

Abstract
Ciliary microtubules are subject to post-translational modifications that act as a “Tubulin Code” to regulate motor traffic, binding proteins and stability. In humans, loss of CCP1, a cytosolic carboxypeptidase and tubulin deglutamylating enzyme, causes infantile-onset neurodegeneration. In C. elegans, mutations in ccpp-1, the homolog of CCP1, result in progressive degeneration of neuronal cilia and loss of neuronal function. To identify genes that regulate microtubule glutamylation and ciliary integrity, we performed a forward genetic screen for suppressors of ciliary degeneration in ccpp-1 mutants. We isolated the ttll-5(my38) suppressor, a mutation in a tubulin tyrosine ligase-like glutamylase gene. We show that mutation in the ttll-4, ttll-5, or ttll-11 gene suppressed the hyperglutamylation-induced loss of ciliary dye filling and kinesin-2 mislocalization in ccpp-1 cilia. We also identified the nekl-4(my31) suppressor, an allele affecting the NIMA (Never in Mitosis A)-related kinase NEKL-4/NEK10. In humans, NEK10 mutation causes bronchiectasis, an airway and mucociliary transport disorder caused by defective motile cilia. C. elegans NEKL-4 localizes to the ciliary base but does not localize to cilia, suggesting an indirect role in ciliary processes. This work defines a pathway in which glutamylation, a component of the Tubulin Code, is written by TTLL-4, TTLL-5, and TTLL-11; is erased by CCPP-1; is read by ciliary kinesins; and its downstream effects are modulated by NEKL-4 activity. Identification of regulators of microtubule glutamylation in diverse cellular contexts is important to the development of effective therapies for disorders characterized by changes in microtubule glutamylation. By identifying C. elegans genes important for neuronal and ciliary stability, our work may inform research into the roles of the tubulin code in human ciliopathies and neurodegenerative diseases.
Author summary
Cilia are microtubule-based organelles that play essential roles in human development and health. Ciliopathies are caused by abnormalities in the structure or function of primary cilia, with polycystic kidney disease (PKD) being a common clinical phenotype. As cilia are found on most non-dividing cells in the human body, ciliopathies often display extrarenal manifestations including neurological disorders and retinal degeneration. The Tubulin Code–combinatorial use of tubulin isotypes and post-translational modifications–dictates ciliary structure, motor-based transport, and function. Mutation in the tubulin deglutamylase ccpp-1 (cytosolic carboxypeptidase) results in ciliary hyperglutamylation and degeneration. C. elegans ccpp-1 ciliary degeneration is suppressed by a mutation in any of three TTLL (tubulin tyrosine ligase-like) glutamylase genes, indicating that regulated glutamylation is critically important for ciliary homeostasis. Pathological hyperglutamylation caused by CCP deglutamylase mutations are associated with human retinal degeneration and murine progressive neurodegeneration and sperm immotility. ccpp-1 ciliary degeneration is also suppressed by a mutation in the kinase NEKL-4/NEK10. NEK kinases are implicated in polycystic kidney disease and other ciliopathies and NEKL-4/NEK10 is important for ciliary stability in C. elegans. By identifying C. elegans genes important for neuronal and ciliary stability, “the worm” may inform research into human ciliopathies and neurodegenerative diseases.
Introduction
Cilia and flagella are antenna-like organelles that are conserved from algae to humans and play important roles in sensation, intercellular communication, development, and homeostasis [1]. The core of both cilia and flagella is the axoneme, a conserved microtubule structure with a ring of nine doublets that may surround inner microtubule singlets in motile cilia [1]. The Tubulin Code hypothesis suggests that post-translational modifications, such as glutamylation, acetylation, glycylation, detyrosination, and others, can increase the diversity of microtubules beyond incorporation of different tubulin isotypes for structural and functional specialization [2].
Glutamylation, or addition of side-chains of the amino acid glutamate, of tubulin tails is emerging as an important factor in organizing the microtubule cytoskeleton [3,4]. Microtubules in neurons and cilia can be reversibly glutamylated. Although the function of glutamylation and other post-translational modifications is not completely understood, enzymes that regulate post-translational glutamylation of microtubules are known. Cytosolic carboxypeptidases (CCPs) such as CCP1 deglutamylate microtubules [5,6], opposing the activity of tubulin tyrosine ligase-like (TTLL) enzymes, which add glutamates to tubulin tails [7–9]. Glutamylation side-chains can be as short as a single glutamate, or as long as 20 glutamates [10]. Particular CCP enzymes may reduce the length of glutamate side-chains, and some can remove the branch point glutamate to eliminate glutamylation [5]. Similarly, some TTLL family members initiate glutamate side-chains, while others elongate [9]. TTLLs may also have preferences for α or β tubulin substrates [9]. Therefore, microtubule glutamylation is not uniform, and both side-chain length and tubulin substrate may exert different influences over motors, microtubule-associated proteins (MAPs), or other regulators of microtubule stability.
Glutamylation is essential for neuronal survival and axoneme stability, and a growing list of reports link hyper- or hypo-glutamylation to disease. Dysregulated microtubule glutamylation causes infantile-onset neurodegeneration and the ciliopathy Joubert syndrome in humans, and progressive cerebellar purkinje cell degeneration (pcd) in mice [3,5,11–14]. Defects in axonal transport, which may be regulated by the Tubulin Code, contribute to neurodegenerative diseases including Alzheimer’s disease and Parkinson’s disease [4,15]. Although the importance of regulated glutamylation on ciliary and neuronal microtubules are clearly established, a comprehensive understanding of microtubule glutamylation is necessary to understand and interpret its context-specific effects in different cilia and neuron types.
The nematode C. elegans offers advantages to unraveling the Tubulin Code. Most basic biological processes, including axon development, neuronal transport, and ciliogenesis are conserved between C. elegans and humans [16]. In the worm, cilia are located on the distal most ends of sensory dendrites [17–19], are not essential for viability, and are diverse in terms of the composition and arrangement of microtubules, tubulin post-translational modifications, and motor proteins [2,18,20]. In male-specific ciliated sensory neurons, the CCPP-1 deglutamylase and TTLL-11 glutamylase control ciliary ultrastructure and particular ciliary kinesin motors [21,22]. In amphid and phasmid sensory neurons, loss of CCPP-1 leads to degeneration of ciliary microtubules indicating cell-specific differences in the tubulin code [21,22].
In C. elegans amphid and phasmid neurons, ciliary integrity is easily tested using a dye-filling assay [18]. ccpp-1 mutants are dye-filling defective (Dyf) in adults but not early larval stages, reflecting ultrastructural defects and progressive ciliary microtubule degeneration [21]. Using a candidate gene approach, we found that deletion mutations in ttll-4, ttll-5, or ttll-11 glutamylase genes suppressed the ccpp-1 Dyf phenotype. To identify new molecules and pathways that function in ciliary homeostasis, we performed a genetic screen for mutations that suppress the ccpp-1 Dyf phenotype. We identified mutations in ttll-5 and in the never-in-mitosis kinase gene nekl-4. In humans, TTLL5 mutation causes recessive retinal dystrophy [23,24]. C. elegans NEKL-4 is homologous to human NEK10, which is a kinase specific to ciliated cells and contributes to ciliogenesis in cultured human kidney cells [25]. In humans, NEK10 mutation causes bronchiectasis, a disorder of mucociliary transport in the airway due to defective motile cilia [26]. Here we show that NEKL-4 is expressed in all ciliated neurons but does not localize to cilia, suggesting that NEKL-4 indirectly influences regulation of ciliary stability. This work represents a step toward elucidating the molecular pathways by which glutamylation, as a component of the Tubulin Code, regulates cilia; this, in turn, allows us to better understand ciliopathies and neuronal survival in humans.
Results
Single gene mutations in glutamylases ttll-4, ttll-5, and ttll-11 suppress the ccpp-1 dye-filling defect
ccpp-1 mutation causes hyperglutamylation and progressive ciliary degeneration in amphid and phasmid neuronal cilia ([21]; Fig 1A). Ciliary degeneration is detectable using dye-filling assays, in which C. elegans nematodes are soaked in the fluorescent lipophilic dye DiI (Fig 1B). The dye is only taken up in neurons that have intact sensory cilia; therefore, adult ccpp-1 do not take up this fluorescent dye and display the Dye-filling defective, or “Dyf,” phenotype ([21]; Fig 1C).
Fig 1. Overview of Dye-filling assays and examples of observed phenotypes.

a. Diagram of a C. elegans hermaphrodite highlighting the amphid and phasmid sensory neurons, which absorb dye through their cilia. Inset shows the amphid channel cilia (adapted from O’Hagan et al [22]). b. Illustration of the dye-filling assays. DiI is a lipophilic fluorescent dye that is taken up by the cilia. c. Examples of non-Dyf, partial Dyf, and Dyf phenotypes. Scale = 10μm.
We first tested candidate genes for suppression of the ccpp-1 Dyf phenotype. Glutamylases of the tubulin tyrosine ligase-like TTLL family oppose CCP deglutamylase function [27]. The C. elegans genome encodes five TTLL glutamylases—ttll-4, ttll-5, ttll-9, ttll-11, and ttll-15 [6,28]. We previously showed that ttll-4 mutation suppresses the ccpp-1 Dyf defect, suggesting that TTLL-4 glutamylates microtubules in amphid and phasmid cilia [21]. We therefore tested deletion alleles in the other ttll genes and found that deletion mutations in ttll-5 and ttll-11 suppressed the Dyf defect of ccpp-1 (Fig 2A). The ttll-4 gene encodes two protein isoforms by alternative splicing, TTLL-4A (601 AA) and TTLL-4B (563 AA) (Fig 2B; [29]). Both isoforms contain a TTL domain (amino acid residues 138–476 in TTLL-4A; 100–438 in TTLL-4B) that is conserved among other TTLL genes. To study ttll-4, we used the tm3310 allele, containing a deletion of 137 AAs of the TTL domain (S2 Table), which is predicted to reduce or eliminate glutamylase function. As previously shown [21], ttll-4(tm3310) strongly suppressed the ccpp-1 Dyf phenotype (Fig 2A).
Fig 2. ttll-4, ttll-5 and ttll-11 act as suppressors of the ccpp-1 Dyf phenotype.

a. Suppression of ccpp-1 Dyf phenotype by ttll-4, ttll-5 and ttll-11. All strains marked with Δ are deletion alleles. ttll-11bΔ refers to the ttll-11b(gk482) allele. N of animals per strain is indicated on bars. *** indicates p ≤ 0.0001 by Kruskal-Wallis one-way ANOVA analysis and post hoc Dunn’s multiple comparison test. Black indicates significance relative to wild type and red indicates significance relative to ccpp-1Δ. b. Gene diagrams of ttll-4, ttll-5 and ttll-11, aligned by TTL domain, including alleles used and important features.
The ttll-5 gene also encodes two protein isoforms, TTLL-5A (677 AA) and TTLL-5B (730 AA) (Fig 2B; [29]). The TTL domain in TTLL-5 spans AA residues 67–425 in TTLL-5a and 120–478 in TTLL-5b. We characterized the recessive deletion allele tm3360, in which 49 AA are deleted, including 33 residues that align with the extended TTLL domain of mammalian TTLL5 [9,30]. Although this deletion is predicted to be in-frame, the large missing region of the TTLL domain might abolish or reduce substrate binding and glutamylase function [A. Roll-Mecak, personal communication]. ttll-5(tm3360) also strongly suppressed the ccpp-1 Dyf phenotype in both amphids and phasmids (Fig 2A).
TTLL-11 also has two isoforms, TTLL-11A (607 AA) and TTLL-11B (707 AA) (Fig 2B; [29]). The TTL domain spans residues 24–388 in TTLL-11A and 124–488 in TTLL-11B. The ttll-11(tm4059) allele encodes an in-frame deletion in the TTL domain and is expected to disrupt function of both isoforms [22]. Like ttll-4 and ttll-5 mutations, ttll-11(tm4059) suppressed the ccpp-1 Dyf phenotype in amphids and phasmids (Fig 2A). In contrast, ttll-11b(gk482), which deletes coding sequence from only the long B isoform, did not suppress ccpp-1. We cannot completely rule out the possibility that gk482 reduces expression of both TTLL-11A and TTLL-11B. However, transcriptional GFP reporters suggest that TTLL-11B is expressed in extracellular vesicle-releasing neurons (EVNs) and not amphids [31], whereas TTLL-11A may be expressed in amphids [22]. These data suggest that TTLL-11A opposes the activity of CCPP-1 in amphid and phasmid neurons.
We also tested candidate genes that are not predicted to act directly in opposition to CCPP-1 deglutamylase activity for a role in the suppression of ciliary degeneration due to loss of CCPP-1 function. Because spas-1/Spastin and mei-1/Katanin are implicated in the severing of glutamylated microtubules [32,33], we tested whether mutations in these genes altered ccpp-1 hyperglutamylation-induced ciliary degeneration. Other candidates included the microtubule-depolymerizing kinesin-13 KLP-7 and the MAP kinase PMK-3, which interact with the deglutamylases CCPP-1 and CCPP-6 to regulate axon regrowth after laser-severing of C. elegans PLM touch receptor neurons [34]. Mutations in these and several other candidate genes failed to suppress the ccpp-1 Dyf phenotype (S1 Fig), suggesting that other unidentified molecules modulate ciliary degeneration due to loss of CCPP-1 loss of function.
A forward genetic screen for suppressors of the ccpp-1 dye-filling defect recovered alleles affecting the glutamylase TTLL-5 and the NIMA-related kinase NEKL-4
To identify genes that act with ccpp-1 to regulate ciliary stability, we conducted a genetic screen for suppressors of the ccpp-1(ok1821) Dyf phenotype. We mutagenized >1000 P0 ccpp-1 mutants, passaged them to homozygose recessive mutations, performed dye-filling assays on F2 adults, and screened for non-Dyf animals (Fig 3A). Mutant lines were maintained and retested for suppression of the Dyf phenotype. From >100,000 mutagenized haploid genomes, we isolated 11 viable lines that displayed suppression of the ccpp-1 Dyf phenotype when retested (S1 Table).
Fig 3. ccpp-1 suppressor screen design and ccpp-1 Dyf suppressors identified.

a. Diagram of EMS mutagenesis screen for ccpp-1 Dyf suppressors (sup). b. Outcrossed single-gene suppressors identified from mutagenesis screen and sequenced. Names of strains and alleles are indicated, as well as the genes identified through sequencing. c. Suppression of ccpp-1 Dyf phenotype by nekl-4. N of animals per strain is indicated on bars. *** indicates p ≤ 0.0001 by Kruskal-Wallis one-way ANOVA analysis and post hoc Dunn’s multiple comparison test. Black indicates significance relative to wild type and red indicates significance relative to ccpp-1Δ. d. Diagram of nekl-4 gene with alleles and important features indicated. e. Protein diagrams of NEKL-4 and its human homolog NEK10.
The my38 allele recovered in our screen suppressed the Dyf phenotype of ccpp-1 in the amphid neurons in the head (Fig 2A; Fig 3B). We identified my38 as a missense mutation in the ttll-5 glutamylase gene. my38 encodes an L338F substitution, located in the TTL domain (Fig 2B), that is predicted to affect ATP binding [A. Roll-Mecak, personal communication]. Therefore, our screen was able to identify a new allele of one of our candidate suppressor genes. This is important both as a validation of our screening method as well as a way to study the effects of different protein alterations on a phenotype.
Whole genome sequencing identified that our my31 suppressor allele encodes a nonsense mutation in exon 5 of the nekl-4 gene that produces NEKL-4(W199X) (Fig 3B; S2 Table; [29]). nekl-4(my31) moderately suppressed ccpp-1 Dyf in both amphid and phasmid neurons and was non-Dyf as a single mutant (Fig 3C). nekl-4 encodes a 981 AA putative serine-threonine kinase (Fig 3D). Prosite and InterPro scans detected a kinase domain (residues 453–725), and two armadillo (ARM) repeats (residues 200–281); ARM repeats are a series of alpha helices which can be indicative of protein-protein interactions [35]. Using EMBOSS epestfind [36], we also identified a PEST domain, important for proteolytic degradation (residues 748–764) [37]. NEKL-4 is a homolog of human NEK10, a protein required for ciliogenesis and associated with ciliopathies ([25,26]; Fig 3E). NEK10 also contains ARM repeats (residues 137–348 and 387–471), a kinase domain (residues 519–791), and a PEST domain (residues 896–921). However, the NEK10 kinase domain is flanked by coiled-coil domains and contains a putative tyrosine kinase active site not found in NEKL-4. NEKL-4 identity ranges from 29.41% (compared with NEK10 isoform 3) to 48.54% (NEK10 isoforms 5, 6 and 7). The NEK10 and NEKL-4 kinase domain is 52.50% identical by BLAST ([38]; S2 Fig).
To confirm that my31 was a mutation in the nekl-4 gene, we performed transgenic rescue experiments by germline injection of the wild-type genomic copy of nekl-4 (injected @1.5ng/μL) into ccpp-1;nekl-4(my31) mutants, and restored the ccpp-1 Dyf phenotype (Fig 3C). We also characterized the deletion allele tm4910, which removes portions of exon 6 and exon 7 and replaces part of the kinase domain, including the putative ATP-binding lysine residue, with a glutamine residue. nekl-4(tm4910) single mutants are non-Dyf. nekl-4(my31) and nekl-4(tm4910) exhibited similar suppression of ccpp-1 Dyf defects in amphid and phasmid neurons. Because both alleles were phenotypically indistinguishable and are likely loss-of-function alleles, we continued with the nekl-4(tm4910) deletion allele for ease of genotyping.
NEKL-4 is expressed in ciliated neurons but does not localize to cilia
We examined NEKL-4 expression pattern and subcellular localization using an extrachromosomal nekl-4p::nekl-4::gfp reporter transgene and endogenously tagged nekl-4::mneongreen and nekl-4::mscarlet CRISPR reporters (Fig 4A). Extrachromosomal NEKL-4::GFP, driven by an endogenous promoter of 1609 base pairs before the start codon, localized in the axons, cell bodies, and dendrites of the ciliated neurons in the head of the worm, as well as the phasmid and PQR neurons in the tail, and was excluded from nuclei (Fig 4B–4C). NEKL-4::GFP localized to the distal dendrite and the base of sensory cilia, but was not observed in the cilium. In the dendrites of some neurons, NEKL-4::GFP accumulated in patches ranging from 2–5 μm in length. Overexpression of a nekl-4p::nekl-4::gfp transgene (injected at a concentration of 13 ng/μL) resulted in a Dyf phenotype (Fig 3A), suggesting that appropriate expression level or activity of NEKL-4 is important to ciliary stability.
Fig 4. NEKL-4 is localized to ciliated neurons and is not enriched in the cilium.

a. Diagram of a C. elegans hermaphrodite. Imaged areas are boxed. b-c. Localization of NEKL-4::GFP, overexpressed by an extrachromosomal array. Scale = 10μm. d-e. Localization of NEKL-4::mNeonGreen, expressed by inserting the fluorescent tag at the 3’ end of the endogenous nekl-4 gene using CRISPR/Cas9. Scale = 10μm. f-g. Localization of NEKL-4::mScarlet to the distal dendrite with NPHP-1::CFP (transition zone) and CHE-13::YFP (axoneme) as reference points. Scale = 10μm. h. Schematic of a phasmid cilium showing NEKL-4, NPHP-1, and CHE-13 localization.
We therefore examined the expression and localization of endogenous NEKL-4 by using CRISPR/Cas9 [39,40] to engineer nekl-4::mneongreen and nekl-4::mscarlet strains (Fig 4D and 4E). These CRISPR-generated reporters use the nekl-4 locus as well as a short flexible linker, and have significantly dimmer fluorescence than the extrachromosomal nekl-4p::nekl-4::gfp array. With both extrachromosomal and CRISPR reporters, NEKL-4 was observed in the same neurons and similar subcellular locations.
With the CRISPR-generated nekl-4::mneongreen reporter, we were able to resolve fine structures in the cell bodies and dendrites. In the amphid cell bodies, NEKL-4::mNG appeared as thin filaments. A similar pattern was seen in the outer labial (OL), inner labial (IL), and cephalic (CEP) cell bodies, though the filaments appeared to be thicker and brighter. The CRISPR-generated NEKL-4::mNG reporter also localized in discrete, discontinuous patches in the dendrites. This pattern continued to the distal dendrite, terminating before entering the cilium of any neurons. We observed NEKL-4::mNG movement in the distal dendrite (Movie S1) and localization to the cell bodies and dendrites of the ray neurons in the male tail (S3 Fig). The intricate localization pattern and movement of NEKL-4::mNG suggests association with organelles or cytoskeletal components.
Because a nekl-4::gfp reporter was previously reported [41] to localize in phasmid cilia, we examined colocalization of NEKL-4::mScarlet with CHE-13/IFT57, an IFT-B component [42], and NPHP-1, a transition zone protein, to confirm NEKL-4 exclusion from the cilium. NEKL-4::mSC did not show significant overlap with NPHP-1::CFP or CHE-13::YFP, and was mainly restricted to the distal dendrite and periciliary membrane compartment (PCMC) (Fig 4F and 4G). We hypothesize that the discrepancy between our results and those of Yi et al [41] regarding NEKL-4 localization could be due to the higher injection concentration of their construct (50ng/μL vs. 13ng/μL for our extrachromosomal reporter), and the use of the dyf-1 promoter as opposed to the endogenous nekl-4 promoter that we utilized for both our extrachromosomal reporter and CRISPR markers. From our data, we conclude that NEKL-4 functions in ciliated neurons, although the protein itself does not localize to the cilium (Fig 4H).
ttll-4(tm3310) and ttll-5(tm3360) partially suppress ccpp-1 glutamylation defects. ttll-11 activity is essential for glutamylation but not ciliogenesis
To analyze the effect of our suppressor mutations on microtubule glutamylation, we conducted indirect immunofluorescence experiments. We used the monoclonal antibody GT335, which recognizes the branch point of glutamate side-chains [43]. In wild type, GT335 labels the cilia of amphid, labial (OL, IL), and cephalic (CEP) neurons (Fig 5A; S4 Fig). Labial and cephalic cilia are seen at the anterior-most tip of the animal, surrounding the buccal cavity. GT335 staining of labial and cephalic cilia appeared as singular axonemes [44]. In amphid channel cilia, GT335 specifically stains the doublet region middle segments that appear as two triangular bundles located posterior to the labial and cephalic cilia [44]. In wild type animals, GT335 strongly stained labial and cephalic cilia and amphid ciliary middle segments (Fig 5B). In the ttll-4(tm3310) mutants, we observed a reduction in the percentage of animals with amphid, labial and cephalic staining with GT335, indicating that mutations in TTLL-4 affect glutamylation (Fig 5C). In the ttll-5(my38) and ttll-5(tm3360) single mutants, we observed normal GT335 staining (Fig 5C). The ttll-11(tm4059) mutation, which affects both TTLL-11 isoforms, abolished GT335 staining, whereas ttll-11b(gk482) showed normal GT335 staining in amphid middle segments (Fig 5B and 5C).
Fig 5. Effects of mutations on glutamylation of amphid, labial and cephalic (LC) cilia.

a. Diagram of the cilia of the C. elegans nose. Red indicates areas that are strained by the GT335 antibody, indicating glutamylation (adapted from O’Hagan et al [22]). MS = amphid middle segments; LC = labial and cephalic cilia. b. Examples of GT335 antibody staining phenotypes. Scale = 10μm. c. Percentage of worms with GT335 stained amphids and/or LC cilia. ccpp-1Δ; nekl-4(my31) was rescued with a nekl-4p::nekl-4::gfp plasmid @ 1.5ng/μL. N of animals per strain is indicated.
We next examined the effect of the ttll gene suppressor mutations on GT335 staining in the ccpp-1 mutant background. We analyzed both the presence of GT335 staining and the appearance of staining to examine subtle differences in glutamylation patterns. We observed a variation in the GT335 staining pattern of some mutants: the amphids were stained but the pattern was abnormal. We classified this staining pattern as an “abnormal amphid” phenotype (Fig 5B). We observed abnormal phenotypes in ciliary glutamylation in ttll mutants and suppressor strains. In some mutants, including ccpp-1, labial and cephalic cilia stained, while amphid cilia did not, consistent with cell-specific regulation of the Tubulin Code [2,22,34,45] (Fig 5B). In ccpp-1 mutant amphid channel cilia, GT335 staining was often undetectable, and occasionally detectable but abnormal (Fig 5B). We observed some restoration of GT335 staining in ampid channel cilia in 15% of the ccpp-1;ttll-4 double mutant animals (Fig 5C). In ccpp-1;ttll-5 double mutants, we observed a higher incidence of abnormal amphid middle segments compared to the ccpp-1 single mutant (Fig 5C). In the ccpp-1;ttll-11(tm4059) double mutants, GT335 staining was undetectable (Fig 5C). ccpp-1;ttll-11b(gk482) mutants, in contrast, had some restoration of amphid and labial and cephalic GT335 staining (Fig 5C).
We also used a polyE antibody, which detects side-chains of 3 or more glutamates [5]. PolyE staining of wild-type males strongly labelled amphid middle segments and labial cilia, suggesting that microtubules in these cilia are decorated with side chains of at least 3 glutamates. Mutations in ttll-4 and ttll-5 did not detectably reduce polyE staining. The ttll-11(tm4059) mutation, however, abolished polyE staining (S5 Fig). Our results on the effect of the suppressor mutations on polyE staining in ccpp-1 mutants closely matched our observations with GT335. In ccpp-1 mutants, amphid channel cilia polyE staining was rarely observed (in 10% of animals) and more often appeared abnormal (S5 Fig). In ccpp-1;ttll-5(tm3360) and ccpp-1;ttll-11b(gk482) double mutant animals, we observed increases in the incidence of polyE stained amphid cilia (S5 Fig). We note that the absence of GT335 and polyE staining indicates an absence of glutamylation, but not necessarily an absence of MTs. Taking into account our antibody staining results, Dyf phenotype suppression, and previously published transmission electron microscopy data [21], we propose that our data indicates that loss of microtubule glutamylase activity suppresses microtubule degeneration due to loss of CCPP-1 function, and that microtubule glutamylation is not essential for dye filling or ciliogenesis. Overall, our data suggests that TTLL-11 is essential for adding the initiating “branch point” glutamate and that TTLL-4 and TTLL-5 are not essential for initiating glutamate side-chains. TTLL-4 and TTLL-5 might act primarily as elongases, but loss of their function was not sufficient to reduce the chain length to less than 3 glutamates.
nekl-4(my31), but not nekl-4(tm4910), mildly suppresses ccpp-1 glutamylation defects
We also examined whether NEKL-4 affects microtubule glutamylation. nekl-4(tm4910) did not affect GT335 staining, indicating NEKL-4 is not essential for initiating glutamate side-chains (Fig 5C). Unlike ttll-4 and ttll-5 mutations, nekl-4(tm4910) did not suppress the abnormal GT335 staining in ccpp-1. Curiously, although nekl-4(my31) and nekl-4(tm4910) phenocopied each other in ccpp-1 Dyf suppression, their effects on ccpp-1 GT335 phenotype were different, in that nekl-4(my31) showed a partial suppression of the ccpp-1 GT335 phenotype (Fig 5C). nekl-4(my31) also mildly suppressed the ccpp-1 polyE phenotype (S5 Fig). In transgenic animals, introduction of a nekl-4(+) genomic construct rescued ccpp-1;nekl-4(my31) amphid GT335 staining to appear similar to ccpp-1 alone (Fig 5C). Based on our antibody staining data, we conclude that NEKL-4 may act downstream or independent of CCPP-1-mediated deglutamylation.
ttll-5 and ttll-11 suppress heterotrimeric kinesin-II span shortening in ccpp-1 mutants
C. elegans amphid and phasmid cilia are built by the cooperative action of heterotrimeric kinesin-II and homodimeric OSM-3/KIF17 [46,47]. Heterotrimeric kinesin-II travels along the doublet-containing middle segment region, which makes up approximately half of the length of the amphid channel and phasmid cilia and correlates with GT335 staining ([46,47]; Fig 6A). We therefore examined the middle segment localization of KAP-1::GFP, the non-motor subunit of heterotrimeric kinesin-II ([46]; Fig 6B). In wild-type phasmid cilia, the span of KAP-1::GFP was approximately 4μm (Fig 6C). In osm-3(p802) mutants that lack distal singlet microtubules, we observed a similarly reduced KAP-1::GFP span of 2μm [18,20,44]. In ccpp-1 mutants, the KAP-1::GFP span was significantly reduced to approximately 2μm. We considered the following possibilities to explain the shortened KAP-1::GFP span in ccpp-1: (i) microtubule loss in ccpp-1 mutants, or (ii) altered association with OSM-3 in the ‘handover zone’, which may be needed for heterotrimeric kinesin-II to travel the appropriate distance [48]. Both of these possibilities are consistent with roles for the Tubulin Code in regulating microtubule stability and motor properties. To further understand the mechanism of suppression of the ccpp-1 Dyf phenotype, we examined KAP-1::GFP spans in suppressor mutations in wild type and ccpp-1 backgrounds. KAP-1::GFP spans were similar to wild type in ttll-4, ttll-5, ttll-11, and nekl-4(tm4910) single mutants suggesting that the single mutants alone did not not cause microtubule loss, or affect kinesin-II interactions with OSM-3 (Fig 6C). The former conclusion is consistent with a lack of Dyf phenotypes in ttll-4, ttll-5, ttll-11, and nekl-4(tm4910) mutants. Mutations in ttll-5 and ttll-11 that suppressed the Dyf phenotype of ccpp-1 also suppressed the KAP-1::GFP span shortening of ccpp-1 (Fig 6C). Importantly, suppression of KAP-1::GFP span shortening of ccpp-1 by ttll-11 confirmed the presence of ciliary MTs despite the lack of antibody staining in ccpp-1;ttll-11(tm4059) double mutants. nekl-4(tm4910) did not suppress the ccpp-1 KAP-1::GFP span shortening (Fig 6C). Our data suggests that reduction of microtubule glutamylation rescues ciliary degeneration caused due to CCPP-1 loss of function by suppressing ciliary microtubule loss, altering motor protein-IFT particle interactions, or both. Additionally, our data on nekl-4(tm4910) revealed the presence of a mechanism for ccpp-1 Dyf suppression that does not involve detectable changes in microtubule glutamylation or heterotrimeric kinesin-II location.
Fig 6. Mutations in TTLL genes suppress ccpp-1 defects in KAP-1 middle segment localization, but do not suppress distal segment defects in osm-3(p802) mutants.

a. Diagram of the C. elegans hermaphrodite phasmid cilia. MS = phasmid middle segments. b. Examples of KAP-1::GFP localization in the phasmid cilia. Arrows = individual phasmid cilia. Scale = 5μm. c. Length of KAP-1::GFP span in the phasmid neurons. Mean ± SEM. *** indicates p ≤ 0.0001 by Kruskal-Wallis one-way ANOVA analysis and post hoc Dunn’s multiple comparison test. n of cilia per strain is indicated. d. Dye-filling assay showing lack of osm-3(p802) suppression by ttll-4, ttll-5, ttll-11 or nekl-4. N of animals per strain is indicated.
In addition to heterotrimeric kinesin-II, homodimeric OSM-3/KIF17 kinesin-2 is important for ciliogenesis in amphid and phasmid neurons [18,49,50]. A previous study indicated that nekl-4 genetically interacts with osm-3 [41]. nekl-4(cas396), which affects the kinase domain, partially suppresses the Dyf phenotype and distal segment loss of osm-3(sa125) mutants [41]. The osm-3(sa125) G444E missense mutation is proposed to relieve the autoinhibition of the coiled-coil stalk of OSM-3 by fixing its hinge region in an extended conformation, but severely impairs its motility. OSM-3(G444E) can still associate with heterotrimeric kinesin-II [41,50]. We examined genetic interactions between the nekl-4(tm4910) deletion allele and the osm-3(p802) allele, which phenocopies the osm-3(sa125) amphid cilia truncation phenotype [18,41]. The osm-3(p802) null mutation was not suppressed by nekl-4(tm4910), indicating that interactions between nekl-4 and osm-3 are allele-specific, and may depend on the physical or functional interactions between OSM-3 and NEKL-4 (Fig 6D).
Discussion
We used a forward genetic screen and candidate approach to identify genes that act with the ccpp-1 deglutamylase to control ciliary stability. We identified tubulin code writers and effectors of ciliary stability as modulators of degenerative phenotypes associated with CCPP-1 loss. Our data provides insight into how glutamylation is precisely controlled to maintain ciliary structure and function and to prevent neurodegeneration. Ciliary degeneration in ccpp-1 mutants can be suppressed by altering microtubule glutamylation through the loss of function of tubulin glutamylating enzymes TTLL-4, TTLL-5, or TTLL-11 (refer to model in Fig 7). Our findings are in agreement with studies in Chlamydomonas, where reducing microtubule glutamylation has a stabilizing effect on axonemal defective mutants ([51,52]; for a summary of phenotypes, refer to S3 Table).
Fig 7. Schematic of TTLL-4, TTLL-5, TTLL-11, CCPP-1, and NEKL-4 interactions and effects on ciliary stability.
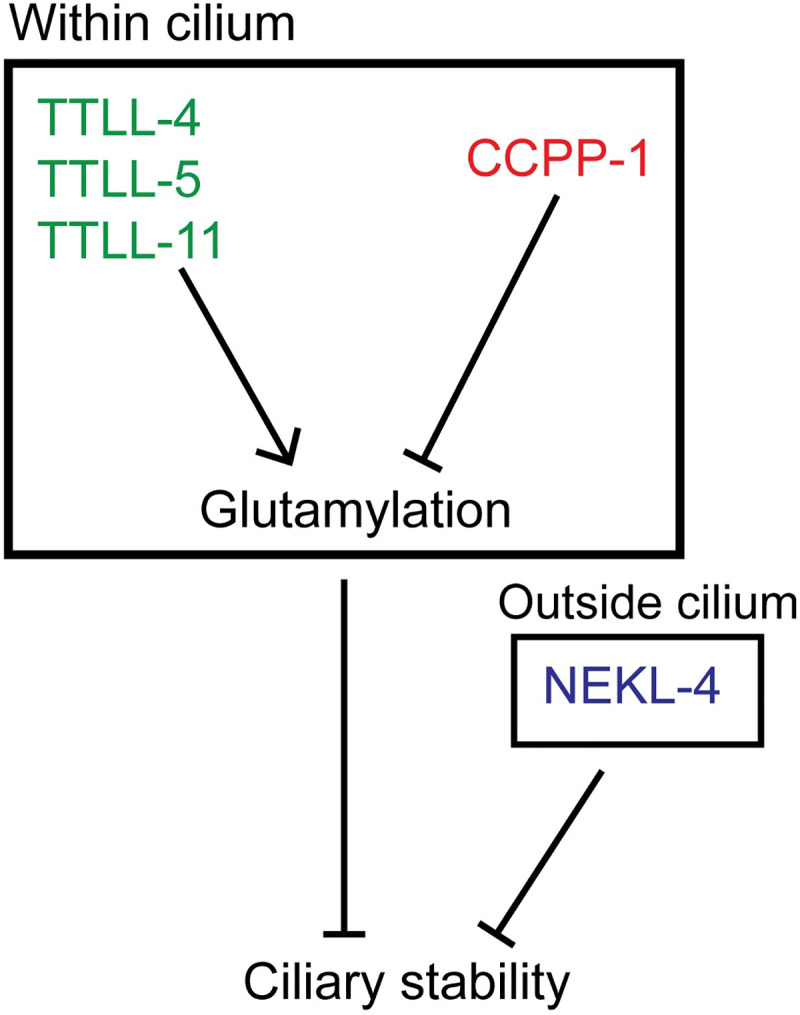
Loss of the NIMA related kinase NEKL-4 opposes the ccpp-1 effect on ciliary stability and may act independently of microtubule glutamylation state. Loss of NEKL-4 function suppresses the ccpp-1 Dyf phenotype while animals overexpressing NEKL-4::GFP are Dyf. Our findings are consistent with previous reports on the roles of NIMA kinases in regulating axonemal stability [52–54]. nekl-4(tm4910) does not strongly affect GT335 staining, arguing against a role for NEKL-4 in the regulation of a glutamylase or deglutamylase enzyme, although we cannot completely rule out this possibility. NEKL-4 is also unlikely to be a reader of ciliary glutamylation because it is largely absent from cilia. Future studies should prioritize ultrastructural analysis by performing transmission electron microscopy to characterize the effect of suppressor mutations on ciliary microtubule loss in ccpp-1 mutants.
How does NEKL-4 regulate ciliary stability despite not localizing to cilia? Several processes including IFT loading/unloading, flagellar length sensing, and ciliary entry of proteins depend on the phosphorylation state of kinesin-II and other IFT components at the base of the cilium [55–58]. Potential substrates of the NEKL-4 human homolog NEK10 include kinesins, IFT components, and other ciliary proteins that have homologs in C. elegans ([26,59]; S4 Table). In human bronchial epithelial cell (HBEC) culture, NEK10 mutation caused a reduction in phosphopeptides of gene products including KIF13B, KIF19, IFT140, IFT46, IFT88, and MAK [26]. These genes and their C. elegans orthologs klp-4, klp-13, che-11, dyf-6, osm-5, and dyf-5 are involved with microtubule dynamics, IFT, and ciliary structure [41,60–68]. NEKL-4 may function in the distal dendrite or periciliary membrane compartment to phosphorylate IFT-related proteins before ciliary entry to indirectly modulate IFT.
NEKs may also regulate endocytosis. C. elegans NEKL-2 and NEKL-3 control clathrin-mediated endocytosis and trafficking in non-ciliated cells [69–71]. NEKL-4 localizes to the periciliary membrane compartment, an active site of endocytosis, a process necessary for regulating ciliary membrane volume, ciliary protein levels, and IFT [72]. Consistent with this idea, a reduction in endocytosis can rescue the Dyf defect of a dominant active G protein α subunit (gpa-3QL) mutant [73].
What could be the mechanism by which loss of CCPP-1 results in ciliary instability? Our data argue against a role for microtubule severing by katanin (MEI-1) and spastin (SPAS-1) in modulating the loss of ciliary stability in ccpp-1 mutants. Our results also did not support a role for other known effectors of microtubule glutamylation—the MAP kinase PMK-3 and the microtubule-depolymerizing kinesin-13 (KLP-7), in the degradation of ciliary microtubules in the absence of CCPP-1 function. Previous studies in Chlamydomonas revealed a link between tubulin turnover and both tubulin polyglutamylation [74] and NEK kinase activity [52]. Changes in IFT due to loss of CCPP-1 function could affect tubulin transport [75], or the binding of structural MAPs [76,77] could also mediate ccpp-1 loss induced loss of ciliary stability. Identification of readers of the tubulin code in amphid and phasmid neurons could identify molecules and mechanisms that modulate ciliary degeneration in response to changes in microtubule glutamylation.
NIMA-related kinases play major roles in microtubule structure regulation and processes such as mitosis, ciliogenesis, and post-translational modifications [54,78–80]. Consequently, mutations of NEKs may result in cancer [81–83] and ciliopathies, such as polycystic kidney disease [26,80,84]. Many NEKs regulate ciliary disassembly through promotion of microtubule instability, such as Tetrahymena Nrk2, Nrk17, and Nrk30 [54], Chlamydomonas FA2, CNK2, and CNK11 [52,85,86], and human NEK7 [53]. Our data on the effects of NEKL-4 on ciliary stability offers insight into mucociliary disease associated with mutations in human NEK10. Our data also expands upon an evolutionarily-conserved link between molecules involved in microtubule glutamylation and NEK kinases [52,87]. Our studies provide a model for the exploration of this poorly-understood link between NEK kinases and microtubule glutamylation, and identify NEKs as a modulator of the degenerative effects associated with dysregulated levels of microtubule glutamylation. The data shown in this work will inform future studies on neurodegenerative disorders and ciliopathies associated with mutations in microtubule glutamylation enzymes and NEK kinases.
Materials and methods
C. elegans strains and maintenance
Nematodes were cultured on Nematode Growth Media (NGM) agar plates containing a lawn of OP50 E. coli and incubated at 20°C, as previously described [88]. Strains used are listed in S5 Table. Some strains contained him-5(e1490) and/or myIs1[pkd-2::gfp+unc-122p::gfp] pkd-2(sy606), which were considered wild type.
ccpp-1 EMS mutagenesis screen
To identify the pathway by which ccpp-1 mutations cause the Dyf phenotype [21], we performed a forward genetic screen for suppressors of the Dyf defect of ccpp-1 mutants. Ethyl methanesulfonate (EMS; Sigma Aldrich Cat. M0880) was used to mutagenize ccpp-1(ok1821) mutants {either Strain PT2168—ccpp-1(ok1821);myIs1[pkd-2::gfp + unc-122p::gfp] pkd-2(sy606);him-5(e1490) or OE4160—ccpp-1(ok1821)}. L4 larvae were suspended in 1 ml of M9 buffer [89] with a final concentration of 47 mM EMS for four hours, before washing three times in M9 and depositing worm suspension onto an NGM plate seeded with OP50 E. coli to recover. We then transferred 10 or 20 of these mutagenized P0 hermaphrodites to new NGM plates and allowed them to lay eggs on 100mm NGM plates seeded with OP50 for one day or two days before removing them. When the F1 hermaphrodites reached adulthood, we synchronized the age of F2 progeny by hypochlorite treatment of gravid adult F1 animals [89] and replating embryos on NGM plates with OP50. We then incubated plates for four days at 20°C or five days at 15°C (to allow the age-synchronized F2 progeny to reach adulthood) before washing them off plates using M9 to be dye-filled using the method described below. ccpp-1 mutants without suppressor mutations typically are completely Dye-filling defective (Dyf) by adulthood. Therefore, to isolate suppressor mutants, we picked F2 adult animals that were non-Dyf or incompletely Dyf. We isolated up to five suppressor mutant isolates corresponding to a single P0 plate to allow for some lethality, but in most cases kept only a single suppressor from each P0 plate, except as noted in S1 Table.
In total, we examined more than 100,000 haploid genomes. We isolated 93 non-Dyf or partially Dyf F2 animals, of which 79 either died, were sterile, or were Dyf (not suppressed) in a subsequent secondary dye-filling assays to confirm the phenotype. 14 isolates were viable and produced non-Dyf or partial Dyf progeny in secondary screens, of which we consider 11 to be independent isolates. S1 Table is based on the qualitative analysis where amphid and phasmid neurons were not scored separately.
Whole-genome sequencing
To facilitate identification of suppressor mutations, whole-genome sequencing (WGS) was performed using the strategy described in Doitsidou et al [90]. Mutants were crossed with PT3037, a strain in which ccpp-1 had been introgressed into the Hawaiian wild isolate genomic background by backcrossing ten times into CB4856. From crosses of PT3068 (my31) X PT3037, at least 20 suppressed F2s were isolated; these F2s were pooled for WGS to identify genomic intervals that lacked PT3037-derived single-nucleotide polymorphisms (SNPs); novel SNPs within those intervals potentially represent the suppressor mutation.
Adapter-ligated libraries were prepared from genomic DNA using the manufacturer’s protocol (Illumina, San Diego, CA) from PT3037 and the pooled suppressed F2s. A HiSeq 2000 instrument (Illumina, San Diego, CA) was used to sequence the genomic libraries. To identify the my31 mutation, we used a pipeline described in Wang et al [91]. Briefly, the pipeline was as follows: BFAST for alignment [92], SAMtools for variant calling [93], and ANNOVAR for annotation [94]. We compared sequence data with C. elegans genome version WS220 to identify mutations. To identify mapping intervals that might contain the my31 suppressor, we plotted Hawaiian SNP density against chromosome position. Non-Hawaiian SNPs that were also identified in PT3037 were removed as background. Candidate mutations for my31 were identified as novel homozygous non-synomymous coding variants within the mapping interval.
To facilitate identification of suppressor mutations, mutants were crossed with PT3037 (a strain in which ccpp-1 had been introgressed into the Hawaiian wild isolate genomic background by backcrossing ten times into CB4856). From crosses of PT3068 (my31) X PT3037, at least 20 suppressed F2s were isolated; these F2s were pooled for whole genome sequencing using the strategy described in Doitsidou et al [90] to identify genomic intervals containing N2 single nucleotide polymorphisms (SNPs) as potentially containing the suppressor mutation.
Adapter-ligated libraries were prepared from genomic DNA using the manufacturer’s protocol (Illumina, San Diego, CA) from PT3037 and the pooled suppressed F2s. A HiSeq 2000 instrument (Illumina, San Diego, CA) was used to sequence the genomic libraries. To identify the my31 mutation, we used a pipeline described in Huang et al [95]. Briefly, the pipeline was as follows: BFAST for alignment [92], SAMtools for variant calling [93], and ANNOVAR for annotation [94]. We compared sequence data with C. elegans genome version WS220 to identify mutations. To identify N2 intervals that might contain the my31 suppressor, we plotted Hawaiian SNP density against chromosome position. Non-Hawaiian SNPs that were also identified in PT3037 were removed as background.
Protein domain identification, homology analysis and alignments
Sequences of ttll-4, ttll-5, ttll-11, and nekl-4 were obtained from WormBase [29] and protein sequences were obtained from WormBase or UniProt [96]. Structural predictions and protein domain information were generated with InterPro [97] and ScanProsite [98]. The PEST proteolytic degradation site in NEKL-4 was identified using EMBOSS epestfind [36]. NEKL-4 homology to NEK10 was predicted using protein BLAST [99] and human NEK10 and C. elegans NEKL-4 kinase domains were aligned using T-COFFEE Expresso [30]. All scans and queries described above were performed using default settings. Gene diagrams were created with the WormWeb Exon-Intron graphic maker [100], and protein diagrams were created with DOG 2.0 software [101,102]. The NEK10/NEKL-4 kinase domain alignment figure was generated with BoxShade [103] from the T-COFFEE sequence alignment.
Dye-filling assays
Three days prior to performing dye-filling assays, healthy, non-crowded plates containing many gravid hermaphrodite animals were washed and bleached to synchronize the worms. Day 1 and Day 2 adults were washed from the plate with 1mL M9 buffer and transferred to 1.5mL tubes. A stock solution of 2.5mg/mL DiI (Thermo Fisher Cat. D282) was added to a 1:1000 dilution and worms were incubated with gentle shaking for 30 min at room temperature. Prior to scoring, worms were briefly spun at 2000RPM and plated on new seeded NGM plates for approximately 1hr to excrete excess dye. The presence and intensity of dye in the amphid and phasmid neurons was visually scored using a dissecting microscope. Worms scored as non-Dyf were similar in brightness to wild type, and Dyf worms had no staining. Partial Dyf worms had dye present, but distinguishably less bright than wild type. Kruskal-Wallis one-way ANOVA analysis (where non-Dyf = 2, partial Dyf = 1. and Dyf = 0) and posthoc Dunn’s multiple comparison test were performed in Prism (Graphpad Software).
Antibody staining
We performed immunofluorescence microscopy using the protocol described in O’Hagan et al [22]. We synchronized animals by bleaching and fixed as Day 1 adults. Fixation was accomplished by washing animals from 3 NGM plates using M9 buffer, then washing animals in a 15 ml conical tube 3 more times with M9 over one hour. Worms were chilled on ice before washing in ice-cold Ruvkun buffer (80 mM KCl, 20 mM NaCl, 10 mM EGTA, 5 mM spermidine-HCl, 15 mM Pipes, pH 7.4 and 25% methanol) plus 2% formaldehyde in 1.5ml centrifuge tubes. The tubes were immersed in liquid nitrogen and melted under tap water to crack the worms’ cuticles. Worms were then washed twice with Tris-Triton buffer (100 mM Tris-HCl, pH 7.4, 1% Triton X-100 and 1 mM EDTA), suspended in Tris-Triton buffer+1% β-mercaptoethanol, and incubated overnight at 37°C. The next day, worms were washed with 1X BO3 buffer (50 mM H3BO3, 25 mM NaOH) + 0.01% Triton, and suspended in 1X BO3 + 0.01% Triton buffer + 10 mM DTT for 15 minutes with gentle agitation at room temperature. Worms were then washed with 1X BO3 buffer (50 mM H3BO3, 25 mM NaOH) + 0.01% Triton, and suspended in 1X BO3 + 0.01% Triton buffer + 0.3% H2O2 for 15 minutes with gentle agitation at room temperature. After washing once with 1X BO3 + 0.01% Triton buffer, worms were washed for 15 minutes in Antibody buffer B (1X PBS, 0.1% BSA, 0.5% Triton X-100, 0.05% sodium azide, 1mM EDTA) with gentle agitation at room temperature. Fixed worms were stored in Antibody buffer A (1X PBS, 1% BSA, 0.5% Triton X-100, 0.05% sodium azide, 1mM EDTA) at 4°C for up to one month before antibody staining. Animals were stained overnight at room temperature with a 1:450 dilution (in Antibody Buffer A) of GT335 (Adipogen Cat. AG-20B-0020-C100), a monoclonal antibody which binds the branch point of both monoglutamylated and polyglutamylated substrates [43], or polyE (Adipogen Cat. AG-25B-0030-C050), a polyclonal antibody which only binds polyglutamylated substrates (3 or more glutamate residues) [5]. Stained worms were washed with several changes of Antibody B Buffer with gentle agitation at room temperature over several hours. After rinsing with Antibody Buffer A, Alexa-fluor 568-conjugated donkey anti-mouse secondary antibody (Invitrogen Cat. A10037) was added at a final dilution of 1:2500 and incubated for 2 hours at room temperature with gentle agitation. Worms were then washed with several changes of Antibody Buffer B over several hours before mounting on 2% agarose pads for imaging.
We observed variation between and within individual GT335 staining experiments with respect to pattern and intensity. Environmental factors may cause changes in levels of ciliary glutamylation as measured by GT335 intensity [104]. It is therefore possible that small variations in experimental techniques or day-to-day lab conditions could have impacted our results. However, the GT335 staining phenotype of each mutant was generally consistent across trials.
Confocal imaging
Day 1 adult hermaphrodites were anaesthetized with 10 mM levamisole and mounted on 4% agarose pads for imaging at room temperature. Confocal imaging was performed with a Zeiss LSM 880 inverted microscope with an Airyscan superresolution module using a LSM T-PMT detector and ZenBlack software (Carl Zeiss Microscopy, Oberkochen, Germany). Laser intensity was adjusted to avoid saturated pixels. Images were acquired using a 63x/1.4 Oil Plan-Apochromat objective in Airyscan Fast mode and deconvolved using Airyscan processing. Image files were imported into Fiji/ImageJ [105] with the BioFormats Importer plugin for linear adjustment of contrast and creation of maximum intensity projections, and Adobe Photoshop CS5 was used to trace the outlines of the worm if needed. Images were placed in Adobe Illustrator CS5 for figure assembly.
KAP-1::GFP localization analysis
Nematodes were mounted as above. Widefield images were acquired on a Zeiss Axio Observer with Colibri 7 LEDs and ZenBlue software (Carl Zeiss Microscopy, Oberkochen, Germany) using a Photometrics Prime 95B sCMOS camera (Teledyne Photometrics, Tucson, AZ). A 100x/1.4 Oil Plan-Apochromat objective was used for imaging the phasmid cilia of Day 1 hermaphrodite animals. Images were imported into Fiji/ImageJ [105] and the length of KAP-1::GFP labeling was measured for each cilium using maximum intensity projections. Length was measured starting at the transition zone, which is distinct and bright, and ending when fluorescence was no longer visible. Kruskal-Wallis one-way ANOVA analysis and posthoc Dunn’s multiple comparison test were performed in Prism (Graphpad Software).
We also wanted to examine if ttll-4 suppressed the ccpp-1 KAP-1::GFP span defect, but were unable to obtain a viable ccpp-1;ttll-4;Ex[kap-1::gfp +pRF4] strain, possibly due to synthetic lethality with the overexpressed kap-1::gfp reporter.
Plasmid construction
For creation of pRO139 (nekl-4p::nekl-4::gfp), nekl-4 plus a 1609bp upstream promoter region was amplified from genomic DNA using primers with homology to pPD95.75 containing gfp. The stop codon for nekl-4 was removed using the reverse primer. pPD95.75 was amplified using primers with homology to nekl-4. Fragments were joined using Gibson assembly [106].
| genomic nekl-4 amplification (forward) GATACGCTAACAACTTGGAAATGAAATagtagctggatgacgactgg |
| genomic nekl-4 amplification (reverse) ggtcctcctgaaaatgttctatgttatgTCcCTTCGCTGCTGGATTTTC |
| pPD95.75 amplification (forward) GAAAATCCAGCAGCGAAGgGAcataacatagaacattttcaggaggacc |
| pPD95.75 amplification (reverse) ccagtcgtcatccagctactATTTCATTTCCAAGTTGTTAGCGTATC |
CRISPR constructs
For creation of the nekl-4::mneongreen and nekl-4::mscarlet-I endogenous CRISPR tags, we followed the Mello Lab protocol [107] and used pdsDNA [108] that included a short flexible linker sequence between the 3’ end of nekl-4 and the start of mneongreen or mscarlet-I. The guide sequence was designed using CRISPOR [109,110] and silent mutation sites for PAM modification and diagnosis were located using WatCut silent mutation scanning [111]. Reagents used are as follows. dg357 was a gift from Dominique A. Glauser and was used in accordance with the C. elegans group license with AlleleBiotech. pSEM90 was a gift from Thomas Boulin.
| mNeonGreen amplification from | |
| dg357 (forward) for donor | TGTCAGCGTGCATTGAATGTTTGATTGCAGAAAATCCA |
| GCCGCTAAAGGAGGTGGCGGATCTGGAGGTGGAGGCTCTG GAGGAGGTGGATCTATGGTGTCGAAGGGAGAAG | |
| mNeonGreen amplification from | |
| dg357 (reverse) for donor | |
| TATACAAAAAAACAGTATATACAATTTAGCATATGCTACTTGTAGAGTTCATCCATTC | |
| mScarlet-I amplification from | |
| pSEM90 (forward) for donor | |
| TGTCAGCGTGCATTGAATGTTTGATTGCAGAAAATCCAGCCG CTAAAGGAGGTGGCGGATCTGGAGGTGGAGGCTCTGGAGGAGGTGGATCTATGGTCAGCAAGGGAGAGG | |
| mScarlet-I amplification from | |
| pSEM90 (reverse) for donor | |
| TATACAAAAAAACAGTATATACAATTTAGCATATGCTACTTGTAGAGCTCGTCCATTCC | |
| crRNA (reverse strand) | AGCAUAUGUCACUUCGCUGC |
Supporting information
For experiments involving the mei-1(or1178) temperature-sensitive allele, eggs from plates kept at the permissive temperature (15°C; [112]) were picked to fresh plates and shifted to 25°C after hatching for 4 days before scoring.
(TIF)
(TIF)
a-b. Localization of NEKL-4::mNeonGreen in adult males. Tail is flat against the coverslip in a. to visualize ray dendrites. Tail in b. is folded but the filamentous pattern in ray cell bodies is visible. c-d. Localization of NEKL-4::mNeonGreen in L4 molt males. Both images are different sections from the same z-stack. Arrows indicate ray cell bodies, arrowheads indicate ray dendrites (numbered). Scale = 10μm.
(TIF)
Scale bar = 10μm.
(TIF)
a. Example images of polyE stained cilia in the nose. The polyE polyclonal antibody stained amphid ciliary middle segments (MS) and labial and cephalic cilia (LC), similar to GT335. Abnormal polyE staining was visible in some strains containing the ccpp-1 deletion mutation. Loss of TTLL-11 abolished polyE staining, but in the absence of TTLL-4 or TTLL-5, polyE staining remained. Scale = 10μm. b. Quantification of polyE staining phenotypes by genotype; number of animals examined indicated at right.
(TIF)
Time lapse of a single z slice with widefield microscope. Images acquired at 4.34 fps and shown at 20 fps.
(MP4)
Strains contain the allele mentioned and the ccpp-1(ok1821) deletion. At least 20 animals were tested for Dye filling for each strain. Although a single P0 plate produced my42 and my43, we considered them independent isolates because their phenotypes are different from one another.
(XLSX)
GKO; Gene Knockout Consortium. CGC; Caenorhabditis Genetics Center. NBRP; National Bio-Resource Project.
(XLSX)
+ indicates any degree of suppression of the ccpp-1 phenotype,—indicates no suppression of the ccpp-1 phenotype, n/a indicates not tested.
(XLSX)
Based on phosphoproteomics data for NEK10 from Chivukula et al, Fig 4F [26]. Original list contains genes with phosphopeptides depleted more than twofold upon NEK10 deletion in human bronchial epithelial cell (HBEC) culture. This list contains genes with known C. elegans homologs from the following relevant classes in the NEK10 list: axonemal dyneins and assembly factors, kinesins, intraflagellar transport, ciliary length control.
(XLSX)
All strains were generated in the Barr laboratory except CB1490 (Hodgkin laboratory), ROH11 and ROH12 (O’Hagan laboratory).
(XLSX)
All strains were generated in the Barr laboratory except BS3383, which was obtained from the Caenorhabditis Genetics Center (CGC).
(XLSX)
(DOCX)
Acknowledgments
We thank members of the Barr lab, the Rutgers University C. elegans community, and K. M. P.’s thesis committee Marc Gartenberg, Monica Driscoll, and Bonnie Firestein for advice and helpful feedback; Antonina Roll-Mecak for insight on TTLL-5 structure; and Helen Ushakov and Gloria Androwski for technical assistance. We thank Dominique A. Glauser and Thomas Boulin for plasmids. We also thank WormBase (U41 HG002223) and WormAtlas (R24 OD010943) for online resources. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440), or by the National Bio-Resource Project of the MEXT, Japan.
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
This work was funded by the New Jersey Commission for Spinal Cord Research (NJCSCR) CSCR15IRG014 to R. O.; NIH DK116606 to M. M. B.; NIH Institutional Research and Academic Career Development Award (IRACDA) K12GM093854 to J. D. W; and the Intramural Research Program of the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (H.S. and A.G.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Reiter JF, Leroux MR. Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol. 2017;18: 533–547. 10.1038/nrm.2017.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Verhey KJ, Gaertig J. The tubulin code. Cell Cycle. 2007;6: 2152–2160. 10.4161/cc.6.17.4633 [DOI] [PubMed] [Google Scholar]
- 3.Magiera MM, Bodakuntla S, Žiak J, Lacomme S, Marques Sousa P, Leboucher S, et al. Excessive tubulin polyglutamylation causes neurodegeneration and perturbs neuronal transport. EMBO J. 2018;37 10.15252/embj.2018100440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bodakuntla S, Schnitzler A, Villablanca C, Gonzalez-Billault C, Bieche I, Janke C, et al. Tubulin polyglutamylation is a general traffic control mechanism in hippocampal neurons. J Cell Sci. 2020. 10.1242/jcs.241802 [DOI] [PubMed] [Google Scholar]
- 5.Rogowski K, van Dijk J, Magiera MM, Bosc C, Deloulme J-C, Bosson A, et al. A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell. 2010;143: 564–578. 10.1016/j.cell.2010.10.014 [DOI] [PubMed] [Google Scholar]
- 6.Kimura Y, Kurabe N, Ikegami K, Tsutsumi K, Konishi Y, Kaplan OI, et al. Identification of tubulin deglutamylase among Caenorhabditis elegans and mammalian cytosolic carboxypeptidases (CCPs). J Biol Chem. 2010;285: 22936–22941. 10.1074/jbc.C110.128280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Janke C, Rogowski K, Wloga D, Regnard C, Kajava AV, Strub J-M, et al. Tubulin polyglutamylase enzymes are members of the TTL domain protein family. Science. 2005;308: 1758–1762. 10.1126/science.1113010 [DOI] [PubMed] [Google Scholar]
- 8.Ikegami K, Mukai M, Tsuchida J-I, Heier RL, Macgregor GR, Setou M. TTLL7 is a mammalian beta-tubulin polyglutamylase required for growth of MAP2-positive neurites. J Biol Chem. 2006;281: 30707–30716. 10.1074/jbc.M603984200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Dijk J, Rogowski K, Miro J, Lacroix B, Eddé B, Janke C. A targeted multienzyme mechanism for selective microtubule polyglutamylation. Mol Cell. 2007;26: 437–448. 10.1016/j.molcel.2007.04.012 [DOI] [PubMed] [Google Scholar]
- 10.Garnham CP, Roll-Mecak A. The chemical complexity of cellular microtubules: tubulin post-translational modification enzymes and their roles in tuning microtubule functions. Cytoskeleton. 2012;69: 442–463. Available: 10.1002/cm.21027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mullen RJ. Site of pcd gene action and Purkinje cell mosaicism in cerebella of chimaeric mice. Nature. 1977;270: 245–247. 10.1038/270245a0 [DOI] [PubMed] [Google Scholar]
- 12.Landis SC, Mullen RJ. The development and degeneration of Purkinje cells in pcd mutant mice. J Comp Neurol. 1978;177: 125–143. 10.1002/cne.901770109 [DOI] [PubMed] [Google Scholar]
- 13.Fernandez-Gonzalez A. Purkinje cell degeneration (pcd) Phenotypes Caused by Mutations in the Axotomy-Induced Gene, Nna1. Science. 2002. pp. 1904–1906. 10.1126/science.1068912 [DOI] [PubMed] [Google Scholar]
- 14.Lee JE, Silhavy JL, Zaki MS, Schroth J, Bielas SL, Marsh SE, et al. CEP41 is mutated in Joubert syndrome and is required for tubulin glutamylation at the cilium. Nat Genet. 2012;44: 193–199. 10.1038/ng.1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brady ST, Morfini GA. Regulation of motor proteins, axonal transport deficits and adult-onset neurodegenerative diseases. Neurobiol Dis. 2017;105: 273–282. 10.1016/j.nbd.2017.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alexander AG, Marfil V, Li C. Use of Caenorhabditis elegans as a model to study Alzheimer’s disease and other neurodegenerative diseases. Front Genet. 2014;5: 279 10.3389/fgene.2014.00279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ward S, Thomson N, White JG, Brenner S. Electron microscopical reconstruction of the anterior sensory anatomy of the nematodecaenorhabditis elegans. The Journal of Comparative Neurology. 1975. pp. 313–337. 10.1002/cne.901600305 [DOI] [PubMed] [Google Scholar]
- 18.Perkins LA, Hedgecock EM, Nichol Thomson J, Culotti JG. Mutant sensory cilia in the nematode Caenorhabditis elegans. Developmental Biology. 1986. pp. 456–487. 10.1016/0012-1606(86)90314-3 [DOI] [PubMed] [Google Scholar]
- 19.Ware RW, Clark D, Crossland K, Russell RL. The nerve ring of the nematodeCaenorhabditis elegans: Sensory input and motor output. The Journal of Comparative Neurology. 1975. pp. 71–110. 10.1002/cne.901610107 [DOI] [PubMed] [Google Scholar]
- 20.Evans JE, Snow JJ, Gunnarson AL, Ou G, Stahlberg H, McDonald KL, et al. Functional modulation of IFT kinesins extends the sensory repertoire of ciliated neurons in Caenorhabditis elegans. The Journal of Cell Biology. 2006. pp. 663–669. 10.1083/jcb.200509115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Hagan R, Piasecki BP, Silva M, Phirke P, Nguyen KCQ, Hall DH, et al. The tubulin deglutamylase CCPP-1 regulates the function and stability of sensory cilia in C. elegans. Curr Biol. 2011;21: 1685–1694. 10.1016/j.cub.2011.08.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Hagan R, Silva M, Nguyen KCQ, Zhang W, Bellotti S, Ramadan YH, et al. Glutamylation Regulates Transport, Specializes Function, and Sculpts the Structure of Cilia. Curr Biol. 2017;27: 3430–3441.e6. 10.1016/j.cub.2017.09.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sergouniotis PI, Chakarova C, Murphy C, Becker M, Lenassi E, Arno G, et al. Biallelic variants in TTLL5, encoding a tubulin glutamylase, cause retinal dystrophy. Am J Hum Genet. 2014;94: 760–769. 10.1016/j.ajhg.2014.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun X, Park JH, Gumerson J, Wu Z, Swaroop A, Qian H, et al. Loss of RPGR glutamylation underlies the pathogenic mechanism of retinal dystrophy caused by TTLL5 mutations. Proc Natl Acad Sci U S A. 2016;113: E2925–34. 10.1073/pnas.1523201113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Porpora M, Sauchella S, Rinaldi L, Delle Donne R, Sepe M, Torres-Quesada O, et al. Counterregulation of cAMP-directed kinase activities controls ciliogenesis. Nat Commun. 2018;9: 1224 10.1038/s41467-018-03643-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chivukula RR, Montoro DT, Leung HM, Yang J, Shamseldin HE, Taylor MS, et al. A human ciliopathy reveals essential functions for NEK10 in airway mucociliary clearance. Nature Medicine. 2020. 10.1038/s41591-019-0730-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bompard G, van Dijk J, Cau J, Lannay Y, Marcellin G, Lawera A, et al. CSAP Acts as a Regulator of TTLL-Mediated Microtubule Glutamylation. Cell Rep. 2018;25: 2866–2877.e5. 10.1016/j.celrep.2018.10.095 [DOI] [PubMed] [Google Scholar]
- 28.Chawla DG, Shah RV, Barth ZK, Lee JD, Badecker KE, Naik A, et al. Caenorhabditis elegans glutamylating enzymes function redundantly in male mating. Biol Open. 2016;5: 1290–1298. 10.1242/bio.017442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.WormBase: Nematode Information Resource. [cited 27 Jan 2020]. Available: https://wormbase.org//
- 30.T-Coffee Server. [cited 27 Jan 2020]. Available: http://tcoffee.crg.cat/apps/tcoffee/do:expresso
- 31.Wang J, Barr MM. Ciliary Extracellular Vesicles: Txt Msg Organelles. Cell Mol Neurobiol. 2016;36: 449–457. 10.1007/s10571-016-0345-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valenstein ML, Roll-Mecak A. Graded Control of Microtubule Severing by Tubulin Glutamylation. Cell. 2016;164: 911–921. 10.1016/j.cell.2016.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharma N, Bryant J, Wloga D, Donaldson R, Davis RC, Jerka-Dziadosz M, et al. Katanin regulates dynamics of microtubules and biogenesis of motile cilia. J Cell Biol. 2007;178: 1065–1079. 10.1083/jcb.200704021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghosh-Roy A, Goncharov A, Jin Y, Chisholm AD. Kinesin-13 and tubulin posttranslational modifications regulate microtubule growth in axon regeneration. Dev Cell. 2012;23: 716–728. 10.1016/j.devcel.2012.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hatzfeld M. The armadillo family of structural proteins. Int Rev Cytol. 1999;186: 179–224. 10.1016/s0074-7696(08)61054-2 [DOI] [PubMed] [Google Scholar]
- 36.EMBOSS: epestfind. [cited 27 Jan 2020]. Available: http://emboss.bioinformatics.nl/cgi-bin/emboss/epestfind
- 37.Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science. 1986. pp. 364–368. 10.1126/science.2876518 [DOI] [PubMed] [Google Scholar]
- 38.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215: 403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 39.Paix A, Folkmann A, Seydoux G. Precision genome editing using CRISPR-Cas9 and linear repair templates in C. elegans. Methods. 2017. pp. 86–93. 10.1016/j.ymeth.2017.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dickinson DJ, Goldstein B. CRISPR-Based Methods for Caenorhabditis elegans Genome Engineering. Genetics. 2016. pp. 885–901. 10.1534/genetics.115.182162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yi P, Xie C, Ou G. The kinases male germ cell-associated kinase and cell cycle-related kinase regulate kinesin-2 motility in Caenorhabditis elegans neuronal cilia. Traffic. 2018;19: 522–535. 10.1111/tra.12572 [DOI] [PubMed] [Google Scholar]
- 42.Haycraft CJ, Schafer JC, Zhang Q, Taulman PD, Yoder BK. Identification of CHE-13, a novel intraflagellar transport protein required for cilia formation. Exp Cell Res. 2003;284: 251–263. 10.1016/s0014-4827(02)00089-7 [DOI] [PubMed] [Google Scholar]
- 43.Wolff A, de Néchaud B, Chillet D, Mazarguil H, Desbruyères E, Audebert S, et al. Distribution of glutamylated alpha and beta-tubulin in mouse tissues using a specific monoclonal antibody, GT335. Eur J Cell Biol. 1992;59: 425–432. Available: https://www.ncbi.nlm.nih.gov/pubmed/1493808 [PubMed] [Google Scholar]
- 44.Pathak N, Obara T, Mangos S, Liu Y, Drummond IA. The Zebrafish fleer Gene Encodes an Essential Regulator of Cilia Tubulin Polyglutamylation. Molecular Biology of the Cell. 2007. pp. 4353–4364. 10.1091/mbc.e07-06-0537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silva M, Morsci N, Nguyen KCQ, Rizvi A, Rongo C, Hall DH, et al. Cell-Specific α-Tubulin Isotype Regulates Ciliary Microtubule Ultrastructure, Intraflagellar Transport, and Extracellular Vesicle Biology. Curr Biol. 2017;27: 968–980. 10.1016/j.cub.2017.02.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scholey JM, Ou G, Snow J, Gunnarson A. Intraflagellar transport motors in Caenorhabditis elegans neurons. Biochemical Society Transactions. 2004. pp. 682–684. 10.1042/BST0320682 [DOI] [PubMed] [Google Scholar]
- 47.Ou G, Blacque OE, Snow JJ, Leroux MR, Scholey JM. Functional coordination of intraflagellar transport motors. Nature. 2005. pp. 583–587. 10.1038/nature03818 [DOI] [PubMed] [Google Scholar]
- 48.Prevo B, Mangeol P, Oswald F, Scholey JM, Peterman EJG. Functional differentiation of cooperating kinesin-2 motors orchestrates cargo import and transport in C. elegans cilia. Nat Cell Biol. 2015;17: 1536–1545. 10.1038/ncb3263 [DOI] [PubMed] [Google Scholar]
- 49.Hao L, Efimenko E, Swoboda P, Scholey JM. The retrograde IFT machinery of C. elegans cilia: two IFT dynein complexes? PLoS One. 2011;6: e20995 10.1371/journal.pone.0020995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Imanishi M, Endres NF, Gennerich A, Vale RD. Autoinhibition regulates the motility of the C. elegans intraflagellar transport motor OSM-3. The Journal of Cell Biology. 2006. pp. 931–937. 10.1083/jcb.200605179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kubo T, Yanagisawa H-A, Yagi T, Hirono M, Kamiya R. Tubulin polyglutamylation regulates axonemal motility by modulating activities of inner-arm dyneins. Curr Biol. 2010;20: 441–445. 10.1016/j.cub.2009.12.058 [DOI] [PubMed] [Google Scholar]
- 52.Lin H, Zhang Z, Guo S, Chen F, Kessler JM, Wang YM, et al. A NIMA-Related Kinase Suppresses the Flagellar Instability Associated with the Loss of Multiple Axonemal Structures. PLoS Genet. 2015;11: e1005508 10.1371/journal.pgen.1005508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cohen S, Aizer A, Shav-Tal Y, Yanai A, Motro B. Nek7 kinase accelerates microtubule dynamic instability. Biochim Biophys Acta. 2013;1833: 1104–1113. 10.1016/j.bbamcr.2012.12.021 [DOI] [PubMed] [Google Scholar]
- 54.Wloga D, Camba A, Rogowski K, Manning G, Jerka-Dziadosz M, Gaertig J. Members of the NIMA-related Kinase Family Promote Disassembly of Cilia by Multiple Mechanisms. Molecular Biology of the Cell. 2006. pp. 2799–2810. 10.1091/mbc.e05-05-0450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chaya T, Omori Y, Kuwahara R, Furukawa T. ICK is essential for cell type-specific ciliogenesis and the regulation of ciliary transport. EMBO J. 2014;33: 1227–1242. 10.1002/embj.201488175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Luo M, Cao M, Kan Y, Li G, Snell W, Pan J. The phosphorylation state of an aurora-like kinase marks the length of growing flagella in Chlamydomonas. Curr Biol. 2011;21: 586–591. 10.1016/j.cub.2011.02.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pan J, Wang Q, Snell WJ. An aurora kinase is essential for flagellar disassembly in Chlamydomonas. Dev Cell. 2004;6: 445–451. 10.1016/s1534-5807(04)00064-4 [DOI] [PubMed] [Google Scholar]
- 58.Liang Y, Pang Y, Wu Q, Hu Z, Han X, Xu Y, et al. FLA8/KIF3B phosphorylation regulates kinesin-II interaction with IFT-B to control IFT entry and turnaround. Dev Cell. 2014;30: 585–597. 10.1016/j.devcel.2014.07.019 [DOI] [PubMed] [Google Scholar]
- 59.van de Kooij B, Creixell P, van Vlimmeren A, Joughin BA, Miller CJ, Haider N, et al. Comprehensive substrate specificity profiling of the human Nek kinome reveals unexpected signaling outputs. Elife. 2019;8 10.7554/eLife.44635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Monteiro MI, Ahlawat S, Kowalski JR, Malkin E, Koushika SP, Juo P. The kinesin-3 family motor KLP-4 regulates anterograde trafficking of GLR-1 glutamate receptors in the ventral nerve cord of Caenorhabditis elegans. Mol Biol Cell. 2012;23: 3647–3662. 10.1091/mbc.E12-04-0334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lacroix B, Bourdages KG, Dorn JF, Ihara S, Sherwood DR, Maddox PS, et al. In situ imaging in C. elegans reveals developmental regulation of microtubule dynamics. Dev Cell. 2014;29: 203–216. 10.1016/j.devcel.2014.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ou G, Koga M, Blacque OE, Murayama T, Ohshima Y, Schafer JC, et al. Sensory ciliogenesis in Caenorhabditis elegans: assignment of IFT components into distinct modules based on transport and phenotypic profiles. Mol Biol Cell. 2007;18: 1554–1569. 10.1091/mbc.e06-09-0805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bell LR, Stone S, Yochem J, Shaw JE, Herman RK. The molecular identities of the Caenorhabditis elegans intraflagellar transport genes dyf-6, daf-10 and osm-1. Genetics. 2006;173: 1275–1286. 10.1534/genetics.106.056721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Qin H, Rosenbaum JL, Barr MM. An autosomal recessive polycystic kidney disease gene homolog is involved in intraflagellar transport in C. elegans ciliated sensory neurons. Curr Biol. 2001;11: 457–461. 10.1016/s0960-9822(01)00122-1 [DOI] [PubMed] [Google Scholar]
- 65.Bae Y-K, Qin H, Knobel KM, Hu J, Rosenbaum JL, Barr MM. General and cell-type specific mechanisms target TRPP2/PKD-2 to cilia. Development. 2006;133: 3859–3870. 10.1242/dev.02555 [DOI] [PubMed] [Google Scholar]
- 66.Chen N, Mah A, Blacque OE, Chu J, Phgora K, Bakhoum MW, et al. Identification of ciliary and ciliopathy genes in Caenorhabditis elegans through comparative genomics. Genome Biol. 2006;7: R126 10.1186/gb-2006-7-12-r126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Burghoorn J, Dekkers MPJ, Rademakers S, de Jong T, Willemsen R, Jansen G. Mutation of the MAP kinase DYF-5 affects docking and undocking of kinesin-2 motors and reduces their speed in the cilia of Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2007;104: 7157–7162. 10.1073/pnas.0606974104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maurya AK, Rogers T, Sengupta P. A CCRK and a MAK Kinase Modulate Cilia Branching and Length via Regulation of Axonemal Microtubule Dynamics in Caenorhabditis elegans. Current Biology. 2019. pp. 1286–1300.e4. 10.1016/j.cub.2019.02.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Joseph BB, Wang Y, Edeen P, Lažetić V, Grant BD, Fay DS. Control of clathrin-mediated endocytosis by NIMA family kinases. PLoS Genet. 2020;16: e1008633 10.1371/journal.pgen.1008633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lažetić V, Joseph BB, Bernazzani SM, Fay DS. Actin organization and endocytic trafficking are controlled by a network linking NIMA-related kinases to the CDC-42-SID-3/ACK1 pathway. PLOS Genetics. 2018. p. e1007313 10.1371/journal.pgen.1007313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lažetić V, Fay DS. Conserved Ankyrin Repeat Proteins and Their NIMA Kinase Partners Regulate Extracellular Matrix Remodeling and Intracellular Trafficking in Caenorhabditis elegans. Genetics. 2017;205: 273–293. 10.1534/genetics.116.194464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kaplan OI, Doroquez DB, Cevik S, Bowie RV, Clarke L, Sanders AAWM, et al. Endocytosis Genes Facilitate Protein and Membrane Transport in C. elegans Sensory Cilia. Curr Biol. 2012;22: 451–460. 10.1016/j.cub.2012.01.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vaart A van der, van der Vaart A, Rademakers S, Jansen G. DLK-1/p38 MAP Kinase Signaling Controls Cilium Length by Regulating RAB-5 Mediated Endocytosis in Caenorhabditis elegans. PLOS Genetics. 2015. p. e1005733 10.1371/journal.pgen.1005733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kubo T, Hirono M, Aikawa T, Kamiya R, Witman GB. Reduced tubulin polyglutamylation suppresses flagellar shortness in Chlamydomonas. Mol Biol Cell. 2015;26: 2810–2822. 10.1091/mbc.E15-03-0182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hao L, Thein M, Brust-Mascher I, Civelekoglu-Scholey G, Lu Y, Acar S, et al. Intraflagellar transport delivers tubulin isotypes to sensory cilium middle and distal segments. Nat Cell Biol. 2011;13: 790–798. 10.1038/ncb2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Das A, Dickinson D, Wood C, Goldstein B, Slep KC. The CHE-12 protein family uses a TOG domain array to regulate microtubules in the primary cilium. MOLECULAR BIOLOGY OF THE CELL. AMER SOC CELL BIOLOGY 8120 WOODMONT AVE, STE 750, BETHESDA, MD 20814–2755 USA; 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sanders A, Cevik S, Kida K, Bowie R, Blacque O. Investigation of a novel cilia-related gene K04F10.2/KIAA0556 in C. elegans. Cilia. 2012. 10.1186/2046-2530-1-s1-p43 [DOI] [Google Scholar]
- 78.Chang J, Baloh RH, Milbrandt J. The NIMA-family kinase Nek3 regulates microtubule acetylation in neurons. J Cell Sci. 2009;122: 2274–2282. 10.1242/jcs.048975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.O’regan L, Blot J, Fry AM. Mitotic regulation by NIMA-related kinases. Cell Div. 2007;2: 25 10.1186/1747-1028-2-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jackson PK. Nek8 couples renal ciliopathies to DNA damage and checkpoint control. Molecular cell. 2013. pp. 407–408. 10.1016/j.molcel.2013.08.013 [DOI] [PubMed] [Google Scholar]
- 81.Bryan MS, Argos M, Andrulis IL, Hopper JL, Chang-Claude J, Malone KE, et al. Germline Variation and Breast Cancer Incidence: A Gene-Based Association Study and Whole-Genome Prediction of Early-Onset Breast Cancer. Cancer Epidemiol Biomarkers Prev. 2018;27: 1057–1064. 10.1158/1055-9965.EPI-17-1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Keller BM, McCarthy AM, Chen J, Armstrong K, Conant EF, Domchek SM, et al. Associations between breast density and a panel of single nucleotide polymorphisms linked to breast cancer risk: a cohort study with digital mammography. BMC Cancer. 2015;15: 143 10.1186/s12885-015-1159-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.García-Díez I, Hernández-Muñoz I, Hernández-Ruiz E, Nonell L, Puigdecanet E, Bódalo-Torruella M, et al. Transcriptome and cytogenetic profiling analysis of matched in situ/invasive cutaneous squamous cell carcinomas from immunocompetent patients. Genes Chromosomes Cancer. 2019;58: 164–174. Available: https://onlinelibrary.wiley.com/doi/abs/10.1002/gcc.22712 10.1002/gcc.22712 [DOI] [PubMed] [Google Scholar]
- 84.Casey JP, Brennan K, Scheidel N, McGettigan P, Lavin PT, Carter S, et al. Recessive NEK9 mutation causes a lethal skeletal dysplasia with evidence of cell cycle and ciliary defects. Hum Mol Genet. 2016;25: 1824–1835. 10.1093/hmg/ddw054 [DOI] [PubMed] [Google Scholar]
- 85.Mahjoub MR, Montpetit B, Zhao L, Finst RJ, Goh B, Kim AC, et al. The FA2 gene of Chlamydomonas encodes a NIMA family kinase with roles in cell cycle progression and microtubule severing during deflagellation. J Cell Sci. 2002;115: 1759–1768. Available: https://www.ncbi.nlm.nih.gov/pubmed/11950892 [DOI] [PubMed] [Google Scholar]
- 86.Bradley BA. A NIMA-related kinase, Cnk2p, regulates both flagellar length and cell size in Chlamydomonas. Journal of Cell Science. 2005. pp. 3317–3326. 10.1242/jcs.02455 [DOI] [PubMed] [Google Scholar]
- 87.Westermann S, Weber K. Identification of CfNek, a novel member of the NIMA family of cell cycle regulators, as a polypeptide copurifying with tubulin polyglutamylation activity in Crithidia. J Cell Sci. 2002;115: 5003–5012. 10.1242/jcs.00170 [DOI] [PubMed] [Google Scholar]
- 88.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77: 71–94. Available: https://www.ncbi.nlm.nih.gov/pubmed/4366476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.WormBook. [cited 27 Jan 2020]. Available: http://wormbook.org/
- 90.Doitsidou M, Jarriault S, Poole RJ. Next-Generation Sequencing-Based Approaches for Mutation Mapping and Identification in Caenorhabditis elegans. Genetics. 2016;204: 451–474. 10.1534/genetics.115.186197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang Y, Wang JT, Rasoloson D, Stitzel ML, O’ Connell KF, Smith HE, et al. Identification of suppressors of mbk-2/DYRK by whole-genome sequencing. G3. 2014;4: 231–241. 10.1534/g3.113.009126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Homer N. Bfast: Blat-like fast accurate search tool. 2009. Available: https://vcru.wisc.edu/simonlab/bioinformatics/programs/bfast/bfast-book.pdf [Google Scholar]
- 93.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009. pp. 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38: e164 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huang S, Holt J, -Y. Kao C, McMillan L, Wang W. A novel multi-alignment pipeline for high-throughput sequencing data. Database. 2014. pp. bau057–bau057. 10.1093/database/bau057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.UniProt. [cited 27 Jan 2020]. Available: https://www.uniprot.org/
- 97.interpro7-client. [cited 27 Jan 2020]. Available: https://www.ebi.ac.uk/interpro/
- 98.ScanProsite. [cited 27 Jan 2020]. Available: https://prosite.expasy.org/scanprosite/
- 99.Protein BLAST: search protein databases using a protein query. [cited 27 Jan 2020]. Available: https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins
- 100.WormWeb.org—Exon-Intron Graphic Maker—by nikhil bhatla. [cited 27 Jan 2020]. Available: http://wormweb.org/exonintron
- 101.DOG 2.0—Protein Domain Structure Visualization. [cited 27 Jan 2020]. Available: http://dog.biocuckoo.org/
- 102.Ren J, Wen L, Gao X, Jin C, Xue Y, Yao X. DOG 1.0: illustrator of protein domain structures. Cell Res. 2009;19: 271–273. 10.1038/cr.2009.6 [DOI] [PubMed] [Google Scholar]
- 103.BoxShade Server. [cited 27 Jan 2020]. Available: https://embnet.vital-it.ch/software/BOX_form.html
- 104.Kimura Y, Tsutsumi K, Konno A, Ikegami K, Hameed S, Kaneko T, et al. Environmental responsiveness of tubulin glutamylation in sensory cilia is regulated by the p38 MAPK pathway. Sci Rep. 2018;8: 8392 10.1038/s41598-018-26694-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.ImageJ. [cited 27 Jan 2020]. Available: https://imagej.net/Welcome
- 106.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA 3rd, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6: 343–345. 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
- 107.Dokshin GA, Ghanta KS, Piscopo KM, Mello CC. Robust Genome Editing with Short Single-Stranded and Long, Partially Single-Stranded DNA Donors in Caenorhabditis elegans. Genetics. 2018;210: 781–787. 10.1534/genetics.118.301532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Paix A, Folkmann A, Goldman DH, Kulaga H, Grzelak MJ, Rasoloson D, et al. Precision genome editing using synthesis-dependent repair of Cas9-induced DNA breaks. Proc Natl Acad Sci U S A. 2017;114: E10745–E10754. 10.1073/pnas.1711979114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Haeussler M, Schönig K, Eckert H, Eschstruth A, Mianné J, Renaud J-B, et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17: 148 10.1186/s13059-016-1012-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.CRISPOR. [cited 27 Jan 2020]. Available: http://crispor.tefor.net/
- 111.WatCut: An on-line tool for restriction analysis, silent mutation scanning, SNP-RFLP analysis. [cited 27 Jan 2020]. Available: http://watcut.uwaterloo.ca/template.php?act=silent_new
- 112.Connolly AA, Osterberg V, Christensen S, Price M, Lu C, Chicas-Cruz K, et al. Caenorhabditis elegans oocyte meiotic spindle pole assembly requires microtubule severing and the calponin homology domain protein ASPM-1. Mol Biol Cell. 2014;25: 1298–1311. 10.1091/mbc.E13-11-0687 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
For experiments involving the mei-1(or1178) temperature-sensitive allele, eggs from plates kept at the permissive temperature (15°C; [112]) were picked to fresh plates and shifted to 25°C after hatching for 4 days before scoring.
(TIF)
(TIF)
a-b. Localization of NEKL-4::mNeonGreen in adult males. Tail is flat against the coverslip in a. to visualize ray dendrites. Tail in b. is folded but the filamentous pattern in ray cell bodies is visible. c-d. Localization of NEKL-4::mNeonGreen in L4 molt males. Both images are different sections from the same z-stack. Arrows indicate ray cell bodies, arrowheads indicate ray dendrites (numbered). Scale = 10μm.
(TIF)
Scale bar = 10μm.
(TIF)
a. Example images of polyE stained cilia in the nose. The polyE polyclonal antibody stained amphid ciliary middle segments (MS) and labial and cephalic cilia (LC), similar to GT335. Abnormal polyE staining was visible in some strains containing the ccpp-1 deletion mutation. Loss of TTLL-11 abolished polyE staining, but in the absence of TTLL-4 or TTLL-5, polyE staining remained. Scale = 10μm. b. Quantification of polyE staining phenotypes by genotype; number of animals examined indicated at right.
(TIF)
Time lapse of a single z slice with widefield microscope. Images acquired at 4.34 fps and shown at 20 fps.
(MP4)
Strains contain the allele mentioned and the ccpp-1(ok1821) deletion. At least 20 animals were tested for Dye filling for each strain. Although a single P0 plate produced my42 and my43, we considered them independent isolates because their phenotypes are different from one another.
(XLSX)
GKO; Gene Knockout Consortium. CGC; Caenorhabditis Genetics Center. NBRP; National Bio-Resource Project.
(XLSX)
+ indicates any degree of suppression of the ccpp-1 phenotype,—indicates no suppression of the ccpp-1 phenotype, n/a indicates not tested.
(XLSX)
Based on phosphoproteomics data for NEK10 from Chivukula et al, Fig 4F [26]. Original list contains genes with phosphopeptides depleted more than twofold upon NEK10 deletion in human bronchial epithelial cell (HBEC) culture. This list contains genes with known C. elegans homologs from the following relevant classes in the NEK10 list: axonemal dyneins and assembly factors, kinesins, intraflagellar transport, ciliary length control.
(XLSX)
All strains were generated in the Barr laboratory except CB1490 (Hodgkin laboratory), ROH11 and ROH12 (O’Hagan laboratory).
(XLSX)
All strains were generated in the Barr laboratory except BS3383, which was obtained from the Caenorhabditis Genetics Center (CGC).
(XLSX)
(DOCX)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.