

Abstract
The NLRC4 inflammasome assembles in response to detection of bacterial invasion, and NLRC4 activation leads to the production of IL-1β and IL-18 together with pyroptosis-mediated cell death. Missense activating mutations in NLRC4 cause autoinflammatory disorders whose symptoms are distinctly dependent on the site of mutation and other aspects of the genetic background. To determine the involvement of IL-1β and IL-18 in the inflammation induced by NLRC4 mutation, we depleted IL-1β, IL-18, or both cytokines in Nlrc4-transgenic mice in which mutant Nlrc4 is expressed under the MHC class II promoter (Nlrc4-H443P-Tg mice). The deletion of the Il1b or Il18 gene in Nlrc4-H443P-Tg mice reduced the neutrophil numbers in the spleen, and mice with deletion of both genes had an equivalent number of neutrophils compared to wild-type mice. Deletion of Il1b ameliorated but did not eliminate bone marrow hyperplasia, while mice deficient in Il18 showed no bone marrow hyperplasia. In contrast, tail bone deformity remained in the presence of Il18 deficiency, but Il1b deficiency completely abolished bone deformity. The decreased bone density in Nlrc4-H443P-Tg mice was counteracted by Il1b but not Il18 deficiency. Our results demonstrate the distinct effects of IL-1β and IL-18 on NLRC4-induced inflammation among tissues, which suggests that blockers for each cytokine should be utilized depending on the site of inflammation.
Keywords: autoinflammation, NLRC4, interleukin-1β, interleukin-18, bone
Introduction
Inflammasomes are composed of an assembler protein such as a nucleotide-binding domain-containing proteins, a leucine-rich family protein, ASC and caspase-1, and inflammasome stimulation leads to the maturation and secretion of the IL-1β and IL-18 cytokines (1–3). Inflammasome activation is induced by pathogen-associated molecular patterns as well as damage-associated molecular patterns. The activation of the inflammasome is accompanied by pyroptosis-mediated cell death, which causes burst secretion of IL-1β and IL-18 (1, 4).
Hyperactivation of inflammasomes causes several types of diseases called autoinflammatory disorders (5–8). NLRP3 mutations were initially reported in cryopyrin-associated periodic fever syndrome (CAPS), which includes familial cold autoinflammatory syndrome, Muckle-Wells syndrome and neonatal-onset multisystem inflammatory disease (7). The NLRC4 inflammasome is activated by flagellin and two components of the type III secretion system, the rod and needle proteins, which directly interact with NAIP proteins in a receptor–ligand fashion (9–14). Once the NAIP protein binds its specific ligand, it can bind to NLRC4, leading to NLRC4 oligomerization (13, 14). Hyperactivation of NLRC4 by genetic mutation causes autoinflammatory disorders characterized by CAPS, enterocolitis, or macrophage activation syndrome (15–18), which we hereafter call NLRC4-dysregulated diseases. Patients with mutations in NLRC4 exhibit increases in IL-18 and IL-1β compared to patients with diseases associated with mutations in NLRP3. Indeed, blockade of IL-18 by IL-18BP ameliorates disease severity in patients with NLRC4 mutations (19, 20).
Here, we assessed the contributions of IL-1β and IL-18 to NLRC4-dysregulated disorders. Mice that harbor a hyperactive Nlrc4 gene spontaneously develop inflammation in the skin, liver and bone with an increased number of neutrophils in the spleen (15). As reported here, deletion of the Il1b gene partially ameliorated bone marrow inflammation and reduced serum IL-18. The deletion of Il18 completely abolished bone marrow inflammation. In contrast, Il1b but not Il18 deficiency abolished tail bone deformity and reduced bone density. These data demonstrate the distinct roles of IL-1β and IL-18 in NLRC4-dysregulated diseases and reveal that blockade of both IL-18 and IL-1β is needed to completely suppress inflammation.
Materials and Methods
Mice
The Nlrc4-H443P-Tg mice, Il1b-deficient mice, and Il18-deficient mice have been previously reported (15, 21, 22). All mice were maintained under specific pathogen-free conditions in the animal facilities at Tokushima University, Japan. All experiments were performed in accordance with institutional guidelines and the animal care research committee at Tokushima University.
ELISA
Cytokines in the serum were analyzed using a Mouse G-CSF Quantikine ELISA kit (catalog #MCS00) and mouse IL-18/IL-1F4 ELISA kit (catalog #7625) according to the manufacturer’s instructions (R&D Systems, MN, USA).
Flow Cytometry
Spleen single-cell suspensions were obtained and treated with RBC lysis buffer. Cells were then incubated with rat anti-mouse CD16/CD32 Ab followed by mAbs specific for extracellular markers. Fluorochrome-conjugated monoclonal antibodies specific for mouse TCRβ (H57-597), CD19 (1D3), CD11b (M1/70), and Gr-1 (RB6-8C5) were purchased from BioLegend (San Diego, CA, USA). Neutrophils were identified as CD11b+Gr-1high cells. Data were collected on a FACS Canto II (BD Biosciences) flow cytometer and analyzed using FACS Diva (BD Biosciences) or FlowJo (Tree Star, OR, USA) software.
Histology
Ear skin samples were collected and fixed in 10% formalin solution. Bone tissues were fixed in 10% formalin and then demineralized in 10% EDTA. The samples were sectioned and stained with H&E and evaluated with respect to cell influx and edema (0, no influx or edema; 1, mild influx and edema; 2, moderate influx and edema; and 3, severe influx and edema) and bone marrow hyperplasia and bone deformity (0, no hyperplasia or deformity; 1, mild hyperplasia and deformity; 2, moderate hyperplasia and deformity; and 3, severe hyperplasia and deformity) by semiquantitative examination.
Computed Tomography
The left foot and tail were analyzed in vivo with microcomputerized tomography at high resolution. The bone density of lower limbs was calculated using Latheta software (Latheta LCT-200, Hitachi Aloka Medical, Tokyo, Japan) 3D morphology reconstruction was performed using 3D Slicer (version 4.10.2). The 3D sections of paw and tail evaluated with respect to bone deformity (0, no deformity, 1, mild; 2, moderate; and 3, severe deformity) by semiquantitative examination.
Statistical Analysis
For all experiments, the significant differences between groups were calculated using Student’s t-test for unpaired data or one-way ANOVA. Differences were considered significant when p < 0.05.
Results
Both IL-1β and IL-18 Are Required for Inflammation in Nlrc4-H443P-Tg Mice
To assess the involvement of IL-1β and IL-18 in Nlrc4-H443P-Tg mice, we crossed Nlrc4-H443P-Tg mice with Il1b- or Il18-deficient (Nlrc4-H443P-Tg/Il1b−/− or Nlrc4-H443P-Tg/Il18−/−, respectively) mice or with mice deficient in both genes (Nlrc4-H443P-Tg/Il1b−/−Il18−/−). We have previously reported that Nlrc4-H443P-Tg mice have an increased number of CD11b+Gr-1high neutrophils in the spleen (15). We first evaluated the number of CD11b+Gr-1high neutrophils in the spleens of Nlrc4-H443P-Tg/Il1b−/−, Nlrc4-H443P-Tg/Il18−/−, and Nlrc4-H443P-Tg/Il1b−/−Il18−/− mice at 8 weeks ( Figure 1A ). The number of CD11b+Gr-1high neutrophils at 8 weeks was comparable among control, Nlrc4-H443P-Tg/Il1b−/−, Nlrc4-H443P-Tg/Il18−/−, and Nlrc4-H443P-Tg/Il1b−/−Il18−/− mice ( Figure 1A ). An increase in CD11b+Gr-1high neutrophils was detected in Nlrc4-H443P-Tg mice at 20 weeks ( Figure 1B ). The number of CD11b+Gr-1high neutrophils was reduced in both Nlrc4-H443P-Tg/Il1b−/− and Nlrc4-H443P-Tg/Il18−/− mice, and the number in Nlrc4-H443P-Tg/Il1b−/−Il18−/− mice was almost equivalent to that in wild-type mice ( Figure 1B ). The number of T cells and B cells are also increased in Nlrc4-H443P-Tg mice, which was reduced in the absence of IL-1β and IL-18 ( Figure 1B ). In further experiments, to analyze the effect of IL-1β and IL-18, we used mice at the age of 20 weeks.
Figure 1.

IL-1β– and Il-18–mediated inflammation in Nlrc4-H443P-Tg mice. (A) Total neutrophil numbers in the spleens of control, Nlrc4-H443P-Tg, Nlrc4-H443P-Tg/Il1b−/−, Nlrc4-H443P-Tg/Il18−/−, and Nlrc4-H443P-Tg/Il1b−/−Il18−/− mice at the age of 8 and weeks were determined (N = 8 in each group). The neutrophils were defined as CD11b+Gr-1high cells. The data are shown as the mean ± SD. *P < 0.05; **P < 0.01. (B) Total neutrophil, T cells or B cells numbers in the spleens of control, Nlrc4-H443P-Tg, Nlrc4-H443P-Tg/Il1b−/−, Nlrc4-H443P-Tg/Il18−/−, and Nlrc4-H443P-Tg/Il1b−/−Il18−/− mice at the age of 20 weeks were determined (N = 8 in each group). The data are shown as the mean ± SD. *P < 0.05; **P < 0.01. Serum (C) IL-18 was measured in control, Nlrc4-H443P-Tg, Nlrc4-H443P-Tg/Il1b−/− and Nlrc4-H443P-Tg/Il18−/− mice at the age of 20 weeks (N = 5 in each group), and serum (D) G-CSF was measured in control, Nlrc4-H443P-Tg, Nlrc4-H443P-Tg/Il1b−/−, Nlrc4-H443P-Tg/Il18−/−, and Nlrc4-H443P-Tg/Il1b−/−Il18−/− mice at the age of 20 weeks (N = 5 in each group). Data are shown as the mean ± SD. **P < 0.01.
We next tested the serum levels of cytokines in mice at the age of 20 weeks. IL-18 was increased in the Nlrc4-H443P-Tg mice, and Il1b deficiency reduced the level ( Figure 1C ), indicating that the increase in IL-18 is partially dependent on IL-1β. The serum G-CSF level was also increased in the Nlrc4-H443P-Tg mice, and deficiency in either Il1b or Il18 reduced the level, whereas deletion of both led to a level equivalent to that in control mice ( Figure 1D ).
Blockade of IL-1β or IL-18 Ameliorates Inflammation in Nlrc4-H443P-Tg Mice
We next analyzed the histology of ear skin, joint and bone marrow samples from control, Nlrc4-H443P-Tg, Nlrc4-H443P-Tg/Il1b−/−, Nlrc4-H443P-Tg/Il18−/−, and Nlrc4-H443P-Tg/Il1b−/−Il18−/− mice ( Figures 2A, B ). The Nlrc4-H443P-Tg mice showed massive infiltration of immune cells in the ear skin, joint and bone marrow samples. The skin in Nlrc4-H443P-Tg mice was thickened because of edema compared with that in wild-type mice. Deletion of either Il1b or Il18 ameliorated cell infiltration in the skin and joints ( Figures 2A, B ). In contrast, deletion of Il18 almost completely abolished hyperplasia in the bone marrow, while mice deficient in Il1b still exhibited moderate hyperplasia in the bone marrow. Deletion of both Il1b and Il18 completely abolished inflammation in any tissues ( Figures 2A, B ). These data indicate that both IL-1β and IL-18 are responsible for inflammation in Nlrc4-H443P-Tg mice and that IL-18 has a greater impact on bone marrow inflammation than IL-1β.
Figure 2.
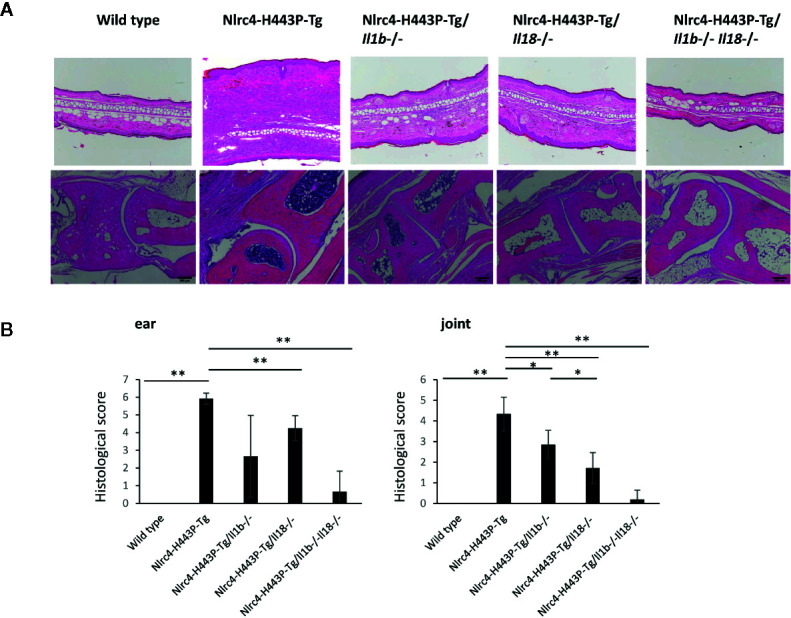
IL-1β– and Il-18–mediated inflammation in the skin and joints of Nlrc4-H443P-Tg mice. (A) Histological sections of ear skin and ankle joints in control, Nlrc4-H443P-Tg, Nlrc4-H443P-Tg/Il1b−/−, Nlrc4-H443P-Tg/Il18−/−, and Nlrc4-H443P-Tg/Il1b−/−Il18−/− mice at the age of 20 weeks were stained with hematoxylin and eosin. (B) The histological scores were evaluated in the ear skin and ankle joints in in control, Nlrc4-H443P-Tg, Nlrc4-H443P-Tg/Il1b−/−, Nlrc4-H443P-Tg/Il18−/−, and Nlrc4-H443P-Tg/Il1b−/−Il18−/− mice at the age of 20 weeks (N = 8 in each group). Data are shown as the mean ± SD. *P < 0.05, **P < 0.01.
Blockade of IL-1β Is More Effective Than That of IL-18 in Treating Bone Erosion in Nlrc4-H443P-Tg Mice
We assessed the shapes of bones in the left foot and tail using μCT analysis to evaluate the effect of IL-1β or IL18 on the bone phenotype of Nlrc4-H443P-Tg mice ( Figure 3 ). Bone deformity was also observed in vertebrae (data not shown). Nlrc4-H443P-Tg mice developed severe deformities of the left foot and tail with narrowing of the joint cavity. Deficiency in Il1b or Il18 completely diminished the deformity in the foot bones and joints. In contrast, mice deficient in Il18 showed mild deformity in the tail bones, while Il1b-deficient mice exhibited no tail bone deformity ( Figure 3 ).
Figure 3.

IL-1β– and Il-18–mediated bone deformity in Nlrc4-H443P-Tg mice. The tail and left foot in control, Nlrc4-H443P-Tg, Nlrc4-H443P-Tg/Il1b−/−, Nlrc4-H443P-Tg/Il18−/−, and Nlrc4-H443P-Tg/Il1b−/−Il18−/− mice at the age of 20 weeks were evaluated with μCT. Red arrows, bone deformity regions (N = 5 in each group). The scores were evaluated as shown in the Materials and Methods in control, Nlrc4-H443P-Tg, Nlrc4-H443P-Tg/Il1b−/−, Nlrc4-H443P-Tg/Il18−/−, and Nlrc4-H443P-Tg/Il1b−/−Il18−/− mice at the age of 20 weeks (N = 8 in each group). Data are shown as the mean ± SD. **P < 0.01.
Bone density was also decreased in Nlrc4-H443P-Tg mice ( Figure 4 ). Il1b deficiency completely rescued the reduced bone density in Nlrc4-H443P-Tg mice, while Il18 deficiency partially reversed this reduction ( Figure 4 ). These data suggest that IL-1β has more important roles in bone phenotypes than IL-18 in NLRC4-dysregulated diseases.
Figure 4.

Rescue of decreased bone density by deficiency of Il1b or Il18 in Nlrc4-H443P-Tg mice. Bone density in control, Nlrc4-H443P-Tg, Nlrc4-H443P-Tg/Il1b−/−, Nlrc4-H443P-Tg/Il18−/−, and Nlrc4-H443P-Tg/Il1b−/−Il18−/− mice at the age of 20 weeks was evaluated with μCT (N = 10 in each group). The data are shown as the mean ± SD. *P < 0.05, **P < 0.01.
Discussion
Heterozygous mutations in NLRC4 cause several types of autoinflammatory disorders. For instance, NLRC4 mutations cause CAPS-like syndrome, including cold-induced autoinflammatory syndrome and NOMID (15, 18). The clinical phenotypes of CAPS-like syndrome caused by NLRC4 mutations are indistinguishable from those caused by NLRP3 mutations. NLRC4 mutations also cause infantile enteritis and macrophage activation syndrome (16, 17). One of the characteristics of NLRC4-dysregulated diseases is high serum IL-18, and IL-18 blockade is effective in treating macrophage activation syndrome caused by NLRC4 mutation (19, 20). However, the roles of IL-18 and IL-1β in NLRC4-dysregulated diseases are not fully understood, and thus, we tested the roles of each cytokine by using mice in which a mutant Nlrc4 is expressed under the MHC class II promoter in combination with Il1b and Il18 deficiency. Our data demonstrated the distinct roles of IL-1β and IL-18 in inflammation in the joints and skin, suggesting that the efficacy of blockers of each cytokine depends on the regions of inflammation.
Blockade of IL-1β and Il-18 is effective for ameliorating inflammation in Nlrc4-H443P-Tg mice, although the contribution of each cytokine to inflammation is distinct among tissues. Induced deficiency of Il18 is more effective than that of Il1b in suppressing bone marrow hyperplasia, and Il1b deficiency better inhibited bone deformity and decreases in bone density than Il18 deficiency. One possibility to explain the distinct effects of these cytokines in tissues would be differences in the cell types that respond to IL-1β or IL-18. In any case, our data suggest that the blockade of both IL-1β and IL-18 is required to completely suppress inflammation in NLRC4-dysregulated diseases. In addition, Nlrc4-H443P-Tg mice with deletion of both Il1b and Il18 do not show any inflammation or any increase in neutrophils, suggesting a small contribution of pyroptosis-mediated cell death and cell death-associated damage-associated molecular patterns to inflammation in NLRC4-dysregulated diseases.
NOMID patients exhibit bone deformity accompanied by joint inflammation (23, 24). Nlrc4-H443P-Tg mice also show severe bone deformity at the age of 20 weeks. Bone deformity is caused by IL-1β and IL-18, but blockade of IL-18 is not enough to suppress the deformity. On the other hand, recent studies have indicated that the blockade of IL-18 is effective in treating macrophage activation syndrome caused by NLRC4 mutation that is refractory to blockade of IL-1β. Our data suggest that bone phenotypes in NLRC4-associated NOMID might be treated with a combination of blockers of IL-1β and IL-18. Regarding the contribution of IL-1β and IL-18 to bone deformity, a previous study demonstrated that IL-18 upregulates membrane-bound receptor activator of nuclear factor kappa B ligand (RANKL) expression and soluble RANKL production, thus increasing the ratio of RANKL/osteoprotegerin, suggesting an effect of IL-18 on the induction of osteoclast formation and bone resorption (25). IL-1β is involved in osteoclast differentiation in part through the induction of TRAF-6 downstream of the IL-1β pathway (26). Further studies are required to clarify not only the molecular mechanism of IL-1β-mediated bone deformity but also the roles of IL-18 in this context.
Several gene-modified mice were reported to investigate the molecular mechanisms of autoinflammatory disorders including CAPS. Pyrin-knock-in mice harboring mutant human B30.2 domains exhibited spontaneous bone marrow-dependent inflammation similar to that seen in human familial Mediterranean fever (27). This inflammation was completely abrogated in the absence of the IL-1 receptor or the adaptor molecule ASC. Nlrp3-knock-in mice demonstrated early mortality mediated by myeloid cells, which was only partially dependent on IL-1β (28). Deletion of Il18r in Nlrp3-knock-in mice resulted in partial phenotypic rescue in skin and visceral disease and reduced serum cytokines (29). Another strain of Nlrp3-knock-in mice exhibited skin inflammation with neutrophil infiltration and a Th17 cytokine-dominant response, which was suppressed in the absence of the IL-1 receptor (30). The comparison of our Nlrc4-H443P-Tg mice with the pyrin- or Nlrp3-knock-in mice side by side would be interesting to understand the effect of each cytokine on organ pathology induced by NLRC4 mutations.
In summary, our data demonstrate the crucial contributions of IL-1β and IL-18 to NLRC4-dysregulated diseases but reveal that the two cytokines have distinct roles depending on the tissue. These data suggest that blockers of IL-1β and IL-18 should be utilized depending on the site of tissue inflammation in NLRC4-dysregulated diseases.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by animal care research committee at Tokushima University.
Author Contributions
YS and KY designed the studies. YS, KO, SI T, and HA analyzed the data. YS and KY wrote the paper. KY supervised all studies. All authors contributed to the article and approved the submitted version.
Funding
The research is supported by The Research Cluster Program on Immunological Diseases, Tokushima University
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank C. Kinouchi, K. Takahashi, and A. Kitamura for their technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.591713/full#supplementary-material
Gating strategy for detecting neutrophils. Total spleen cells were stained with 7AAD, anti-CD11b and Gr-1 antibodies. In the singlet and 7AAD negative populations, CD11b+Gr-1high cells were defined as neutrophils.
References
- 1. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol (2009) 27:229–65. 10.1146/annurev.immunol.021908.132715 [DOI] [PubMed] [Google Scholar]
- 2. Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol (2010) 10:826–37. 10.1038/nri2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16:407–20. 10.1038/nri.2016.58 [DOI] [PubMed] [Google Scholar]
- 4. Kesavardhana S, Malireddi RKS, Kanneganti TD. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu Rev Immunol (2020) 38:567–95. 10.1146/annurev-immunol-073119-095439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aksentijevich I, Kastner DL. Genetics of monogenic autoinflammatory diseases: past successes, future challenges. Nat Rev Rheumatol (2011) 7:469–78. 10.1038/nrrheum.2011.94 [DOI] [PubMed] [Google Scholar]
- 6. de Jesus AA, Canna SW, Liu Y, Goldbach-Mansky R. Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu Rev Immunol (2015) 33:823–74. 10.1146/annurev-immunol-032414-112227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Manthiram K, Zhou Q, Aksentijevich I, Kastner DL. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat Immunol (2017) 18:832–42. 10.1038/ni.3777 [DOI] [PubMed] [Google Scholar]
- 8. Gattorno M, Hofer M, Federici S, Vanoni F, Bovis F, Aksentijevich I, et al. Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis (2019) 78:1025–32. 10.1136/annrheumdis-2019-215048 [DOI] [PubMed] [Google Scholar]
- 9. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature (2011) 477:592–5. 10.1038/nature10394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature (2011) 477:596–600. 10.1038/nature10510 [DOI] [PubMed] [Google Scholar]
- 11. Yang J, Zhao Y, Shi J, Shao F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci U.S.A. (2013) 110:14408–13. 10.1073/pnas.1306376110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao Y, Shi J, Shi X, Wang Y, Wang F, Shao F. Genetic functions of the NAIP family of inflammasome receptors for bacterial ligands in mice. J Exp Med (2016) 213:647–56. 10.1084/jem.20160006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fusco WG, Duncan JA. Novel aspects of the assembly and activation of inflammasomes with focus on the NLRC4 inflammasome. Int Immunol (2018) 30:183–93. 10.1093/intimm/dxy009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Duncan JA, Canna SW. The NLRC4 Inflammasome. Immunol Rev (2018) 281:115–23. 10.1111/imr.12607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kitamura A, Sasaki Y, Abe T, Kano H, Yasutomo K. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J Exp Med (2014) 211:2385–96. 10.1084/jem.20141091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet (2014) 46:1140–6. 10.1038/ng.3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Romberg N, Al Moussawi K, Nelson-Williams C, Stiegler AL, Loring E, Choi M, et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet (2014) 46:1135–9. 10.1038/ng.3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kawasaki Y, Oda H, Ito J, Niwa A, Tanaka T, Hijikata A, et al. Identification of a High-Frequency Somatic NLRC4 Mutation as a Cause of Autoinflammation by Pluripotent Cell-Based Phenotype Dissection. Arthritis Rheumatol (2017) 69:447–59. 10.1002/art.39960 [DOI] [PubMed] [Google Scholar]
- 19. Novick D, Dinarello CA. IL-18 binding protein reverses the life-threatening hyperinflammation of a baby with the NLRC4 mutation. J Allergy Clin Immunol (2017) 140:316. 10.1016/j.jaci.2017.02.037 [DOI] [PubMed] [Google Scholar]
- 20. Canna SW, Girard C, Malle L, de Jesus A, Romberg N, Kelsen J, et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J Allergy Clin Immunol (2017) 139:1698–701. 10.1016/j.jaci.2016.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Horai R, Asano M, Sudo K, Kanuka H, Suzuki M, Nishihara M, et al. Production of mice deficient in genes for interleukin (IL)-1alpha, IL-1beta, IL-1alpha/beta, and IL-1 receptor antagonist shows that IL-1beta is crucial in turpentine-induced fever development and glucocorticoid secretion. J Exp Med (1998) 187:1463–75. 10.1084/jem.187.9.1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takeda K, Tsutsui H, Yoshimoto T, Adachi O, Yoshida N, Kishimoto T, et al. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity (1998) 8:383–90. 10.1016/s1074-7613(00)80543-9 [DOI] [PubMed] [Google Scholar]
- 23. Ozen S, Bilginer Y. A clinical guide to autoinflammatory diseases: familial Mediterranean fever and next-of-kin. Nat Rev Rheumatol (2014) 10:135–47. 10.1038/nrrheum.2013.174 [DOI] [PubMed] [Google Scholar]
- 24. Kuemmerle-Deschner JB, Ozen S, Tyrrell PN, Kone-Paut I, Goldbach-Mansky R, Lachmann H, et al. Diagnostic criteria for cryopyrin-associated periodic syndrome (CAPS). Ann Rheum Dis (2017) 76:942–7. 10.1136/annrheumdis-2016-209686 [DOI] [PubMed] [Google Scholar]
- 25. Zhang W, Cong XL, Qin YH, He ZW, He DY. Dai S.M. IL-18 upregulates the production of key regulators of osteoclastogenesis from fibroblast-like synoviocytes in rheumatoid arthritis. Inflammation (2013) 36:103–9. 10.1007/s10753-012-9524-8 [DOI] [PubMed] [Google Scholar]
- 26. Choe JY, Park KY, Kim SK. Monosodium Urate in the Presence of RANKL Promotes Osteoclast Formation through Activation of c-Jun N-Terminal Kinase. Mediators Inflammation (2015) 2015:597512. 10.1155/2015/597512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chae JJ, Cho YH, Lee GS, Cheng J, Liu PP, Feigenbaum L, et al. Gain-of-function Pyrin mutations induce NLRP3 protein-independent interleukin-1beta activation and severe autoinflammation in mice. Immunity (2011) 34:755–68. 10.1016/j.immuni.2011.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brydges SD, Mueller JL, McGeough MD, Pena CA, Misaghi A, Gandhi C, et al. Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity (2009) 30:875–87. 10.1016/j.immuni.2009.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brydges SD, Broderick L, McGeough MD, Pena CA, Mueller JL, Hoffman HM. Divergence of IL-1, IL-18, and cell death in NLRP3 inflammasomopathies. J Clin Invest (2013) 123:4695–705. 10.1172/JCI71543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Meng G, Zhang F, Fuss I, Kitani A, Strober W. A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity (2009) 30:860–74. 10.1016/j.immuni.2009.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gating strategy for detecting neutrophils. Total spleen cells were stained with 7AAD, anti-CD11b and Gr-1 antibodies. In the singlet and 7AAD negative populations, CD11b+Gr-1high cells were defined as neutrophils.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.