

Abstract
5-Hydroxytryptamine receptor 2A (HTR2A) is a central regulator of fetal brain development and cognitive function in adults. However, the roles of HTR2A in the cardiovascular system are not fully understood. Here in this study, we explored the function of HTR2A in cardiac hypertrophy. Significantly, the expression levels of HTR2A mRNA and protein levels were upregulated in hypertrophic hearts of human patients. Besides, the expression of HTR2A was also upregulated in isoproterenol (ISO)-induced cardiac hypertrophy in the mouse. Next, the expression of HTR2A was knocked down with shRNA or overexpressed with adenovirus in neonatal rat cardiomyocytes, and ISO was used to induce cardiomyocyte hypertrophy. We showed that HTR2A knockdown repressed ISO-induced cardiomyocyte hypertrophy, which was demonstrated by decreased cardiomyocyte size and repressed expression of hypertrophic fetal genes (e.g., myosin heavy chain beta (β-Mhc), atrial natriuretic peptide (Anp), and brain natriuretic peptide (Bnp)). By contrast, HTR2A overexpression promoted cardiomyocyte hypertrophy. Of note, we observed that HTR2A promoted the activation (phosphorylation) of AKT-mTOR (mammalian target of rapamycin) signaling in cardiomyocytes, and repression of AKT-mTOR with perifosine or rapamycin blocked the effects of HTR2A on cardiomyocyte hypertrophy. Finally, we showed that HTR2A regulated AKT-mTOR signaling through activating the PI3K-PDK1 pathway, and inhibition of either PI3K or PDK1 blocked the roles of HTR2A in regulating AKT-mTOR signaling and cardiomyocyte hypertrophy. Altogether, these findings demonstrated that HTR2A activated PI3K-PDK1-AKT-mTOR signaling and promoted cardiac hypertrophy.
Keywords: Cardiac hypertrophy, HTR2A, PI3K, PDK1, Akt, mTOR
Introduction
Cardiac hypertrophy is the fundamental biological process underlying cardiac diseases, including arrhythmia and heart failure (Nakamura and Sadoshima 2018). Cardiac hypertrophy includes physiological and pathological types, and the latter one is irreversible and contributes to cardiac remodeling and functional decline (Shimizu and Minamino 2016). Physiologically hypertrophic growth of cardiomyocytes is observed during postnatal heart development and duration of pregnancy, which can significantly improve and maintain cardiac function and homeostasis (Chen et al. 2020). By contrast, pathologically hypertrophic growth is triggered by pathological stresses, and uncontrolled cardiac hypertrophy leads to cardiac dysfunction, such as arrhythmia and heart failure (Tang et al. 2020). Various pathological stresses can induce the hypertrophic growth of cardiomyocytes, including hypertension, myocardial infarction, and neuroendocrine factors (e.g., endothelin 1 (ET-1), isoproterenol (ISO), and angiotensin II (Ang II)) (Nakamura and Sadoshima 2018; Shimizu and Minamino 2016; Rohini et al. 2010). For instance, the neuroendocrine factors can bind to the seven-transmembrane G protein-coupled receptors (GPCRs) on the cardiomyocyte membrane to trigger the activation of intracellular signaling pathways and drive the hypertrophy growth program within cardiomyocytes (Nakamura and Sadoshima 2018).
Various intracellular signaling pathways and factors contribute to the development of cardiac hypertrophy (Rohini et al. 2010). These factors include signaling transducers (e.g., phosphatidylinositol 3-kinase (PI3K), AKT, and mitogen-activated protein kinase (MAPK)) (Rohini et al. 2010; Aoyagi and Matsui 2011), metabolic regulators (e.g., AMP-dependent protein kinase (AMPK), NAD+-dependent Sirtuins, mammalian target of rapamycin (mTOR), the Forkhead box subfamily O (FOXO)) (Sciarretta et al. 2018; Tang et al. 2017; Kemi et al. 2008), and histone regulators (e.g., histone deacetylase (HDAC) and demethylases) (Nakamura and Sadoshima 2018; Shimizu and Minamino 2016). For instance, the neuroendocrine factors and insulin, as well as an insulin-like growth factor (IGFs), can bind their receptors and activate PI3K, which can activate AKT in a phosphoinositide-dependent protein kinase-1 (PDK1)-dependent and -independent manner (Aoyagi and Matsui 2011; Zhou et al. 2018). The activation of AKT can trigger the phosphorylation and inhibition of tuberous sclerosis complex (TSC)1/TSC2, the upstream repressor of mTOR (Sciarretta et al. 2018). Activated mTOR is a key orchestrator that determines cell size partially via activating ribosomal protein S6 kinase 1 (S6K1) to promote de novo protein synthesis (Sciarretta et al. 2018; Edinger and Thompson 2002). Previous studies have reported the critical roles of mTOR in cardiac hypertrophy, remodeling, and heart failure (Sciarretta et al. 2018; Xu and Brink 2016; Shende et al. 2016; Ren et al. 2018).
5-Hydroxytryptamine (5-HT) receptor 2A (HTR2A) is one of the receptors of 5-HT, a neurotransmitter with many roles. HTR2A is a central regulator for the development of the fetal brain and cognitive function of adults (Paquette and Marsit 2014). For instance, the methylation of the HTR2A gene can be modulated by biological and environmental factors and is closely correlated with the key measures of neurodevelopment (Paquette et al. 2013). Besides, HTR1A participates in the early stage of binocular visual cortex plasticity, whereas HTR2A and HTR3A contribute to the late cross-modal recruitment of the medial monocular visual cortex (Lombaert et al. 2018). The roles of HTR2A in other organs are also implicated. One such example is that the genetic polymorphism of HTR2A-1438G/A is associated with smoking and chronic obstructive pulmonary disease in human patients (Genis-Mendoza et al. 2019). Recently, in a community-based cohort study, Koh and colleagues reported that genetic variations in the HTR2A gene were correlated with the risk of hypertension (Choi et al. 2020). Besides, Lairez and colleagues reported the potential roles of HTR2A-CAMKII-HDAC4 in cardiac hypertrophy (Lairez et al. 2013). However, the roles and underlying mechanisms of HTR2A in cardiac hypertrophy are not fully understood.
In this study, we investigated the roles and underlying mechanisms of HTR2A in cardiac hypertrophy. HTR2A was overexpressed in hypertrophic hearts of humans and mice. HTR2A overexpression promoted cardiac hypertrophy through activating the PI3K-PDK1 signaling to trigger AKT-mTOR activation. Therefore, our findings showed that HTR2A was a critical regulator of cardiac hypertrophy.
Materials and methods
Human heart samples
In this study, human hypertrophic heart tissues (n = 6) and control heart tissues (n = 6) were collected at The Second Hospital of Hebei Medical University from May 2016 to January 2019. Hypertrophic heart specimens (left ventricle) were obtained from hypertrophic (failing) hearts that were removed during orthotopic heart transplantation. Control samples (left ventricle) were obtained intraoperatively from non-hypertrophic healthy hearts undergoing ventricular corrective surgery. Hypertrophy was diagnosed by histological analysis and heart function analysis as well as the expression of hypertrophic genes. The written form of consent was signed by each of the patients. The study design and experimental protocol were approved by the Ethics Committee of Clinical Research of Hebei Medical University.
Animal model of cardiac hypertrophy
The animal model of cardiac hypertrophy was induced in the mouse by ISO (Sigma) treatment using a previously described protocol (Cai et al. 2017). Briefly, 8–12-week-old male C57BL/6 mice were treated with ISO (50 mg/kg/day) via subcutaneous chronic infusion with Alzet minipump 2004 for 28 days. The study design and experimental protocol of animal studies were approved by the Ethics Committee of Animal Research of Hebei Medical University.
Quantitative real-time PCR
The total RNA was extracted and purified from heart tissue or cardiomyocytes with TRIZOL reagent (Invitrogen). Then, the first-strand cDNA was synthesized with 1-μg total RNA by using the high-capacity cDNA Reverse Transcription Kit (ThermoFisher, # 4368814). Quantitative real-time PCR was next performed to analyze the mRNA expression levels of target genes using the SYBR Green II quantitative real-time PCR KIT (QIAGEN). The forward and reverse primers used for quantitative real-time PCR are shown in Table 1. The double delta CT method was applied to analyze the quantitative real-time PCR (qRT-PCR) data using GAPDH as a house-keeping control gene.
Table 1.
Primers used for quantitative real-time PCR
| Gene symbol | Forward primer (5′-3′) | Reverse primer (5′-3′) | Ref |
|---|---|---|---|
| Human ANP | AAGAAAGCACACCAACGCAG | GATGGTGACTTCCTCGCCTC | Primerbank |
| Human BNP | CTGATCCGGTCCATCTTCCT | TGGAAACGTCCGGGTTACAG | Primerbank |
| Human β-MHC | CCAAGTTCACTCACATCCATCA | AGTGGCAATAAAAGGGGTAGC | Primerbank |
| Human HTR2A | CTTTGTGCAGTCTGGATTTACCT | ACTGATATGGTCCAAACAGCAAT | Designed in-house |
| Human GAPDH | GGCTGTTGTCATACTTCTCATGG | GGAGCGAGATCCCTCCAAAAT | Primerbank |
| Mouse Anp | TCTTCCTCGTCTTGGCCTTT | CCAGGTGGTCTAGCAGGTTC | (Luo et al. 2017) |
| Mouse Bnp | TGGGAGGTCACTCCTATCCT | GGCCATTTCCTCCGACTTT | (Luo et al. 2017) |
| Mouse β-Mhc | CGGACCTTGGAAGACCAGAT | GACAGCTCCCCATTCTCTGT | (Luo et al. 2017) |
| Mouse HTR2A | TAATGCAATTAGGTGACGACTCG | GAGGCTTCGGAAGTGTTAGCA | Designed in-house |
| Mouse Gapdh | TGTAGACCATGTAGTTGAGGTCA | AGGTCGGTGTGAACGGATTTG | (Luo et al. 2017) |
| Rat Anp | GAAGATGCCGGTAGAAGATGAG | AGAGCCCTCAGTTTGCTTTTC | (Luo et al. 2017) |
| Rat Bnp | CTGGAGACTGGCTAGGACTTC | GGTGCTGCCCCAGATGATT | (Luo et al. 2017) |
| Rat β-Mhc | GCCCCAAATGCAGCCAT | CGCTCAGTCATGGCGGAT | (Luo et al. 2017) |
| Rat HTR2A | GCTGGGTTTCCTTGTCATGC | ACAGATATGGTCCACACGGC | Designed in-house |
| Rat Gapdh | TGACAACTCCCTCAAGATTGTCA | GGCATGGACTGTGGTCATGA | (Luo et al. 2017) |
Western blot
The total protein was isolated from heart tissues or cardiomyocytes using the RIPA lysis buffer (Beyotime, #P0013K) supplied with a proteinase inhibitor cocktail (Sigma). For extraction of proteins from tissues, the tissues were cut and attrited in a glass burnisher within cold RIPA buffer, followed by RIPA lysis for 30 min at 4 °C. Thirty (30) micrograms of total protein was subjected to SDS-PAGE separation, and western blot was performed with the standard protocol described previously (Gong et al. 2020). SDS-PAGE Electrophoresis Buffer (#P0014A) and Western Transfer Buffer (#P0021A) were purchased from Beyotime. The primary anti-HTR2A (#ab66049, 1:1,000) and anti-GAPDH (#ab9485, 1:1,000) antibodies were purchased from Abcam. The primary anti-pPI3K (#17366, 1:1,000), anti-PI3K (#4257, 1:1,000), anti-pPDK1 (#3438, 1:1,000), anti-PDK1 (#3062, 1:1,000), anti-pAKT (#4060, 1:1,000), anti-AKT (#9272, 1:1,000), anti-pmTOR (#5536, 1:1,000), anti-mTOR (#2983, 1:1,000), anti-pS6K1 (#97596, 1:1,000), and anti-S6K1 (#9202, 1:1,000) antibodies were purchased from Cell Signaling Technology. The secondary antibodies (#G1213, #G1214) and ECL kit (#G2014) were purchased from Servicebio.
Isolation and culture of rat cardiomyocytes
Neonatal rat cardiomyocytes were isolated from 1- to 3-day-old Sprague-Dawley rats and used as previously described (Tang et al. 2016). The isolated cardiomyocytes were cultured in DMEM (Millipore) with 10% fetal bovine serum (FBS, ThermoFisher), penicillin/streptomycin (1,000 U/ml each; Gibco) at 37 °C, and 5% CO2. Besides, 100-mM 5-Bromo-2-deoxyuridine (Sigma) was supplied in the culture medium to repress the growth of cardiac fibroblasts. Forty-eight (48) hours later, the culture medium was replaced and the cells were used for further experiments.
Cardiomyocyte hypertrophy and treatment of drugs
To induce hypertrophy, the cardiomyocytes were starved for a serum for 24 h and followed by ISO treatment (50 μM) for an additional 48 h. Cardiomyocyte size was analyzed using the ImageJ (Fiji) software. A total of >100 cardiomyocytes were analyzed per sample, and the average cardiomyocyte size per sample was used for quantification, and three independent experiments were involved. mTOR inhibitor rapamycin, AKT inhibitor perifosine, PDK1 inhibitor OSU-03012, and PI3K inhibitor LY294002 were purchased from Selleck Chemicals and used to the treatment of cardiomyocytes. The detailed concentrations were shown in the figure legends.
Adenovirus packaging and gene silence/overexpression
To knock down the expression of HTR2A in rat cardiomyocytes, short-hairpin RNA (shRNA) targeting HTR2A (shHTR2A) was applied. shHTR2A (5′-GCTCAATTCCAAACTCCTTAA-3′) and control shRNA (shCtrl, 5′-GTTCACCGTAGTTCCGTTC-3′) were cloned into the adenovirus expressing plasmid pAdtrack with a U6 promoter. For overexpression of HRT2A, rat HTR2A (NM_017254.1) was cloned into the adenovirus expressing plasmid pAdtrack with a CMV promoter. The adenovirus was prepared in HEK293A cells using a protocol described previously (Jia et al. 2014). The cardiomyocytes were infected with the corresponding adenoviruses at a multiplicity of infection of 100 particles per cell for 24 h and were used for subsequent experiments with ISO with/without kinase inhibitors for an additional 48 h. Kinase inhibitors were added at the same time as ISO treatment.
Statistical analysis
The values were expressed as mean ± SD. All the experiments were repeated for at least three independent times. For analysis of the difference between the two groups, standard Student’s t test was applied. For analysis of the difference among the four groups, two-way ANOVA with Tukey post hoc test was performed. The statistical analysis was performed with GraphPad Prism version 8.0 and P values less than 0.05 were considered significant.
Results
HTR2A expression level is increased in hypertrophic hearts
HTR2A is the receptor for 5-HT and participates in the neuron system. The roles of HTR2A in the cardiovascular system were not known. To investigate the role of HTR2A in cardiac hypertrophy, we analyzed the expression of HTR2A in hypertrophic heart tissues in humans. We collected six hypertrophic heart tissues and six control heart tissues from humans. The overexpression of hypertrophy-associated fetal genes (ANP, BNP, and β-MHC) was verified in hypertrophic human hearts (Fig. 1a). The qRT-PCR and western blot analyses showed that HTR2A mRNA and protein levels were significantly upregulated in human hypertrophic hearts compared with the control hearts (Fig. 1b–c). Next, we tested the expression change of HTR2A in mouse hearts with hypertrophy. Cardiac hypertrophy in mice was induced by chronic treatment of ISO (50 mg/kg/day) for 4 weeks. Indeed, the expression of hypertrophy-associated marker genes (Anp, Bnp, and β-Mhc) was induced by ISO treatment in mouse hearts (Fig. 1d). Notably, the qRT-PCR and western blot results demonstrated that the mRNA and protein levels of HTR2A were increased in mouse hypertrophic hearts induced by ISO treatment (Fig. 1e–f). Taken together, these results collectively demonstrated that HTR2A expression was overexpressed in hypertrophic hearts in humans and mice.
Fig. 1.

HTR2A is overexpressed in human and mouse hypertrophic hearts. a mRNA level of marker genes of hypertrophy in human hearts. The RNAs from control and hypertrophic heart tissues of human patients were used for mRNA analysis with qRT-PCR (n = 6 in each group). ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide; β-MHC, myosin heavy chain beta. b mRNA level of HTR2A in human hearts from control donors and patients with cardiac hypertrophy (n = 6 in each group). HTR2A, 5-hydroxytryptamine receptor 2A. c The protein level of HTR2A in human hearts from control donors and patients with cardiac hypertrophy (n = 4 in each group). d mRNA level of marker genes of hypertrophy in mouse hearts. Cardiac hypertrophy was induced in male C57BL/6J mice by chronic treatment of isoproterenol (ISO, 50 mg/kg/day) for 28 days. mRNA levels of hypertrophic marker genes were analyzed with qRT-PCR (n = 5 in each group). e mRNA level of HTR2A in mouse control and hypertrophic hearts (n = 5 in each group). f The protein level of HTR2A in mouse control and hypertrophic hearts (n = 4 in each group). **P < 0.01 analyzed by using the Student’s t test
HTR2A promotes cardiomyocyte hypertrophy
HTR2A overexpression in cardiac hypertrophy implicated its involvement in cardiomyocyte hypertrophy. To test the roles of HTR2A in regulating cardiomyocyte hypertrophy, we knocked down HTR2A in neonatal rat cardiomyocytes with adenovirus-mediated shRNA (Fig. 2a and b ). Hypertrophy in rat cardiomyocyte was induced by ISO treatment (50 μM) for 48 h. ISO treatment induced the increase in cardiomyocyte size and overexpression of hypertrophic fetal genes such as Anp, Bnp, and β-Mhc (Fig. 2c and d ). Interestingly, HTR2A knockdown repressed ISO-induced hypertrophic growth of rat cardiomyocytes (Fig. 2c and d ). We also overexpressed rat HTR2A in rat cardiomyocytes with adenovirus, which was verified by qRT-PCR and western blot (Fig. 2e and f ). By contrast, HTR2A overexpression promoted ISO-induced cardiomyocyte hypertrophy, which was demonstrated by the increased cardiomyocyte size and enhanced expression of Anp, Bnp, and β-Mhc (Fig. 2g and h ). Taken together, these findings collectively supported that HTR2A promoted the hypertrophic growth of cardiomyocytes.
Fig. 2.

HTR2A promotes ISO-induced cardiomyocyte hypertrophy. a, b Short-hairpin RNA (shRNA) knockdown of HTR2A in cardiomyocytes. Rat cardiomyocytes were infected with adenovirus expressing shHTR2A (Ad-shHTR2A) or the control shRNA (Ad-shCtrl) for 48 h. Then, the mRNA (a) and protein (b) levels were analyzed with qRT-PCR and western blot respectively (n = 3 in each group). c, d The knockdown of HTR2A inhibits cardiomyocyte hypertrophy induced by ISO. Rat cardiomyocytes with/without HTR2A knockdown were subjected to hypertrophy induction with ISO (50 μM) treatment for 48 h. Cardiomyocyte size was quantified with the ImageJ software (c), and the expression of hypertrophy-associated fetal genes was analyzed with qRT-PCR (d, n = 3 in each group). e, f Overexpression of rat HTR2A in cardiomyocytes. Rat cardiomyocytes were infected with adenovirus overexpressing rat HTR2A gene (Ad-HTR2A) or control adenovirus (Ad-Ctrl) for 48 h. Then, the mRNA (e) and protein (f) levels were analyzed with qRT-PCR and western blot respectively (n = 3 in each group). g, h Overexpression of HTR2A promotes cardiomyocyte hypertrophy induced by ISO. Rat cardiomyocytes with/without HTR2A overexpression were subjected to hypertrophy induction with ISO (50 μM) treatment for 48 h. Cardiomyocyte size was quantified with the ImageJ software (g), and the expression of hypertrophy-associated fetal genes was analyzed with qRT-PCR (h, n = 3 in each group). **P < 0.01 analyzed by using the Student’s t test (a, e) or two-way ANOVA followed by the Tukey post hoc test (c, d, g, h)
HTR2A promotes AKT-mTOR signaling to regulate cardiomyocyte hypertrophy
The above results demonstrated that HTR2A overexpression in hypertrophic hearts contributed to cardiomyocyte hypertrophy. The next question was how HTR2A promoted cardiomyocyte hypertrophy? The AKT-mTOR is a key intracellular signaling pathway controlling protein synthesis and cardiomyocyte hypertrophy via activating the protein S6K1 (Xu and Brink 2016). Indeed, we observed that the phosphorylations of Akt, mTOR, and S6K1 were induced by ISO treatment. Interestingly, HTR2A knockdown repressed ISO-induced phosphorylations of AKT, mTOR, and S6K1 in rat cardiomyocytes (Fig. 3a). By contrast, HTR2A overexpression facilitated ISO-mediated phosphorylations of AKT, mTOR, and S6K1 (Fig. 3b). These findings demonstrated that HTR2A was an upstream activator for the AKT-mTOR-S6K1 signaling pathway. Next, we tested whether AKT and mTOR critically contributed to HTR2A function during cardiomyocyte hypertrophy. We treated the cardiomyocytes with AKT inhibitor perifosine or mTOR inhibitor rapamycin. We observed that either perifosine or rapamycin repressed the effects of HTR2A on the increase in cardiomyocyte size and the overexpression of hypertrophic fetal genes in ISO-induced hypertrophic cardiomyocytes (Fig. 3c–f). Collectively, we identified HTR2A as an upstream activator of the AKT-mTOR-S6K1 signaling pathway, which essentially participated in the function of HTR2A in cardiomyocyte hypertrophy.
Fig. 3.

HTR2A promotes AKT-mTOR-S6K1 signaling activation. a HTR2A knockdown represses AKT-mTOR-S6K1 signaling activated by ISO in rat cardiomyocytes. Cardiomyocytes with/without HTR2A knockdown were subjected to ISO treatment (50 μM) for 24 h. mTOR, mammalian target of rapamycin; S6K1, p70 ribosomal protein S6 kinase 1. Representative western blot and quantitative results are shown (n = 3). b HTR2A overexpression promotes AKT-mTOR-S6K1 signaling activated by ISO in rat cardiomyocytes. Cardiomyocytes with/without HTR2A overexpression were subjected to ISO treatment (50 μM) for 24 h. Representative western blot and quantitative results are shown (n = 3). c, d Inhibition of AKT represses HTR2A function in cardiomyocyte hypertrophy. Rat cardiomyocytes with/without HTR2A overexpression were subjected to hypertrophy induction with ISO (50 μM) treatment for 48 h. The cardiomyocytes were also treated with/without AKT inhibitor perifosine (1 μM). Cardiomyocyte size was quantified with the ImageJ software (c), and the expression of hypertrophy-associated fetal genes was analyzed with qRT-PCR (d, n = 3 in each group). e, f Inhibition of mTOR represses HTR2A function in cardiomyocyte hypertrophy. Rat cardiomyocytes with/without HTR2A overexpression were subjected to hypertrophy induction with ISO (50 μM) treatment for 48 h. The cardiomyocytes were also treated with/without AKT inhibitor rapamycin (100 nM). Cardiomyocyte size was quantified with the ImageJ software (e), and the expression of hypertrophy-associated fetal genes was analyzed with qRT-PCR (f, n = 3 in each group). **P < 0.01 analyzed by two-way ANOVA followed by the Tukey post hoc test. Cardiomyocyte culture sets were performed on three different dates. Western blot was performed for each experiment set, and representative western blot results were shown
HTR2A activates AKT-mTOR via PI3K-PDK1 during cardiomyocyte hypertrophy
Next, we investigated the mechanism by which HTR2A activated AKT-mTOR. The AKT is activated by PI3K in a PDK1-dependent or -independent manner during cardiac hypertrophy (Edinger and Thompson 2002). Indeed, we observed that ISO treatment induced the phosphorylations of PI3K and PDK1. Interestingly, the results showed that HTR2A deficiency reduced the phosphorylations of PI3K and PDK1, whereas HTR2A overexpression led to enhance phosphorylations of PI3K and PDK1 in cardiomyocytes, implicating that HTR2A promoted the activation of PI3K and PDK1 (Fig. 4a and b ). To investigate whether HTR2A regulated PI3K to activate AKT-mTOR signaling, we inhibited PI3K with LY294002, and the results showed that the LY294003 treatment repressed HTR2A-mediated activation of AKT-mTOR signaling (Fig. 4c). Besides, LY294003-mediated inhibition of PI3K repressed the pro-hypertrophic role of HTR2A in ISO-induced cardiomyocyte hypertrophy (Fig. 4d and e ). Similarly, we also inhibited PDK1 with inhibitor OSU-03012. The results also showed that the OSU-03012 treatment inhibited HTR2A function in activating the AKT-mTOR signaling pathway and promoting cardiomyocyte hypertrophy (Fig. 4f–h). Taken together, these findings demonstrated that HTR2A activated PI3K-PDK1 signaling to promote the activation of AKT-mTOR-S6K1 signaling and subsequently facilitated cardiomyocyte hypertrophy.
Fig. 4.

PI3K-PDK1-mediated HTR2A activation on the AKT-mTOR pathway. a HTR2A knockdown represses PI3K-PDK1 signaling. Cardiomyocytes with/without HTR2A knockdown were subjected to ISO treatment (50 μM) for 24 h. Representative western blot and quantitative results are shown (n = 3). b HTR2A overexpression promotes PI3K-PDK1 signaling. Cardiomyocytes with/without HTR2A overexpression were subjected to ISO treatment (50 μM) for 24 h. Representative western blot and quantitative results are shown (n = 3). c PI3K inhibition blocks HTR2A function in regulating AKT-mTOR signaling in cardiomyocytes. Rat cardiomyocytes with/without HTR2A overexpression were subjected to hypertrophy induction with ISO (50 μM) treatment for 24 h. The cardiomyocytes were also treated with/without PI3K inhibitor LY294002 (500 nM). d, e PI3K inhibition blocks HTR2A function in cardiomyocyte size. Rat cardiomyocytes with/without HTR2A overexpression were subjected to hypertrophy induction with ISO (50 μM) treatment for 48 h. The cardiomyocytes were also treated with/without PI3K inhibitor LY294002 (500 nM). Cardiomyocyte size was quantified with the ImageJ software (d), and the expression of hypertrophy-associated fetal genes was analyzed with qRT-PCR (e, n = 3 in each group). f PDK1 inhibition blocks HTR2A function in AKT-mTOR signaling. Rat cardiomyocytes with/without HTR2A overexpression were subjected to hypertrophy induction with ISO (50 μM) treatment for 24 h. The cardiomyocytes were also treated with/without PDK1 inhibitor OSU-03012 (2 μM). Representative western blot and quantitative results are shown (n = 3). g, h Rat cardiomyocytes with/without HTR2A overexpression were subjected to hypertrophy induction with ISO (50 μM) treatment for 48 h. The cardiomyocytes were also treated with/without PDK1 inhibitor OSU-03012 (2 μM). Cardiomyocyte size was quantified with the ImageJ software (h), and the expression of hypertrophy-associated fetal genes was analyzed with qRT-PCR (h, n = 3 in each group). **P < 0.01 analyzed by two-way ANOVA followed by the Tukey post hoc test. Cardiomyocyte culture sets were performed on three different dates. Western blot was performed for each experiment set, and representative western blot results were shown
Discussion
In the present study, we identified HTR2A as an important regulator for cardiac hypertrophy. The expression of HTR2A was increased in hypertrophic heart tissues in humans and mice. Our gene silencing and overexpression experiments demonstrated that HTR2A acted as a pro-hypertrophic factor. HTR2A promoted the PI3K-PDK1 signaling pathway and contributed to cardiac hypertrophy in an AKT-mTOR-dependent manner (Fig. 5).
Fig. 5.
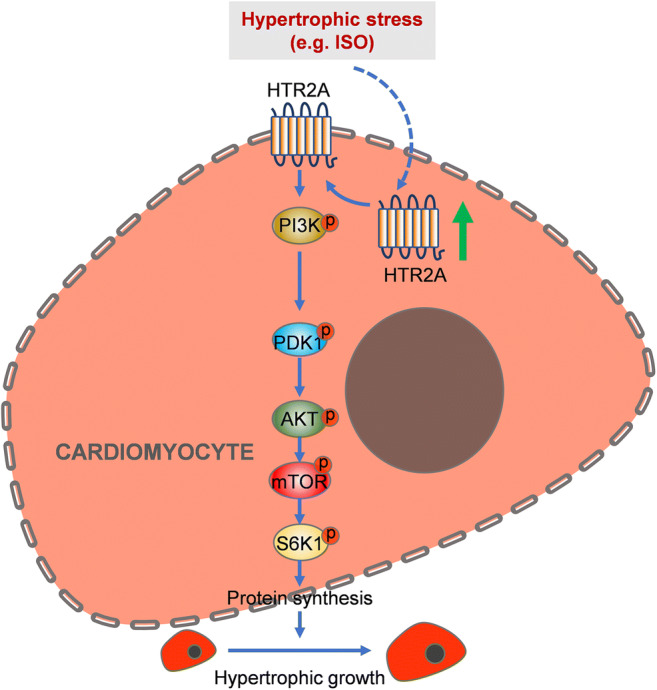
Illustrator showing HRT2A function during cardiac hypertrophy. Upon hypertrophic stress, HTR2A expression is upregulated, which leads to the activation of PI3K. Activated PI3K promotes the activation of AKT in a PDK1-dependent manner. Finally, mTOR and S6K1 signalings are activated by AKT, which results in increased protein synthesis and hypertrophic growth of cardiomyocytes
HTR2A is a GPCR protein that essentially participates in brain development in fetal and functions in homeostasis in adults (Paquette and Marsit 2014). The potential involvement of HTR2A and its ligand serotonin (5-HT) was also implicated in the cardiovascular system. Low platelet level of 5-HT concentration is a common hallmark of both gestational hypertension without proteinuria and pre-eclampsia (Sabolovic Rudman et al. 2015). Gene polymorphisms of HTR2A are also associated with metabolic syndrome, central adiposity, and blood pressure (Choi et al. 2020; Sabolovic Rudman et al. 2015; Halder et al. 2007). The roles of HTR2A in cardiac hypertrophy remain to be defined. Here in this work, we identified the roles of HTR2A in cardiac hypertrophy in humans and mice. Our data collectively showed that the expression of HTR2A was highly expressed in hypertrophic hearts in humans and mice, implicating the conserved roles of HTR2A in cardiac hypertrophy across species. Our adenovirus-mediated knockdown and overexpression data implicated that HTR2A promoted cardiomyocyte hypertrophy using an in vitro model of cardiomyocyte hypertrophy. Previous work showed that HTR2A knockout in mice prevented cardiac fibrosis (Ayme-Dietrich et al. 2015). These studies collectively demonstrated that the HTR2A high expression contributed to cardiac remodeling, including cardiac fibrosis and hypertrophy. To this point, HTR2A may be a promising target for the treatment of cardiac remodeling. However, the mechanism by which HTR2A was upregulated during cardiac hypertrophy remains unknown. This is one of the limitations of this study. Epigenetic regulation (e.g., DNA methylation) of HTR2A is critically for controlling the expression of HTR2A in physiological and pathological conditions (Paquette and Marsit 2014; Hranilovic et al. 2016). As thus, HTR2A may be regulated by epigenetic modification. Further work is still needed to address how HTR2A expression is upregulated by hypertrophy.
The AKT-mTOR is a central regulator of cardiac hypertrophy. Activated AKT can phosphorylate the TSC1/TSC2 complex and inhibit the activation of this complex to trigger the activation of mTOR (Xu and Brink 2016). mTOR serves as a key controller of cell size partially via promoting de novo protein synthesis via phosphorylating the ribosomal protein S6K1 (Sciarretta et al. 2018). Indeed, we also observed the activation of the AKT-mTOR-S6K1 signaling pathway in ISO-induced hypertrophic hearts. Interestingly, HTR2A knockdown repressed, while HTR2A overexpression promoted the ISO-induced phosphorylation of the components of the AKT-mTOR-S6K1 pathway. Importantly, we inhibited AKT with perifosine or inhibited mTOR with rapamycin and observed that inhibition of the AKT-mTOR signaling pathway blocked the effects of HTR2A overexpression on ISO-induced cardiomyocyte hypertrophy. Therefore, we identified HTR2A as a novel upstream activator of the AKT-mTOR-S6K1 signaling pathway, which critically contributed to the roles of HTR2A in cardiac hypertrophy (Fig. 5).
Finally, we also explored the mechanism underlying HTR2A-mediated activation of the AKT-mTOR-S6K1 signaling pathway. Many previous studies have established the notion that PI3K can activate AKT in a PDK1-dependent or -independent manner (Aoyagi and Matsui 2011). Besides, either PI3K or PDK1 acts as a pro-hypertrophic factor during cardiac hypertrophy (Edinger and Thompson 2002). Indeed, we observed that the PI3K-PDK1 signaling pathway was activated by the ISO treatment. Of note, the activation of the PI3K-PDK1 signaling pathway was repressed by HTR2A knockdown and promoted by HTR2A overexpression. Importantly, we observed that treatment with either PI3K inhibitor or PDK1 inhibitor can repress HTR2A-mediated activation of AKT-mTOR signaling in ISO-induced hypertrophic hearts. These findings demonstrated that HTR2A promoted the activation of AKT-mTOR-S6K1 signaling pathways to the regulation of PI3K and PDK1, which are involved in AKT activation. Additionally, inhibition of PI3K or PDK1 blocked the pro-hypertrophic function of HTR2A in ISO-induced cardiomyocyte hypertrophy (Fig. 5).
In summary, we identified that HTR2A acts as a novel upstream activator for AKT-mTOR signaling via a PI3K-PDK1-dependent manner, and through which, HTR2A functions as a pro-hypertrophic factor in cardiac hypertrophy across species. Therefore, HTR2A may be a novel target for the treatment of cardiac remodeling and heart failure.
Author contributions
Weinian Gao and Shuguang Zhao designed the study and wrote the manuscript. Weinian Gao performed most of the experiments with help from Na Guo. Ziying Chen and Wenli Zhang collected human samples. Fang Yan and Hongjuan Liao performed the animal study. Kui Chi isolated cardiomyocytes.
Funding information
This study was supported by the Health Commission of Hebei Province (20190058).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics declarations
All animal experiments were performed according to the terms of the Animal Committee of Hebei Medical University (#2018-M051).
Ethical approval
Ethics approval for the collection of human samples was obtained from the Committee of Hebei Medical University (#2018-P047).
Informed consent
All the participants expressed informed consent.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Aoyagi T, Matsui T. Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr Pharm Des. 2011;17:1818–1824. doi: 10.2174/138161211796390976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayme-Dietrich E, Marzak H, Lawson R, Mokni W, Wendling O, Combe R, Becker J, El Fertak L, Champy MF, Matz R, Andriantsitohaina R, Doly S, Boutourlinsky K, Maroteaux L, Monassier L. Contribution of serotonin to cardiac remodeling associated with hypertensive diastolic ventricular dysfunction in rats. J Hypertens. 2015;33:2310–2321. doi: 10.1097/HJH.0000000000000695. [DOI] [PubMed] [Google Scholar]
- L. Cai, G. Fan, F. Wang, S. Liu, T. Li, X. Cong, J. Chun, X. Chen,(2017) Protective role for LPA3 in cardiac hypertrophy induced by myocardial infarction but not by isoproterenol, Front. Physiol., 8. [DOI] [PMC free article] [PubMed]
- Chen X-F, Chen X, Tang X. Short-chain fatty acid, acylation and cardiovascular diseases. Clin Sci. 2020;134:657–676. doi: 10.1042/CS20200128. [DOI] [PubMed] [Google Scholar]
- Choi JR, Jeon M, Koh SB. Association between serotonin 2A receptor (HTR2A) genetic variations and risk of hypertension in a community-based cohort study. BMC Med Genet. 2020;21:5. doi: 10.1186/s12881-019-0927-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edinger AL, Thompson CB. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol Biol Cell. 2002;13:2276–2288. doi: 10.1091/mbc.01-12-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genis-Mendoza AD, Ruiz-Ramos D, Lopez-Narvaez ML, Tovilla-Zarate CA, Rosa Garcia A, Cortes Meda G, Martinez-Magana JJ, Gonzalez-Castro TB, Juarez-Rojop IE, Nicolini H. Genetic association analysis of 5-HTR2A gene variants in eating disorders in a Mexican population. Brain Behav. 2019;9:e01286. doi: 10.1002/brb3.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong H, Tai H, Huang N, Xiao P, Mo C, Wang X, Han X, Zhou J, Chen H, Tang X, Zhao T, Xu W, Gong C, Zhang G, Yang Y, Wang S, Xiao H. Nrf2-SHP cascade-mediated STAT3 inactivation contributes to AMPK-driven protection against endotoxic inflammation. Front Immunol. 2020;11:414. doi: 10.3389/fimmu.2020.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder I, Muldoon MF, Ferrell RE, Manuck SB. Serotonin receptor 2A (HTR2A) gene polymorphisms are associated with blood pressure, central adiposity, and the metabolic syndrome. Metab Syndr Relat Disord. 2007;5:323–330. doi: 10.1089/met.2007.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hranilovic D, Blazevic S, Stefulj J, Zill P. DNA methylation analysis of HTR2A regulatory region in leukocytes of autistic subjects. Autism Res. 2016;9:204–209. doi: 10.1002/aur.1519. [DOI] [PubMed] [Google Scholar]
- Jia Y-Y, Lu J, Huang Y, Liu G, Gao P, Wan Y-Z, Zhang R, Zhang Z-Q, Yang R-F, Tang X, Xu J, Wang X, Chen H-Z, Liu D-P. The involvement of NFAT transcriptional activity suppression in SIRT1-mediated inhibition of COX-2 expression induced by PMA/ionomycin. PLoS One. 2014;9:e97999. doi: 10.1371/journal.pone.0097999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemi OJ, Ceci M, Wisloff U, Grimaldi S, Gallo P, Smith GL, Condorelli G, Ellingsen O. Activation or inactivation of cardiac Akt/mTOR signaling diverges physiological from pathological hypertrophy. J Cell Physiol. 2008;214:316–321. doi: 10.1002/jcp.21197. [DOI] [PubMed] [Google Scholar]
- Lairez O, Cognet T, Schaak S, Calise D, Guilbeau-Frugier C, Parini A, Mialet-Perez J. Role of serotonin 5-HT2A receptors in the development of cardiac hypertrophy in response to aortic constriction in mice. J Neural Transm (Vienna) 2013;120:927–935. doi: 10.1007/s00702-013-1011-3. [DOI] [PubMed] [Google Scholar]
- Lombaert N, Hennes M, Gilissen S, Schevenels G, Aerts L, Vanlaer R, Geenen L, Van Eeckhaut A, Smolders I, Nys J, Arckens L. 5-HTR2A and 5-HTR3A but not 5-HTR1A antagonism impairs the cross-modal reactivation of deprived visual cortex in adulthood. Mol Brain. 2018;11:65. doi: 10.1186/s13041-018-0404-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y-X, Tang X, An X-Z, Xie X-M, Chen X-F, Zhao X, Hao D-L, Chen H-Z, Liu D-P. Sirt4 accelerates Ang II-induced pathological cardiac hypertrophy by inhibiting manganese superoxide dismutase activity. Eur Heart J. 2017;38:1389–1398. doi: 10.1093/eurheartj/ehx493.P6486. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15:387–407. doi: 10.1038/s41569-018-0007-y. [DOI] [PubMed] [Google Scholar]
- Paquette AG, Marsit CJ. The developmental basis of epigenetic regulation of HTR2A and psychiatric outcomes. J Cell Biochem. 2014;115:2065–2072. doi: 10.1002/jcb.24883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquette AG, Lesseur C, Armstrong DA, Koestler DC, Appleton AA, Lester BM, Marsit CJ. Placental HTR2A methylation is associated with infant neurobehavioral outcomes. Epigenetics. 2013;8:796–801. doi: 10.4161/epi.25358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Sowers JR, Zhang Y. Metabolic stress, autophagy, and cardiovascular aging: from pathophysiology to therapeutics. Trends Endocrinol Metab. 2018;29:699–711. doi: 10.1016/j.tem.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohini A, Agrawal N, Koyani CN, Singh R. Molecular targets and regulators of cardiac hypertrophy. Pharmacol Res. 2010;61:269–280. doi: 10.1016/j.phrs.2009.11.012. [DOI] [PubMed] [Google Scholar]
- Sabolovic Rudman S, Mustapic M, Kosec V, Pivac N, Rudman F, Muck-Seler D. Serotonin risk factors for the development of hypertension in pregnancy. Arch Gynecol Obstet. 2015;291:779–785. doi: 10.1007/s00404-014-3461-8. [DOI] [PubMed] [Google Scholar]
- Sciarretta S, Forte M, Frati G, Sadoshima J. New insights into the role of mTOR signaling in the cardiovascular system. Circ Res. 2018;122:489–505. doi: 10.1161/CIRCRESAHA.117.311147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shende P, Xu L, Morandi C, Pentassuglia L, Heim P, Lebboukh S, Berthonneche C, Pedrazzini T, Kaufmann BA, Hall MN. Cardiac mTOR complex 2 preserves ventricular function in pressure-overload hypertrophy. Cardiovasc Res. 2016;109:103–114. doi: 10.1093/cvr/cvv252. [DOI] [PubMed] [Google Scholar]
- Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. 2016;97:245–262. doi: 10.1016/j.yjmcc.2016.06.001. [DOI] [PubMed] [Google Scholar]
- Tang X, Ma H, Han L, Zheng W, Lu Y-B, Chen X-F, Liang S-T, Wei G-H, Zhang Z-Q, Chen H-Z, Liu D-P. SIRT1 deacetylates the cardiac transcription factor Nkx2.5 and inhibits its transcriptional activity. Sci Rep. 2016;6:36576. doi: 10.1038/srep36576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Chen X-F, Chen H-Z, Liu D-P. Mitochondrial Sirtuins in cardiometabolic diseases. Clin Sci. 2017;131:2063–2078. doi: 10.1042/CS20160685. [DOI] [PubMed] [Google Scholar]
- X. Tang, P.-H. Li, H.-Z. Chen,(2020) Cardiomyocyte senescence and cellular communications within myocardial microenvironment, Front. Endocrinol. (Lausanne). [DOI] [PMC free article] [PubMed]
- Xu L, Brink M. mTOR, cardiomyocytes and inflammation in cardiac hypertrophy. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 2016;1863:1894–1903. doi: 10.1016/j.bbamcr.2016.01.003. [DOI] [PubMed] [Google Scholar]
- Zhou S, Tang X, Chen HZ. Sirtuins and insulin resistance. Front Endocrinol (Lausanne) 2018;9:748. doi: 10.3389/fendo.2018.00748. [DOI] [PMC free article] [PubMed] [Google Scholar]