

Abstract
Transient receptor potential ankyrin 1 (TRPA1), the non-selective cation channel, was found that can mediate the generation of multiple sclerosis, while the mechanism is still controversial. Lysophosphatidylcholine (LPC) is a critical trigger of multiple sclerosis which results from the syndrome of neuronal inflammation and demyelination. In this work, we suggested that TRPA1 can mediate the LPC-induced oxidative stress and cytotoxicity in OLN-93 oligodendrocyte. The expression of TRPA1 in OLN-93 was detected by using quantitative real-time PCR (qRT-PCR) and immunofluorescence. The calcium overload induced by LPC via TRPA1 was detected by calcium imaging. The mechanism of LPC-induced mitochondrial reactive oxygen species (mtROS) generation, mitochondria membrane depolarization, nitric oxide (NO) increase, and development of superoxide production via TRPA1 was verified by using confocal imaging. The cell injury elicited by LPC via TRPA1 was confirmed by both CCK-8 and LDH cytotoxicity detection. These results indicate that TRPA1 plays an important role of the LPC-induced oxidative stress and cell damage in OLN-93 oligodendrocyte. Therefore, inhibition of TRPA1 may protect the LPC-induced demyelination.
Keywords: TRPA1, LPC, ROS, Oligodendrocyte, NO
Introduction
Multiple sclerosis is a kind of chronically neuronal disease, with the syndrome of neuronal inflammation, demyelination, and neurodegeneration (Trapp and Nave 2008). The previous studies confirmed that multiple sclerosis is a primary autoimmune disease resulting from myelin-induced lymphocytes activation, which mediates the downstream immune responses and leads to the secondary macrophage invasion and demyelination (Compston and Coles 2008; Saghy et al. 2016). However, there are some clinical findings indicated that some patients present the primary oligodendrocyte apoptosis rather than autoimmune reaction in the early stage of multiple sclerosis (Barnett and Prineas 2004), which demonstrates the heterogeneity about the pathogenesis of multiple sclerosis. In this study, we used the rat oligodendrocyte cell line OLN-93 as a cell model to investigate the mechanism of oligodendrocyte damage. OLN-93, a cell line acquired from autogenously transformed rat brain oligodendrocyte cultures, is widely used in the study of migration, oxidative stress, or apoptosis of oligodendrocyte (Liu et al. 2018; Brand et al. 2010; Gerstner et al. 2006).
Lysophosphatidylcholine (LPC), a phospholipid generated by phospholipase A2 (Kougias et al. 2006), is used to induce the syndrome of demyelination in experimental setting (Jarjour et al. 2012). In the body, LPC is a kind of endogenous mediator which mainly participates in various diseases including the development of multiple sclerosis (Kalyvas et al. 2009; Plemel et al. 2018). The previous study found that the concentration of LPC increases in the cerebrospinal fluid of multiple sclerosis patients (Pieragostino et al. 2015). Although the important role of LPC was broad verified in the generation of multiple sclerosis, the precise mechanism of LPC-induced demyelination has still been unclear. From the data of previous research, LPC could elicit the calcium overload in neuron and increase the inflammation and cytotoxicity in microglia and astroglia as well as the death of oligodendrocyte, all of them resulting in the nervous tissue severely injury (Plemel et al. 2018; Freeman et al. 2017; Fressinaud et al. 1996). It was reported that LPC can induce demyelination via calcium mobilization, NOD-like receptor 3 (NLRP3) which is a critical inflammasome and caspase-1 which can induce the release of proinflammatory cytokine including IL-1β (Freeman et al. 2017). In this process, lipopolysaccharide (LPS), a potent trigger of the innate immune response, stimulates the expression of NLRP3 and pro-caspase-1 via Toll-like receptor 4 (TLR4) and then LPC promotes activation of NLRP3 and caspase-1 which are mediated by calcium influx (Freeman et al. 2017; Lehnardt et al. 2002). However, it has been unclear which receptor could be activated by LPC in oligodendrocyte and mediates the oligodendrocyte injury.
The transient receptor potential (TRP) channel family is a sort of non-selective cation channel, which has broad expression and various physiological function in different cells (Li 2017). Transient receptor potential ankyrin 1 (TRPA1), the member of TRP family, can be activated by the extracellular irritants and mediates the redox reaction and inflammation in various cells (Andersson et al. 2008; Tian et al. 2020). Some studies indicated that TRPA1 may contribute to the development of multiple sclerosis, which regulates the calcium overload, mitochondrial damage, and astroglia-dependent oligodendrocyte apoptosis (Saghy et al. 2016; Silva et al. 2018; Bolcskei et al. 2018; Hamilton et al. 2016). The TRPA1 knockout mice appear the relieved syndrome in multiple sclerosis (Bolcskei et al. 2018). Some previous data showed that in all sorts of cells, LPC can elicit calcium influx through different TRP channels. In monocytes, TRPV1 and TRPC6 may be involved in the process of activation elicited by LPC (Schilling and Eder 2009). In microglial, LPC induces the calcium influx via TRPM2 (Jeong et al. 2017). In our previous study, we found that LPC could induce the calcium overload and inflammation via TRPA1 in THP-1-derived macrophage (Tian et al. 2020), while in non-immunocytes, especially in nervous tissue, whether LPC elicits activation via TRPA1 has still been unknown. In this study, we supposed that LPC can directly induce the OLN-93 oligodendrocyte injury via TRPA1.
The redox level is a critical mediator in the process of demyelination (Islam 2017). The major oxidative productions are reactive oxygen and reactive nitric oxide species (RNS), which cause the mitochondrial injury and further cytotoxicity (Angelova and Abramov 2018; Ghasemi et al. 2018). Nitric oxide synthetase (NOS) is the main enzyme involved in the generation of nitric oxide (NO), which has three forms, including inducible (iNOS), endothelial (eNOS), and neuronal (nNOS) nitric oxide synthetase (Yao et al. 2010). In neuronal system, iNOS and nNOS are the main enzymes to generate NO, while only nNOS can be mediated by calcium influx (Ignarro 1999). nNOS was found that participates in the process of demyelination (Yao et al. 2010). Despite LPC can promote the expression of nNOS in dorsal root ganglion (DRG) (Wang et al. 2013), it has been unclear that nNOS can be activated by LPC in oligodendrocyte. In addition, given that TRPA1 can mediate phosphorylation of nNOS (Sinharoy et al. 2015), in this study, we speculated that both nNOS and TRPA1 are required for the generation of NO induced by LPC in OLN-93 oligodendrocyte. Beside NO, mitochondrial reactive oxygen species (mtROS) is another vital trigger in multiple sclerosis (Fetisova et al. 2017). Our early study showed that LPC elicits the increase of mitochondrial ROS and mitochondrial damage in macrophage (Tian et al. 2020). However, whether mtROS could be induced by LPC in oligodendrocyte has still been unknown. So, we made a speculation that LPC can induce the generation of mtROS and mitochondrial membrane depolarization, which would further mediate the cytotoxicity of OLN-93 oligodendrocyte.
Therefore, the target of this study was to investigate the role of TRPA1 in the process of LPC-induced oligodendrocyte oxidative stress and damage by analyzing the change of calcium influx, mitochondrial ROS, mitochondrial membrane potential, intracellular NO concentration, superoxide production, and cell vitality elicited by LPC in OLN-93 oligodendrocyte.
Materials and methods
Chemicals
L-alpha-lysophosphatidylcholine (LPC, L39888) and lipopolysaccharide (LPS from Escherichia coli 055: B5, L26331) were purchased from ABCONE (Shanghai, China). DMEM culture medium (C11965500BT, Gibco), ProLong Live Antifade Reagent, for live cell imaging (P36975), and MitoSOX (M36008) were obtained from Thermo Fisher (MA, USA). MitoTEMPO as a selective mitochondrial ROS antagonist (APEC4234) was purchased from ApexBio (Houston, USA). Spermidine as a selective nNOS antagonist (S0010), goat serum (C0265), DAPI (C1005), Hoechst 33342 (C1022), 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethylbenzimidazol-carbocyanine iodide (JC-1, C2006), 3-Amino,4-aminomethyl-2',7'-difluorescein, diacetate (DAF-FM DA, S0019), and dihydroethidium (DHE, S0063) were purchased from Beyotime (Shanghai, China). AP-18 as a selective TRPA1 antagonist (GC18020), HC030031 as a selective TRPA1 inhibitor (GC15947), and Cell Counting Kit-8 (CCK-8, GK10001) were obtained from GLPBIO (Montclair, USA). GdCl3 as a non-selective cation channel broad spectrum inhibitor (HWG25664) was purchased from HWRK (Beijing, China). Ruthenium red as a TRPV pan-inhibitor (RR, R817195) was obtained from Macklin (Shanghai, China). A967079 as a selective TRPA1 antagonist (HY-108463) was purchased from MedChemExpress (NJ, USA). Fetal bovine serum (FBS, S601P-500) was purchased from Sera Pro (Bavaria, Germany). Ac-YVAD-cmk as a selective caspase-1 antagonist and Triton X-100 (T8787), as well as Tween-20 (P1379), were purchased from Sigma (St. Louis, USA). Tetramethyl Rhodamine Methyl Ester (TMRM, T4058) was purchased from US EVERBRIGHT Inc. (Suzhou, China). Fluo 4-AM (40704ES50) and Rhod 2-AM (40776ES50) were purchased from Yeasen (Shanghai, China). All of antagonists used in this study are listed in Table 1.
Table 1.
All of antagonists used in this study
| Antagonist | Function |
|---|---|
| AP-18 | A selective TRPA1 antagonist |
| HC030031 | A selective TRPA1 antagonist |
| A967079 | A selective TRPA1 antagonist |
| GdCl3 | A non-selective cation channel broad spectrum inhibitor |
| Ruthenium red | A TRPV pan-inhibitor |
| MitoTEMPO | A mtROS inhibitor |
| Ac-YVAD-cmk | A caspase-1 inhibitor |
| Spermidine | A nNOS selective inhibitor |
Cell culture
The rat oligodendrocyte cell line OLN-93 was purchased from iCell Bioscience company (iCell-r023, Shanghai, China). OLN-93 oligodendrocyte was maintained in DMEM containing 10% FBS overnight for subsequent experiments. The OLN-93 cells were cultured in 5% CO2 at 37 °C and used for subsequent experiments until 80% confluence.
RNA isolation and quantitative real-time PCR measurement
RNA was extracted by RNAeasy™ Animal RNA Isolation Kit with Spin Column (R0024, Beyotime), and 1.0 μg RNA was used for cDNA synthesis with HiScript® II Reverse Transcriptase (R223-01, Vazyme, Nanjing, China) according to the manufacturer’s protocol. Quantitative real-time PCR (qRT-PCR) was measured with 2× RealStar Green Power Mixture (A311-10, GenStar, Beijing, China) using a CFX96 Touch Real-Time PCR Detection System (Bio-Rad, California, USA). Rat GAPDH (rGAPDH) is the reference gene for OLN-93 oligodendrocyte. The qPCR primer sequences used in this study were as follows: rTRPA1, 5’-TGTGTCCGTTCATTCCAAAAGC-3’ (F) and 5’-CAAAAGCCTTGTGTCGCTGA-3’ (R); rGAPDH, 5’-CATCATCTCCGCCCCTTCC-3’ (F) and 5’-GTTGTCATGGATGACCTTGGC-3’ (R).
Immunofluorescence
The OLN-93 cells were plated in a 4-chamber coverglass dish (In Vitro Scientific, Hangzhou, China) at a density of 2 × 105 cells per chamber. After washing thrice with PBS, the cells were fixed in 4% paraformaldehyde at RT for 15 min. After that, they were washed thrice with PBS and permeabilized with 0.1% Triton X-100 in PBS for 5 min at RT. Then, the cells were blocked with 10% goat serum diluted in 0.1% tween-20/PBS for 1 h at RT and incubated overnight at 4 °C with anti-myelin basic protein primary antibody (1:100; ab209328; Abcam, Cambridge, MA, USA), anti-TRPA1 primary antibody (1:200; OmnimAbs, Alhambra, CA, USA), and anti-nNOS primary antibody (1:200; ab76067; Abcam). After washing three times with PBS, the cells were incubated with Alexa Fluor 594-conjugated goat anti-human IgG secondary antibody (1:100; bs-0297G-AF594; Bioss, Beijing, China) and Alexa Fluor 488-conjugated goat anti-rabbit IgG secondary antibody (1:100; SA00006-2; Proteintech, Rosemont, IL, USA) for 2 h at RT in the dark. Then, the cells were loaded in DAPI staining solution for 15 min and washed thrice with PBS before mounted with anti-fade mounting medium. The images were collected using IX73 fluorescence microscope (Olympus, Tokyo, Japan) or LSM800 confocal microscope (Carl Zeiss Microscopy GmbH, Oberkochen, Germany).
Ca2+ imaging
The calcium indicators Fluo 4-AM and Rhod 2-AM can be used for cytoplasmic and mitochondrial calcium influx detection, respectively. We used two kinds of TRPA1 selective antagonists AP-18 and A967079 as well as TRPV pan-inhibitor ruthenium red (RR) and non-selective cation channel broad spectrum inhibitor GdCl3 because they can reveal the role of TRPA1, TRPV channels, and non-selective cation channels in the process of LPC-induced calcium influx in OLN-93. We performed Ca2+ imaging by using Fluo 4-AM and Rhod 2-AM following the manufacturer's instructions. OLN-93 cell line was plated in 4-chamber coverglass dishes at a density of 2 × 105 cells per chamber and incubated with 2.5 μM Fluo 4-AM and 2.5 μM Rhod 2-AM simultaneously for 45 min at 37 °C. L-alpha-lysophosphatidylcholine (LPC) from egg yolk diluted in Hank’s or D-Hank’s solution and LPC combined with AP-18, A967079 and RR and GdCl3 were added to the final concentrations of 30 μM LPC, 10 μM AP-18, 10 μM A967079, 10 μM RR, and 100 μM GdCl3 in the coverslip dishes after baseline recording in drug-free Hank’s or D-Hank’s solution for 30 s. Confocal images were obtained (LSM800; Carl Zeiss Microscopy GmbH) at 1-s intervals for 3 min. For simultaneously detecting cytoplasmic and mitochondrial Ca2+ dynamics, the stained cells were excited at 488 nm (for Fluo 4-AM) and 561 nm (for Rhod 2-AM), respectively. The fluorescence intensity was measured by ImageJ software (Molecular Devices, San Jose, CA, USA).
Mitochondrial ROS measurement
LPC-induced mitochondrial ROS (mtROS) was measured by fluorescence microscopy in OLN-93 oligodendrocyte. We used two kinds of TRPA1 selective antagonists HC030031 and A967079 because they can reveal the role of TRPA1 in the process of LPC-induced mtROS generation in OLN-93. To detect the level of mtROS by confocal microscopy, the cells were cultured in a 4-chamber coverglass dish at a density of 2 × 105 cells per chamber. Following treatment with selective TRPA1 antagonist 10 μM HC030031 or 10 μM A967079 for 30 min, the cells were treated with 30 μM LPC for 1 h, incubated with 2.5 μM MitoSOX or 10 min at 37 °C, and then by washed thrice with Hank’s solution. The live cells were loaded in Hoechst 33342 staining solution at 37 °C for 30 min and then washed thrice with PBS and incubated in Hank’s solution containing live antifade reagent. The images were collected using the LSM710 confocal microscope (Carl Zeiss Microscopy GmbH).
Mitochondrial membrane potential
The fluorochrome JC-1 (5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethylbenzimidazol-carbocyanine iodide) can be used to detect the variation of mitochondrial membrane potential (∆Ψm). OLN-93 oligodendrocyte was cultured in a 4-chamber coverglass dish at a density of 2 × 105 cells per chamber. We used toll-like receptor 4 activator lipopolysaccharide (LPS) because it can induce the expression of NLRP3 and pro-caspase-1 which are involved in the LPC-elicited mitochondrial injury. We used TRPA1 selective antagonist A967079 because they can reveal the role of TRPA1 in the process of LPC-induced mitochondrial membrane depolarization in OLN-93. The cells were pretreated with 100 ng/mL LPS for 12 h. After that, the cells were incubated with 10 μM A967079 for 30 min before treatment with 30 μM LPC for 1 h 30 min. After loading with JC-1 dye following the manufacturer’s protocol, the variation of fluorescence intensity in live cells was detected by using the LSM800 confocal microscope. TMRM is another fluorescent dye to detect the ∆Ψm. For TMRM recording, OLN-93 cells cultured in a 4-chambered coverglass dish at a density of 2.0 × 105. After preprocessed with 100 ng/mL LPS for 12 h and washed with Hank’s solution thrice, the cells were loaded with 30 nM TMRM diluted in Hank’s solution at 37 °C for 30 min. The images were recorded at 30-s interval for 19 min 30 s for drugs treatment after basal recording for 30 s by using the LSM800 confocal microscope. The fluorescence intensity was measured by ImageJ software.
Nitric oxide measurement
The fluorochrome DAF-FM DA (3-Amino,4-aminomethyl-2',7'-difluorescein, diacetate) is a nitric oxide (NO) selective probe. LPC-induced generation of NO was detected by fluorescence microscopy in OLN-93 oligodendrocyte. We used TRPA1 selective antagonist A967079 and nNOS selective inhibitor spermidine because they can reveal the role of TRPA1 and nNOS in the process of LPC-induced NO generation in OLN-93. To detect the level of NO by confocal microscopy, the cells were cultured in a 4-chamber coverglass dish at a density of 2 × 105 cells per chamber. Following treatment with 100 μM spermidine or 10 μM A967079 for 30 min, the cells were treated with 30 μM LPC for 2 h, loaded with 5 μM DAF-FM DA at 37 °C for 30 min. The live cells were loaded in Hoechst 33342 staining solution at 37 °C for 30 min and then washed once with PBS and incubated in Hank’s solution containing live antifade reagent. The images were collected using the LSM800 confocal microscope.
Superoxide measurements
LPC-induced superoxide was detected by fluorescence microscopy in OLN-93 oligodendrocyte. We used LPS because it can reveal whether LPS elicits superoxide production directly. We used TRPA1 selective antagonist A967079 because they can reveal the role of TRPA1 in the process of LPC-induced superoxide production generation in OLN-93. To detect the level of superoxide by confocal microscopy, the cells were cultured in a 4-chamber coverglass dish at a density of 2 × 105 cells per chamber. The cells were pretreated with 100 ng/mL LPS for 12 h. Following treatment with 10 μM A967079 for 30 min, the cells were treated with 30 μM LPC for 2 h, loaded with 5 μM dihydroethidium (DHE) at 37 °C for 30 min. The live cells were loaded in Hoechst 33342 staining solution at 37 °C for 20 min and then washed once with PBS and incubated in Hank’s solution containing live antifade reagent. The images were collected using the LSM710 confocal microscope.
CCK-8 assay
We used LPS because it can induce the expression of NLRP3 and pro-caspase-1 which are involved in the LPC-elicited cytotoxicity. We used TRPA1 selective antagonist A967079 because they can reveal the role of TRPA1 in the process of LPC-induced cytotoxicity in OLN-93. OLN-93 cell line was plated in a 96-well plate at a density of 8.0 × 104 cells/well, pretreated with 100 ng/mL LPS for 16 h at 37 °C, and then washed thrice with PBS. After that, the cells were incubated in FBS-free DMEM treated with LPC (ranging from 1 to 100 μM) for 3 h at 37 °C or A967079 (ranging from 5 to 30 μM) for 30 min before 30 μM LPC stimulation. Then, the CCK-8 reagent was added into the supernatant for 2 h at 37 °C following the manufacturer’s instructions. The absorbance was recorded at 450 nm by using a multimode reader (Berthold, Bad Wildbad, Germany).
LDH assay
The release of lactate dehydrogenase (LDH) was detected by using the LDH Cytotoxicity Assay Kit (C0016, Beyotime). We used TRPA1 selective antagonist AP-18 because they can reveal the role of TRPA1 in the process of LPC-induced cytotoxicity in OLN-93. OLN-93 cell line was plated in a 96-well plate at a density of 5.0 × 104 cells/well and pretreated with 100 ng/mL LPS for 16 h. Following treatment with AP-18 (ranging from 0.5 to 30 μM), mtROS inhibitor 10 μM mitoTEMPO, caspase-1 inhibitor 30 μM Ac-YVAD-cmk, 100 μM GdCl3, or 100 μM spermidine for 30 min, the cells were treated with 50 μM LPC for 3 h in FBS-free DMEM. The cells were also treated with different doses of LPC (ranging from 10 to 100 μM) for 3 h at 37 °C. After that, the supernatants were mixed with LDH reaction agent and incubated for 30 min at RT away from light, and then the absorbance recorded at 490 nm by using a multimode reader.
Statistical analysis
By using GraphPad Prism 8 Software (San Diego, CA, USA), the data were analyzed and presented as mean ± SEM. Statistical significance test was assessed using Holm-Sidak’s multiple comparisons test of one-way ANOVA.
Results
TRPA1 mediates the LPC-induced calcium influx in OLN-93 oligodendrocyte
To investigate the expression of TRPA1 in OLN-93 oligodendrocyte, we performed the qRT-PCR and immunofluorescence respectively. The data of TRPA1 mRNA transcription (Fig. 1a) and the cytoplasmic expression of TRPA1 (Fig. 1b) show the constitutive expression of TRPA1 in OLN-93 cell line. Next, we demonstrated that LPC also activates the calcium influx through TRPA1 in OLN-93 cell line (Fig. 2a). On the basis of results of calcium imaging (Fig. 2b, c), the TRPA1 selective antagonists AP-18 and A967079 partly inhibit the calcium influx induced by LPC, which is similar to the result treated with TRPV pan-antagonist ruthenium red (RR). However, the broad-spectrum cation channel inhibitor GdCl3 nearly inhibits the trend of calcium influx induced by LPC, which seems that there may have other cation channels participating in the LPC-elicited activation in oligodendrocyte.
Fig. 1.

The constitutive expression of TRPA1 in OLN-93 oligodendrocyte. a The transcription of Trpa1 mRNA in OLN-93 cell line was measured by qRT-PCR. Gapdh mRNA was used as a reference gene. b Immunofluorescence staining of TRPA1 in OLN-93 oligodendrocyte. DAPI (blue), MBP-AF594 (red), and TRPA1-AF488 (green) are shown. The scale bar represents 50 μm
Fig. 2.

TRPA1 mediates LPC-induced cytoplasmic and mitochondrial calcium influx in OLN-93 oligodendrocyte. a OLN-93 cell line loaded with cytoplasmic and mitochondrial calcium indicators fluo 4-AM and Rhod 2-AM simultaneously. OLN-93 oligodendrocyte was treated with 30 μM LPC with or without Ca2+ or together with TRPA1 inhibitors 10 μM A967079, 10 μM AP-18, and 10 μM TRPV pan-inhibitor RR and 100 μM non-selective cation channel broad spectrum inhibitor GdCl3, which was imagined by confocal microscopy. Scale bar, 50 μm. b LPC elicits lasting cytoplasmic and mitochondrial calcium dynamic changes via TRPA1 in OLN-93 oligodendrocyte. Real-time measurement of calcium ion dynamics elicited by 30 μM LPC. The data acquired by using confocal microscopy was presented. The mean value of 15 cells was represented by each line. c LPC elicits similar variation of cytoplasmic and mitochondrial calcium influx via TRPA1. The quantification of average variation of both cytoplasmic and mitochondrial calcium influx was presented (n = 15). Data are shown as mean ± SEM for three independent experiments. ΔF/F0, fluorescence intensity ratios; ****p < 0.0001 vs LPC
TRPA1 mediates the generation of mtROS and mitochondrial membrane depolarization induced by LPC in OLN-93 oligodendrocyte
The generation of mtROS is the major trigger in the process of mitochondrial damage and development of multiple sclerosis (Fetisova et al. 2017). In this study, we found that LPC can induce mtROS increase and mitochondrial membrane depolarization via TRPA1 in OLN-93 cell line. According to the results (Fig. 3), A967079 or HC030031, the selective inhibitors of TRPA1 obviously decline the trend of mtROS generation compared to the group elicited by LPC only, which implies that LPC can elicit the generation of mtROS via TRPA1 in OLN-93 oligodendrocyte.
Fig. 3.

LPC elicits generation of mitochondrial ROS (mtROS) via TRPA1 in OLN-93 oligodendrocyte. a TRPA1 could mediate the generation of mtROS elicited by LPC in OLN-93 cell line. By using the mtROS-specific probe MitoSOX (red), the data shown indicated the rise of mtROS resulted from treatment of 30 μM LPC for 1 h, while this trend was inhibited when the cells were treated with TRPA1 inhibitors A967079 10 μM, or HC030031 10 μM for 30 min before the induction of LPC. The live cell’s nucleus was stained with Hoechst 33342 (blue). The scale bar represents 50 μm. b The quantification of each sample was presented by mean optical density (MOD). The data were quantified by calculating the ratio between the optical density (red) of single cell and the area of individual cell (n = 30). Data are shown as mean ± SEM for two independent experiments. ****p < 0.0001 vs LPC
Furthermore, we detected whether LPC induces mitochondrial membrane depolarization via TRPA1 by using JC-1 and TMRM to measure the variation of mitochondrial membrane potential (∆Ψm). The JC-1 has two status including accumulated or single status. In normal cells, accumulated JC-1 in mitochondria emits red fluorescence, while in injured cells, JC-1 is freed into cytoplasm due to mitochondrial membrane depolarization, which emits green fluorescence (Cossarizza et al. 1993). According to the results (Fig. 4a, b), in comparison with the control group, the density of red fluorescence in LPC treated group greatly decreases. However, TRPA1 antagonist A967079-treated group inhibits the trend of red fluorescence decrease, which suggests that TRPA1 may be involved in the process of LPC-induced mitochondrial membrane depolarization in OLN-93 oligodendrocyte. In addition, in order to investigate the dynamics of ∆Ψm induced by LPC following the treated-time extension, we perform TMRM to detect the variation of ∆Ψm during the period of LPC or LPC together with TRPA1 antagonist treated. The results of TMRM indicated that inhibition of TRPA1 could significantly relieve the level of mitochondrial membrane depolarization induced by LPC in a time-dependent way (Fig. 4c).
Fig. 4.

LPC elicits mitochondrial membrane depolarization through TRPA1 in OLN-93 oligodendrocyte. a TRPA1 could mediate mitochondrial membrane depolarization elicited by LPC in OLN-93 cell line. The decrease of JC-1 red fluorescence density indicated the decline of mitochondrial membrane potential (∆Ψm) resulted from the treatment of 30 μM LPC for 1 h 30 min and this trend can be inhibited by TRPA1 antagonist A967079 10 μM. The scale bar represents 50 μm. b The quantification of each sample was presented by mean optical density (MOD). The data were quantified by calculating the ratio between the red fluorescence MOD and the green fluorescence MOD of all cells. Data are shown as mean ± SEM for two independent experiments. ***p < 0.001, ****p < 0.0001 vs LPC. c Change of ∆Ψm elicited by LPC was measured by TMRM. Real-time measurement of ∆Ψm dynamics induced by 30 μM LPC or 10 μM A967079 combined with 30 μM LPC by using confocal microscopy after preprocessed with 100 ng/mL LPS for 12 h. Data are shown as mean ± SEM for three independent experiments
TRPA1 and nNOS mediate the generation of NO and superoxide production elicited by LPC in OLN-93 oligodendrocyte
As previously reported, the increase of NO concentration may lead to the cell death in ischemia/reperfusion-treated OLN-93 oligodendrocyte due to the development of superoxide production (Nadjafi et al. 2015). LPC was found that can induce the generation of superoxide production in endothelial cells, mediated by nNOS (Campos-Mota et al. 2017). In this study, the constitutive expression of nNOS was detected in OLN-93 cell line (Fig. 5a). According to the results of NO detection using NO-specific fluorescent probe DAF-FM DA (Fig. 5b, c), LPC could induce the elevation of NO, which was inhibited by TRPA1 and nNOS selective inhibitor. Furthermore, we performed the superoxide production detection by using superoxide production fluorescent dye DHE. On the basis of the data (Fig. 6), we suggested that LPC widely elicits the generation of superoxide production in OLN-93 oligodendrocyte, while it can be inhibited by TRPA1 antagonist, which seems that TRPA1 may play a critical role in the generation of mtROS and reactive nitrogen species (RNS) induced by LPC.
Fig. 5.

LPC elicits generation of nitric oxide (NO) through TRPA1 in OLN-93 oligodendrocyte. a Immunofluorescence staining of nNOS in OLN-93 oligodendrocyte. DAPI (blue), MBP-AF594 (red), and nNOS-AF488 (green) are shown. The scale bar represents 50 μm. b TRPA1 could mediate the generation of NO elicited by LPC in OLN-93 cell line. By using the NO-specific probe DAF-FM DA (green), the data shown indicated the rise of NO resulted from treatment of 30 μM LPC for 2 h, while this trend was inhibited when the cells were treated with TRPA1 antagonist A967079 10 μM and nNOS inhibitor spermidine 100 μM for 30 min before the supplement of LPC. The live cell’s nucleus was stained with Hoechst 33342 (blue). The scale bar represents 50 μm. c The quantification of each sample was presented by mean optical density (MOD). The data were quantified by calculating the ratio between the optical density (green) of single cell and the area of individual cell (n = 30). Data are shown as mean ± SEM for two independent experiments. ****p < 0.0001 vs LPC
Fig. 6.

LPC elicits generation of superoxide production through TRPA1 in OLN-93 oligodendrocyte. a TRPA1 could mediate the generation of superoxide production elicited by LPC in OLN-93 cell line. By using the superoxide production-specific probe DHE (red), the data shown indicated the rise of superoxide production resulted from treatment of 30 μM LPC for 2 h, while this trend was inhibited when the cells were treated with TRPA1 antagonist A967079 10 μM for 30 min before the supplement of LPC. The live cell’s nucleus was stained with Hoechst 33342 (blue). The scale bar represents 50 μm. b The data were quantified by calculating the ratio between the optical density (red) of single cell and the area of individual cell (n = 30). Data are shown as mean ± SEM for two independent experiments. ****p < 0.0001 vs LPC
TRPA1 mediates the cytotoxicity induced by LPC in OLN-93 oligodendrocyte
To confirm the role of TRPA1 in LPC-induced OLN-93 oligodendrocyte damage, we performed CCK-8 and LDH cytotoxicity detection in OLN-93 cell line. According to the results (Fig. 7), LPC leads to the increase of cell death in OLN-93 cell line in a dose-dependent way, which can be inhibited by treating TRPA1 selective antagonists AP-18 or A967079 and non-selective cation channel broad spectrum inhibitor GdCl3. GdCl3 shows greater effect to suppress the LPC-induced cytotoxicity than that treated with TRPA1 inhibitor (Fig. 7b). In addition, using mtROS selective antagonist mitoTEMPO, caspase-1 selective antagonist Ac-YVAD-cmk and nNOS selective antagonist spermidine also inhibit the progress of cytotoxicity in OLN-93 oligodendrocyte (Fig. 7b), which seems that LPC may elicit cytotoxicity through mtROS and nNOS-induced oxidative stress as well as caspase-1 mediated inflammatory injury.
Fig. 7.
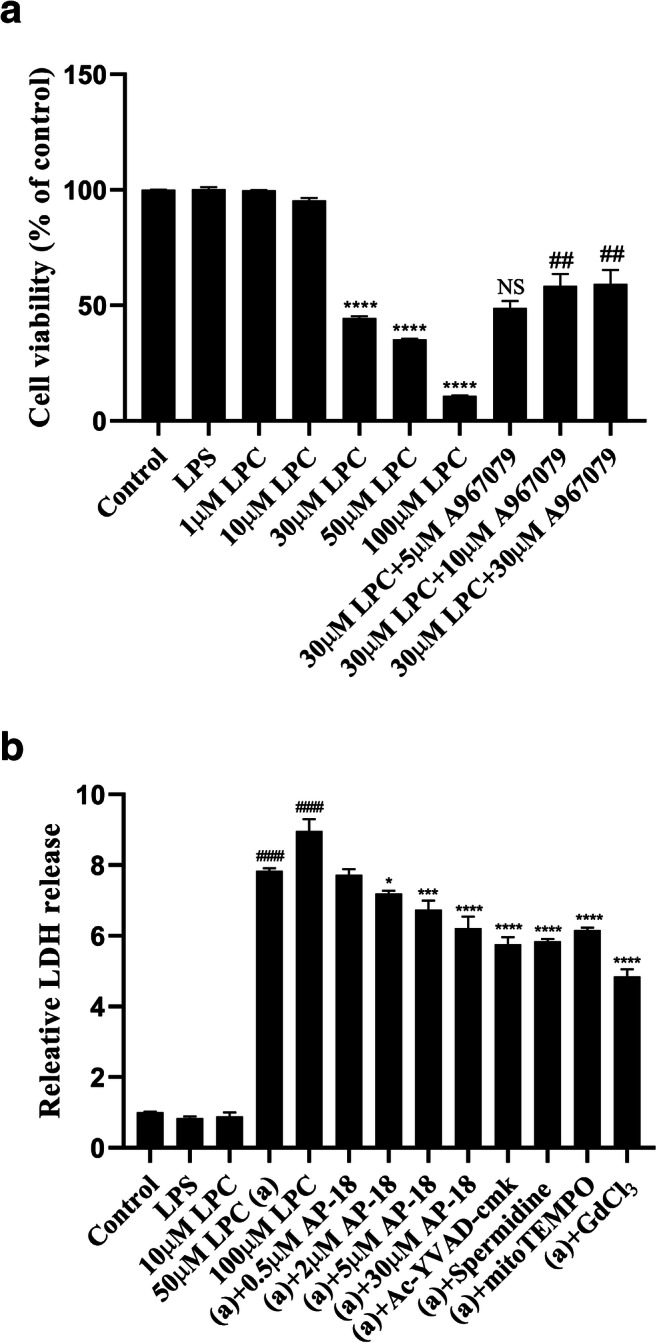
LPC elicits cytotoxicity via TRPA1 in OLN-93 oligodendrocyte. a TRPA1 mediates LPC-induced cytotoxicity in OLN-93 cell line. OLN-93 oligodendrocyte was treated with LPC (ranging from 1 to 50 μM) for 3 h or treated with TRPA1 antagonist A967079 (ranging from 5 to 30 μM) for 30 min before the treatment of 30 μM LPC and then mixed with CCK-8 for 2 h. Data are shown as mean ± SEM (ns equals no significance, ****p < 0.0001 vs LPS, ##p < 0.01 vs 30 μM LPC, n = 3). b LPC elicited LDH release via TRPA1, nNOS, mtROS, and caspase-1 mediation. OLN-93 oligodendrocyte was treated with LPC (ranging from 1 to 50 μM) for 3 h or treated with AP-18 (ranging from 0.5 to 30 μM), Ac-YVAD-cmk 30 μM, mitoTEMPO 10 μM, spermidine 100 μM, and GdCl3 100 μM for 30 min before the induction of 50 μM LPC for 3 h. Data are shown as mean ± SEM (*p < 0.05, ***p < 0.001, ****p < 0.0001 vs 50 μM LPC, ####p < 0.0001 vs LPS, n = 3)
Discussion
In this study, we firstly demonstrated that TRPA1 could mediate the LPC-induced calcium overload, generation of mtROS, mitochondrial membrane depolarization, NO increasing, development of superoxide production, and cytotoxicity in OLN-93 oligodendrocyte.
The expression of TRPA1 in oligodendrocyte is hardly reported. Our study firstly identified that TRPA1 expresses in rat OLN-93 oligodendrocyte by detecting both messenger RNA (mRNA) transcription and protein expression. In the previous study, TRPA1 could be induced by proton, which leads to the calcium overload and further cytotoxicity in cerebellum oligodendrocyte of mice (Hamilton et al. 2016). In our study, we demonstrated that inhibition of TRPA1 can greatly decrease the calcium influx elicited by LPC in OLN-93 oligodendrocyte while the calcium rise is nearly inhibited by voltage-gated cation channel antagonist GdCl2, which seems that there may be other channels causing the LPC-induced calcium influx. LPC is a critically lipid which mediates various physiological function (Soga et al. 2005). During the generation of multiple sclerosis, the concentration of LPC in the blood is significantly increasing, which results in the calcium influx, inflammation, and oligodendrocyte damage (Plemel et al. 2018; Pieragostino et al. 2015; Fressinaud et al. 1996). Some studies showed that LPC mediates activation through receptors, including G protein couple receptor (GPCR) and TRP ion channel (Murakami et al. 2004; Monet et al. 2009), while there has been unclear about the special receptor directly interacting with LPC. Other study found that LPC induces demyelination via the process of cellular membrane disrupt since its lipid disrupting properties (Plemel et al. 2018). Considering the character of LPC-elicited cytotoxicity, including calcium overload, NLRP3-dependent inflammation (Freeman et al. 2017; Li et al. 2016), it is more likely that LPC induces the activation of some receptors which mediate the downstream signal pathway and further induce broad stress reaction and cell damaging. TRPA1 is considered as a replied target to prevent the development of multiple sclerosis (Mihai et al. 2019). In our study, we have a newly find that TRPA1 is required for the LPC-induced oxidative stress and cytotoxicity in OLN-93 oligodendrocyte, which further verifies the ideal about the important relation between TRPA1 and LPC-induced cytotoxicity reported in our early study (Tian et al. 2020). The data of this study indicated that TRPA1 may be involved in the process of LPC-elicited oxidative stress and cytotoxicity in non-immunocytes, especially in glial cell. The previous study suggested that TRPA1 in astrocyte could indirectly mediate the oligodendrocyte apoptosis via calcium influx in the cuprizone-induced multiple sclerosis model (Saghy et al. 2016). Taken together, the mechanism of TRPA1 mediating demyelination may vary in different type of cells.
In this study, we firstly reported that LPC could induce generation of mtROS and nitric oxide (NO) in OLN-93 oligodendrocyte and both of them can be inhibited by TRPA1 selective antagonists, which implied that TRPA1 may play a vital role in the process of LPC-induced oxidative stress in OLN-93 oligodendrocyte. The imbalance of the redox is the major trigger of many neurologic diseases including Parkinson’s disease, Alzheimer disease, amyotrophic lateral sclerosis, and multiple sclerosis (Islam 2017). Two major oxidative productions, reactive oxygen species (ROS) and reactive nitrogen species (RNS), can lead to mitochondrial injury and cell death (Angelova and Abramov 2018; Ghasemi et al. 2018). The high concentration of nitric oxide (NO) would damage the cells by producing peroxynitrite, causing nitrosylation of thiol groups and nitration of tyrosine of normal protein, which widely damage the physiological function of normal protein (Yao et al. 2010). Our data indicated that LPC induces NO increase and generation of superoxide production via TRPA1 in OLN-93 oligodendrocyte. NO is a critical inflammatory mediator which would induce extensively immunological reaction, including neutrophil activation and oxidative stress (Guzik et al. 2003). The previous study suggested that LPC can promote the expression of nNOS in dorsal root ganglion (DRG) (Wang et al. 2013). Furthermore, TRPA1 also contributes to the generation of NO and phosphorylation of nNOS (Sinharoy et al. 2015). However, the relation between nNOS and LPC in oligodendrocyte is still unknown. In our study, we demonstrated that nNOS can mediate the generation of NO induced by LPC, which implies that TRPA1 may mediate the activation of nNOS in OLN-93 oligodendrocyte though the details about their interaction need further study.
Considering the fact about LPC-induced development of inflammation and inflammatory injury in the progress of demyelination (Freeman et al. 2017), we confirmed the role of TRPA1 in the process of LPC elicited inflammatory injury in OLN-93. In our study, we performed the priming of inflammation in OLN-93 by treating cells with LPS in a suitable concentration which would not elicit superoxide production increasing or cytotoxicity in OLN-93. In fact, LPS could not induce OLN-93 injury in low concentration (100 ng/mL), but in higher concentration (1 μg/mL), LPS elicits cytotoxicity in OLN-93 through increasing the levels of ROS, NO, and tumor necrosis factor α (TNF-α) (Askari and Shafiee-Nick 2019). Caspase-1 is the major trigger of pyroptosis which is the process of inflammation-induced programmed cell death (Shi et al. 2017). The previous study showed that caspase-1 is involved in the development of pyroptosis in oligodendrocyte (McKenzie et al. 2018). In this study, we further suggested that LPC can induce the oligodendrocyte injury via caspase-1. In addition, both nNOS and mtROS are the major mediators to induce the cytotoxicity of oligodendrocyte and nNOS can mediate mitochondrial damage in oligodendrocyte (Yao et al. 2010; Fetisova et al. 2017; Yao et al. 2012). In our study, we found that LPC-induced cytotoxicity can be alleviated by inhibition of nNOS, mtROS, and TRPA1 in OLN-93 oligodendrocyte, which implies that TRPA1 is required for the process of demyelination by mediating the generation of mtROS and activation of nNOS.
For the current study, we used three kinds of TRPA1 selective antagonists (HC030031, A967079, and AP-18) in order to identify the critical role of TRPA1 in the process of LPC-induced oxidative stress or cytotoxicity and minimize the possibility of false positive due to the non-specificity of single drug. AP-18, a compound of oxime, is a potent and selective TRPA1 antagonist which blocks the rat TRPA1 with an IC50 of 4.5 μM for the inhibition of cinnamaldehyde (CA), an agonist of TRPA1, evoked calcium influx (Strassmaier and Bakthavatchalam 2011). A967079 is a very similar analog of AP18 (Doihara et al. 2009). Compared to AP-18, the IC50 of A967079 improves over 20-fold for rat TRPA1, which is up to 0.289 μM for the inhibition of allyl isothiocyanate (AITC), an agonist of TRPA1, evoked calcium influx (Strassmaier and Bakthavatchalam 2011). HC030031, a compound of acetamide, is another selective TRPA1 antagonist which was acquired from a small molecule compound library screen. HC030031 blocks the human TRPA1 with an IC50 of 6.2 μM for the inhibition of AITC-induced calcium influx (McNamara et al. 2007). From the data of this study, the TRPA1 antagonists (HC030031, A967079, AP-18) or their analogs could be used as potential drugs to further perform in the prospective clinical or pre-clinical research about oligodendrocyte injury.
In summary, in this study, we firstly demonstrated that TRPA1 can mediate LPC-induced calcium overload, generation of mtROS, mitochondrial membrane depolarization, NO increase, development of superoxide production, and cytotoxicity in OLN-93 oligodendrocyte, which reveals that TRPA1 may play an important role in LPC-induced oxidative stress and oligodendrocyte damage. These results also provide a viewpoint that TRPA1 can mediate oligodendrocyte injury directly, and inhibition of TRPA1 in oligodendrocyte may alleviate the syndrome of demyelination.
Funding information
This study was funded by the National Natural Science Foundation of China (31671211), Science and Technology Planning Project of Guangdong Province, China (2017B030314056), Frontier Research Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory (2018GZR110105020), and Guangdong Provincial Natural Science Foundation (2017A030313757 and 2016A030313170).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Andersson DA, Gentry C, Moss S, Bevan S. Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J Neurosci. 2008;28:2485–2494. doi: 10.1523/JNEUROSCI.5369-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelova PR, Abramov AY. Role of mitochondrial ROS in the brain: from physiology to neurodegeneration. FEBS Lett. 2018;592:692–702. doi: 10.1002/1873-3468.12964. [DOI] [PubMed] [Google Scholar]
- Askari VR, Shafiee-Nick R. Promising neuroprotective effects of beta-caryophyllene against LPS-induced oligodendrocyte toxicity: a mechanistic study. Biochem Pharmacol. 2019;159:154–171. doi: 10.1016/j.bcp.2018.12.001. [DOI] [PubMed] [Google Scholar]
- Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol. 2004;55:458–468. doi: 10.1002/ana.20016. [DOI] [PubMed] [Google Scholar]
- Bolcskei K, Kriszta G, Saghy E, Payrits M, Sipos E, Vranesics A, Berente Z, Abraham H, Acs P, Komoly S, Pinter E. Behavioural alterations and morphological changes are attenuated by the lack of TRPA1 receptors in the cuprizone-induced demyelination model in mice. J Neuroimmunol. 2018;320:1–10. doi: 10.1016/j.jneuroim.2018.03.020. [DOI] [PubMed] [Google Scholar]
- Brand A, Bauer NG, Hallott A, Goldbaum O, Ghebremeskel K, Reifen R, Richter-Landsberg C. Membrane lipid modification by polyunsaturated fatty acids sensitizes oligodendroglial OLN-93 cells against oxidative stress and promotes up-regulation of heme oxygenase-1 (HSP32) J Neurochem. 2010;113:465–476. doi: 10.1111/j.1471-4159.2010.06611.x. [DOI] [PubMed] [Google Scholar]
- Campos-Mota GP, Navia-Pelaez JM, Araujo-Souza JC, Stergiopulos N, Capettini LSA. Role of ERK1/2 activation and nNOS uncoupling on endothelial dysfunction induced by lysophosphatidylcholine. Atherosclerosis. 2017;258:108–118. doi: 10.1016/j.atherosclerosis.2016.11.022. [DOI] [PubMed] [Google Scholar]
- Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/s0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- Cossarizza A, Baccarani-Contri M, Kalashnikova G, Franceschi C. A new method for the cytofluorimetric analysis of mitochondrial membrane potential using the J-aggregate forming lipophilic cation 5,5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazolcarbocyanine iodide (JC-1) Biochem Biophys Res Commun. 1993;197:40–45. doi: 10.1006/bbrc.1993.2438. [DOI] [PubMed] [Google Scholar]
- Doihara H, Nozawa K, Kojima R, Kawabata-Shoda E, Yokoyama T, Ito H. QGP-1 cells release 5-HT via TRPA1 activation: a model of human enterochromaffin cells. Mol Cell Biochem. 2009;331:239–245. doi: 10.1007/s11010-009-0165-7. [DOI] [PubMed] [Google Scholar]
- Fetisova E, Chernyak B, Korshunova G, Muntyan M, Skulachev V. Mitochondria-targeted antioxidants as a prospective therapeutic strategy for multiple sclerosis. Curr Med Chem. 2017;24:2086–2114. doi: 10.2174/0929867324666170316114452. [DOI] [PubMed] [Google Scholar]
- Freeman L, Guo H, David CN, Brickey WJ, Jha S, Ting JP. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med. 2017;214:1351–1370. doi: 10.1084/jem.20150237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fressinaud C, Vallat JM, Pouplard-Barthelaix A (1996) Platelet-derived growth factor partly prevents chemically induced oligodendrocyte death and improves myelin-like membranes repair in vitro. Glia 16:40–50. 10.1002/(sici)1098-1136(199601)16:1<40::aid-glia5>3.0.co;2-f [DOI] [PubMed]
- Gerstner B, Buhrer C, Rheinlander C, Polley O, Schuller A, Berns M, Obladen M, Felderhoff-Mueser U. Maturation-dependent oligodendrocyte apoptosis caused by hyperoxia. J Neurosci Res. 2006;84:306–315. doi: 10.1002/jnr.20880. [DOI] [PubMed] [Google Scholar]
- Ghasemi M, Mayasi Y, Hannoun A, Eslami SM, Carandang R. Nitric Oxide and mitochondrial function in neurological diseases. Neuroscience. 2018;376:48–71. doi: 10.1016/j.neuroscience.2018.02.017. [DOI] [PubMed] [Google Scholar]
- Guzik TJ, Korbut R, Adamek-Guzik T. Nitric oxide and superoxide in inflammation and immune regulation. J Physiol Pharmacol. 2003;54:469–487. [PubMed] [Google Scholar]
- Hamilton NB, Kolodziejczyk K, Kougioumtzidou E, Attwell D. Proton-gated Ca(2+)-permeable TRP channels damage myelin in conditions mimicking ischaemia. Nature. 2016;529:523–527. doi: 10.1038/nature16519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignarro LJ. Nitric oxide: a unique endogenous signaling molecule in vascular biology. Biosci Rep. 1999;19:51–71. doi: 10.1023/a:1020150124721. [DOI] [PubMed] [Google Scholar]
- Islam MT. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol Res. 2017;39:73–82. doi: 10.1080/01616412.2016.1251711. [DOI] [PubMed] [Google Scholar]
- Jarjour AA, Zhang H, Bauer N, Ffrench-Constant C, Williams A. In vitro modeling of central nervous system myelination and remyelination. Glia. 2012;60:1–12. doi: 10.1002/glia.21231. [DOI] [PubMed] [Google Scholar]
- Jeong HJ, Kim YH, Lee YS, Jung SJ, Oh SB. TRPM2 contributes to LPC-induced intracellular Ca2+ influx and microglial activation. Biochem Biophys Res Commun. 2017;485:301–306. doi: 10.1016/j.bbrc.2017.02.087. [DOI] [PubMed] [Google Scholar]
- Kalyvas A, Baskakis C, Magrioti V, Constantinou-Kokotou V, Stephens D, Lopez-Vales R, Lu JQ, Yong VW, Dennis EA, Kokotos G, David S. Differing roles for members of the phospholipase A2 superfamily in experimental autoimmune encephalomyelitis. Brain. 2009;132:1221–1235. doi: 10.1093/brain/awp002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kougias P, Chai H, Lin PH, Lumsden AB, Yao Q, Chen C. Lysophosphatidylcholine and secretory phospholipase A2 in vascular disease: mediators of endothelial dysfunction and atherosclerosis. Med Sci Monit. 2006;12:Ra5–R16. [PubMed] [Google Scholar]
- Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE, Rosenberg PA, Volpe JJ, Vartanian T. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J Neurosci. 2002;22:2478–2486. doi: 10.1523/jneurosci.22-07-02478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. TRP channel classification. Adv Exp Med Biol. 2017;976:1–8. doi: 10.1007/978-94-024-1088-4_1. [DOI] [PubMed] [Google Scholar]
- Li X, Fang P, Li Y, Kuo YM, Andrews AJ, Nanayakkara G, Johnson C, Fu H, Shan H, Du F, Hoffman NE, Yu D, Eguchi S, Madesh M, Koch WJ, Sun J, Jiang X, Wang H, Yang X. Mitochondrial reactive oxygen species mediate lysophosphatidylcholine-induced endothelial cell activation. Arterioscler Thromb Vasc Biol. 2016;36:1090–1100. doi: 10.1161/atvbaha.115.306964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Guo W, Zhou H, Tang L, Feng S, Zhong JH, Zhou XF. proBDNF inhibits the proliferation and migration of OLN93 oligodendrocytes. Mol Med Rep. 2018;18:3809–3817. doi: 10.3892/mmr.2018.9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie BA, Mamik MK, Saito LB, Boghozian R, Monaco MC, Major EO, Lu JQ, Branton WG, Power C. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc Natl Acad Sci U S A. 2018;115:E6065–e6074. doi: 10.1073/pnas.1722041115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara CR, Mandel-Brehm J, Bautista DM, Siemens J, Deranian KL, Zhao M, Hayward NJ, Chong JA, Julius D, Moran MM, Fanger CM. TRPA1 mediates formalin-induced pain. Proc Natl Acad Sci U S A. 2007;104:13525–13530. doi: 10.1073/pnas.0705924104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihai DP, Nitulescu GM, Ion GND, Ciotu CI, Chirita C, Negres S. Computational Drug repurposing algorithm targeting TRPA1 calcium channel as a potential therapeutic solution for multiple sclerosis. Pharmaceutics. 2019;11:446. doi: 10.3390/pharmaceutics11090446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monet M, Gkika D, Lehen'kyi V, Pourtier A, Vanden Abeele F, Bidaux G, Juvin V, Rassendren F, Humez S, Prevarsakaya N. Lysophospholipids stimulate prostate cancer cell migration via TRPV2 channel activation. Biochim Biophys Acta. 2009;1793:528–539. doi: 10.1016/j.bbamcr.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Murakami N, Yokomizo T, Okuno T, Shimizu T. G2A is a proton-sensing G-protein-coupled receptor antagonized by lysophosphatidylcholine. J Biol Chem. 2004;279:42484–42491. doi: 10.1074/jbc.M406561200. [DOI] [PubMed] [Google Scholar]
- Nadjafi S, Ebrahimi SA, Rahbar-Roshandel N. Noscapine protects OLN-93 oligodendrocytes from ischemia-reperfusion damage: calcium and nitric oxide involvement. Acta Physiol Hung. 2015;102:351–362. doi: 10.1556/036.102.2015.4.2. [DOI] [PubMed] [Google Scholar]
- Pieragostino D, D'Alessandro M, di Ioia M, Rossi C, Zucchelli M, Urbani A, Di Ilio C, Lugaresi A, Sacchetta P, Del Boccio P. An integrated metabolomics approach for the research of new cerebrospinal fluid biomarkers of multiple sclerosis. Mol BioSyst. 2015;11:1563–1572. doi: 10.1039/c4mb00700j. [DOI] [PubMed] [Google Scholar]
- Plemel JR, Michaels NJ, Weishaupt N, Caprariello AV, Keough MB, Rogers JA, Yukseloglu A, Lim J, Patel VV, Rawji KS, Jensen SK, Teo W, Heyne B, Whitehead SN, Stys PK, Yong VW. Mechanisms of lysophosphatidylcholine-induced demyelination: a primary lipid disrupting myelinopathy. Glia. 2018;66:327–347. doi: 10.1002/glia.23245. [DOI] [PubMed] [Google Scholar]
- Saghy E, Sipos E, Acs P, Bolcskei K, Pohoczky K, Kemeny A, Sandor Z, Szoke E, Setalo G, Jr, Komoly S, Pinter E. TRPA1 deficiency is protective in cuprizone-induced demyelination—a new target against oligodendrocyte apoptosis. Glia. 2016;64:2166–2180. doi: 10.1002/glia.23051. [DOI] [PubMed] [Google Scholar]
- Schilling T, Eder C. Non-selective cation channel activity is required for lysophosphatidylcholine-induced monocyte migration. J Cell Physiol. 2009;221:325–334. doi: 10.1002/jcp.21857. [DOI] [PubMed] [Google Scholar]
- Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42:245–254. doi: 10.1016/j.tibs.2016.10.004. [DOI] [PubMed] [Google Scholar]
- Silva RBM, Greggio S, Venturin GT, da Costa JC, Gomez MV, Campos MM. Beneficial effects of the calcium channel blocker CTK 01512-2 in a mouse model of multiple sclerosis. Mol Neurobiol. 2018;55:9307–9327. doi: 10.1007/s12035-018-1049-1. [DOI] [PubMed] [Google Scholar]
- Sinharoy P, Zhang H, Sinha S, Prudner BC, Bratz IN, Damron DS. Propofol restores TRPV1 sensitivity via a TRPA1-, nitric oxide synthase-dependent activation of PKCepsilon. Pharmacol Res Perspect. 2015;3:e00153. doi: 10.1002/prp2.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soga T, Ohishi T, Matsui T, Saito T, Matsumoto M, Takasaki J, Matsumoto S, Kamohara M, Hiyama H, Yoshida S, Momose K, Ueda Y, Matsushime H, Kobori M, Furuichi K. Lysophosphatidylcholine enhances glucose-dependent insulin secretion via an orphan G-protein-coupled receptor. Biochem Biophys Res Commun. 2005;326:744–751. doi: 10.1016/j.bbrc.2004.11.120. [DOI] [PubMed] [Google Scholar]
- Strassmaier T, Bakthavatchalam R. Transient receptor potential A1 modulators. Curr Top Med Chem. 2011;11:2227–2236. doi: 10.2174/156802611796904915. [DOI] [PubMed] [Google Scholar]
- Tian C, Huang R, Tang F, Lin Z, Cheng N, Han X, Li S, Zhou P, Deng S, Huang H, Zhao H, Xu J, Li Z. Transient receptor potential ankyrin 1 contributes to lysophosphatidylcholine-induced intracellular calcium regulation and THP-1-derived macrophage activation. J Membr Biol. 2020;253:43–55. doi: 10.1007/s00232-019-00104-2. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci. 2008;31:247–269. doi: 10.1146/annurev.neuro.30.051606.094313. [DOI] [PubMed] [Google Scholar]
- Wang HY, Tsai YJ, Chen SH, Lin CT, Lue JH. Lysophosphatidylcholine causes neuropathic pain via the increase of neuronal nitric oxide synthase in the dorsal root ganglion and cuneate nucleus. Pharmacol Biochem Behav. 2013;106:47–56. doi: 10.1016/j.pbb.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Yao S, Pandey P, Ljunggren-Rose A, Sriram S. LPS mediated injury to oligodendrocytes is mediated by the activation of nNOS: relevance to human demyelinating disease. Nitric Oxide. 2010;22:197–204. doi: 10.1016/j.niox.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Yao SY, Natarajan C, Sriram S. nNOS mediated mitochondrial injury in LPS stimulated oligodendrocytes. Mitochondrion. 2012;12:336–344. doi: 10.1016/j.mito.2012.01.002. [DOI] [PubMed] [Google Scholar]