7, 3917–3923. 10.1039/b509242f. [DOI] [PubMed] [Google Scholar]
- Stanton J. F.; et al. ACES II; Quantum Theory Project: Gainesville, Florida, 2007.
- Śmiga S.; Della Sala F.; Buksztel A.; Grabowski I.; Fabiano E. Accurate Kohn-Sham ionization potentials from scaled-opposite-spin second-order optimized effective potential methods. J. Comput. Chem. 2016, 37, 2081–2090. 10.1002/jcc.24436s. [DOI] [PubMed] [Google Scholar]
- Grabowski I.; Fabiano E.; Teale A. M.; Śmiga S.; Buksztel A.; Sala F. D. Orbital-dependent second-order scaled-opposite-spin correlation functionals in the optimized effective potential method. J. Chem. Phys. 2014, 141, 024113. 10.1063/1.4887097. [DOI] [PubMed] [Google Scholar]
- Fabiano E.; Śmiga S.; Giarrusso S.; Daas T. J.; Della Sala F.; Grabowski I.; Gori-Giorgi P. Investigation of the Exchange-Correlation Potentials of Functionals Based on the Adiabatic Connection Interpolation. J. Chem. Theory Comput. 2019, 15, 1006–1015. 10.1021/acs.jctc.8b01037. [DOI] [PubMed] [Google Scholar]
- Görling A. New KS method for molecules based on an exchange charge density generating the exact local KS exchange potential. Phys. Rev. Lett. 1999, 83, 5459–5462. 10.1103/physrevlett.83.5459. [DOI] [Google Scholar]
- Ivanov S.; Hirata S.; Bartlett R. J. Exact exchange treatment for molecules in finite-basis-set Kohn-Sham theory. Phys. Rev. Lett. 1999, 83, 5455–5458. 10.1103/physrevlett.83.5455. [DOI] [Google Scholar]
- Hirata S.; Ivanov S.; Grabowski I.; Bartlett R. J.; Burke K.; Talman J. D. Can optimized effective potentials be determined uniquely?. J. Chem. Phys. 2001, 115, 1635–1649. 10.1063/1.1381013. [DOI] [Google Scholar]
- Ivanov S.; Hirata S.; Bartlett R. J. Finite-basis-set optimized effective potential exchange-only method. J. Chem. Phys. 2002, 116, 1269–1276. 10.1063/1.1427712. [DOI] [Google Scholar]
- Widmark P.-O.; Malmqvist P. k.; Roos B. r. O. Density matrix averaged atomic natural orbital (ANO) basis sets for correlated molecular wave functions. Theor. Chim. Acta 1990, 77, 291–306. 10.1007/bf01120130. [DOI] [Google Scholar]
- Peterson K. A.; Dunning T. H. Accurate correlation consistent basis sets for molecular core-valence correlation effects: The second row atoms Al-Ar, and the first row atoms B-Ne revisited. J. Chem. Phys. 2002, 117, 10548–10560. 10.1063/1.1520138. [DOI] [Google Scholar]
- Dunning T. H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. 10.1063/1.456153. [DOI] [Google Scholar]
- Herzberg G.Spectra of Diatomic Molecules; Van Nostrand Reinhold Co.: Princeton. N.J., 1970. [Google Scholar]
- Boucier S.Spectroscopic Data Relative to Diatomic Molecules. Tables of Constants and Numerical Data; Pergainon Press: Elmsford, N.Y., 1970; Vol. 17. [Google Scholar]
- Callonion J. H.; Hirota E.; Kuchitsu K. .; Lafferty W. J.; Maki A. G.; Pote C. S.. Numerical Data and Function Relationships in Science and Technology. In Structure Data on Free Polyatomic Molecules; Springer-Verlag: West Berlin, 1976; Vol. 7. [Google Scholar]
- Bartlett R. J.; Grabowski I.; Hirata S.; Ivanov S. The exchange-correlation potential inab initiodensity functional theory. J. Chem. Phys. 2005, 122, 034104. 10.1063/1.1809605. [DOI] [PubMed] [Google Scholar]
- Śmiga S.; Marusiak V.; Grabowski I.; Fabiano E. The ab initio density functional theory applied for spin-polarized calculations. J. Chem. Phys. 2020, 152, 054109. [DOI] [PubMed] [Google Scholar]
- Raghavachari K.; Trucks G. W.; Pople J. A.; Head-Gordon M. A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 1989, 157, 479–483. 10.1016/s0009-2614(89)87395-6. [DOI] [Google Scholar]
- Handy N. C.; Schaefer H. F. On the evaluation of analytic energy derivatives for correlated wave functions. J. Chem. Phys. 1984, 81, 5031–5033. 10.1063/1.447489. [DOI] [Google Scholar]
- Rice J. E.; Zenser T. V. On the efficient evaluation of analytic energy gradients. Chem. Phys. Lett. 1985, 122, 585–590. 10.1016/0009-2614(85)87275-4. [DOI] [Google Scholar]
- Davis R. J.Analytical evaluation of gradients in coupled-cluster and many-body perturbation theory. In Geometrical Derivatives of Energy Surfaces and Molecular Properties; Jørgensen P., Simons J., Eds.; Reidel: Dordecht, The Netherlands, 1986; pp 35–61. [Google Scholar]
- Salter E. A.; Trucks G. W.; Bartlett R. J. Analytic energy derivatives in many-body methods. I. First derivatives. J. Chem. Phys. 1989, 90, 1752–1766. 10.1063/1.456069. [DOI] [Google Scholar]
- Helgaker T.; Jørgensen P. Configuration-interaction energy derivatives in a fully variational formulation. Theor. Chim. Acta 1989, 75, 111–127. 10.1007/bf00527713. [DOI] [Google Scholar]
- Jørgensen P.; Helgaker T. Møller-Plesset energy derivatives. J. Chem. Phys. 1988, 89, 1560–1570. 10.1063/1.455152. [DOI] [Google Scholar]
- Koch H.; Jensen H. J. r. A.; Jørgensen P.; Helgaker T.; Scuseria G. E.; Schaefer H. F. III Coupled cluster energy derivatives. Analytic Hessian for the closed-shell coupled cluster singles and doubles wave function: Theory and applications. J. Chem. Phys. 1990, 92, 4924. 10.1063/1.457710. [DOI] [Google Scholar]
- Hald K.; Halkier A.; Jørgensen P.; Coriani S.; Hättig C.; Helgaker T. A Lagrangian, integral-density direct formulation and implementation of the analytic CCSD and CCSD(T) gradients. J. Chem. Phys. 2003, 118, 2985. 10.1063/1.1531106. [DOI] [Google Scholar]
- Śmiga S.; Buksztel A.; Grabowski I.. Chapter 7 - Density-Dependent Exchange-Correlation Potentials Derived From highly Accurate Ab initio Calculations. In Proceedings of MEST 2012: Electronic Structure Methods with Applications to Experimental Chemistry; Hoggan P., Ed.; Advances in Quantum Chemistry; Academic Press, 2014; Vol. 68; pp 125–151.
- Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Ernzerhof M.; Burke K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. 10.1063/1.472933. [DOI] [Google Scholar]
- Tao J.; Staroverov V. N.; Scuseria G. E.; Perdew J. P. Exact-exchange energy density in the gauge of a semilocal density-functional approximation. Phys. Rev. A 2008, 77, 012509. 10.1103/physreva.77.012509. [DOI] [Google Scholar]
- Iikura H.; Tsuneda T.; Yanai T.; Hirao K. A long-range correction scheme for generalized-gradient-approximation exchange functionals. J. Chem. Phys. 2001, 115, 3540–3544. 10.1063/1.1383587. [DOI] [Google Scholar]
- Tawada Y.; Tsuneda T.; Yanagisawa S.; Yanai T.; Hirao K. A long-range-corrected time-dependent density functional theory. J. Chem. Phys. 2004, 120, 8425–8433. 10.1063/1.1688752. [DOI] [PubMed] [Google Scholar]
- Gerber I. C.; Ángyán J. G.; Marsman M.; Kresse G. Range separated hybrid density functional with long-range Hartree-Fock exchange applied to solids. J. Chem. Phys. 2007, 127, 054101. 10.1063/1.2759209. [DOI] [PubMed] [Google Scholar]
- Constantin L. A. Correlation energy functionals from adiabatic connection formalism. Phys. Rev. B 2019, 99, 085117. 10.1103/physrevb.99.085117. [DOI] [Google Scholar]
- Neese F. Prediction of molecular properties and molecular spectroscopy with density functional theory: From fundamental theory to exchange-coupling. Coord. Chem. Rev. 2009, 253, 526–563. 10.1016/j.ccr.2008.05.014. [DOI] [Google Scholar]
- Bartlett R. J.; Schweigert I. V.; Lotrich V. F. Ab initio DFT: getting the right answer for the right reason. J. Mol. Struct.: THEOCHEM 2006, 771, 1–8. 10.1016/j.theochem.2006.02.004. [DOI] [Google Scholar]
- Gaiduk A. P.; Staroverov V. N. How to tell when a model Kohn-Sham potential is not a functional derivative. J. Chem. Phys. 2009, 131, 044107. 10.1063/1.3176515. [DOI] [PubMed] [Google Scholar]
- Karolewski A.; Armiento R.; Kümmel S. Electronic excitations and the Becke-Johnson potential: The need for and the problem of transforming model potentials to functional derivatives. Phys. Rev. A 2013, 88, 052519. 10.1103/physreva.88.052519. [DOI] [Google Scholar]
- Becke A. D.; Johnson E. R. A simple effective potential for exchange. J. Chem. Phys. 2006, 124, 221101. 10.1063/1.2213970. [DOI] [PubMed] [Google Scholar]
- Tran F.; Blaha P. Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys. Rev. Lett. 2009, 102, 226401. 10.1103/physrevlett.102.226401. [DOI] [PubMed] [Google Scholar]
- Patra A.; Jana S.; Myneni H.; Samal P. Laplacian free and asymptotic corrected semilocal exchange potential applied to the band gap of solids. Phys. Chem. Chem. Phys. 2019, 21, 19639–19650. 10.1039/c9cp03356d. [DOI] [PubMed] [Google Scholar]
- Bylander D. M.; Kleinman L. Energy Gaps and Cohesive Energy of Ge from the Optimized Effective Potential. Phys. Rev. Lett. 1995, 74, 3660–3663. 10.1103/physrevlett.74.3660. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Staroverov V. N.; Tao J.; Scuseria G. E. Density functional with full exact exchange, balanced nonlocality of correlation, and constraint satisfaction. Phys. Rev. A 2008, 78, 052513. 10.1103/physreva.78.052513. [DOI] [Google Scholar]
- Becke A. D.; Johnson E. R. A unified density-functional treatment of dynamical, nondynamical, and dispersion correlations. J. Chem. Phys. 2007, 127, 124108. 10.1063/1.2768530. [DOI] [PubMed] [Google Scholar]
- Fabiano E.; Trevisanutto P. E.; Terentjevs A.; Constantin L. A. Generalized gradient approximation correlation energy functionals based on the uniform electron gas with gap model. J. Chem. Theory Comput. 2014, 10, 2016–2026. 10.1021/ct500073b. [DOI] [PubMed] [Google Scholar]
- Constantin L. A.; Fabiano E.; Sala F. D. Spin-dependent gradient correction for more accurate atomization energies of molecules. J. Chem. Phys. 2012, 137, 194105. 10.1063/1.4766324. [DOI] [PubMed] [Google Scholar]
- Kananenka A. A.; Kohut S. V.; Gaiduk A. P.; Ryabinkin I. G.; Staroverov V. N. Efficient construction of exchange and correlation potentials by inverting the Kohn-Sham equations. J. Chem. Phys. 2013, 139, 074112. 10.1063/1.4817942. [DOI] [PubMed] [Google Scholar]
- Yang Z.-h.; Peng H.; Sun J.; Perdew J. P. More realistic band gaps from meta-generalized gradient approximations: Only in a generalized Kohn-Sham scheme. Phys. Rev. B 2016, 93, 205205. 10.1103/physrevb.93.205205. [DOI] [Google Scholar]
- Medvedev M. G.; Bushmarinov I. S.; Sun J.; Perdew J. P.; Lyssenko K. A. Density functional theory is straying from the path toward the exact functional. Science 2017, 355, 49–52. 10.1126/science.aah5975. [DOI] [PubMed] [Google Scholar]
- The reference results are obtained using method from ref (41) and CCSD(T) density. The computational setup is identical as in refs (9) and (48).
- Pearson R. G. Chemical hardness and density functional theory. J. Chem. Sci. 2005, 117, 369–377. 10.1007/bf02708340. [DOI] [Google Scholar]
- Hodgson M. J. P.; Kraisler E.; Schild A.; Gross E. K. U. How Interatomic Steps in the Exact Kohn-Sham Potential Relate to Derivative Discontinuities of the Energy. J. Phys. Chem. Lett. 2017, 8, 5974–5980. 10.1021/acs.jpclett.7b02615. [DOI] [PubMed] [Google Scholar]
- Shishkin M.; Marsman M.; Kresse G. Accurate quasiparticle spectra from self-consistent GW calculations with vertex corrections. Phys. Rev. Lett. 2007, 99, 246403. 10.1103/physrevlett.99.246403. [DOI] [PubMed] [Google Scholar]
- Krukau A. V.; Vydrov O. A.; Izmaylov A. F.; Scuseria G. E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. 10.1063/1.2404663. [DOI] [PubMed] [Google Scholar]
- Jana S.; Patra A.; Constantin L. A.; Samal P. Screened range-separated hybrid by balancing the compact and slowly varying density regimes: Satisfaction of local density linear response. J. Chem. Phys. 2020, 152, 044111. 10.1063/1.5131530. [DOI] [PubMed] [Google Scholar]
- Patra B.; Jana S.; Constantin L. A.; Samal P. Relevance of the Pauli kinetic energy density for semilocal functionals. Phys. Rev. B 2019, 100, 155140. 10.1103/physrevb.100.155140. [DOI] [Google Scholar]
- Armiento R.; Kümmel S. Orbital localization, charge transfer, and band gaps in semilocal density-functional theory. Phys. Rev. Lett. 2013, 111, 036402. 10.1103/physrevlett.111.036402. [DOI] [PubMed] [Google Scholar]
- Cohen A. J.; Mori-Sánchez P.; Yang W. Challenges for Density Functional Theory. Chem. Rev. 2012, 112, 289–320. 10.1021/cr200107z. [DOI] [PubMed] [Google Scholar]
- Faber F. A.; Hutchison L.; Huang B.; Gilmer J.; Schoenholz S. S.; Dahl G. E.; Vinyals O.; Kearnes S.; Riley P. F.; Von Lilienfeld O. A. Prediction errors of molecular machine learning models lower than hybrid DFT error. J. Chem. Theory Comput. 2017, 13, 5255–5264. 10.1021/acs.jctc.7b00577. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Perspective: Fifty years of density-functional theory in chemical physics. J. Chem. Phys. 2014, 140, 18A301. 10.1063/1.4869598. [DOI] [PubMed] [Google Scholar]
- Yang W.; Parr R. G.; Lee C. Various functionals for the kinetic energy density of an atom or molecule. Phys. Rev. A 1986, 34, 4586. 10.1103/physreva.34.4586. [DOI] [PubMed] [Google Scholar]
- Śmiga S.; Fabiano E.; Constantin L. A.; Della Sala F. Laplacian-dependent models of the kinetic energy density: Applications in subsystem density functional theory with meta-generalized gradient approximation functionals. J. Chem. Phys. 2017, 146, 064105. 10.1063/1.4975092. [DOI] [PubMed] [Google Scholar]
- Śmiga S.; Constantin L. A.; Della Sala F.; Fabiano E. The Role of the Reduced Laplacian Renormalization in the Kinetic Energy Functional Development. Computation 2019, 7, 65. 10.3390/computation7040065. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.