

Abstract
Chlamydia trachomatis is an obligate intracellular pathogen that has been historically difficult to genetically manipulate. Definitive progress in elucidating the mechanisms that C. trachomatis use to create and maintain a privileged intracellular niche has been limited due to a lack of genetic tools. Fortunately, there have recently been several new advances in genetic manipulation techniques. Among these is the development of fluorescence-reported allelic exchange mutagenesis (FRAEM). This method allows targeted gene deletion coupled with insertion of a selection cassette encoding antibiotic resistance and green fluorescent protein (GFP). Reliance on this strategy can be complicated when targeting genes within polycistronic operons due to the potential of polar effects on downstream genes. Floxed cassette allelic exchange mutagenesis (FLAEM), the protocol for which is described here, was developed to alleviate cassette-induced polar effects. FLAEM utilizes Cre-loxP genome editing to remove the selection cassette after targeted deletion by allelic exchange. The resulting strains contain markerless gene deletions of one or more coding sequences. This technique facilitates direct assessment of gene function and expands the repertoire of tools for genetic manipulation in C. trachomatis.
Keywords: Cre recombinase, FLAEM, FRAEM, Chlamydia, mutagenesis, allelic exchange, allelic recombination
SUMMARY:
Described here is a method for targeted, markerless gene deletion in Chlamydia trachomatis using floxed cassette allelic exchange mutagenesis, FLAEM.
INTRODUCTION:
Chlamydia trachomatis is the leading cause of bacterial sexually transmitted disease and represents a significant burden to human health. Over 100 million people are infected every year with C. trachomatis1. Approximately 70% of the infections in women are asymptomatic despite detrimental reproductive health effects, such as pelvic inflammatory disease, ectopic pregnancy, and/or infertility. Disease sequela are directly related to immunopathology initiated by C. trachomatis infection2. An efficacious vaccine has yet to be developed; therefore, understanding the function of bacterial virulence factors and other bacterial gene products is an important and urgent research question.
As intracellular bacteria, host cell invasion, intracellular replication, release of progeny, and evasion of host immunological responses are critical processes. C. trachomatis forms a parasitophorous membrane bound vacuole, termed an inclusion, for intracellular development. Establishment of the inclusion and many other critical processes are achieved by secretion of effector proteins via a type III secretion system (T3SS)3. Elucidating the functions of these secreted effectors was limited for many years due to the genetic intractability of C. trachomatis. Unlike E. coli, many classical cloning techniques are not applicable to Chlamydia. A few major limitations involve transformation efficiency, lack of counterselection reporters such as sacB, and plasmid maintenance. Whereas E. coli plasmids can generally be maintained indefinitely with an origin of replication and appropriate selective pressure, C. trachomatis plasmids requires an additional eight open reading frames (pgp1-8) for maintenance that are found on the native pL2 plasmid within the L2 serovar4.
In recent years there have been multiple genetic tools generated that accommodate Chlamydia’s unique biology, yet there are still limitations5-7. Chemical mutagenesis by ethyl methanesulfonate (EMS) treatment can introduce missense mutations, or (less frequently) can result in nucleotide transitions introducing a premature stop codon to yield a nonsense mutation8. Transposon insertion is efficient for gene disruption, but current technology in Chlamydia research is laborious and time-consuming9. Both EMS treatment and transposon mutagenesis techniques generate random mutations and require rigorous screening methods to isolate mutant strains. A method to disrupt genes by insertion of group II introns (e.g., TargeTron) allows for directed mutagenesis; however, this method is limited by efficiency, and the insertion site is not always properly predicted10.
Fluorescence-reported allelic exchange mutagenesis (FRAEM) is a strategy used for targeted gene deletion coupled with insertion of a selection cassette providing antibiotic resistance and a fluorescence reporter11. Yet, FRAEM is complicated by the potential of cassette-induced polar effects on downstream genes, especially when targeting genes within polycistronic operons. Floxed cassette allelic exchange mutagenesis (FLAEM) is a novel genetic approach developed to alleviate the cassette-induced polar effects previously observed with the FRAEM selection cassette12. FLAEM utilizes Cre-loxP genome editing to remove the selection cassette and restore expression of downstream genes. The selection cassette containing antibiotic resistance and green fluorescent protein (GFP) is reengineered with flanking loxP sites. These loxP sites can recombine in the presence of Cre recombinase and result in excision of the cassette from the genome13. This strategy has been shown to alleviate cassette induced polar effects when targeting tmeA for deletion12,14.
Both FRAEM and FLAEM methods utilize the same suicide vector, pSUmC 4.0, which can be conditionally maintained through inducible expression of pgp6. Expression of pgp6 has previously been shown to be necessary for plasmid retention and is therefore leveraged to control plasmid maintenance11,15. When C. trachomatis is grown in media supplemented with anhydrous tetracycline (aTc) to induce pgp6 expression, the vector is maintained. In the absence of aTc, the vector is lost. Targeted gene deletion is achieved through allelic exchange of the gene for the selection cassette. The 3 kb regions directly upstream and downstream of the targeted gene serve as homology arms for recombination. These arms are cloned into the pSUmC 4.0 vector flanking the selection cassette. Successful C. trachomatis transformation and recombination events are observed through fluorescence reporting. Expression of mCherry on the vector backbone and gfp within the selection cassette yield red and green fluorescent inclusions. Once aTc is removed from culture media, green-only inclusions indicate successful recombination events with the loss of the suicide vector and integration of the selection cassette into the bacterial genome.
FLAEM represents an extension of FRAEM via subsequent transformation of a Cre recombinase-expressing vector, pSU-Cre, into the newly created mutant strain. Cre recombinase facilitates recombination between loxP sites and excision of the selection cassette. Recombination events are indicated via fluorescence reporting. The pSU-Cre vector encodes mCherry; thus, successful transformation is indicated by addition of red fluorescence to gfp-expressing inclusions. Cultivation in the absence of selective pressure for the cassette results in Cre-mediated recombination at the loxP sites, and loss of the cassette is indicated by red-only inclusions. As with pSUmC-4.0, inducible expression of pgp6 is used to conditionally maintain pSU-Cre. Once aTc and antibiotic selection are removed, the plasmid is cured, and the resulting markerless deletion strain is nonfluorescent. This method addresses the issue of cassette-induced polar effects.
PROTOCOL:
1. Design and assembly of pSUmC-4.0 with homology arms specific to the gene of interest
1.1 Identify ~3 kb regions directly upstream and downstream of the gene targeted for deletion to serve as the 5’ and 3’ homology arms for homologous recombination (Figure 1).
Figure 1: Schematic representation for identifying 5’ and 3’ homology arms from genomic DNA and PCR screening for their insertion into pSUmC-4.0.
(A) A gene targeted for deletion from the C. trachomatis genome is represented by the blue arrow. The 3 kb regions identified as the 5’ and 3’ homology arms for allelic recombination are bracketed in purple and red, respectively. (B,C) Insertion of the 5’ and 3’ homology arms into pSUmC-4.0 is determined by PCR screening. The Sall and Sbfl restriction enzyme sites are shown flanking the aadA (black arrow) and gfp (green arrow) selection cassette which is flanked by loxP sites (yellow squares) on the pSUmC-4.0 vector. (B) No insertion is determined by a 1 kb PCR product when 5’ screening primers (purple arrow heads) are used to PCR amplify across the Sall site, or 3’ screening primers (red arrow heads) are used to amplify across the SbfI site. (C) Successful insertion is determined by 3 kb PCR products under the same conditions. 5’ and 3’ homology arms are represented by the purple and red brackets, respectively.
1.2 Design PCR primers to 1) amplify the 3 kb 5’ homology arm from chlamydial genomic DNA and 2) contain a 15–30 bp overhang specific to pSUmC-4.0 when digested at the Sall restriction enzyme site. Ensure that the primer regions overlapping with pSUmC-4.0 have melting temperatures of 55 °C and that hairpin melting temperatures for the entire primer are <35 °C.
1.3 Design PCR primers to 1) amplify the 3 kb 3’ homology arm from chlamydial genomic DNA and 2) contain a 15–30 bp overhang specific to pSUmC-4.0 when digested at the SbfI restriction enzyme site. Ensure that the primer regions overlapping with pSUmC-4.0 have melting temperatures of 55 °C and that hairpin melting temperatures for the entire primer are <35 °C.
1.4 Design PCR screening primers that will amplify the insertion of each homology arm into the pSUmC-4.0 vector after a DNA assembly reaction16. Design these primers to anneal ~500 bp outside the restriction sites to allow easy visualization of a PCR product even if the insertion was unsuccessful (Figure 1).
1.5 Amplify the 3 kb 5’ and 3’ homology arms from freshly extracted chlamydial genomic DNA by PCR in one to two reactions to yield enough fragment for later DNA assembly. Follow the DNA polymerase manufacturer’s protocol for specific reaction set-up and cycling conditions.
1.6 Verify that both PCR products are the correct size and that the reactions do not contain any contaminating bands. Run 5 μL of each PCR reaction on a 1% DNA agarose gel.
1.7 Purify and concentrate the PCR fragments by phenol/chloroform extraction and ethanol precipitation.
1.7.1 Pool all 3’ PCR reactions together in a 1.5 mL tube and bring to a final volume of 200 μL with nuclease-free H2O. Repeat this process for the 5’ PCR reactions.
1.7.2 Add 200 μL of phenol to each tube and vortex for 30–60 s. Spin each tube in a microcentrifuge at ≥21,000 x g for 20 min at room temperature (RT).
1.7.3 Transfer the aqueous phase top layer of each tube into a new 1.5 mL tube, add one-tenth the volume of 3 M sodium acetate to each tube, then flick to mix.
1.7.4 Add 3 volumes of 100% ethanol to each tube and flick to mix. Incubate both tubes at −80 °C for 20 min.
1.7.5 Pellet the DNA by spinning each tube at ≥21,000 x g for 5 min at RT. Discard the supernatant.
1.7.6 Wash the DNA pellet with 100 μL of 70 % ethanol. Spin each tube at ≥21,000 x g for 5 min at RT. Discard the supernatant.
1.7.7 Wash the DNA pellet again with 100 μL of 100% ethanol. Spin each tube at ≥21,000 for 5 min at RT. Discard the supernatant.
1.7.8 Let the pellet fully air-dry, then gently resuspend the DNA in 12 μL of nuclease-free H2O by flicking to mix.
1.8 Digest 500 ng of pSUmC-4.0 vector in a 20 μL reaction with 1 unit of Sall and 1 unit of SbfI according to the manufacture’s protocol. Incubate the reaction for approximately 3 h or overnight to ensure complete digestion of the vector.
1.9 Purify and concentrate the digested backbone by phenol/chloroform extraction and ethanol precipitation as described earlier (step 1.7.1–1.7.9).
1.10 Combine the digested pSUmC-4.0 vector and both the 5’ and 3’ homology arm PCR products together at a ratio of 0.01 pmol pSUmC-4.0 to 0.09 pmol each homology arm. Assemble the fragments in a DNA assembly reaction according to the manufacture’s protocol.
1.11 Transform 50 μL of electrocompetent E. coli with 1 μL of the DNA assembly reaction. Plate the transformed E. coli onto LB agar plates containing 100 μg/mL spectinomycin by spreading 150 μL per plate. Incubate the plates at 37 °C overnight.
1.12 The next day, screen colonies for green and red fluorescence using an epifluorescence inverted microscope. A total of 5–30 colonies per agar plate is expected.
1.13 Pick 5–10 colonies to inoculate 4 mL of LB broth supplemented with 100 μg/mL spectinomycin. Incubate cultures at 37 °C overnight with shaking at 250 rpm.
1.14 Using the overnight E. coli cultures, perform small-scale DNA purification. Elute the DNA with 50 μL of nuclease-free H2O.
1.15 Confirm the insertion of each homology arm into the pSUmC-4.0 vector using the PCR screening primers (designed in step 1.4) to amplify each insertion. If DNA assembly is successful, a ~4 kb PCR product should be observed in a 1% DNA agarose gel for both the 5’ and 3’ arm regions. A ~1 kb PCR product indicates a lack of insertion.
NOTE: If the insertion of the homology arms is unsuccessful in a dual fragment assembly reaction, perform two assembly reactions to insert the 5’ arm then 3’ arm, individually. Digest the vector with only Sall before assembling with the 5’ arm, then digest the vector with SbfI to assemble the 3’ arm.
1.16 Transform dam−/dcm− competent E. coli with 200 ng of pSUmc-4.0 assembled with both homology arms. Plate the transformed bacteria onto LB agar plates containing 100 μg/mL spectinomycin by spreading 25 μL per plate. Incubate the plates at 37 °C overnight. A total of 20–200 colonies are expected.
1.17 Screen the plates for red and green fluorescent colonies as described in step 1.13.
1.18 Set up a culture for a large-scale DNA purification by inoculating 100 mL of LB broth containing 100 μg/mL spectinomycin with red and green colonies. Incubate the culture at 37 °C overnight with shaking at 250 rpm.
1.19 Harvest the culture and purify the DNA using a large-scale DNA purification.
1.19.1 Resuspend the purified plasmid DNA in 100 μL of NF-H2O. A total DNA yield of at least 2 μg/μL is ideal for C. trachomatis transformation.
1.19.2 Sequence the 5’ and 3’ homology arm regions to ensure correct assembly into pSUmC-4.0 before transforming C. trachomatis.
2. Transformation of C. trachomatis with pSUmC 4.0 + homology arms for gene deletion by allelic exchange.
2.1 Seed McCoy cells, mouse fibroblasts standardly used for cultivating Chlamydia, into two 6 well plates (1 x 106 cells/well) with growth media #1. Incubate the plates at 37 °C with 5% CO2 until the monolayer is confluent (approximately 24 h).
2.2 In a 1.5 mL tube, add a volume of C. trachomatis WT serovar L2 elementary bodies (EBs) that is sufficient to infect 12 wells of a 6 well plate at an MOI of 2. Pellet the EBs at >20,000 x g for 30 min, then discard the supernatant.
2.3 Initiate chlamydial transformation by gently resuspending the EBs in 600 μL of CaCl2 buffer, and then add 12 μg of pSUmC-4.0 + homology arms to the tube. Gently mix the solution by flicking. Incubate the tube at RTfor 30 min and flick to mix every 10 min.
2.4 Add the transformation solution to 24 mL of Hanks’ balanced salt solution (HBSS) and mix by gently pipetting. Transfer 2 mL of the inoculum to each well of confluent McCoy cells in the 6 well plates and infect by centrifugation at 900 x g for 1 h at 20 °C.
2.5 Remove the inoculum and replace with 2 mL/well RPMI growth media #2. Incubate the plates at 37 °C with 5% CO2.
2.6 After a minimum of 7 h, but no more than 12 h, replace the media with 2 mL/well of selection media #1. Continue the incubation at 37 °C for 48 h from the time of infection.
2.7 Harvest and passage the bacteria onto a fresh monolayer of McCoy cells.
2.7.1 Using a cell scraper, gently scrape the McCoy monolayer to lift the cells into the media. Transfer the material from each well into a 2 mL tube. Pellet the cell material at >20,000 x g in a microcentrifuge for 30 min at 4 °C.
2.7.2 Remove and discard the supernatant. Resuspend the cell pellet in 1 mL of HBSS by gently pipetting.
2.7.3 Pellet the cell debris at 200 x g for 5 min at 4 °C. Transfer the supernatant onto a fresh well of confluent McCoy cells. Add an additional 1 mL of HBSS to each well for a total volume of 2 mL/well.
2.7.4 Infect by centrifugation at 900 x g for 1 h at 20 °C. Replace the inoculum with selection media #1 immediately following infection.
2.8 Continue passaging the bacteria onto a fresh monolayer of McCoy cells (section 2.7) every 48 h for three or more passages until red and green inclusions are detected using an epifluorescence inverted microscope 24 h post-infection (24 hpi).
2.9 Once red and green inclusions are detected, continue passaging the monolayer (section 2.7) using selection media #2.
2.10 Continue passaging the monolayer until green-only inclusions are detected 24 hpi (passage #3 or later), which indicates the loss of the pSUmC-4.0 vector and incorporation of the loxP selection cassette into the genome.
2.11 Choose one well of green-only inclusions to enrich by passaging. Harvest and freeze any other wells that also contain green-only inclusions in sucrose-phosphate-glutamate buffer (SPG).
2.11.1 To enrich green-only inclusions, passage the monolayer from one well of a 6 well plate as done in step 2.7, but after pelleting the cell debris (step 2.7.3), transfer the supernatant into a conical with 12 mL of HBSS. Use this inoculum to infect two 6 well plates by adding 2 mL per well, then spin the plates at 900 x g for 60 min at 20 °C.
2.11.2 To freeze additional wells, harvest the monolayer as in step 2.7.1, but resuspend the pellets in 1 mL of SPG instead of HBSS. Pellet the cell debris (step 2.7.3) and transfer the supernatant into a 1.5 mL cryotube. Freeze the material at −80 °C.
2.12 After enriching green-only inclusions to a MOI of 0.5–1.0 (one to two more passages after detection), harvest the monolayers into SPG as described in step 2.11.2. Obtain aliquots of 50 μL in 1.5 mL tubes and freeze at −80 °C.
2.13 Titer green-only bacteria to determine inclusion, forming units/μL, according to a standard titration protocol17.
3. Clonal isolation of green-only C. trachomatis deletion mutant containing the loxP flanked selection cassette by limiting dilution.
3.1 Seed a 384 well tissue culture plate with 50 μL of McCoy cells at ~2000 cells/well in growth media #1. Incubate at 37 °C with 5 % CO2 for ~24 h or until confluent.
3.2 Dilute an aliquot of green-only bacteria in HBSS to achieve ~50 EBs per 20 mL. Transfer the diluted inoculum to a reservoir tray.
3.2.1. Remove the media from the 384 well plate by firmly flicking the entire plate upside down into a waste container. Using a multichannel pipette, add 50 μL of C. trachomatis inoculum to each well. Infect by centrifugation at 900 x g for 1 h at 20 °C.
NOTE: Be mindful not to flick so hard that the monolayer detaches. If cells are over-confluent, they will detach more easily.
3.3 Remove the inoculum by flicking and replace with 50 μL/well growth media #2. Incubate plates at 37 °C with 5% CO2.
NOTE: At this stage, integration of the selection cassette into the genome is stable, so antibiotic selection with spectinomycin is no longer required.
3.4 After incubating the plate for 5–12 days, identify individual wells with green fluorescent inclusions using an epifluorescence inverted microscope or a high content screening platform.
3.5 Harvest wells with green-only inclusions by scraping the monolayer with a p-10 tip and transferring the entire contents of the well into a tube containing 2 mL of HBSS. Gently mix and apply the 2 mL of inoculum to a fresh confluent McCoy monolayer in a 6 well plate for infection.
NOTE: When identifying individual wells with inclusions, the inclusions will be in a cluster due to expansion from the initial inclusion. If a well has multiple clusters, it is likely that more than one EB initially infected that well. These wells should not be harvested, and in some cases, multiple rounds of clonal isolation by serial dilution may be necessary.
3.6 Infect the 6 well plate by centrifugation at 900 x g for 1 h at 20 °C. Replace the inoculum with 2 mL/well growth media #2. Incubate at 37 °C with 5 % CO2 for 48 h.
3.7 Repeat steps 2.11–2.13 to enrich, freeze, and titer clonal populations.
3.8 Confirm the deletion of targeted genes from clonally isolated C. trachomatis using qPCR as previously described12.
4. Transformation of C. trachomatis FRAEM mutant with pSU-Cre to initiate removal of loxP flanked selection cassette
4.1 Repeat the transformation process in a 6 well plate as previously described (steps 2.1–2.8) using the clonally isolated C. trachomatis FRAEM mutant, pSU-Cre vector, and selection media #3.
NOTE: A successful transformation is indicated by inclusions that are red and green fluorescent.
4.1.1 Passage the monolayer until red-only inclusions are detected using an epifluorescence inverted microscope, which indicates loss of the selection cassette (two to three passages). Continue passaging the monolayer until red-only inclusions are enriched to a MOI of ~0.5.
4.2 Clonally isolate red-only inclusions by limiting dilution in a 384 well plate as previously described (steps 3.1-3.8); however, use selection media #3 for all steps.
4.3 Enrich clonal populations by passaging until an MOI of 0.1. Once enriched, replace section media with growth media #2 to initiate loss of the pSU-Cre vector (approximately three to four passages).
4.4 Clonally isolate nonfluorescent bacteria (steps 3.1–3.8); however, manually scan the plate with a brightfield microscope to detect inclusions. Enrich and freeze bacteria (step 2.11 – 2.12).
4.5 Screen for the mutant strain using quantitative real-time PCR, Western blotting, and whole genome DNA Sequencing as previously described12.
REPRESENTATIVE RESULTS:
The method for markerless gene deletion in C. trachomatis using FLAEM is reliant upon careful cloning and transformation techniques. Successful allelic recombination is an essential first step and requires the identification and insertion of homology arms into the pSUmC-4.0 cloning vector (Figure 1). An essential second step for markerless gene deletion is removal of the fluorescence reporter and antibiotic selection cassette by Cre-lox genome editing, represented in Figure 2. The vectors used to accomplish each of these steps are annotated in Figure 3. Figure 4 shows a schematic representation of the transformation strategy to generate a markerless deletion mutant when starting with wild-type C. trachomatis.
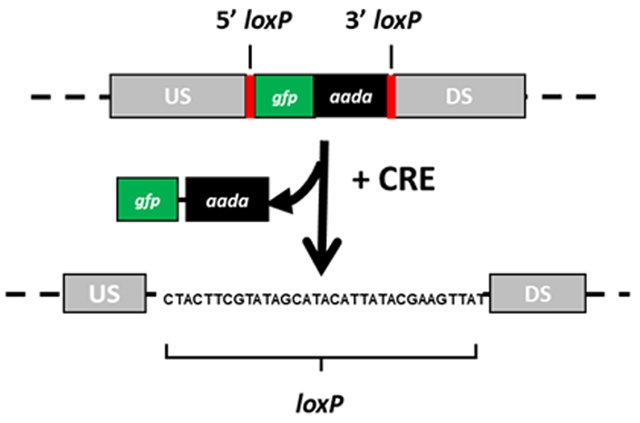
Figure 2: Schematic representation of Cre-lox mediated recombination to remove the gfp-aadA selection cassette.
In the presence of Cre recombinase, the loxP sites (yellow) recombine, resulting in excision of the gfp-aadA selection cassette (green and black squares). The resulting locus is shown containing one remaining loxP scar sequence. Upstream (US) and downstream (DS) regions are shown in gray. This figure has been modified from Keb et al.12.
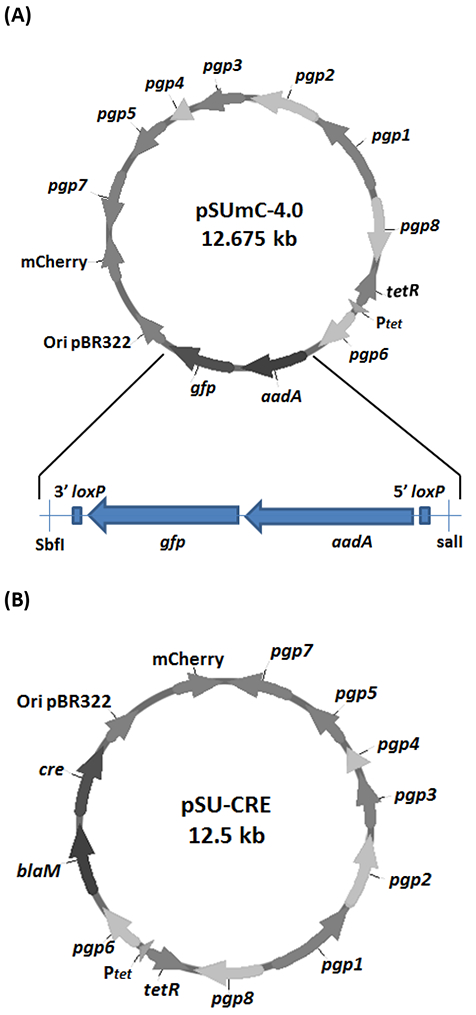
Figure 3: Genetic organization of pSUmC-4.0 and pSU-CRE.
For controllable maintenance of the vectors in C. trachomatis, pgp6 is engineered downstream of a tetracycline-inducible promoter. The remaining pgp genes are downstream of their native Chlamydial promoters, and mCherry is constitutively expressed on both vectors. (A) pSUmC-4.0 contains a cassette encoding constitutively expressed aadA and gfp genes for antibiotic and fluorescence selection capability, respectively. LoxP sites for Cre mediated recombination as well as restriction enzyme sites for the insertion of gene specific homology arms flank the selection cassette. (B) pSU-CRE contains a similar backbone to pSUmC-4.0; yet, instead of the recombination cassette, blaM and cre are constitutively expressed for antibiotic selectivity and CRE recombinase generation, respectively. These figures have been modified from Keb et al.12.
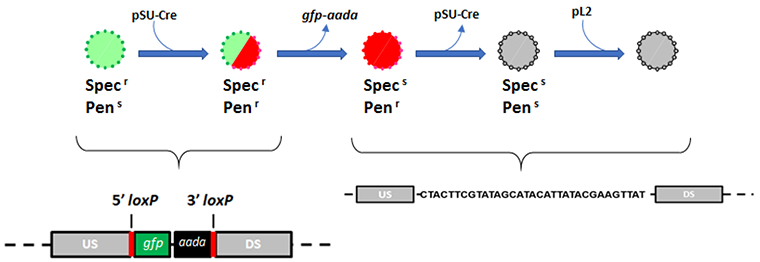
Figure 4: Schematic representation of the FLAEM transformation strategy used to create a markerless deletion mutant.
The general FLAEM method is represented here where Wild-type C. trachomatis is sequentially transformed and serially passaged to generate a markerless deletion mutant. Transformation steps are represented by the addition of small arrows, and the loss of genetic elements is represented by the subtraction of small arrows. C. trachomatis intermediates (circles) are represented with antibiotic sensitivities (penicillin-resistant or penicillin-sensitive = PenGr or PenGs; spectinomycin-resistant or spectinomycin-sensitive = Specr or Specs) and fluorescence-reporting qualities (green = gfp +, red = mCherry +, gray = no fluorescence). Schematic representations of the gene locus at each step are shown below. This figure has been modified from Keb et al.12.
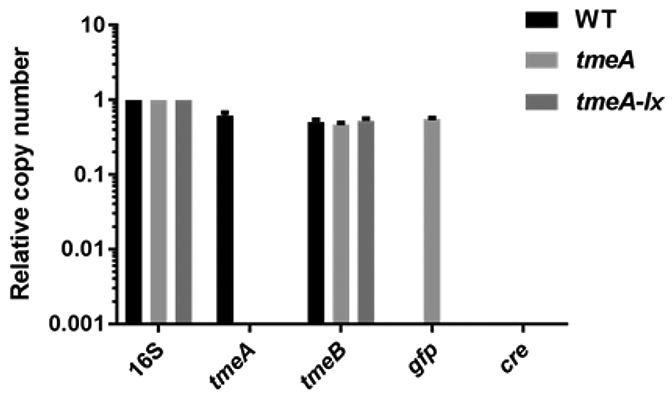
Representative data is shown in Figure 5 and Figure 6, in which tmeA is targeted for gene deletion. The C. trachomatis tmeA deletion strain is generated using FRAEM, and the C. trachomatis tmeA-lx strain is generated using FLAEM. Both mutant strains contain a deletion of the tmeA locus; however, tmeA-lx does not contain the selection cassette, as indicated by the absence of gfp DNA shown in Figure 5. The tmeA mutant strain has decreased expression of tmeB, shown in Figure 6 by mRNA and protein levels. When FLAEM is utilized to generate the tmeA-lx mutant strain, Figure 6 shows that expression of tmeB is restored.
Figure 5: FRAEM and FLAEM are used to generate tmeA gene deletions.
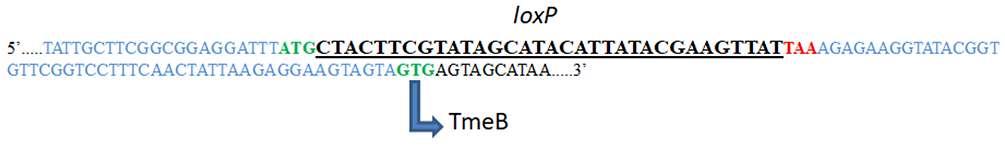
(A) FRAEM was used to generate tmeA, and FLAEM was used to generate tmeA-lx. Relative DNA copy numbers of tmeA and downstream tmeB; gfp contained on the pSUmC-4.0 selection cassette; and cre contained on pSU-CRE vector are all shown. McCoy cells infected with equal inclusion forming units of WT, L2 tmeA, or L2Rif tmeA-lx C. trachomatis were harvested at 24 hpi, and DNA was extracted for qPCR. Relative copy numbers were assessed by signal normalization to chlamydial 16sRNA. ND = none detected. (B) The sequenced tmeAB locus from L2Rif tmeA-lx is shown with a single remaining loxP scar sequence (underlined). The flanking regions of DNA are shown in blue, the start codons are shown in green, and the TmeA stop codon is shown in red. The non-canonical start codon for TmeB is also depicted. These figures have been modified from Keb et al.12.
Figure 6: Excision of selection cassette using FLAEM restores expression of tmeB when targeting tmeA and does not disrupt downstream genes.
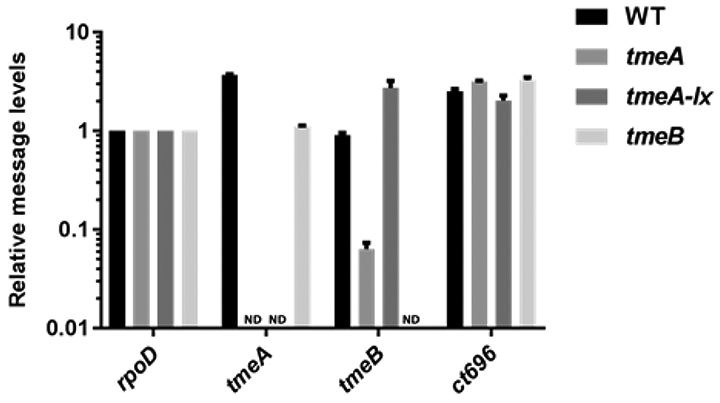
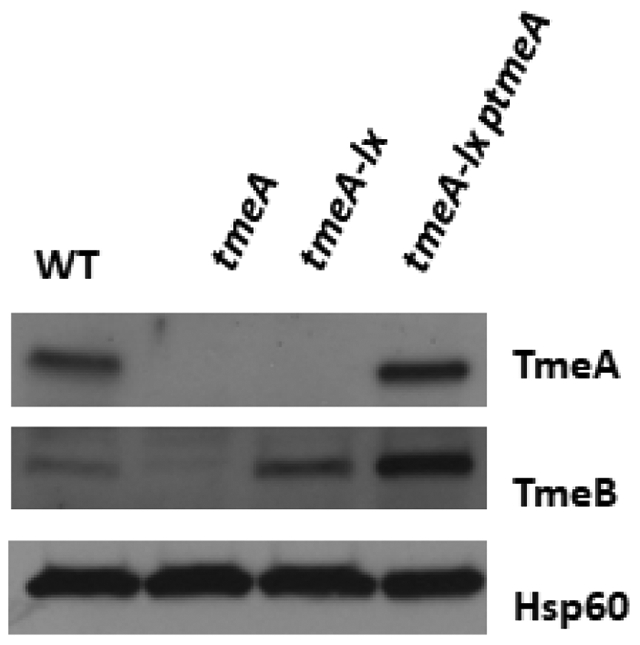
(A) Relative mRNA level of C. trachomatis tmeA and tmeB mutant strains. The presence of transcripts downstream of tmeA was determined by reverse transcriptase (RT) quantitative PCR. The region immediately downstream of the tmeA/B operon encodes ct696. Total RNA was isolated at 24 hpi from McCoy cells infected at an MOI of 1 with WT, L2 tmeA, L2 tmeB, or L2Rif tmeA-lx. Transcripts for tmeA, tmeB, and ct696 were detected by qRT-PCR. Signals are presented after normalization to rpoD. ND = none detected. (B) Western blot for presence of TmeA and TmeB in C. trachomatis mutant strains. Equal quantities of whole-culture material from 24 hpi cultures infected with equal inclusion forming units of WT, L2 tmeA, L2Rif tmeA-lx, or L2Rif tmeA-lx ptmeA were probed in immunoblots for TmeA and TmeB. Hsp60 was used as a loading control, and proteins were visualized by chemiluminescence. Figure has been modified from Keb et al.12.
DISCUSSION:
The protocol described here for the generation of markerless gene deletions in C. trachomatis by FLAEM allows targeted deletion of nonessential genes and eliminates cassette-induced polar effects. The protocol relies upon careful design of 5’ and 3’ homology arms inserted into the pSUmC 4.0 suicide vector, efficient transformation of C. trachomatis, and careful screening of isolated mutant strains. Successful genome engineering via this method results in bacteria that are nonfluorescent and contain a single loxP scar sequence at the site of targeted gene deletion. Furthermore, this method has the potential to be adapted for sequential targeting of genes within the same C. trachomatis strain.
Careful design of the 5’ and 3’ homology arms and insertion into the suicide vector is the first and most critical step of the cloning process. It has been found that 3 kb homology arms provide the most efficient allelic recombination. While insertion of these arms generates a vector that is large and sometimes difficult to work with, there has been less success with shorter arms, with ~2 kb representing a minimum for success. Utilization of the Sall and Sbfl restriction sites provides a convenient one-step construction reaction when performing DNA assembly.
Other cloning methods, such as insertion PCR, have also been effective in inserting homology arms, but they introduce greater possibility of PCR-based errors. Interestingly, it has been observed that cloning of the 5’ arm is comparatively more efficient than the 3’ arm. If issue arise, it is recommended to divide the DNA assembly into two reactions and construct the vector in a stepwise manner. Then, it is advised to digest the vector backbone with SbfI, insert the 3’ arm first, then digest the vector with Sall and insert the 5’ arm in a second DNA assembly reaction.
When amplifying PCR fragments for the homology arms, it is also recommended to use freshly purified C. trachomatis genomic DNA that has not been previously frozen. This limits the possibility of DNA shearing and increases efficiency for generating larger amplicons. If amplifying homology arms from another vector, the purified PCR product will need to be DpnI restriction enzyme treated before proceeding to DNA assembly to reduce background colonies during E. coli transformation.
Transformation is another critical step of this protocol in which issues may arise. If the transformation with pSUmC 4.0 is successful, green and red inclusions should be visible by passage #3; however, it can take several more passages before transformants become enriched. Like other mutagenesis approaches, this method is limited to targeting of nonessential genes, yet transformation should still be readily accomplished. Long-term passaging of transformants in the absence of allelic exchange, indicated by retention of red fluorescence, may indicate that deletion of the targeted gene is a lethal event.
Because transformation vectors for C. trachomatis contain the same origin of replication as the native pL2, it is not uncommon for the native plasmid to be cured after multiple (>5) passages. The pgp genes on the pSUmC vectors are in a different order as compared to the native pL2; therefore, PCR can be used to detect the presence of pL2 in an isolated strain by amplifying the region spanning pgp7–pgp8. If the native plasmid is lost in final deletion mutant, it will need to be reintroduced before conducting developmental studies. It has been have found success in utilizing lateral gene transfer (LGT) to reintroduce the pL2 plasmid19.
In many cases, early deletion mutants with the selection cassette still contain the pL2 plasmid. We have utilized these strains for LGT with success to reintroduce pL2 and avoid repair of the deleted gene. LGT can also be utilized as a secondary method for transformation when introducing pSU-Cre. In some cases, LGT was more efficient than classical CaCl2 transformation. In instances where LGT is utilized to introduce pSU-Cre, it important to start with C. trachomatis that express a rpoB allele, which confers rifampin resistance prior to allelic exchange and insertion of the spectinomycin selection cassette12,19. Starting with rifampin resistant bacteria allows selection after co-culture.
FLAEM enables reverse-genetic approaches with targeted deletion of entire coding sequences, compared to other genetic methods that rely on random mutagenesis or insertion of nucleotide sequences to achieve gene disruption. FLAEM is essentially an extension of FRAEM, as it allows removal of the selection cassette and eliminates previously observed cassette-induced polar effects. This method also creates the opportunity to generate multiple gene deletions in a single C. trachomatis strain.
Multiple mutations can be generated via two different mechanisms. In the first, FLAEM can be used to generate a markerless gene mutation and sequentially used to target another gene in the same strain. The first deletion mutant can be retransformed with the pSUmC 4.0 vector containing homology arms specific for secondary gene of interest. In this case, the protocol should be repeated in the same manner as the first deletion and repeated multiple times to target genes sequentially. In the second mechanism, the markerless deletion mutant isolated after FLAEM can be co-infected with a deletion mutant that still contains a selection cassette. Through LGT, the second deletion is acquired and can be selected for. When using this approach, additional mutations are limited by the number of unique selection cassettes left in the genome. Commonly used antibiotics used as selective pressure during transformation are limited for C. trachomatis, but they do include penicillin, chloramphenicol, and spectinomycin. Removing the selection cassette by Cre-loxP genome editing reduces the need to use multiple antibiotics for selective pressure. Deleting multiple gene sequences at the same time is beneficial when studying proteins with related functions or biological processes that may have multiple key players.
ACKNOWLEDGMENTS:
This work was supported by Public Health Service grants from the National Institute of Health, NIAID (grants A1065530 and Al124649), to K.A. Fields.
Footnotes
DISCLOSURES:
The authors have no conflicts of interest to disclose.
REFERENCES:
- 1.Global Incidence and Prevalence of Selected Curable Sexually Transmitted Infections: 2008: World Health Organization, Department of Reproductive Health and Research, 2012 ISBN 978 92 4 1503839. Sexual and Reproductive Health Matters. 20, 207–208 (2012). [Google Scholar]
- 2.Stephen RS The Cellular Paradigm of Chlamydial Pathogenesis. Trends in Microbiology. 11, 44–51 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Mueller KE, Plano GV, Fields KA New Frontiers in Type III Secretion Biology: the Chlamydia Perspective. Infection and Immunity. 82, 2–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seth-Smith HMB et al. Co-evolution of genomes and plasmids within Chlamydia trachomatis and the emergence in Sweden of a new variant strain. BMC Genomics. 10, 239 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brothwell JA, Muramatsu MK, Zhong G, Nelson DE Advances and obstacles in genetic dissection of chlamydial virulence. Current Topics in Microbiology and Immunology. 412, 133–158 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McClure EE et al. Engineering of obligate Intracellular bacteria: progress, challenges and paradigms. Nature Reviews Microbiology. 15, 544–558 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rahnama M, Fields KA Transformation of Chlamydia: current approaches and impact on our understanding of chlamydial infection biology. Microbes and Infection. 20 (7–8), 445–450 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kari L et al. Generation of targeted Chlamydia trachomatis null mutants. Proceedings of the National Academy of Sciences of the United States of America. 108, 7189–7193 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.LaBrie SD et al. Transposon Mutagenesis in Chlmaydia trachomatis Identifies CT339 as a ComEC Homolog Important for DNA Uptake and Later Gene Transfer. mBio. 10 (4), e01343–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson CM, Fisher DJ Site-specific, insertional inactivation of incA in Chlamydia trachomatis using a group II intron. PLoS One. 8, e83989 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mueller KE, Wolf K, Fields KA Gene Deletion by Fluorescence-Reported Allelic Exchange Mutagenesis in Chlamydia trachomatis. mBio. 7, e01817–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keb G, Hayman R, Fields KA Floxed cassette Allelic Exchange Mutagenesis Enables Markerless Gene Deletion in Chlamydia trachomatis and can Reverse Cassette-Induced Polar Effects. Journal of Bacteriology. 200, e00479–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yarmolinsky M, Hoess R THe legacy of Nat Sternberg: the genesis of Cre-lox technology. Annual Review of Virology. 2, 25–40 (2015). [DOI] [PubMed] [Google Scholar]
- 14.McKuen MJ, Mueller KE, Bae YS, Fields KA Fluorescence-Reported Allelic Exchange Mutagenesis Reveals a Role for Chlamydia trachomatis TmeA in Invasion that is Independent of Host AHNAK. Infection and Immunity. 85 (12), e00640–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song L et al. Chlamydia trachomatis Plasmid-Encoded Pgp4 is a Transcriptional Regulator of Virulence-Associated Genes. Infection and Immunity. 81 (3), 636–44 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silayeva O, Barnes AC Gibson Assembly facilitates bacterial allelic exchange mutagenesis. Journal of Microbiological Methods. 144, 157–163 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Hackstadt T, Scidmore M, Rockey D Lipid Metabolism in Chlamydia trachomatis-Infected Cells: Directed Trafficking of Golgi-Derived Sphingolipids to the Chlamydial Inclusion. Proceedings of the National Academy of Sciences of the United States of America. 92 (11), 4877–4881 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeffrey BM, Suchland RJ, Eriksen SG, Sandoz KM, Rockey DD Genomic and phenotypic characterization of in vitro-generated Chlamydia trachomatis recombinants. BMC Microbiology. 13, 142 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suchland RJ, Bourillon A, Denamur E, Stamm WE, Rothstein DM Rifampin-resistant RNA polymerase mutants of Chlamydia trachomatis remain susceptible to the ansamycin rifalazil. Antimicrobial Agents Chemotherapy. 49, 1120–1126 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]