

Abstract
The recombinant adeno-associated virus (AAV) vector is one of the most utilized viral vectors in gene therapy due to its robust, long-term in vivo transgene expression and low toxicity. One major hurdle for clinical AAV applications is large-scale manufacturing. In this regard, the baculovirus-based AAV production system is highly attractive due to its scalability and predictable biosafety. Here, we describe a simple method to improve the baculovirus-based AAV production using the ExpiSf Baculovirus Expression System with a chemically defined medium for suspension culture of high-density ExpiSf9 cells. Baculovirus-infected ExpiSf9 cells produced up to 5 × 1011 genome copies of highly purified AAV vectors per 1 mL of suspension culture, which is up to a 19-fold higher yield than the titers we obtained from the conventional Sf9 cell-based system. When mice were administered the same dose of AAV vectors, we saw comparable transduction efficiency and biodistributions between the vectors made in ExpiSf9 and Sf9 cells. Thus, the ExpiSf Baculovirus Expression System would support facile and scalable AAV manufacturing amenable for preclinical and clinical applications.
Keywords: AAV, baculovirus, insect cell
Graphical Abstract
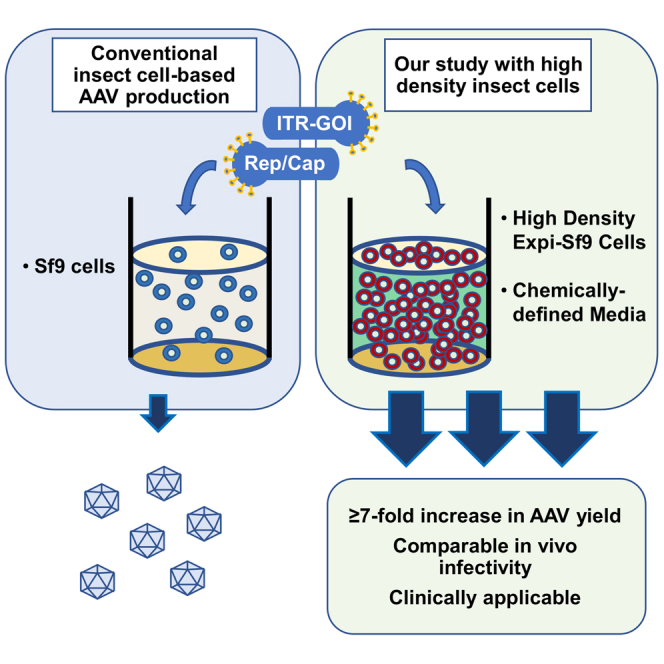
Large-scale manufacturing is a major challenge for preclinical and clinical AAV vector applications. Ikeda and colleagues describe a simple method using a high-density insect cell-based system to improve AAV vector yield up to 19-fold higher than the conventional Sf9 cell-based system, which would facilitate scalable AAV vector manufacturing.
Introduction
Adeno-associated virus (AAV)-based vectors have been established as a safe and robust gene transfer vehicle for efficient transgene delivery in vitro and in vivo.1 The recent US Food and Drug Administration (FDA) approvals of clinical AAV products, Luxturna and Zolgensma, further represent the clinical success of this technology.2,3 On the other hand, Luxturna and Zolgensma are currently the two most expensive drugs on the market, with the cost of large-scale manufacturing of AAV vectors being one of the major contributors.
Traditionally, helper-free AAV vectors have been made by transient transfection of human embryonic kidney (HEK)293 cells with three plasmids carrying the following: (1) AAV genome transfer vector, (2) AAV Cap and Rep genes encoding the capsid and replication proteins, and (3) adenoviral helper functions.4,5 Although this system is convenient for optimizing AAV vectors at laboratory scale, large-scale manufacturing is challenging, mainly due to the transfection process.6
One option to overcome the scalability issue is to use the well-established baculovirus expression system that has been used to produce various recombinant proteins in insect cells, including clinical-grade products, like Cervarix and Flublok.7, 8, 9 Several factors make this system highly attractive as a clinically applicable, large-scale AAV vector-production platform.10 First, infection of insect cells with baculoviral vectors carrying the AAV vector genome and AAV Cap and Rep proteins allows for transfection-free AAV vector production. Second, Spodoptera frugiperda Sf9 cells are grown in suspension in serum-free media and can reach higher cell densities than HEK293 cells. Third, Sf9 cells do not support the replication of potentially contaminating human agents. Lastly, baculoviruses cannot replicate in humans. These characteristics increase the biosafety profile of Sf9-derived AAV vectors for preclinical and clinical applications. Indeed, this insect cell-based expression system was used for the AAV gene-therapy drug Glybera, which was approved by the European Medicines Agency (EMA) in 2012.11
The use of baculoviruses to produce infectious AAV in insect cells was reported by Dr. Kotin’s group12 in 2002. Briefly, Sf9 cells were infected with three different recombinant baculoviruses to produce AAV2: one containing the AAV2 Rep gene, another containing the AAV2 Cap gene, and the last containing the transgene cassette flanked by the AAV2 inverted terminal repeat (ITR) elements. In the following years, further modifications were made to both simplify and improve the stability of the system by placing the Rep and Cap genes into one dual cassette and removing homologous regions in the Rep construct.13 This improved system uses attenuated start codons for both VP1 and Rep78 to utilize “leaky scanning” of the ribosome and promote translation of the downstream products.14, 15, 16 Other modifications were also made in between the Rep78 and Rep52 start codons.
Recently, Thermo Fisher Scientific developed the ExpiSf Baculovirus Expression System, which is the first-ever, clinically applicable, chemically defined (CD) insect protein expression system, which can achieve a 3× higher protein yield.17 In this study, we assessed AAV production in the ExpiSf Baculovirus Expression System. With the same baculoviral stocks, we infected both Sf9 and ExpiSf9 cells with the same multiplicity of infection (MOI) to compare the differences in AAV vector yield. In addition, we characterized the purified AAVs from both cell lines in vitro and in vivo for their transduction efficiency.
Results
Generation of a Baculovirus and Sf9 Cell-Based AAV Production System
We designed and generated baculovirus constructs as shown in Figure 1A. Specifically, our two baculoviral vectors harbor either the transgene cassette flanked by the AAV2 ITRs or the AAV2 Rep (Rep2) and Cap genes of various serotypes with attenuated start codons for both VP1 and Rep78 to utilize leaky scanning of the ribosome and promote translation of the downstream products, as previously reported.13 After expansion of the baculoviruses in Sf9 cells, we obtained titers of approximately 1–2 × 108 plaque forming units (PFUs) per milliliter.
Figure 1.

Design of Constructs and Confirmation of Infectivity
(A) The packaging construct (top) containing both the AAV2 Rep and Cap genes from AAV2, AAV8, or AAV9 were cloned into a pFastBac Dual vector. The Cap cassette is driven by the p10 promoter and contains the herpes simplex virus (HSV) thymidine kinase (tk) polyadenylation signal (pA). The Rep cassette is driven by the polyhedrin promoter (pH) and contains the simian virus 40 (SV40) pA. The transgene construct (bottom) contains either the ZsGreen or luciferase gene, which is driven by the cytomegalovirus (CMV) promoter. It also contains a beta-globin (B-globin) intron and a human growth hormone (hGH) pA signal. Lastly, the transgene cassette is flanked by the AAV2 inverted terminal repeat (ITR). (B) Purified AAV2 containing the ZsGreen transgene cassette was used to infect 293T cells at an MOI of 200. The cells were then imaged for ZsGreen expression 48 h postinfection. (C) Purified baculovirus containing the ZsGreen transgene cassette only was used to infect 293T cells at an MOI of 200. The cells were then imaged for ZsGreen expression 48 h postinfection.
Next, we verified that the rescued baculoviruses had the capacity of AAV vector production. 3 days after infection of Sf9 cells with the Rep- and Cap-carrying and AAV2 vector genome-carrying baculoviruses at MOIs of 1 for both, we harvested the cells. The cell-associated AAV vectors were purified through density gradient ultracentrifugation.18 When we infected HEK293T cells with the purified AAV2 containing the ZsGreen transgene cassette, we found ZsGreen transduction (Figure 1B). In order to rule out the possibility that the ZsGreen expression may be coming from the transgene-containing baculovirus itself, we infected Sf9 cells with the ZsGreen-containing baculovirus only. After 72 h postinfection, we harvested the cells and purified the cell-associated baculovirus in a similar fashion as above. We then infected HEK293T cells with the purified baculovirus containing ZsGreen with the same MOI as above and found no ZsGreen transduction (Figure 1C). Thus, we demonstrated successful AAV vector production from our baculoviral expression system.
Optimization of AAV Production in ExpiSf9 Cells
We next evaluated the potential of using high-density ExpiSf9 cells for improved AAV vector production. To optimize the AAV production conditions in ExpiSf9 cells, we determined the impact of cell density, AAV vector harvest time, and baculoviral MOIs on AAV vector yields. Overall, our results showed that the 72-h time point, a cell density of 5 × 106 cells/mL (Figure 2A, left panel), and an MOI of 1 per baculovirus (Figure 2B, left panel) were optimal. Lower cell densities and higher MOI were associated with reduced cell viabilities (Figures 2A and 2B, right panels). Although the AAV genome copy titers were the highest at 96 h postinfection for MOIs ≥5 (Figure 2C, left panel), the in vitro infectivity of the AAVs declined after 72 h (Figure 2D). Data comparing the 24-, 72-, and 96-h harvest times with varying cell densities are shown in Figure S1.
Figure 2.

Optimization of AAV Production in ExpiSf Expression System
(A) Infection with varying cell densities (cells/mL) at the time of infection was tested using an MOI of 1 per baculovirus (left panel). qPCR was used to measure the viral genomes per milliliter (vg/mL) of each sample. Percentage of viable cells (% viability) was monitored during the optimization of the cell density (right panel). (B) Infection with varying MOIs of the baculoviruses was tested using a cell density of 5 × 106 cells/mL (left panel). qPCR was used to measure the titers (vg/mL). Percentage of viable cells (% viability) was monitored during the optimization of the MOI (right panel). (C) Infection with varying MOIs of the baculoviruses was tested using a cell density of 5 × 106 cells/mL (left panel). Samples were taken at 48 h, 72 h, and 96 h postinfection. qPCR was used to measure the titers (vg/mL). Percentage of viable cells (% viability) was monitored during the optimization of the MOI and harvest time (right panel). For statistical analysis, each condition was compared to the 72-h MOI of 1 condition. (D) Crude cell lysates of AAV2 produced with varying baculoviral MOIs were harvested at 48 h, 72 h, and 96 h postinfection. The infectivities were assessed by transducing Ad293 cells with a MOI of 100. The percentage of ZsGreen-positive cells was measured by flow cytometry. Statistical analysis was performed within each MOI group and was compared to its 72-h time point. ∗p < 0.05, ∗∗p < 0.005.
Significant Increase in AAV Vector Production in ExpiSf9 Cells
Next, we compared the AAV vector productivity in Sf9 and ExpiSf9 cells using the same baculoviral stocks. 3 days after baculoviral infection at MOIs of 1, we harvested cells, and the cell-associated AAV vectors were then purified with two rounds of ultracentrifugation on an iodixanol gradient.18 Measurement of the AAV genome copy numbers by qPCR demonstrated a significant 7- to 19-fold increase in AAV vector yields when produced in the ExpiSf system as compared to Sf9 cells (Figure 3A). We also determined the infectivity of the purified, ZsGreen-expressing AAV vectors via flow cytometry. When Ad293 cells were infected by AAV vectors at an MOI of 1,000 for AAV2 and 100,000 for AAV8 and AAV9, all three tested serotypes showed comparable ZsGreen transduction efficiency, with marginally reduced infectivity with ExpiSf9-derived vectors for AAV8 and AAV9 (Figure 3B).
Figure 3.

Viral Genome Titers and Infectivity of Purified AAV Preparations
(A) Purified AAV preparations of various serotypes were produced using either the Bac-to-Bac System in Sf9 cells or the ExpiSf expression system in ExpiSf9 cells, and total viral genomes obtained from 300 mL of culture were measured using qPCR. (B) The infectivity of the purified AAV preparations produced in either Sf9 or ExpiSf9 cells was assessed by measuring the percentage of ZsGreen-positive cells by flow cytometry. ∗p < 0.05. (C) The infectivity of the purified AAV preparations produced in Sf9, ExpiSf9, or Ad293 cells was assessed by measuring the percentage of ZsGreen-positive cells by flow cytometry. ∗p < 0.05. (D) AAV9 yields from the three cell lines tested are shown in terms of viral genomes per cell (vg/c) along with the standard deviation (SD).
In addition to comparing the two insect cell-based systems, we also compared the in vitro infectivity with mammalian cell-derived AAV9. The results showed the Ad293-derived AAV9 had slightly lower infectivity when compared to the insect cell-derived AAV9 (Figure 3C). Comparisons of the yields per cell of the three cell lines tested are shown in Figure 3D.
Characterization of the VP1, VP2, and VP3 Ratios of Sf9- and ExpiSf9-Derived AAV Particles
When Sf9- and ExpiSf9-derived AAV8 and AAV9 vectors were analyzed by SDS-PAGE, we found comparable VP1, VP2, and VP3 expression between the vectors from the two cell lines (Figure 4A). To further characterize the AAV capsid ratios of the purified AAV preparations, capillary electrophoresis sodium dodecyl sulfate (CE-SDS) was used to analyze the VP1, VP2, and VP3 ratios of our purified AAV9 preparations from both Sf9 and ExpiSf9 cells. The results showed that the capsid ratios of AAV9 produced in the two cell lines were indeed similar (Figure 4B).
Figure 4.

Sodium Dodecyl Sulfate (SDS)-PAGE and Capsid Ratio Analysis of AAV9 Produced in Sf9 and ExpiSf9 Cells Using Capillary Electrophoresis SDS
(A) Purified AAV8 and AAV9 from both systems were assessed by SDS-PAGE gel stained with SYPRO Red. Lane 1, AAV8 produced in Sf9 cells (~2.6 × 1011 vg); lane 2, AAV8 produced in ExpiSf9 cells (~2.6 × 1011 vg); lane 3, AAV9 produced in Sf9 cells (~1.0 × 1011 vg); lane 4, AAV9 produced in ExpiSf9 cells (~1.0 × 1011 vg). (B) Chromatogram showing the overlay of the AAV9 samples produced in either Sf9 (red) or ExpiSf9 (blue) cells. Bottom table shows the number of VP1, VP2, and VP3 proteins per AAV9 particle produced in either Sf9 or ExpiSf9 cells.
Analytical Ultracentrifugation of Purified AAV9 Preparations from ExpiSf9 Cells Identifies Predominantly Full Capsids
In order to further assess the quality of the purified AAV vector preparations, we analyzed the amount of full and empty AAV capsids of AAV9 produced in the ExpiSf system by analytical ultracentrifugation (AUC).19 As a control, we generated empty AAV9 capsid particles by infecting ExpiSf9 cells with the Rep2 and AAV9 Cap baculovirus alone. We first analyzed purified empty AAV9 capsids on the AUC using interference optics to find the lower sedimentation coefficient boundary (Figure 5A). Next, we ran AAV9 preparations made with coinfection of baculoviral vector with ZsGreen (Figure 5B). The results showed that the AAV9 preparation was predominately full, with the percentage of full AAV particles being approximately 75%.
Figure 5.

Analytical Ultracentrifugation-Sedimentation Velocity Profiles of AAV9 Produced in ExpiSf9 Cells and Alkaline Gel Electrophoresis of Insect Cell-Derived AAV9 Genomes
(A) Empty AAV9 capsid was analyzed by AUC in order to identify the lower boundary. (B) Once the lower empty capsid boundary was set, AAV9 containing the transgene cassette was run. Each peak was then integrated using the SEDFIT software to determine the percentage of full viral particles. The sedimentation coefficient on the x axis is in Svedberg units (S). The y axis represents the concentration (C) as a function of S. (C) Diagram showing the sequencing coverage of the recombinant AAV9 genome. Bottom table shows the percentages of the AAV genome in the recombinant AAV9 preparations. (D) Sf9- and ExpiSf9-derived AAV9 ZsGreen genomes were isolated and run on an alkaline gel. Lane 1, DNA ladder; lane 2, Sf9-derived AAV9 ZsGreen genome; lane 3, ExpiSf9-derived AAV9 ZsGreen genome. 500 ng of DNA was loaded on the left gel, and 7,500 ng of DNA was loaded on the right gel. The predicted genome size is ~2.6 kilobase (kb) pairs.
To check the genome integrity of our AAV preparations produced in ExpiSf9 cells, we used next-generation sequencing (NGS) to characterize AAV9 (Figure 5C). The results showed that our AAV9 vectors contain the intact AAV genome, with no major truncations or insertions. In addition, we isolated the DNA from AAV9 derived from either Sf9 or ExpiSf9 cells and ran an alkaline gel. We found similar band patterns for both samples, with the most prominent band being the predicted monomeric single-stranded DNA (ssDNA) genome size of ~2.6 kilobase (kb) pairs. We also detected the dimeric form in both samples (~5.2 kb) (Figure 5D, left panel). To detect for any major truncated forms of AAV genomic DNA, we also loaded the wells with high amounts of DNA (Figure 5D, right panel). We predominantly saw monomeric ssDNA and dimeric forms and no major truncated forms, which supported our NGS results (Figure 5C).
The ExpiSf9-Derived AAV9 Vector Shows In Vivo Transduction Efficiency and Biodistribution Comparable to the Sf9-Derived AAV9 Vector
Finally, we tested the transduction efficiency of luciferase-expressing AAV9 vectors from the two cell lines in mice. We infected C57BL/6 mice intravenously with the AAV9 vectors at 2 × 1011 vector genomes per mouse. The noninvasive In Vivo Imaging System (IVIS) was used to visualize luciferase expression at days 7 and 14 post-transduction. We saw no significant differences in luciferase signal intensities and distributions at day 7 (Figure S2). At day 14, we found marginally higher levels and wider distributions of luciferase expression with the ExpiSf9-derived vector (Figure 6A, middle panel) than those of the Sf9-derived vector (Figure 6A, upper panel). The total flux was slightly higher in mice treated with ExpiSf9-derived AAV9 (Figure 6B). To further determine the biodistributions of the AAV9 vectors, we sacrificed the mice 3 weeks post-transduction and assessed the luciferase expression ex vivo (Figure 6C), which showed similar luciferase signal biodistributions between the two Sf9 and ExpiSf9 groups. Although luciferase signals in individual tissues were generally higher in the ExpiSf9 group, the biodistribution between the two groups was comparable, with major luciferase signals seen in the liver, heart, pancreas, and skeletal muscles (Figure 6D, upper panel). This similarity in biodistribution between the two groups was further confirmed with qPCR analysis of AAV genome copy numbers from DNA extracted from various tissues (Figure 6D, lower panel), although ExpiSf9-derived AAV9 had statistically higher amounts of AAV genomes in the liver, pancreas, kidney, and skeletal muscles and trended higher in the heart, lung, and spleen.
Figure 6.

Luciferase Expression and Biodistribution in Mice
Mice were tail-vein injected with AAV9 containing luciferase produced from Sf9, ExpiSf9, or Ad293 cells. (A) The mice were imaged by the In Vivo Imaging System (IVIS) at day 14, with the Sf9-derived AAV9 group (top), ExpiSf9-derived AAV9 group (middle), and Ad293-derived group (bottom). (B) In vivo luciferase signals were quantitated, and the average total flux (photons per second [p/s]) for each group of five mice is shown. ∗∗p < 0.005 and ∗∗∗p < 0.0005. (C) Ex vivo images of the mice organs were also taken at day 21, with the Sf9-derived AAV9 group (top), ExpiSf9-derived AAV9 group (middle), and Ad293-derived AAV9 group (bottom). (D) Ex vivo luciferase signals were quantitated, and the average total flux (p/s) for each group of two mice is shown (upper panel). qPCR was performed on DNA extracted from the collected mice tissues to quantitate how many copies of AAV viral genomes were present (lower panel). Sf9-derived AAV9 (green), ExpiSf9-derived AAV9 (yellow), and Ad293-derived AAV9 (blue). ∗p < 0.05, comparing Sf9-derived AAV9 to ExpiSf9-derived AAV9; &p < 0.05, comparing ExpiSf9-derived AAV9 to Ad293-derived AAV9; #p < 0.05, comparing Sf9-derived AAV9 to Ad293-derived AAV9.
In addition to comparing the in vivo transduction efficiencies of the two insect cell-based expression systems, we also compared them to the human Ad293 cell-derived AAV9. The results showed that the Ad293-derived AAV9 had higher luciferase signals both in vivo (Figures 6A, bottom panel, 6B, and S2B) and ex vivo (Figures 6C, bottom panel, and 6D, top panel). The qPCR analysis showed similar, increased levels of AAV genomic DNA in the liver of mice treated with Ad293-derived AAV9 (Figure 6D, lower panel). Unexpectedly, we found generally similar levels of AAV DNA in other organs tested for all three expression systems (Figure 6D).
Overall, our results demonstrate the increased AAV vector productivity of ExpiSf9 cells compared to Sf9 cells, with no notable difference in vector infectivity and biodistributions between the AAV9 preparations produced from the two insect expression systems.
Discussion
In this study, we have shown that significantly higher AAV yields can be obtained by using the ExpiSf Baculovirus Expression System with a clinically applicable, CD culture medium. For the three serotypes tested, AAV9 yields increased ~7-fold, AAV2 yields increased ~15-fold, and AAV8 yields increased ~19-fold. Of note, the yields from 300 mL of suspension culture for AAV8 and AAV9 were over or close to 5 × 1013 viral genomes (vgs), which would be sufficient for general preclinical AAV studies. Our data thus demonstrate that the ExpiSf expression system allows facile and scalable manufacturing of AAV vectors in CD medium.
After optimization of AAV vector production in the ExpiSf expression system, we determined the AAV expression conditions of using an MOI of 1 per baculovirus with a cell density of 4–5 × 106 cells per mL at the time of infection, and harvesting at 72 h postinfection was close to optimal. Based on our optimization data, we may be able to obtain similar titers at 48 h postinfection if we use an MOI ≥3. However, we concluded that it would be more economical to maintain the culture for 72 h postinfection in order to conserve our baculoviral stocks. We did not see any difference in AAV infectivity between the 48 and 72 h post-baculovirus infection-harvested AAVs. Although at 96 h postinfection, the MOI ≥5 had higher titers when compared to all other conditions, its in vitro infectivity declined when compared to 72 h. Since insect cell viability dropped to less than 50% at 96 h, it is conceivable that after 72 h postinfection, many defective AAV particles are produced in dying cells.
In order to ensure that the quality of the purified AAV preparations from the two expressions systems was similar, we compared the capsid ratio and infectivity of AAV9. The CE-SDS results showed that the VP1:VP2:VP3 ratios between the two systems were indeed comparable. The in vitro results with ZsGreen-expressing AAV8 and AAV9 vectors showed a slight decrease in infectivity for the ExpiSf9-derived vectors (Figure 3B). In contrast, the in vivo results showed that the ExpiSf9-derived AAV9 had a marginal but statistically significant increase in luciferase signals. In either case, the differences were no more than 2-fold. Thus, we concluded that there is no detrimental impact on the AAV vector infectivity when using the ExpiSf expression system.
When comparing the in vivo transduction of the insect cell-derived AAV9 versus the mammalian cell-derived AAV9, we saw higher signals for the mammalian cell-derived AAV9. This phenomenon may be due to the suboptimal capsid VP1 ratio for the insect cell-derived AAV9 since the capsid proteins may play a role in second-strand synthesis and transcription.20 Indeed, insect cell-based AAV production results in the lower levels of VP1 incorporation in the AAV particles, which results in lower infectivity.12 Our current study confirmed this observation. It is conceivable that improved VP1 incorporation in insect cell-derived AAVs by introduction of an artificial intron21 or an optimized Kozak sequence22 would improve the transduction efficiency of ExpiSf9-derived AAV vectors in vivo.
One major hurdle for AAV gene therapy is producing enough virus for clinical applications. Typically, vector requirements range between 1 × 1015 and 1 × 1016 vector genomes in total per patient (1 × 1013 to 1 × 1014 vector genomes per kilogram).6 In this study, we achieved titers up to 1 × 1014 vector genomes of AAV vectors, highly purified through two rounds of density gradient ultracentrifugation, from 300 mL of suspension ExpiSf9 cell culture. Thus, 3 L to 30 L of ExpiSf9 cell culture would be sufficient to support production of AAV vectors for a single patient, which is still challenging, but feasible, when compared with other insect and mammalian expression systems.
In conclusion, we have shown that use of a high-density insect cell line expression system is a viable option for obtaining high AAV yields. The use of this new expression system would help alleviate the burden of AAV production at lab scale and also potentially be useful for large-scale manufacturing of clinical-grade AAV vectors.
Materials and Methods
Cell Lines and Media
The Sf9 cell line (Thermo Fisher Scientific, Waltham, MA, USA) was cultured in Sf-900 II serum-free medium (SFM; Thermo Fisher Scientific). The ExpiSf9 cell line (Thermo Fisher Scientific) was cultured in ExpiSf CD medium (Thermo Fisher Scientific). Cells were maintained at 28°C, 125 rpm in a Multitron incubator (Infors HT, Annapolis Junction, MD, USA). Both HEK293T (ATCC, Manassas, VA, USA) and Ad293 cell lines (ATCC) were cultured in DMEM (Gibco, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (FBS; Gibco). Cell counts and viabilities were measured with a Vi-CELL XR (Beckman Coulter, Brea, CA, USA).
Plasmid Constructs
For the baculoviral constructs, the transgene cassettes flanked by the AAV2 ITRs were cloned into the pFastBac1 plasmid (Thermo Fisher Scientific). The AAV2 Rep gene and Cap genes of different serotypes were both designed and cloned into the pFastBac Dual (Thermo Fisher Scientific) plasmid, as previously described.12 Recombinant baculoviruses were then produced using the Bac-to-Bac Baculovirus Expression System (Thermo Fisher Scientific). The baculoviral stocks were then amplified to increase both baculoviral titer and volume of stock. The titers for the baculoviral stocks were approximately 1–2 × 108 PFU/mL. The constructs for triple transfection in Ad293 cells were designed as previously described.4,5 The transgene cassette is identical to the one depicted in Figure 1A (bottom construct).
Optimization of ExpiSf9 AAV Production
For optimization experiments, the cells were split at the appropriate cell densities, and the ExpiSf Enhancer reagent (Thermo Fisher Scientific) was added 24 h prior to transduction with baculovirus according to the vendor. Due to the temporary halt in cell division after ExpiSf Enhancer addition and for simplicity, the amount of baculovirus used was based on the cell density at the time of addition of the ExpiSf Enhancer reagent. 1 mL aliquots of each condition were taken at the predefined time points and centrifuged for 5 min at 2,000 rpm in a microcentrifuge. The supernatants were then discarded, and the cell pellets were resuspended in 1 mL of lysis buffer (50 mM Tris-HCl, 150 mM NaCl, pH 8.0). Three freeze/thaws were performed, and the cell debris was spun down for 15 min, 13,000 rpm in a microcentrifuge. The supernatant containing AAV2 was transferred to a fresh microcentrifuge tube and frozen at −80°C.
qPCR for Measuring the AAV Titer
For the AAV samples prepared during the optimization experiments, the viral DNAs for qPCR were prepared using the AAVpro Titration Kit, version (v.)2 (Takara Bio USA, Mountain View, CA, USA). For the iodixanol-purified AAV preparations, the viral DNA was also prepared using the same kit; however, the DNase step was omitted, since the preparations were already Benzonase treated before purification. The prepared viral DNA samples were analyzed using the TaqMan Fast Universal PCR Master Mix (Applied Biosystems, Foster City, CA, USA). All assays were performed on the 7900HT Fast Real-Time PCR instrument (Applied Biosystems) using a Fast Optical 96-well reaction plate (Applied Biosystems). Thermocycler conditions were 1 cycle of 95°C for 120 s, followed by 40 cycles of 95°C for 5 s and 60°C for 30 s. Each reaction contained 10 ng of viral DNA, 1 μM forward primer (5′-GGAACCCCTAGTGATGGAGTT-3′), 1 μM reverse primer (5′-CGGCCTCAGTGAGCGA-3′), and 0.25 μM probe (6-Carboxyfluorescein [FAM] 5′-CACTCCCTCTCTGCGCGCTCG-3′ non fluorescent quencher [NFQ]), which targets the AAV2 ITR. For the detection of AAV genomes in mice tissues, each reaction contained 40 ng of DNA.
Recombinant AAV Production
For AAV production in Sf9 and ExpiSf9 cells, 300 mL culture with a density of 1 × 106 cells/mL or 4–5 × 106 cells/mL was used, respectively. In addition, for AAV production in ExpiSf9 cells, the ExpiSf Enhancer reagent (Thermo Fisher Scientific) was added, according to the vendor’s instructions, 24 h prior to the addition of the baculoviruses. Each 300 mL of culture was infected at an MOI of 1 per baculovirus. The cell pellets were harvested 72 h postinfection, resuspended in 4–6 mL of lysis buffer (50 mM Tris-HCl, 150 mM NaCl, pH 8.0), and frozen at −80°C for purification. For AAV production in Ad293 cells, 1.8e8 cells were plated in a 5-layer CellSTACK (Corning, Corning, NY, USA) and placed in 37°C, 5% CO2 overnight. The next day, the cells were transfected using an in-house-developed method similarly described previously.23 Alternatively, 2.8e8 cells were plated in a 5-layer CellSTACK and placed in 37°C, 5% CO2 for 6 h before transfection. The cell pellets were harvested 72 h postinfection, resuspended in 4–6 mL of lysis buffer (50 mM Tris-HCl, 150 mM NaCl, pH 8.0), and frozen at −80°C for purification.
AAV Purification
The frozen cell pellets were lysed by performing 3 freeze/thaws, followed by addition of ≥500 units of Benzonase (Sigma, St. Louis, MO, USA) and incubation at 37°C for 1 h. The samples were then centrifuged for 30 min, 4,750 rpm, 4°C in an Allegra X-15R centrifuge (Beckman Coulter, Brea, CA, USA). The supernatants were then placed on top of an iodixanol gradient for purification.18 OptiSeal polypropylene centrifuge tubes (Beckman Coulter) were used. The samples were placed into a Ti70 rotor (Beckman Coulter, Brea, CA, USA) and spun at 69,000 rpm for 1 h and 10 min at 17°C. After centrifugation, an 18-gauge syringe (Becton Dickinson, Franklin Lakes, NJ, USA) was used to extract the purified AAV (~5 mL) and was placed into a 50-mL conical tube (VWR, Radnor, PA, USA). 40 mL of Dulbecco’s PBS (DPBS; Sigma, St. Louis, MO, USA), supplemented with 0.001% Pluronic F-68 (Gibco, Gaithersburg, MD, USA), was added to the 50-mL conical tube. After washing a Vivaspin 20, 100-kDa molecular weight cutoff (MWCO) sample concentrator (GE Healthcare, Chicago, IL, USA) with 20 mL of DPBS + 0.001% F-68, the 45 mL of AAV solution was concentrated down to ~2–3 mL. Fresh DPBS was added to bring the volume up to ~20 mL and was concentrated back down to ~2–3 mL. Fresh DPBS was added again up to ~20 mL and concentrated down to ~1 mL and then aliquoted and frozen at −80°C.
Imaging of AAV-Infected Cells
HEK293T cells were plated 1 day before transduction at a cell density of 1 × 105 cells/well in 1 mL total volume in a 24-well plate (Corning, Corning, NY, USA). The next day, with a cell density of ~2 × 105 cells/well, the cells were transduced with the appropriate amount of purified AAV. The cells were then imaged by EVOS (Thermo Fisher Scientific), 48 h postinfection.
SDS-PAGE Stained with SYPRO Red
Samples of purified AAV8 and AAV9 produced in both Sf9 and ExpiSf9 cells were prepared for SDS-PAGE gel by adding the appropriate amount of NuPAGE lithium dodecyl sulfate (LDS) sample buffer 4× and NuPAGE sample-reducing agent 10× (Thermo Fisher Scientific). The samples were then placed at 70°C for 10 min. Equal amounts of AAV were loaded in a NuPAGE 4%–12% Bis-Tris 10-well gel (Thermo Fisher Scientific) for each serotype and run in 2-(N-morpholino)ethanesulfonic acid (MES) buffer (Life Technologies, Carlsbad, CA, USA). After the run, the gel was rinsed in distilled water (diH2O) and incubated in 7.5% acetic acid with SYPRO Red Protein Gel Stain 1:5,000 dilution (Sigma, St. Louis, MO, USA) for 1 h. The gel was then rinsed in 7.5% acetic acid and imaged on a Gel Doc EZ Imager (Bio-Rad, Hercules, CA, USA).
Flow Cytometry for In Vitro Potency
To measure the potency in vitro, the percentage of ZsGreen-positive cells was measured using flow cytometry. Ad293 cells were plated 1 day before transduction at a cell density of 1 × 105 cells/well in 1 mL total volume in a 24-well plate (Corning, Corning, NY, USA). The next day, with a cell density of ~2 × 105 cells/well, the cells were transduced with the appropriate amount of purified AAV. The cells were then run on a LSR II (BD Biosciences, San Jose, CA, USA), 48 h postinfection. Data analysis was done using FlowJo (Becton Dickinson, Franklin Lakes, NJ, USA).
AAV9 Cap Ratio Analysis via CE-SDS
A volume of 50 μL of AAV9 sample (~4 × 1012 vg/mL) was combined with 45 μL sample buffer (100 mM Tris-HCl, pH 9.0/1% SDS) and 5 μL 2-mercaptoethanol (Millipore Sigma, Burlington, MA, USA). The Cap proteins were denatured by incubating the mixture at 75°C for 10 min. The mixture was then added to a Vivaspin 500, 10,000 MWCO sample concentrator (GE Healthcare, Chicago, IL, USA), combined with 400 μL of a mixture of 0.5 mg/mL SDS + 5% 2-mercaptoethanol, and centrifuged at 15,000 × g for 5 min. The collection tube was emptied, and 500 μL of 0.5 mg/mL SDS + 5% 2-mercaptoethanol was added to the Vivaspin sample concentrator before centrifuging at 15,000 × g for 5 min. This process was done twice to desalt the sample. After the final centrifugation, the final volume was brought up to 100 μL by adding 90 μL of water. This 100-μL sample was transferred into a CE autosampler vial for injection. A PA800 Plus Pharmaceutical Analysis System (SCIEX, Framingham, MA, USA) with a photodiode array (PDA) detector was used for the analysis. The 32 Karat software (SCIEX, Framingham, MA, USA) was used to run the instrument, and the data were exported to Empower 3 software (Waters, Milford, MA, USA) for analysis. The instrument method followed was as previously published with the following exceptions: no water preinjection plug was used, and samples were injected for 10 s due to higher viral titers in our samples.24 Peaks were assigned as VP3, VP2, and VP1, based on theoretical mass, compared to a size standard and retention time, as has been shown in the literature.24 The relative percent areas of VP3, VP2, and VP1 were calculated using the % time-corrected area (TCA), determined by CE-SDS and each protein’s molar extinction coefficient (EC). The molar EC for each protein was manually calculated using the amino acid sequence of the VP1, VP2, and VP3 proteins.25 The relative percent area of each protein was then converted to a molar percentage based on the sum of all three VP proteins. Finally, the number of VP proteins was calculated based on the molar percentage and assuming a total of 60 copies of the viral protein per Cap.
Full versus Empty AAV Cap Analysis via AUC
Sedimentation velocity AUC was performed on AAV9 samples using the Optima (Beckman Coulter), according to Burnham et al.19 Briefly, 400 μL of either sample buffer or AAV9 sample was loaded into a two-sector velocity cell. The sample cell was then placed in a 8-hole An-50 Ti rotor (Beckman Coulter) and then placed in the Optima. Radial calibration was then performed at 20°C, and the samples were left in the instrument under vacuum for 1 h to equilibrate. Sedimentation velocity ultracentrifugation was then performed at 20,000 rpm at 20°C. Raleigh interference optics were used. For analysis, the SEDFIT software (NIH, Bethesda, MD, USA) was used to calculate the percentage of full AAV particles via peak integration.
Alkaline Gel Electrophoresis
AAV9 genomes derived from either Sf9 or ExpiSf9 cells were run on an alkaline gel, as previously described.26 Briefly, 1% agarose gel was made in alkaline electrophoresis buffer, which is composed of 50 mM NaOH and 1 mM EDTA. AAV DNA samples were prepared using 6× alkaline sample loading dye (VWR), heated at 95°C for 3 min, and then cooled on ice prior to gel loading. The gel was run in the cold room for 4 h at 50 V. The gel was then gently rocked in 0.1 M Tris-HCl (pH 8.5) buffer for 1 h. The buffer was then discarded and replaced with 0.1 M NaCl buffer containing 0.5 μg/mL of ethidium bromide. The container was protected from light and gently rocked for another 2 h and then visualized on a UV transilluminator.
NGS of AAV9
AAV9 DNA was quantified using the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). Each virus was PCR amplified with AmpliTaq Gold 360 PCR Mastermix (Thermo Fisher Scientific) with 25 μL reaction volumes (forward PCR primer: 5′-AGG GGT TCC TAT CGA TAT CAA-3′; reverse PCR primer: 5′-AGG GGT TCC TAG ATC TAT CG-3′). AAV9 DNA input into PCR was 172 ng. Thermal cycling protocol was the following: 95°C, 10 min; 35 cycles of 95°C for 30 s, 56°C for 30 s, and 72°C for 2.5 min; final extension, 72°C for 7 min.
The ~2.3-kb amplicon was purified with the QIAGEN PCR Purification Kit (QIAGEN, Hilden, Germany) and eluted in 31 μL of water. Purified amplicons were visualized with the 4200 TapeStation System using D5000 ScreenTape (Agilent, Santa Clara, CA, USA). The purified amplicons were size selected using BluePippin (Sage Science, Beverly, MA, USA) with the 0.75% agarose gel cassette (BLF7510) and external marker S1, selected for range mode 1747-2925 bp. The size-selected amplicons were visualized and quantified on the 4200 TapeStation System using D5000 ScreenTape (Agilent).
Approximately 5 ng of each amplicon was used as input into the Nextera XT DNA Library Preparation Kit (Illumina, San Diego, CA, USA). Libraries were prepared and cleaned per standard Illumina protocol. Cleaned libraries were quantified on the QuantStudio 12K Flex Real-Time PCR System (Thermo Fisher Scientific) using the KAPA Library Quantification Kit for Illumina platforms with ROX Low qPCR Mastermix (Roche Sequencing and Life Science, Kapa Biosystems, Wilmington, MA, USA).
Libraries were sequenced on the MiSeq System with the MiSeq Reagent Nano Kit v.2 (Illumina, San Diego, CA, USA) with 251 bp paired-end reads and 1% PhiX spike-in.
To check the sequencing reads for potential truncations and/or insertions, we used the BBSplit program to map the reads to the AAV transgene cassette.27
Assembly of the sequencing reads into a consensus sequence was performed with our NGS-Microbial Surveillance Toolbox (MSTB), a fully automated distributed pipeline that was implemented at AstraZeneca with a common workflow language (CWL) and with a user interface based on the Galaxy bioinformatics workbench. The pipeline extends the Ariba package with the internal assembly step designed to tolerate high local variation of sequencing depth typical for the shotgun Illumina sequencing of viral PCR amplicons. That assembly step performs multiple rounds of read-length filtering after adaptor trimming and digital normalization,27 followed by a de novo assembly with SPAdes28,29 and a polishing step with a Pilon30 package modified to emit ambiguous nucleotides in the FASTA consensus based on majority voting at each base.27, 28, 29, 30 Ariba then selects the assembly with the best coverage of the reference genome. Our pipeline creates an interactive Web report that provides genome browser views to examine minor variants relative to the assembly consensus, as well as to the reference. A manuscript with detailed description of the assembly pipeline and its open-source release is under review.
Animal Studies
All animal experiments were approved by the Institutional Animal Care and Use Committee of AstraZeneca (Gaithersburg, MD, USA). Male C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA) at the age of 4 weeks. On the 5th week, mice were preheated with a heat lamp and injected with 2 × 1011 vector genomes of AAV9 luciferase vectors per mouse through the tail vein using an insulin syringe (Becton Dickinson, Franklin Lakes, NJ, USA). The mice were monitored daily for any possible signs of illness. For in vivo bioluminescence imaging, mice were injected intraperitoneally at 150 mg/kg D-luciferin (Perkin Elmer, Waltham, MA, USA) and followed by gas anesthesia at 2.5% isoflurane. The IVIS (Spectrum; Perkin Elmer) was used to take images every 7 days postinjection (day 0) until the end of the study (day 21). At day 21, the mice were also sacrificed, and ex vivo imaging was performed on selected organs (heart, lungs, liver, pancreas, liver, spleen, and skeletal muscle from the left upper thigh). Mice tissues were frozen in dry ice in a microcentrifuge tube for qPCR analysis. DNA extraction from the tissues were done using the AllPrep DNA/RNA Mini Kit (QIAGEN). Luciferase signals were quantified using LivingImage 4.4 software (Perkin Elmer).
Statistical Analyses
All statistical analyses were performed using GraphPad Prism 8 software. The Student’s t test analysis was used, with a p value <0.05 considered to be statistically significant.
Author Contributions
Conceptualization, J.H.K., S.D.W., and Y.I.; Methodology, J.H.K., A.P., R.A.H., C.R.S., A.T., and Y.I.; Investigation, J.H.K., A.P., R.A.H., C.R.S., and A.T.; Manuscript writing, J.H.K.; Editing, C.L.D., A.E.S., C.G., S.D.W., and Y.I.
Conflicts of Interest
All authors are employees of AstraZeneca and have stock and/or stock options or interests in AstraZeneca.
Acknowledgments
We would like to thank Cuihua Gao, Andrew Garcia, Sandrina Phipps, Radhika Rayanki and Darrick Yu at AstraZeneca for their technical support and helpful discussions.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.09.018.
Supplemental Information
References
- 1.Wang D., Tai P.W.L., Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019;18:358–378. doi: 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.U.S. Food & Drug Administration LUXTURNA. 2018. https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/luxturna
- 3.U.S. Food & Drug Administration ZOLGENSMA. 2020. https://www.fda.gov/vaccines-blood-biologics/zolgensma
- 4.Xiao X., Li J., Samulski R.J. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsushita T., Elliger S., Elliger C., Podsakoff G., Villarreal L., Kurtzman G.J., Iwaki Y., Colosi P. Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther. 1998;5:938–945. doi: 10.1038/sj.gt.3300680. [DOI] [PubMed] [Google Scholar]
- 6.Clément N., Grieger J.C. Manufacturing of recombinant adeno-associated viral vectors for clinical trials. Mol. Ther. Methods Clin. Dev. 2016;3:16002. doi: 10.1038/mtm.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith G.E., Summers M.D., Fraser M.J. Production of human beta interferon in insect cells infected with a baculovirus expression vector. Mol. Cell. Biol. 1983;3:2156–2165. doi: 10.1128/mcb.3.12.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monie A., Hung C.F., Roden R., Wu T.C. Cervarix: a vaccine for the prevention of HPV 16, 18-associated cervical cancer. Biologics. 2008;2:97–105. [PMC free article] [PubMed] [Google Scholar]
- 9.Cox M.M., Hollister J.R. FluBlok, a next generation influenza vaccine manufactured in insect cells. Biologicals. 2009;37:182–189. doi: 10.1016/j.biologicals.2009.02.014. [DOI] [PubMed] [Google Scholar]
- 10.Felberbaum R.S. The baculovirus expression vector system: A commercial manufacturing platform for viral vaccines and gene therapy vectors. Biotechnol. J. 2015;10:702–714. doi: 10.1002/biot.201400438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.European Medicines Agency Glybera. 2017. https://www.ema.europa.eu/en/medicines/human/EPAR/glybera#authorisation-details-section
- 12.Urabe M., Ding C., Kotin R.M. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum. Gene Ther. 2002;13:1935–1943. doi: 10.1089/10430340260355347. [DOI] [PubMed] [Google Scholar]
- 13.Smith R.H., Levy J.R., Kotin R.M. A simplified baculovirus-AAV expression vector system coupled with one-step affinity purification yields high-titer rAAV stocks from insect cells. Mol. Ther. 2009;17:1888–1896. doi: 10.1038/mt.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kozak M. The scanning model for translation: an update. J. Cell Biol. 1989;108:229–241. doi: 10.1083/jcb.108.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kozak M. Bifunctional messenger RNAs in eukaryotes. Cell. 1986;47:481–483. doi: 10.1016/0092-8674(86)90609-4. [DOI] [PubMed] [Google Scholar]
- 16.Kozak M. Initiation of translation in prokaryotes and eukaryotes. Gene. 1999;234:187–208. doi: 10.1016/s0378-1119(99)00210-3. [DOI] [PubMed] [Google Scholar]
- 17.Thermo Fisher Scientific ExpiSf Baculovirus Expression System. 2019. https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-expression/insect-protein-expression/expisf-expression-system.html
- 18.Crosson S.M., Dib P., Smith J.K., Zolotukhin S. Helper-free Production of Laboratory Grade AAV and Purification by Iodixanol Density Gradient Centrifugation. Mol. Ther. Methods Clin. Dev. 2018;10:1–7. doi: 10.1016/j.omtm.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burnham B., Nass S., Kong E., Mattingly M., Woodcock D., Song A., Wadsworth S., Cheng S.H., Scaria A., O’Riordan C.R. Analytical Ultracentrifugation as an Approach to Characterize Recombinant Adeno-Associated Viral Vectors. Hum. Gene Ther. Methods. 2015;26:228–242. doi: 10.1089/hgtb.2015.048. [DOI] [PubMed] [Google Scholar]
- 20.Salganik M., Aydemir F., Nam H.J., McKenna R., Agbandje-McKenna M., Muzyczka N. Adeno-associated virus capsid proteins may play a role in transcription and second-strand synthesis of recombinant genomes. J. Virol. 2014;88:1071–1079. doi: 10.1128/JVI.02093-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen H. Intron splicing-mediated expression of AAV Rep and Cap genes and production of AAV vectors in insect cells. Mol. Ther. 2008;16:924–930. doi: 10.1038/mt.2008.35. [DOI] [PubMed] [Google Scholar]
- 22.Kondratov O., Marsic D., Crosson S.M., Mendez-Gomez H.R., Moskalenko O., Mietzsch M., Heilbronn R., Allison J.R., Green K.B., Agbandje-McKenna M., Zolotukhin S. Direct Head-to-Head Evaluation of Recombinant Adeno-associated Viral Vectors Manufactured in Human versus Insect Cells. Mol. Ther. 2017;25:2661–2675. doi: 10.1016/j.ymthe.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang X., Hartley A.V., Yin Y., Herskowitz J.H., Lah J.J., Ressler K.J. AAV2 production with optimized N/P ratio and PEI-mediated transfection results in low toxicity and high titer for in vitro and in vivo applications. J. Virol. Methods. 2013;193:270–277. doi: 10.1016/j.jviromet.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang C.X., Meagher M.M. Sample Stacking Provides Three Orders of Magnitude Sensitivity Enhancement in SDS Capillary Gel Electrophoresis of Adeno-Associated Virus Capsid Proteins. Anal. Chem. 2017;89:3285–3292. doi: 10.1021/acs.analchem.6b02933. [DOI] [PubMed] [Google Scholar]
- 25.Kuipers B.J., Gruppen H. Prediction of molar extinction coefficients of proteins and peptides using UV absorption of the constituent amino acids at 214 nm to enable quantitative reverse phase high-performance liquid chromatography-mass spectrometry analysis. J. Agric. Food Chem. 2007;55:5445–5451. doi: 10.1021/jf070337l. [DOI] [PubMed] [Google Scholar]
- 26.Kohlbrenner E., Weber T. Production and Characterization of Vectors Based on the Cardiotropic AAV Serotype 9. Methods Mol. Biol. 2017;1521:91–107. doi: 10.1007/978-1-4939-6588-5_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bushnell B., Rood J., Singer E. BBMerge - Accurate paired shotgun read merging via overlap. PLoS ONE. 2017;12:e0185056. doi: 10.1371/journal.pone.0185056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nurk S., Bankevich A., Antipov D., Gurevich A.A., Korobeynikov A., Lapidus A., Prjibelski A.D., Pyshkin A., Sirotkin A., Sirotkin Y. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 2013;20:714–737. doi: 10.1089/cmb.2013.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walker B.J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., Cuomo C.A., Zeng Q., Wortman J., Young S.K., Earl A.M. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.