

Abstract
The Type I Interferons (IFN-Is) are innate antiviral cytokines that include 12 different IFNα subtypes and IFNβ that signal through the IFN-I receptor (IFNAR), inducing hundreds of IFN-stimulated genes (ISGs) that comprise the ‘interferome’. Quantitative differences in IFNAR binding correlate with antiviral activity, but whether IFN-Is exhibit qualitative differences remains controversial. Moreover, the IFN-I response is protective during acute HIV-1 infection, but likely pathogenic during the chronic stages. To gain a deeper understanding of the IFN-I response, we compared the interferomes of IFNα subtypes dominantly-expressed in HIV-1-exposed plasmacytoid dendritic cells (1, 2, 5, 8 and 14) and IFNβ in the earliest cellular targets of HIV-1 infection. Primary gut CD4 T cells from 3 donors were treated for 18 hours ex vivo with individual IFN-Is normalized for IFNAR signaling strength. Of 1,969 IFN-regulated genes, 246 ‘core ISGs’ were induced by all IFN-Is tested. However, many IFN-regulated genes were not shared between the IFNα subtypes despite similar induction of canonical antiviral ISGs such as ISG15, RSAD2 and MX1, formally demonstrating qualitative differences between the IFNα subtypes. Notably, IFNβ induced a broader interferome than the individual IFNα subtypes. Since IFNβ, and not IFNα, is upregulated during chronic HIV-1 infection in the gut, we compared core ISGs and IFNβ-specific ISGs from colon pinch biopsies of HIV-1-uninfected (n = 13) versus age- and gender-matched, antiretroviral-therapy naïve persons with HIV-1 (PWH; n = 19). Core ISGs linked to inflammation, T cell activation and immune exhaustion were elevated in PWH, positively correlated with plasma lipopolysaccharide (LPS) levels and gut IFNβ levels, and negatively correlated with gut CD4 T cell frequencies. In sharp contrast, IFNβ-specific ISGs linked to protein translation and anti-inflammatory responses were significantly downregulated in PWH, negatively correlated with gut IFNβ and LPS, and positively correlated with plasma IL6 and gut CD4 T cell frequencies. Our findings reveal qualitative differences in interferome induction by diverse IFN-Is and suggest potential mechanisms for how IFNβ may drive HIV-1 pathogenesis in the gut.
Author summary
The Type I Interferons (IFN-Is) serve as the first line of defense against viral infections. IFN-Is are evolutionarily diverse, with 12 distinct IFNα subtypes and IFNβ in humans. All IFN-Is bind to the same receptor, but it remains unclear whether distinct IFN-Is will trigger the same set of IFN-stimulated genes. Here, we provide evidence that diverse IFN-Is altered gene expression in gut CD4 T cells in different ways. Specifically, IFNβ induced a broader gene expression profile than individual IFNα subtypes. Genes uniquely induced by IFNβ in gut CD4 T cells ex vivo were downregulated in the gut during chronic HIV-1 infection. This downregulation correlated with markers of inflammation and immune dysfunction. Our data unravel qualitative differences between the IFN-Is and suggest a complex picture of how IFNβ may be driving HIV-1 pathogenesis in the gastrointestinal tract.
Introduction
The type I interferons (IFN-Is) are innate antiviral cytokines that include IFNα (12 different subtypes) and IFNβ [1]. These cytokines significantly inhibited HIV-1 replication in vitro, but human clinical trials with IFNα2 showed only moderate or no inhibitory effects on HIV-1 infection [2, 3]. All IFN-Is bind to the same IFN-I receptor that is composed of two subunits, IFNAR1 and IFNAR2, resulting in phosphorylation of JAK1 and TYK2. This in turn results in the phosphorylation of STAT1 and STAT2 that associate with IRF9 to form the transcriptional activator, ISGF3 [4]. ISGF3 translocates to the nucleus, where it binds to promoters of genes encoding IFN response elements (ISREs), resulting in the induction of hundreds of IFN stimulated genes (ISGs), collectively referred to as the ‘interferome’[5].
Recent studies highlighted IFNα as a potential adjunct to an HIV-1 curative strategy [6–10]. However, almost all HIV-1 clinical trials with IFNα were performed with only one subtype, IFNα2, with mixed results (reviewed in [2, 3]). More recently, IFNα2 treatment of SIV rhesus macaques under antiretroviral therapy did not reduce the latent HIV-1 reservoir [11]. Our group and others reported that IFNα2 only moderately inhibited HIV-1 in humanized mice and primary CD4+ T cells compared to the more potent subtypes IFNα6, IFNα14 and IFNα8 [12–15]. Interestingly, the anti-HIV-1 potency of IFNα subtypes correlated with their binding affinity to IFNAR2, suggesting that the antiviral differences between the IFNα subtypes were due to quantitative differences in IFNAR signaling strength [12, 15, 16]. However, several human IFNα subtypes exhibit strong signals of purifying selection [17], suggesting that these IFNα subtypes may have evolved to have specific, nonredundant functions. Some IFNα subtypes were better at inducing certain immune responses in vivo [13, 18] and may trigger distinct intracellular signaling pathways [19]. Nevertheless, the notion of qualitative differences between IFNα subtypes remains controversial [20, 21]. One reason is that most comparative studies normalized IFN-Is using protein amounts or Units/ml based on inhibition of vesicular stomatitis virus or encephalomyocarditis virus [1]. To unravel qualitative differences between the IFN-Is, it would be important to normalize IFN-Is for quantitative differences in IFNAR signaling strength.
It is widely accepted that IFN-I signaling can prevent acute retrovirus infection. Genetic ablation of IFNAR resulted in higher Friend retrovirus replication in mice co-infected with lactate-dehydrogenase elevating virus, a potent IFN-I inducer [22]. Moreover, IFNAR blockade increased SIV replication in rhesus macaques [23, 24]. Administration of IFNα2 decreased retrovirus replication in mice, monkeys and humans [3, 18, 23]. However, in persistent virus infections, chronic IFN-I stimulation was linked to pathogenic outcomes [25–29]. Although clinical administration of IFNα2 increased the number of low-dose mucosal inoculations needed for breakthrough SIV infection in rhesus macaques, once the infection was established, lower CD4 T cell counts were observed in IFNα2-treated monkeys relative to untreated controls [23]. These findings highlight that HIV-1 infection shares features with ‘interferonopathies’ such as Aicardi-Goutières Syndrome [30], which are currently being targeted through IFN-I blockade strategies (clinicaltrial.gov NCT03921554). In fact, IFNAR blockade during chronic HIV-1 infection in humanized mice restored immune function, leading to better HIV-1 control [31, 32]. Neutralization of (most) IFNα subtypes in SIV-infected rhesus macaques prior to infection increased viral loads as expected, but also decreased subsequent immune activation profiles [24]. By contrast, blockade of IFN-I signaling in chronic SIV-treated and untreated rhesus macaques decreased inflammation profiles associated with ISGs but did not reverse T cell exhaustion or activation [33].
The basis for the protective versus pathogenic effects of IFN-Is remains unclear. One hypothesis is that the initial IFN-I response is protective due to the induction of antiviral factors, whereas chronic stimulation promotes inflammation through other ISGs with sustained, elevated expression. Distinct IFN-Is present in the acute versus chronic stages of persistent viral infection may induce divergent cellular immune responses. Tissue compartmentalization may also play a role. The gut is a critical site not only for early HIV-1 infection, but also in driving chronic immune activation [34]. Epithelial barrier dysfunction, partly due to the loss of Th17 cells, leads to the translocation of enteric bacteria from the gut lumen to the lamina propria and systemic circulation, resulting in chronic immune activation [35–37]. We recently reported increased IFNβ gene expression in the gut, but not the blood, in persons with HIV-1 (PWH) infection compared to age/gender-matched HIV-1 uninfected controls [38]. By contrast, IFNα subtypes were downregulated in PWH, and IFNλ, a type III IFN linked to mucosal immunity in mouse models [39], was undetectable in these samples [38]. These findings suggest that among the diverse IFN-Is, IFNβ may play a dominant role in the gut during chronic HIV-1 infection.
Here, we utilized transcriptomic approaches to evaluate whether the IFNα subtypes and IFNβ exhibit qualitatively different effects on gene expression. We then tracked how interferon-regulated genes were altered during chronic HIV-1 infection in the gut. Our analyses highlight potential mechanisms driven by IFNβ that may influence mucosal HIV-1 pathogenesis.
Results
IFNβ potently inhibits HIV-1 replication in lamina propria mononuclear cells (LPMCs)
The relative anti-HIV-1 activity of IFNβ in primary LPMCs remains unclear, though studies using PBMCs suggest that IFNβ is relatively potent [40]. We previously reported a spectrum of antiviral potencies of the 12 IFNα subtypes against HIV-1 in primary LPMCs [12]. These data were extended to show a 10-fold difference in 50% inhibitory concentrations (IC50) between IFNα14 and IFNα2 [13].
Using the same 6 LPMC donors used to calculate the IC50s of IFNα14 and IFNα2, we titrated the anti-HIV-1 potencies of IFNα1 (weak), IFNα8 (potent) and IFNβ. The IFN-Is were added into LPMC cultures at the time of infection with HIV-1BaL. At 4 d post-infection, the percentage of HIV-1 Gag p24+ cells were evaluated by flow cytometry. Sigmoidal curves were fitted into the average inhibition data for 6 donors, and used to calculate IC50 values (pg/ml protein concentration). We also calculated a metric known as ‘Vres’, which corresponds to the level of residual virus replication at maximal doses of IFN-Is [41].
For comparison, previously reported inhibition curves for IFNα14 and IFNα2 are also shown [13]. IFNα8 showed IC50 concentrations over 10-fold lower than that of IFNα1 (Fig 1). Notably, IFNβ had a similar potency as IFNα8 and IFNα14. We also calculated IC50s for individual donors (S1A Fig) and show significantly lower inhibition by IFNα2 compared to IFNα8 and IFNα14 (S1B Fig). IFNα8 and IFNα14 reduced HIV-1 infection levels to ~30% at the maximal doses tested (10 ng/ml), whereas IFNα1, IFNα2 and IFNβ had higher Vres between 38–42%. However, these differences in Vres were not significant (S1C Fig). These findings validate prior results on the relative anti-HIV-1 activity of these IFNα subtypes and highlight IFNβ as a potent anti-HIV-1 IFN. Quantitative differences were evident, as increasing the dose of weaker IFNα subtypes should enable these cytokines to inhibit HIV-1 just as well as the potent IFNα subtypes.
Fig 1. HIV-1 inhibition curve of IFNβ.
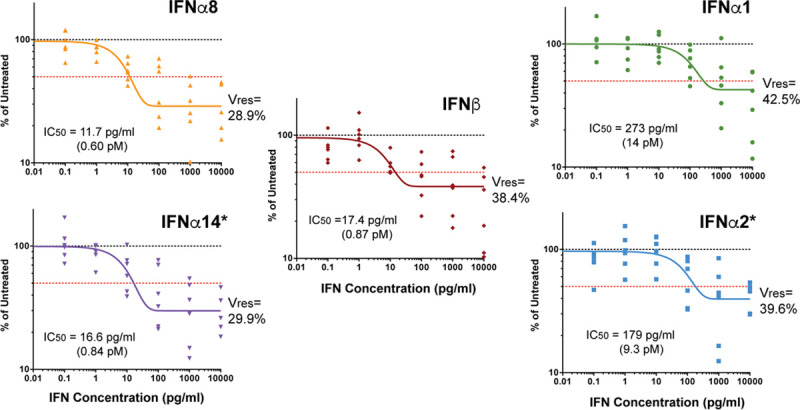
LPMCs (n = 6 donors) were infected with HIV-1BaL then resuspended with various doses of IFN-Is. After 4 d, the frequencies of HIV-1 p24+ cells were evaluated on CD3+CD8- cells via flow cytometry. Data were normalized to mock (untreated) as 100% for each donor. Dose-response curves were generated using a one-phase decay equation in GraphPad Prism 5.0 to determine IC50 values. Vres, the percentage of cells that remain infected relative to the mock at the maximum dose tested, corresponded to plateau values from the decay equation. *Note that data on IFNα14 and IFNα2 were previously published [13].
IFNα subtypes and IFNβ exhibit quantitative differences in ISRE-activity
The stronger anti-HIV-1 potencies of IFNα14 and IFNα8 compared to IFNα2 and IFNα1 were associated with higher ISG induction [12], but it remained unknown how the other IFNα subtypes and IFNβ compare. To test the ISRE signaling activity of IFN-Is, we used a commercially-available iLite assay (see Methods). The iLite cells are human U937 monocytic cell lines transduced with a luciferase reporter downstream of an ISRE from ISG15, a canonical ISG. Serial 10-fold dilutions of the 12 IFNα subtypes and IFNβ were added into the iLite cells and relative light units were measured after 18 h. 50% effective concentrations (EC50) were then calculated from best-fit sigmoidal plots.
Fig 2A highlights the 482-fold EC50 difference in ISRE-activity between IFNα14 and IFNα1. The EC50s of the other IFNα subtypes and IFNβ fell in-between IFNα14 and IFNα1 (Fig 2B). Notably, the ISRE EC50 of the IFNα subtypes significantly correlated with anti-HIV-1 potency data from our previous study (Fig 2C) [12]. A significant positive correlation was also observed between ISRE-activity and the IC50 values from the 5 IFN-Is tested in Fig 1 that includes IFNβ (R2 = 0.87, p = 0.02). Importantly, ISRE EC50 values correlated strongly with published IFNAR2 binding affinity values when comparing the IFNα subtypes (Fig 2D) [16]. However, the inclusion of IFNβ, which was reported to have a higher IFNAR2 binding affinity than multiple IFNα subtypes [42, 43], abolished the correlation (Fig 2E). These data demonstrate that ISRE-activity as measured by the iLite assay can be used to evaluate quantitative differences between the IFN-Is, particularly for the IFNα subtypes.
Fig 2. ISRE-activity of IFN-Is.

IFNα subtypes and IFNβ were titrated in iLite cells, a luciferase reporter cell line encoding the ISG15-ISRE. (A) Determination of ISRE-activity. Serial dilutions of IFN-Is were incubated with iLite cells and luciferase readings were determined after 18 h. In this example, the titration curves for IFNα1 and IFNα14 are shown, showing a 482-fold difference in ISRE EC50 values. (B) ISRE-activity of IFN-Is. EC50 values are shown for all IFN-Is tested. IFNα subtypes that had potent activity against HIV-1 in a previous study are highlighted in black. IFNβ is highlighted in green. Note that the EC50s are negative log values; thus the higher the bar, the less concentration is needed to achieve an EC50. (C) ISRE-activity versus HIV-1 inhibition. HIV-1 inhibition values were previously reported [12] showing % inhibition of HIV-1 p24+ cells relative to mock in LPMC cultures. ISRE-activity versus IFNAR2 binding affinity (D) without or (E) with IFNβ. IFNAR2 binding affinity data were previously reported using surface plasmon resonance. For panels C to E, linear regression curves were plotted using Prism 5.0 and evaluated using Pearson statistics.
Identification of Novel IFN-Regulated Genes (IRGs) based on RNAseq profiling
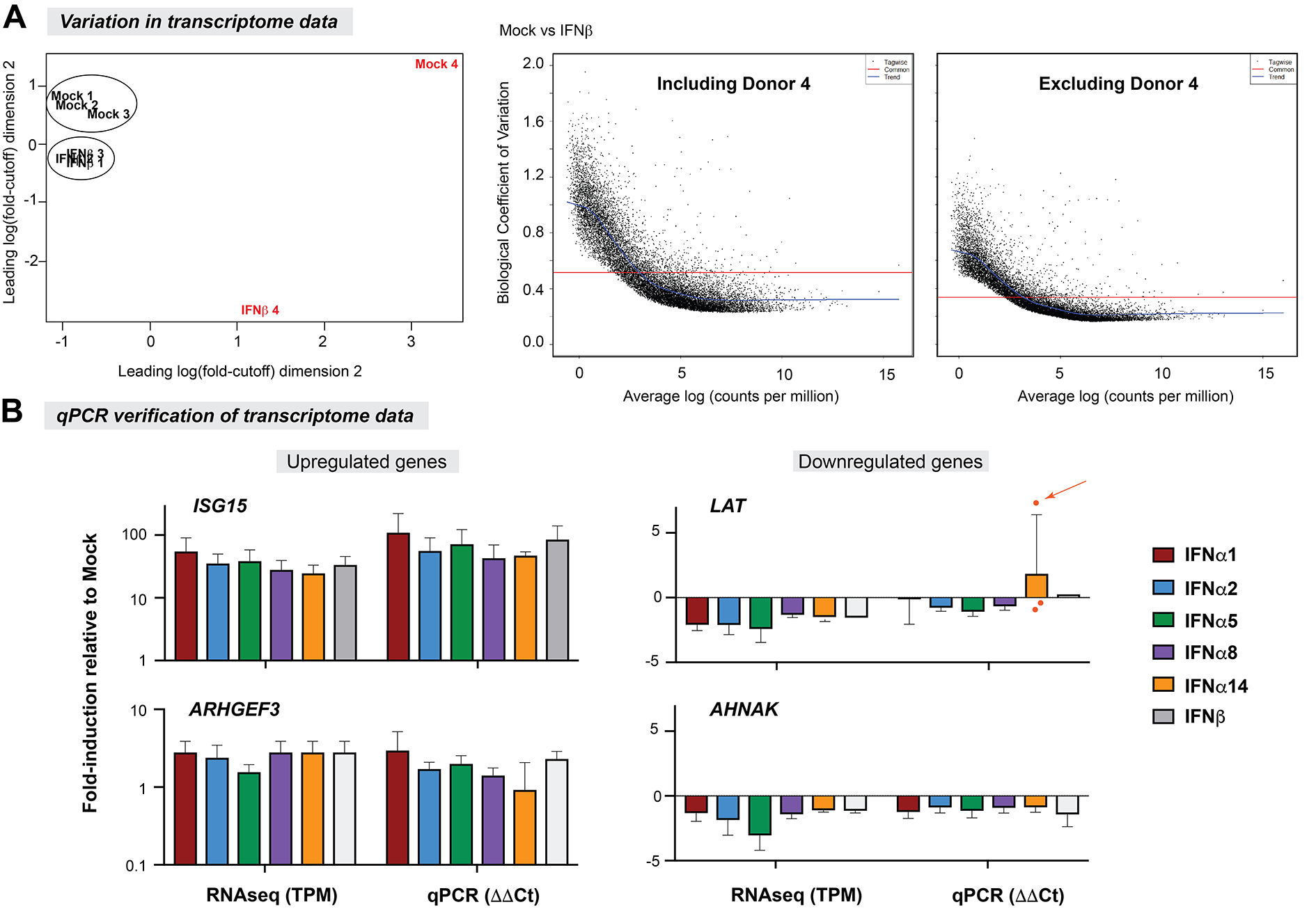
Our data in Figs 1 and 2, as well as data from other studies [12–15], provide strong evidence for quantitative differences among the IFN-Is. To uncouple quantitative versus qualitative differences, we normalized our IFN-I treatments for ISRE-activity (Fig 2B) for unbiased transcriptomics. Purified gut CD4+ T cells from 4 different donors were treated with 100 pg/ml IFNα14, and the other IFN-Is were added at higher concentrations that match the ISRE-activity of this IFNα14 dose (e.g., 48.2 ng/ml IFNα1). Purified CD4+ T cells were evaluated instead of total LPMCs to reduce potential confounders due to cell type heterogeneity in LPMCs when performing RNAseq. CD4+ T cells account for majority of cells in LPMCs (65%) [44] and are the main cell types for the interaction between HIV-1 and antiviral ISGs. Given that only limited numbers of primary LPMCs can be obtained per donor, we selected IFNα1, IFNα2, IFNα5, IFNα8 and IFNα14, as these were highly expressed in HIV-1-exposed primary pDCs in vitro and in PBMCs during chronic HIV-1 infection in vivo [12, 38]. We also selected IFNβ because it is significantly upregulated in the gut during chronic HIV-1 infection [38]. RNA was extracted from IFN-I- and mock-treated gut CD4+ T cells after 18 h for RNA sequencing (RNAseq). Since a full HIV-1 replication cycle takes about 24–48 h [45, 46], the 18 h time point will likely capture ISG induction relevant to IFN-I-mediated HIV-1 inhibition. Gene counts were normalized using transcripts per million (TPM). One donor (donor 4) was removed because of a skewed transcriptome profile based on Principal Component Analyses and Biological Coefficient of Variation plots (S2A Fig). IFN regulated genes (IRGs) were defined based on a 1.5-fold change (FC) cutoff and a False Discovery Rate (FDR) of ≤20% (see Methods). We performed quantitative PCR (qPCR) on 4 IRGs identified via RNAseq: a highly upregulated gene (ISG15; >10-fold relative to mock), a moderately upregulated gene (ARHGEF3; <2-fold), and 2 downregulated genes (LAT, AHNAK; both <5-fold). The qPCR results were consistent with that of RNAseq (S2B Fig).
On average, we obtained 34.6 million (range: 10.4 to 167 million) sequence reads per sample (S1 Table). We first evaluated the transcripts per million (TPM) levels of ISG15, from which the ISRE was genetically linked to luciferase in the iLite assay. All IFNα subtypes tested and IFNβ induced ISG15 to similar levels (Fig 3A, S2B Fig). In fact, the treatment dose used also induced the canonical ISGs RSAD2 (viperin) and MX1 (Fig 3B), as well as OASL, BST2 and APOBEC3G (S3 Table) to similar extents. These data confirmed that the treatments were indeed normalized for ISRE-activity and IFNAR signaling strength. Overall, the 5 IFNα subtypes and IFNβ altered 1,969 genes. Upregulated genes (ISGs) were more prevalent (68–80% of IRGs) than downregulated genes across all IFN-Is (Fig 3C). We next compared our IRG set with IRGs catalogued in the INTERFEROME database [5]. A majority (70.8%) of the observed IRGs were found in the INTERFEROME database (Fig 3D). We then partitioned the data into individual IFN-Is. Strikingly, there were, on average, 2.7-fold more IFNβ-regulated genes than those regulated by any of the individual IFNα subtypes tested (Fig 3C). On average, IFNβ upregulated 2.4-fold and downregulated 3.9-fold more genes than the individual IFNα subtypes (Fig 3C). There was a strong correlation between IFNAR2 binding affinity and the number of IRGs or ISGs (R2>0.94, p<0.01), but these correlations were lost if IFNβ was removed (p<0.05). In addition, our current analysis revealed 578 novel IRGs (S2 Table). Many of these novel IRGs may not encode protein products and/or have tentative gene designations, potentially explaining why these genes are not in the INTERFEROME database. However, some long non-coding RNAs such as NRIR (negative regulator of the interferon response) [47] and BISPR (BST2 interferon stimulated positive regulator) [48] could have critical roles for modulating IFN-I responses. Some repressed protein-coding genes such as CCR9 appeared specific to just one IFNα subtype (S2 Table).
Fig 3. Identification of Novel IRGs.

LPMCs (n = 3 donors) were stimulated with IFNα subtypes and IFNβ normalized to ISG15 ISRE-activity and the transcriptomes were evaluated at 18 h via RNAseq. Sequences were compared against annotated Ensembl genes (version GRCh38). IRGs were defined based on a 1.5-fold change and an FDR cut-off of 20% relative to mock. Induction of (A) ISG15 and (B) RSAD2 and MX1 by IFN-Is based on the RNAseq data. (C) Number of upregulated versus downregulated IRGs. (D) Overlap of gut CD4+ T cell ‘interferomes’ with published IRGs in the INTERFEROME database 2.0 [5].
Qualitative biological differences of IFNα subtypes revealed by transcriptomic profiling
The IFNα subtypes are homologous genes that activate IFNAR, raising questions on whether their differential effects were primarily quantitative. We postulated that if the differences between the IFNα subtypes are mainly quantitative, then we should observe a substantial overlap between the IRGs of cells treated with different IFNα subtypes that were normalized for ISRE-activity. The number of IRGs that overlapped between any two of the 5 tested IFNα subtypes ranged from 59% (IFNα2 vs IFNα5) to 82% (IFNα2 vs IFNα1) (Fig 4A). This included a core set of 266 IRGs altered by all 5 IFNα subtypes tested (Fig 4B; S3 Table). Interestingly, many additional genes appeared to be specific to an IFNα subtype, particularly for IFNα5 and IFNα14 (Fig 4B and S4 Table). To test if the IRGs unique to each IFNα subtype could have been regulated by other IFNα subtypes but excluded due to a stringent FDR cut-off of 20%, we investigated the median FDR of these unique genes against the other 4 IFNα subtypes. This analysis is illustrated in S3 Fig, where the FDR values of each of the 201 IFNα5-specific genes or 257 IFNα14-specific genes (Fig 4B) were plotted against the gene induction datasets for the other IFNα subtypes tested. As shown, majority of the IFNα5 or IFNα14-specific genes had FDR values that were >90% in the other IFNα subtypes. These analyses were expanded on S5 Table, where IFNα-specific IRGs had a median FDR of at least 75%, with most >90%, when tested against genes sets from other IFNα subtypes. Thus, IRGs that were differently expressed by a specific IFNα subtype were unlikely to be significantly altered by the other IFNα subtypes.
Fig 4. Interferome Differences between the IFNα subtypes.

(A) Percentage of shared IRGs. The denominators used for the top diagonal half were the left column, whereas the denominators used for the lower diagonal half were the top rows. For example: 79.0% of IFNα2-regulated genes were found among IFNα1-RGs, whereas 59.7% of IFNα1-RGs were found among IFNα2-RGs. (B) Euler diagram showing the interferome overlap between the 5 IFNα subtypes tested. A core set of 266 IRGs altered by all 5 IFNα subtypes were detected. IFNα-subtype specific genes were highlighted (e.g., 243 for IFNα14, 201 for IFNα5). The total number of IRGs (n = 1,257) include genes not shown that were shared between 2 to 4 IFNα subtypes. (C) Heatmap of IRGs from distinct IFNα subtypes. Highlighted areas in red correspond to core ISGs, whereas those in blue correspond to genes differentially regulated by IFNα5 relative to the four other IFNα subtypes tested. (D) Ingenuity Pathway Analyses of IFNα subtypes, highlighting Z-scores for shared pathways and those predicted to be specific to IFNα5 and IFNα14.
We next evaluated trends as to whether IRGs altered by at least one IFNα subtype (a total of 1,257 genes) were similarly upregulated or downregulated. These comparisons were based on mean fold-induction values that can be visualized in the heatmap (Fig 4C). Surprisingly, a large number of genes (n = 367) that trended to be upregulated by IFNα1, IFNα2, IFNα8 and IFNα14 appeared to be downregulated by IFNα5 (Fig 4C, S6 Table).
As the IRGs were based on an arbitrary cut-off (≥1.5× relative to mock, FDR ≤20%), we next evaluated whether increasing our FC criteria changed the differential expression patterns. At 2×, 2.5× and 3× cut-offs, we still observed genes that were differentially regulated by IFNα5 and IFNα14 (S4 Fig). At a 3× cut-off, we no longer observed the differentially downregulated IFNα5 genes (S5 Fig). Of note, increasing the FC cut-off to ≥2.0 excluded the known ISGs TRIM56, APOBEC3G and NLRC5. For subsequent analyses, we thus utilized a FC cut-off of 1.5.
To determine whether IFNα subtypes induced molecular programs distinct from each other, we subjected the IRGs to Ingenuity Pathway Analyses (IPA) [49]. S7 Table provides a ranked list of activated and inhibited pathways for each IFN-I. As expected, ‘Interferon signaling’ and ‘Pattern Recognition Receptors’ were the top induced pathways predicted for the 5 IFNα subtypes tested (Fig 4D). Death Receptor, NFκB and Inflammasome signaling were also highly induced by all IFNα subtypes (S7 Table). Interestingly, IPA also predicted distinct pathways induced by the IFNα subtypes. CD28 signaling, CTL and NK function, p38 MAPK signaling and sumoylation were predicted for IFNα14 but not the other IFNα subtypes (S7 Table and Fig 4D). Only the IFNα5 interferome was associated with downregulated apoptosis, PPAR and LXR/RXR activation, likely due to some downregulated genes such as CASP9 (S6 Table). These differential predictions provide confirmatory evidence of qualitative biological differences in endpoint effector mechanisms induced by different IFNα subtypes.
IFNβ induces a broader interferome compared to the 5 IFNα subtypes combined
Our data in Fig 3C revealed ~970 more IRGs for IFNβ than the individual IFNα subtypes. We pooled IFNα regulated genes independently of the subtype and compared them to IFNβ-regulated genes. Almost half (46.4%) of IFNβ interferome genes were not regulated by any of the 5 IFNα subtypes (Fig 5A). These included cytokines and cytokine receptor genes such as IL2, IFNGR1, IL21R and TGFB1 (Fig 5A; S8 Table). We compared the IPA results for the IFNα subtypes versus IFNβ regulated genes. IFNβ induced more pathways (Fig 5B) than the IFNα subtypes, such as ‘Th2 pathway’, ‘ERK/MAPK signaling’ and ‘Regulation of actin motility’. These findings indicated that IFNβ induced a broader interferome than IFNα subtypes. Moreover, compared to IFNα, IFNβ likely regulated more cellular pathways in primary gut CD4+ T cells.
Fig 5. IFNβ induces a broader transcriptome than the IFNα subtypes.

(A) Interferome overlap between IFNα subtypes and IFNβ. IRGs from all 5 IFNα subtypes were combined, and compared to that of IRGs from IFNβ. The purple region corresponds to genes shared between the IFNα- and IFNβ-specific interferomes. A few ‘IFNβ-specific genes’ (yellow) were highlighted. (B) Predicted IFNβ pathways. Z-scores of various pathways predicted from IRGs of individual IFNα subtypes and IFNβ were compared. Rows labeled ‘Fig 4D’ corresponds to the ‘core pathways’ in Fig 4D.
Chronic HIV-1 infection in the gut is associated with a strong type I IFN response
We recently reported that during chronic, untreated HIV-1 infection, IFN-I inducible antiretroviral genes APOBEC3G, BST2 and MX2, as well as IFNβ, but not IFNα, were expressed to significantly higher levels when compared to HIV-1 uninfected individuals [38]. To expand on these findings, we performed RNAseq on these gut biopsies to more broadly investigate the nature of the IFN-I response in persons with HIV-1 infection (PWH). Table 1 provides the demographic characteristics of this clinical cohort, which included 19 PWH and 13 HIV-1 uninfected individuals [50]. On average, we processed 41.2 million sequence reads from colon biopsies per subject (Table 1). Filtered data were normalized using Transcripts Per Million (TPM), Trimmed Mean of M values (TMM) [51, 52] and DESeq2 [53] methods. Based on relative log expression plots [54], the TMM method was most efficient at removing unwanted variation (S6 Fig).
Table 1. Clinical Cohort of PWH versus HIV Uninfected Controls for Interferome Profiling.
| Patient | Blood | Colon | Plasma | Colon | Plasma | Plasma | Colon | Colon | Colon | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Code | Sex | Age | CD4a | CD4a | Viral Loadb | HIV RNAb | IL-6c | LPSc | IFNαd | IFNβd | RNAseqe |
| H124 | M | 48 | 400 | 17.21 | 8400 | 96367 | 4.00 | 17.92 | 1.42 | 0.49 | 26582093 |
| H132 | M | 25 | 532 | 23.35 | 26000 | 9766703 | 1.39 | 19.87 | 2.33 | 0.83 | 40017132 |
| H154 | F | 58 | 400 | 10.65 | 22000 | 33674734 | 5.09 | 14.49 | 1.27 | 1.08 | 36371772 |
| H88 | M | 44 | 836 | 13.60 | 133000 | 7711893 | 1.22 | - | 0.70 | 0.32 | 37883074 |
| H217 | M | 22 | 744 | 11.35 | 25200 | 877550 | 0.56 | 12.63 | 1.40 | 0.53 | 28424144 |
| H286 | F | 52 | 693 | 12.34 | 3850 | 5146 | 1.19 | 14.26 | 1.25 | 0.89 | 15826112 |
| H307 | M | 34 | 624 | 9.36 | 9180 | 58572835 | 1.16 | 15.06 | 1.74 | 0.75 | 36680004 |
| H323 | M | 54 | 429 | 15.14 | 9440 | 2638399 | 1.02 | 11.25 | 2.62 | 1.46 | 32581853 |
| H391 | F | 29 | 238 | 5.63 | 196000 | 63106379 | 1.52 | 27.02 | 1.60 | 0.91 | 22519787 |
| H428 | M | 28 | 460 | 13.66 | 25100 | 82064684 | 1.78 | 20.29 | 1.16 | 1.09 | 14458796 |
| H594 | M | 46 | 338 | 7.46 | 88600 | 17181454 | 1.55 | - | 1.63 | 0.85 | 114526468 |
| H622 | M | 33 | 340 | 12.36 | 112000 | 98345366 | 1.96 | 16.85 | 1.30 | 0.88 | 27481534 |
| H648 | M | 31 | 420 | 13.39 | 2880 | 235737 | 0.52 | 11.84 | 2.08 | 0.58 | 40795690 |
| H683 | M | 25 | 504 | 12.43 | 59500 | 228774501 | 0.43 | 19.34 | 1.32 | 0.57 | 14796014 |
| H819 | M | 27 | 527 | 14.50 | 4670 | 42395309 | 0.98 | 15.90 | 1.50 | 0.81 | 27989141 |
| H825 | M | 34 | 364 | 8.81 | 43200 | 2238746 | 0.60 | 16.48 | - | - | 19068317 |
| H839 | F | 39 | 250 | 8.34 | 64900 | 453116 | 3.98 | 19.04 | 1.25 | 0.98 | 27398968 |
| H965 | F | 26 | 221 | 5.21 | 155000 | 17123 | 1.43 | 16.70 | - | - | 7814434 |
| H998 | F | 25 | 782 | 12.77 | 119000 | 163678357 | 0.87 | 8.29 | 1.52 | 0.92 | 158592070 |
| C138 | M | 29 | 728 | 35.48 | - | - | 2.16 | 9.52 | 2.26 | 0.17 | 13485914 |
| C178 | M | 33 | 736 | 35.74 | - | - | 0.51 | 9.80 | 2.15 | 0.22 | 12991623 |
| C255 | M | 34 | 588 | 34.27 | - | - | 0.69 | 14.48 | 3.95 | 0.33 | 36937567 |
| C278 | M | 23 | 532 | 27.52 | - | - | 0.73 | 7.45 | 3.23 | 0.27 | 42178568 |
| C361 | F | 33 | 720 | 34.68 | - | - | 0.19 | 8.68 | 7.80 | 0.26 | 43047608 |
| C404 | F | 29 | 1071 | 27.14 | - | - | 1.35 | 9.70 | 8.92 | 1.03 | 63291613 |
| C493 | F | 28 | 672 | 37.26 | - | - | 0.19 | 5.99 | 2.99 | 0.45 | 24871985 |
| C582 | M | 54 | 976 | 49.89 | - | - | 0.7 | 8.13 | 2.23 | 0.42 | 54886951 |
| C708 | M | 47 | 468 | 32.03 | - | - | 0.27 | 14.11 | 8.96 | 0.47 | 34241151 |
| C716 | M | 27 | 1035 | 26.52 | - | - | 0.47 | 11.78 | 3.90 | 0.30 | 107139307 |
| C914 | F | 43 | 690 | 20.44 | - | - | 1.21 | 8.78 | 4.77 | 0.29 | 62956103 |
| C947 | M | 25 | 480 | 25.18 | - | - | 0.78 | 7.66 | - | - | 21578149 |
| C972 | F | 51 | 1035 | 22.12 | - | - | 0.48 | 11.93 | 1.98 | 0.19 | 71620117 |
a CD4 counts were based on number per μl for the blood, and as a percentage of CD45+ cells in the colon.
b Plasma viral loads were based on copies/ml, whereas colon HIV RNA levels were based on copies per CD4 T cell [40]
c Plasma levels of IL6 and LPS were based on pg/ml.
d Colon IFNα and IFNβ mRNA levels (copies per 104 x GAPDH) were quantified by qPCR as described [33]
e Illumina Sequence Read Counts for each colon biopsy sample following quality control (this study)
We next determined the expression levels in the colon biopsies in vivo of the 266 ‘core IRGs’ that were similarly regulated by the 5 IFNα subtypes and IFNβ in gut CD4+ T cells in vitro. The majority (92%; n = 246) of these core IRGs that passed the RNAseq filter criteria were ISGs (e.g., these genes were induced by the IFN-Is tested, not downregulated). Of these 246 core ISGs, 126 (51%) were significantly altered in HIV-1 infected versus uninfected individuals at 5% FDR (Fig 6A). The majority (89%) of these altered core ISGs were upregulated during chronic HIV-1 infection. These included the antiretroviral genes APOBEC3G, BST2 and MX2, consistent with our previous report [38]. Genes linked to innate sensing, immunomodulation and cell death/proliferation, as well as negative feedback regulation of the IFN-I response, were also expressed at significantly higher levels in PWH (Fig 6B and 6C, S9 Table).
Fig 6. Core ISGs significantly altered in chronic gut HIV-1 infection.

(A) Volcano Plot showing the FDR and fold-change criteria for altered core ISGs. Majority of the core ISGs were upregulated in PWH relative to HIV-uninfected controls. (B) Gene categories of altered core ISGs. (C) TMM values of representative antiviral, innate sensing, negative feedback and immunomodulatory genes. Floating bars correspond to min and max values, with the central line corresponding to the mean.
Inversion of IFNβ-specific ISGs during chronic HIV-1 infection in the gut
Since IFNβ induced a broader interferome than all IFNα subtypes tested (Fig 5A), we evaluated whether ISGs that were unique to IFNβ (IFNβ-specific ISGs) were also induced in the gut during chronic HIV-1 infection. Of the 712 IFNβ-specific IRGs, 57% (n = 406) were ISGs that passed the RNAseq filter criteria (Fig 3C). Nearly a third of these IFNβ-specific ISGs (28%; n = 112) were significantly altered in PWH compared to HIV-1 uninfected controls (Fig 7A). Only a few of the IFNβ-specific ISGs were significantly upregulated in PWH (Fig 7A). By contrast, the vast majority (>90% (n = 102) of the altered IFNβ-specific ISGs were downregulated during chronic HIV-1 infection in the gut (Fig 7A). These repressed IFNβ-specific ISGs are involved in intracellular vesicle trafficking, transcriptional/translational regulation, protein ubiquitination and transport (Fig 7B and 7C; S10 Table). Moreover, several known HIV-1 cofactors such as TSG101, NUP54, and CUL5 were downregulated [55–57]. We again emphasize that all these genes were induced by IFNβ in gut CD4 T cells ex vivo (S8 Table). Given that a great majority of these IFNβ-specific ISGs were downregulated in PWH, we conclude that a significant inversion of IFNβ-specific ISGs was observed during chronic HIV-1 infection in the gut.
Fig 7. Inversion of IFNβ-specific ISGs during chronic HIV-1 infection in the gut.

(A) Volcano Plot showing the FDR and fold-change criteria for altered IFNβ-specific ISGs. Majority of the IFNβ-specific ISGs were downregulated in PWH relative to HIV-uninfected controls. (B) Gene categories of altered IFNβ-specific ISGs. (C) TMM values of representative HIV-1 cofactors as well as immunomodulatory, trafficking/transport and transcriptional/translational regulation genes. Floating bars correspond to min and max values, with the central line corresponding to the mean.
We next investigated potential mechanisms for how these IFNβ-specific ISGs may have been downregulated in the gut in the presence of high IFNβ levels. Two known negative feedback regulators, USP18 [58] and UNC93B1 [59], were induced by all IFN-Is tested ex vivo (S3 Table). Gut IFNβ mRNA levels positively correlated with USP18 and UNC93B1 transcripts (S12 Table). However, none of the IFNβ-specific ISGs altered in PWH correlated with USP18 (S11 and S13 Tables). By contrast, 89% of these IFNβ-specific ISGs negatively correlated with UNC93B1 (S11 and S13 Tables). Thus, one potential mechanism for the inversion IFNβ-specific ISGs in the gut during chronic HIV-1 infection may be the IFNβ-mediated induction of UNC93B1.
Distinct interferomes are linked to immunopathogenic outcomes
The clinical study described in Table 1 involved cohorts where paired blood (PBMCs/plasma) and colon pinch biopsies (up to 30 per donor) were obtained. Colon pinch biopsies (~20) were pooled for same-day flow-based immunophenotyping [50, 60] and the rest were frozen for later histology and transcriptomics. The study obtained comprehensive data including gut and PBMC IFNα and IFNβ transcript levels, plasma and gut viral loads, gut CD4+ T cell percentages (of CD45+ cells), myeloid activation (CD40 MFI in CD1c+ myeloid DCs), blood CD4+ T cell counts, markers of microbial translocation (sCD27, LPS, LTA), inflammation (CRP, IL6), and epithelial barrier dysfunction (iFABP) (Table 1 and S11 Table) [38, 50, 60–63]. We investigated how individual altered core ISGs (n = 126) and IFNβ-specific ISGs (n = 112) correlated with these clinically relevant parameters using linear regression models, after adjusting the data for age and gender. We used a 5% FDR threshold for these associations.
Five clinically relevant parameters–gut IFNβ mRNA, plasma LPS, gut CD4 T cell frequencies, blood CD4 T cell counts and plasma IL6 levels–correlated significantly with the altered core and IFNβ-specific ISGs (Fig 8 and S11 Table). The expression of core and IFNβ-specific ISGs significantly correlated with transcript levels of IFNβ (>90%) rather than IFNα (<1%) (S11 Table). Interestingly, the directionality of the correlations was discordant between these 2 gene sets: higher IFNβ levels correlated with higher expression of 82% of core ISGs, whereas higher IFNβ levels were associated with lower expression of 88% of IFNβ-specific ISGs (Fig 8A). Both core ISGs and IFNβ-specific ISGs were significantly associated with plasma LPS levels, but again with discordant directionalities (Fig 8B). Gut CD4 T cell counts (as a percentage of CD45+ cells) were more significantly associated with core-ISGs (78% of genes negatively correlated) than IFNβ-specific ISGs (50% of genes positively correlated) (Fig 8C). By contrast, blood CD4 T cell counts were more significantly associated with IFNβ-specific ISGs (87% negatively correlated) than core ISGs (Fig 8D). Notably, the lower expression of a substantial fraction of IFNβ-specific ISGs (58%) was associated with higher levels of plasma IL6. None of the core ISGs correlated significantly with plasma IL6 at the 5% FDR cut-off.
Fig 8. Percentages of Altered Core and IFNβ-specific ISGs that correlated with clinically relevant parameters during chronic mucosal HIV-1 infection.

Correlations between the expression of individual core-ISGs (Fig 6A) and IFNβ-specific ISGs (Fig 7A) in gut biopsies from the clinical cohort were determined against (A) gut IFNβ transcripts, (B) plasma LPS levels, (C) gut CD4+ T cell percentages, (D) Blood CD4 T cell counts and (E) Plasma IL6 levels using linear regression models, controlling for age and gender. The clinical cohort included PWH (n = 19) and matched HIV uninfected controls (n = 13). Correlations with FDR ≤ 5% were considered significant; the proportion of those core and beta ISGs are plotted as pie charts, with red, blue and gray depicting positive, negative and no significant correlations, respectively. The top genes in the relevant categories with ≥50% representation are highlighted; a full list is available in S12 Table.
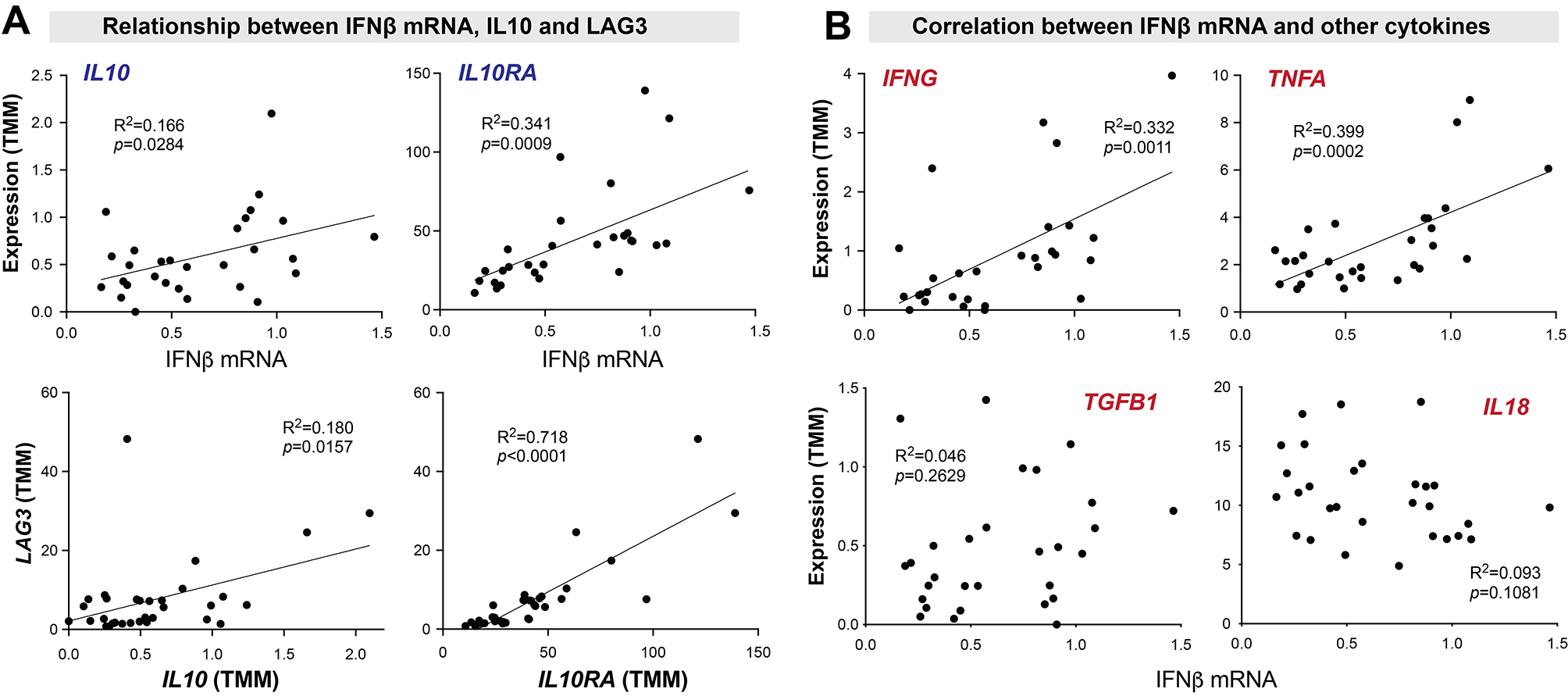
The complete list of genes that correlated with the 5 clinically relevant parameters (gut IFNβ mRNA, plasma LPS, gut CD4 T cell frequencies, blood CD4 T cell count and plasma IL6 levels) are described in S12 Table. We highlight several core ISGs with the highest correlations in Fig 9A, 9B, 9C and 9D (panels in the left half). IFNβ levels in the gut positively correlated with: (1) IRF9, a component of the ISGF3 complex (Fig 9A); (2) CD38, a marker of T cell activation (Fig 9B) [64]; (3) NLRC5, a transcriptional activator of MHC-I [65], which in turn present peptides for CD8+ T cell recognition; (4) PSMB9, a component of the immunoproteasome [66]; and (5) LAG3, a marker of T cell exhaustion [67, 68]. NLRC5 and LAG3 also positively correlated with plasma LPS levels and inversely correlated with the percentage of CD4+ T cells in the gut (Fig 9C and 9D). Since T cell exhaustion in persistent LCMV infection has been linked to the suppressive cytokine IL10 [26], we evaluated if IFNβ mRNA levels correlated with cytokine transcripts in the RNAseq dataset. Notably, IFNβ mRNA levels were associated with increased levels of IL10 and IL10RA, which were in turn correlated with LAG3 (S7A Fig). IFNβ mRNA levels also correlated with transcript levels of TNFα and IFNγ, but not IL18 and TGFβ (S7B Fig). These data suggest that increased IFNβ in the gut of chronic PWH may drive genes associated with sustained ISG expression, antigen processing, T cell activation, inflammation and immune exhaustion.
Fig 9. Core and IFNβ-specific ISGs that correlated with immunopathogenic markers of chronic HIV-1 infection.

(A-D; panels on the left half) Correlations between select Core ISGs (A) IRF9, (B) CD38, (C) NLRC5 and (D) LAG3 and clinically relevant parameters. (E-I; panels on the right half) Correlations between select IFNβ-specific ISGs (E) EIF4H, (F) SMAD4, (G) VIMP, (H) NUP54 and (I) MUS81 and clinically relevant parameters. Expression levels were based on TMM-transformed counts. Linear regression was performed in R statistical package, adjusting for age and gender. A full list of the proportion of core and IFNβ-specific genes that correlated with clinical parameters is presented in S11 Table.
Among the IFNβ-specific ISGs (Fig 9; panels in the right half), higher IFNβ transcript levels negatively correlated with: (1) EIF4H, a eukaryotic translational initiation factor (Fig 9E) [69]; (2) SMAD4, a key regulator of TGFβ [70], which in turn promotes mucosal barrier integrity (Fig 9F); (3) VIMP and SEP15, selenoproteins that regulate protein folding in the endoplasmic reticulum [71, 72] that were linked to anti-inflammatory processes [72]; and (4) two co-factors of HIV-1 Vpr, NUP54 and MUS81 [56, 73]. NUP54 is a nuclear pore component and MUS81 is an endonuclease; both are involved in maintaining genomic DNA integrity [74, 75]. Reduced expression of VIMP and SEP15 were associated with lower gut CD4 T cell percentages (Figs 8 and 9G). Downregulated NUP54 and MUS81 were associated with higher plasma IL6 levels (Fig 9H and 9I), lower gut CD4 T cell percentages (S12 Table) and blood CD4 T cell counts (Fig 9H and 9I). Thus, our analyses link elevated IFNβ levels in the gut of PWH to decreased protein translation and decreased protection against DNA damage, protein misfolding and barrier dysfunction.
Discussion
Two aspects of type I IFN biology that could influence its translational potential remain insufficiently addressed. First, there is an ongoing debate as to whether the biological effects of IFN-Is are primarily due to quantitative differences in IFNAR signaling capacity. For example, would administration of higher doses of weakly antiviral IFN-Is (e.g., IFNα2 for HIV-1 infection) achieve the same in vivo effect as that of more potent IFN-Is (e.g., IFNα14)? Second, the mechanisms governing how and why IFN-Is become pathogenic during chronic HIV-1 infection remains unclear. Which ISGs are responsible for the discordant effects of IFN-Is at distinct phases of infection with persistent viruses? We have undertaken an unbiased transcriptomics approach to gain deeper insights on these questions.
Our group and others reported diverse antiviral potencies of the IFNα subtypes and IFNβ that correlated with their IFNAR binding properties. These data emphasize the contribution of quantitative differences in IFNAR signaling in controlling acute viral infection [12–15, 76]. However, data from multiple in vivo studies also revealed that the IFNα subtypes may have qualitative differences in mediating antiviral immunity (reviewed in [2]). In fact, IFNβ has a significantly higher binding affinity to IFNAR2 than all IFNα subtypes [42, 43] but did not exhibit higher ISRE activity and inhibitory activity against HIV-1. Recently, Schaepler et al utilized saturating concentrations of various IFNα subtypes to inhibit HIV-1 infection in activated PBMCs in vitro. Since 25 canonical ISGs were induced to similar levels at saturating IFN-I doses, the authors concluded that there were no qualitative differences between the IFNα subtypes [15]. We argue that this conclusion is premature, as there are hundreds of ISGs encompassing the interferome [5, 20]. We postulate that if there were no qualitative differences, the interferomes of diverse IFN-Is should have substantial overlap following stimulation of cells with IFN-Is normalized for IFNAR signaling strength.
We utilized a luciferase reporter cell line linked to the ISRE of a canonical ISG, ISG15, to normalize for IFNAR signaling strength. The 6 normalized IFN-Is tested (IFNα1, 2, 5, 8, 14 and IFNβ) induced ISG15 and other antiviral ISGs to similar extents in gut CD4+ T cells as expected. Interestingly, despite normalizing for quantitative differences in IFNAR signaling strength, the overlap between the IFNα subtype ‘interferomes’ were not complete, ranging from 59 to 82%. IFNα14 altered >200 genes not found in any of the other IFNα subtypes. IFNα5 altered >200 additional genes and weakly downregulated genes that were induced by other IFNα subtypes, raising the intriguing possibility that IFNα5 may modulate the overall IFN-I response. While it was possible that these IFNα-subtype specific genes were an artifact of the low numbers of donors used in this study, these genes were not close to statistical significance of being differentially expressed by the other IFNα subtypes, as majority had FDR values >90%. The sample size we used for this work was also counterbalanced by utilizing a homogenous cell subpopulation (purified CD4+ T cells) treated in a controlled fashion in vitro that should decrease overall variability. In fact, >240 canonical ISGs were captured by all IFNs tested including our positive control, ISG15. Interestingly, IFNβ altered nearly three times the number of genes compared to the individual IFNα subtypes we tested. We speculate that the higher binding affinity of IFNβ to the IFNAR2 may contribute in part to the increased gene induction numbers, but it was notable that enhanced binding affinity did not track with ISRE activity. The broader interferome associated with IFNβ was also reported by other groups using PBMCs [77, 78], although it was unclear whether the doses were normalized for IFNAR signaling strength. Altogether, the incomplete overlap between the interferomes strongly suggests that there are qualitative differences between diverse IFN-Is. The underlying molecular mechanisms for differential interferome induction remain unclear. One possibility is that subtle differences in IFNAR binding may have triggered differential phosphorylation of diverse STATs, JAKs and MAPKs. This possibility is currently under investigation.
We speculate that differences in interferome regulation may have consequences for the therapeutic use of IFN-Is in vivo. Clinical administration of IFNα2 and IFNβ in vivo were associated with a multitude of side-effects [79]. One approach to potentially reduce toxicity issues is to utilize IFNα subtypes with a more ‘restricted’ gene regulation signature. IFNα8 is ten times more potent at inhibiting HIV-1 than IFNα2, but did not induce as many genes as IFNα14. Thus, IFNα8 may be a potent anti-HIV-1 therapeutic with limited side-effects. However, IFNα8 did not seem to inhibit HIV-1 when administered in humanized mice as encoded plasmids via hydrodynamic injection [14]. IFN-Is with desirable immunomodulatory properties may also be useful in HIV-1 curative strategies [13], as reactivation of latent HIV-1 was insufficient to reduce the reservoir during antiretroviral therapy [80]. IFN-Is could be potent additive drugs for HIV-1 cure by activating immune cells with latent HIV-1 while stimulating immune responses that can kill infected cells and prevent subsequent infection through intrinsic restriction. IFNα14 strongly induced TRAIL+ NK cells and APOBEC3G-mediated hypermutation compared to IFNα2 in humanized mice [13] and was predicted to induce pathways associated with immune function in gut CD4+ T cells compared to IFNα1, 2, 5 and 8. Thus, IFNα14 could be a viable candidate to pursue for HIV-1 curative strategies. The current data could serve as a useful template to design transcriptome-wide studies that extend to the 7 other IFNα subtypes not studied here, as well as other cell types (DCs, NK cells, CD8+ T cells, B cells) in various tissue compartments. Such follow-up studies may aid in designing focused IFN-I biologicals for various clinical applications.
Our recent study suggested that IFNβ may play an important role during chronic HIV-1 immunopathogenesis in the gut [38]. We observed downregulated IFNα, but elevated IFNβ levels in colon biopsies from PWH, while IFNβ was rarely detected in the PBMCs and plasma of these patients [38]. Since IFNβ induced a broader interferome than the individual IFNα subtypes, we determined how ISGs induced by all IFN-Is tested (‘core ISGs’) versus ISGs specifically induced by IFNβ (‘IFNβ-specific ISGs’) were expressed in the gut biopsies following RNAseq. One limitation of our approach is that colon biopsies encompass other cell types that the interferomes based on gut CD4+ T cells may not capture. Specifically, it is possible that the decreased expression of IFNβ-specific ISGs in PWH relative to uninfected controls may be due to the significant loss CD4+ T cells in PWH. If this was the case, we should have also observed a decrease in core ISG expression in PWH. However, over a hundred core ISGs were upregulated in PWH, correlating significantly with gut IFNβ expression. Alterations in IFNAR expression in mucosal CD4+ T cells during HIV-1 infection may also contribute to our results, but our recent study have not detected significant changes in IFNAR2 expression in mucosal immune cells in PWH [81]. Expanding the interferome analyses to other mucosal cell types may enable deconvolution of the bulk RNAseq data to specific cell types.
Consistent with our previous study [38], many canonical ISGs that include antiviral genes remained upregulated in chronic HIV-1 infection in the gut. Core ISGs positively correlated with IFNβ rather than IFNα transcripts, suggesting that IFNβ drove these responses in the gut. Interestingly, core ISG expression positively correlated with plasma LPS levels. Previously, we showed that many ISGs were upregulated in gut CD4+ T cells following co-incubation with Prevotella stercorea, a gram-negative microbe present in colon tissue of PWH [82]. These findings strengthen a link between microbial translocation, IFNβ, and elevated ISG signatures in the gut during chronic HIV-1 infection. Interestingly, we did not observe correlations between core ISGs and the monocyte activation marker sCD14 or myeloid dendritic cell activation. Other markers such as sCD163 [83, 84] may need to be investigated in future work. IRF9, STAT1 and STAT2 (the components of ISGF3) remained elevated in PWH, suggesting a mechanism for constitutive ISG expression during chronic infection. Notably, IFNβ expression in the gut correlated with gene markers of immune activation and inflammation (CD38, PSMB9, NLRC5, TNFA, IFNG), and exhaustion (LAG3). IFNβ-mediated induction of these immunomodulatory genes may account for how IFNβ contributes to pathogenesis during chronic infection. Specifically, IFNβ levels in the gut were associated with IL10 and IL10RA expression, which in turn correlated with LAG3 levels, suggesting that IFNβ may drive immune exhaustion through the IL10 pathway. It remains to be determined whether these core ISGs are similarly regulated in the systemic circulation, where IFNα subtypes, and not IFNβ, are more prominent [38]. Further, the rationale for why the gut appears primed to express IFNβ remains unclear. A recent study noted that helix 4 of IFNβ may have direct antimicrobial properties [85]. Efforts are ongoing to investigate links between IFNβ levels, IFNβ-specific genes and the altered microbiome in PWH.
Could IFNβ have distinct biological effects that are likely not due to other IFN-Is? In persistent LCMV infection of mice, neutralization of IFNβ, and not IFNα, accelerated virus clearance and improved T cell responses [86]. In the present study, we observed that a substantial fraction of IFNβ-specific ISGs, but not core ISGs, correlated with plasma levels of the inflammatory cytokine IL6. A link between IFNβ and IL6 has previously been reported [87]. Surprisingly, the IFNβ-specific ISGs that correlated with IL6 were downregulated during chronic HIV-1 infection in the gut. The magnitude of downregulation correlated with higher IFNβ levels, suggesting negative feedback control. Among these negative feedback control genes, we identified UNC93B1 as a potential driver for the downregulation of IFNβ-specific ISGs, possibly through the regulation of TLR7 signaling via endosomal trafficking pathways [59, 88]. It is thought that constitutive expression of antiviral ISGs may protect from virus infection [89, 90], but decreased protein translation (EIF4H) in PWH may minimize the contribution of antiviral ISGs that are constitutively expressed. Furthermore, our data suggest a potential link between IFNβ, decreased TGFβ signaling (SMAD4), increased unfolded protein response (VIMP and SEP15) and increased DNA damage (NUP54 and MUS81) in chronic HIV-1 infection. These results raise the possibility that genes downregulated by IFNβ could have important consequences for mucosal HIV-1 pathogenesis. One possibility is that these altered IFNβ-specific genes (such as those that decreased protection from DNA damage) may directly contribute to CD4+ T cell death, in line with previous studies linking IFN-Is and CD4+ T cell depletion [91, 92]. Given the predicted pleiotropic effects of IFNβ, our data also raise concerns in utilizing IFNβ for treating chronic Hepatitis B virus infections that became refractory to IFNα treatment [76, 93, 94]. Notably, IFNβ administered <7 days post-symptom onset may help resolve infection with the novel pandemic coronavirus, SARS-CoV-2 [95]. By contrast, delayed IFNβ treatment in murine coronavirus models exacerbated immune pathology [96, 97]. Based on our studies as well as others, understanding the role of distinct IFNs during the course of infection with diverse pathogenic viruses may yield important therapeutic insights.
The mechanisms for the differential regulation of core versus IFNβ-specific ISGs during chronic HIV-1 infection remain to be determined. Studies in cell lines reveal that unphosphorylated STAT1, STAT2 and IRF9 can assemble into an unphosphorylated ISGF3 (U-ISGF3) complex that could sustain the expression of canonical ISGs after a single stimulation with IFNβ [90, 98]. Constitutively expressed ISGs regulated by U-ISGF3 included antiviral genes such as BST2, APOBEC3G, MX2 that remained elevated during chronic HIV-1 infection. By contrast, ISGs specifically regulated by phosphorylated ISGF3 are more sensitive to negative feedback regulation. The ISGF3/U-ISGF3 model would imply that core ISGs are regulated by U-ISGF3, whereas the IFNβ-specific ISGs are regulated by ISGF3. Investigations on the phosphorylation status of STAT1, STAT2 and IRF9 in chronic HIV-1 infection are underway.
In conclusion, we demonstrate that diverse IFN-Is trigger non-overlapping interferomes in a single cell type, providing strong evidence for qualitative differences between the IFN-Is. Furthermore, conserved and qualitative differences in interferome induction correlated with clinically relevant markers of immunopathogenesis during chronic HIV-1 infection. Further studies on the differences between the IFN-Is and how these cytokines regulate distinct gene expression profiles in various tissue compartments could inform strategies to harness these biologicals and/or block these responses for HIV-1 control.
Materials and methods
Ethics statement
The in vitro studies utilized disaggregated cells from macroscopically normal jejunum tissue that would otherwise be discarded. These tissues were obtained from patients undergoing elective surgery at the University of Colorado Hospital. The patients signed a release form for the unrestricted use of tissues for research following de-identification to laboratory personnel. The Colorado Multiple Institutional Review Board approved the procedures and have given exempt status.
Study participants
Colon pinch biopsies from PWH and age/gender-matched HIV-uninfected controls were obtained from archived samples from a completed, COMIRB-approved clinical study. This clinical study included 24 PWH and 14 HIV-uninfected controls, but archived colon pinch biopsies were available only for 19 PWH and 13 controls, respectively. Study participants signed an informed consent. Clinical parameters that include blood CD4-T cell counts (cells/μl), plasma viral load, tissue HIV RNA (per CD4 T cell), Tissue CD4 T cells (% viable CD45+ cells), IL-6 (pg/ml), CRP (μg/ml), iFABP (pg/ml), sCD27 (U/ml), CD14 (ng/ml), LPS (pg/ml), LTA (optical density), gut IFNα and IFNβ transcripts, were reported previously [38].
ISRE-activity titrations
IFN-Is (PBL Assay Science, Piscataway NJ) were frozen into small aliquots for single-use. iLite Type I IFN Assay Ready cells, purchased from Svar Life Science AB (Malmõ, Sweden, Cat# BM3049) were seeded into a 96-well flat-bottomed cell culture plate and maintained in complete DMEM (with 10% FBS and 1% Penicillin-streptomycin and L-glutamine). Various doses of recombinant IFN-Is were added to corresponding wells. After 18 h, the cells were lysed and luciferase activities were developed using Bright-Glo Luciferase Assay System (Promega, Madison, WI, Cat# E2610) according to manufacturer’s instruction. Luciferase activity was subsequently measured using a Perkin-Elmer Victor X5 plate reader. 50% effective concentrations were computed using dose-response curve in Prism 5.0.
HIV-1 Inhibition Curves of IFN-Is in LPMCs
HIV-1BaL (NIH AIDS Reagent Program Cat# 510) was prepared in MOLT4-CCR5 cells and titrated using an HIV-1 p24 ELISA (Advanced Bioscience Laboratories, Rockville, MD). 10 ng p24 of HIV-1BaL/ 106 LPMCs (n = 6 donors) was spinoculated in the presence or absence of titrated doses of IFNα subtypes and IFNβ (PBL Assay Science). The cells were harvested at 4 d and analyzed by intracellular p24 flow cytometry as previously described [12, 99]. 50% inhibitory concentrations and Vres were calculated using a one-phase decay equation in Prism 5.0 as previously described [12, 13].
IFN-I treatments of gut CD4+ T cells
CD4+ T cells were purified from LPMCs (n = 4 donors) by negative selection using EasySep™ Human CD4+ T Cell Isolation Kit (Stemcell, Vancouver, BC, Canada, Cat# 17952) and followed by flow sorting to reach a purity greater than 99%. 1x106 of these purified CD4+ T cells were treated with mock, 100 pg/ml IFNα14, or an equivalent ISRE-activity of IFNα1, 2, 5, 8 and IFNβ. After 18 h, RNA was extracted using RNeasy Mini Kit (Qiagen, Germany, Cat# 74104). RNAseq libraries were constructed from 500 ng RNA using QuantSeq 3’ mRNAseq Library Prep Kit (Lexogen, Austria, Cat# 016.96). The RNAseq library quality was verified using 2100 BioAnalyzer (Agilent, Santa Clara, CA) before loaded into a HiSeq 2500 by the Genomics and Microarray Sequencing Core facility at the University of Colorado Anschutz Medical Campus.
Quantitative PCR
The 96-well array contained primers for target genes ISG15, ARHGEF3, LAT and AHNAK (S1 Fig), a reverse transcription control, a genomic DNA control and a positive PCR control as well as the housekeeping gene GAPDH (PPH00150F). Ninety-six-well Custom RT2 Profiler PCR Arrays were performed according to the manufacturer’s instructions (Qiagen, Valencia, California, USA) using the Bio-Rad CFX-96 Real-time PCR instrument (Bio-Rad, Hercules, California, USA) and Bio-Rad CFX Manager software (Ver. 3.1). cDNA templates were mixed with ready-to-use RT2 qPCR Master Mixes and 25 μl of the PCR component mix was aliquoted into each well containing predispensed gene-specific primer sets. Each plate was loaded with cDNA from 6 individual samples. Gene expression was normalized according to the average expression level of GAPDH in the same sample.
Transcriptome of colon pinch biopsies
RNA (500 ng) from the colon pinch biopsies were used to prepare the RNAseq libraries. The RNAseq work flow of library construction, quality control, and the subsequent sequencing were similar to that of IFN-I treated gut CD4+ T cells as aforementioned.
RNAseq data pre-processing
RNAseq data were downloaded as FASTQ files and their quality examined and visualized using FastQC (Babraham Institute, https://www.bioinformatics.babraham.ac.uk/projects/fastqc). The removal of Illumina sequencing adapters and the by-base quality screening was performed with cutadapt (https://cutadapt.readthedocs.io/en/stable). The quality screened FASTQ files were mapped to the current human genome assembly GRCh38.p12 using Hisat2 (Johns Hopkins University, https://ccb.jhu.edu/software/hisat2/index.shtml), taking into account the Ensembl ID, gene symbol and gene length. Raw gene expression counts were extracted using featureCounts from the Subread package (Walter+Eliza Hall Institute of Medical Research, http://subread.sourceforge.net).
Differential Expression (DE) Analysis: Interferomes
RNAseq raw counts were obtained for gut CD4 T cells treated with 6 IFN-Is (IFNα1, 2, 5, 8, 14 and IFNβ) and mock from 4 donors. Gene counts were normalized using transcripts per million (TPM). Principal component analyses necessitated removal of one donor as the transcriptome of the mock condition was extremely skewed relative to the other 3 donors (S1A Fig). Using edgeR differential expression analysis, IFN regulated genes (IRGs) were defined based on a 1.5-fold change (FC) cutoff relative to mock and a false-discovery rate (FDR) ≤20%.
DE analysis: Chronic HIV-1 infection
The RNAseq raw counts data had gene-level read counts for 13 HIV-1-uninfected donors and 19 PWH. Lowly expressed genes which have average read counts less than 5 per library were filtered out. As a result, 19890 out of 43297 genes were kept for further analysis and gene counts were normalized using the trimmed mean of M values (TMM) normalization method from edgeR (version 3.24.3) in R (version 3.5.1) with the “estimateDisp” function and its default settings. The TMM method calculates the scaling normalization factor for each sample by accounting for library size and observed counts, while under the assumption that the majority of genes are not DE. In our case, the TMM method provided the best normalization results in terms of removing the unwanted variation, according to the relative log expression (RLE) plots (S6 Fig). Two-group differential expression (DE) analysis was performed to test for differences between healthy controls and PWH, using normalized counts in edgeR with default settings and false discovery rate (FDR) of 5% to control for multiple testing. The DE analysis was done in edgeR by the exact test using the “exactTest” function and its default settings for two-group design. The Benjamini-Hochberg procedure (BH step-up procedure) controlling the FDR was used as the multiple testing correction method in this study.
Correlation of gene sets with clinically relevant data
The normalized RNAseq counts were further transformed by the variance stabilization and bias reduction method called “regularized log transformation” (rlog) from DESeq2 (version 1.22.2) [53]. The transformed counts were then used as the outcome in a multivariate linear regression model. Specifically, the linear regression models were fit in a gene by gene fashion to test the correlation between RNA expression levels and immunopathogenic markers of chronic HIV-1 infection, with one marker included in the model at a time, while adjusting for age and gender. Based on the results of DE analysis, significantly altered genes from core-ISGs and IFNβ-specific ISGs were selected to test the associations with clinical parameters through the above model, separately. Several immunopathogenic markers were used in this part, including gut IFNβ transcript levels, gut CD4 T cell percentages, blood CD4 T cell counts, plasma IL6 levels and plasma LPS levels. Genes were considered as significantly associated with the corresponding clinical parameter with an FDR cutoff at 5%. In addition, the difference in proportions of significant genes in each gene list were tested with Chi-squared test in R, as well as the difference in proportions of positive correlations of each gene list. All the plots were generated by ggplot2 (version 3.1.1) package in R (version 3.5.1).
Accession numbers
Next generation sequencing data were deposited in the Sequence Read Archive PRJNA558974 (interferome dataset) and PRJNA558500 (colon pinch biopsies). These were also deposited in the Gene Expression Omnibus archive with accession numbers GSE156844 (interferome dataset) and GSE156861 (colon pinch biopsies).
Supporting information
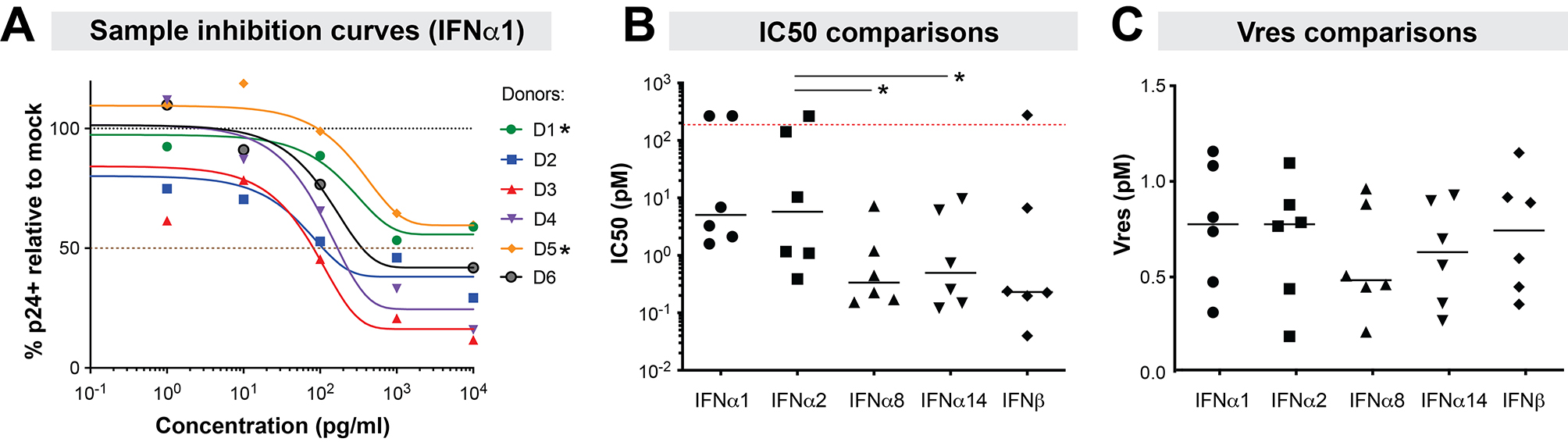
(A) HIV-1 inhibition curves for 6 different donors with IFNα1 based on a one-phase decay equation. Note that 2 donors (asterisk) did not reach 50% inhibition (above the red dashed line). The IC50s for these samples were calculated as 10,000 pg/ml, the highest dose used. (B) IC50s were calculated based on the sigmoidal plot and converted to pM. Each dot corresponds to a different LPMC donor. A few datapoints did not reach an IC50. The difference in IC50 between IFNα2 and either IFNα8 or IFNα14 were significant (*p<0.05) based on 2-tailed Wilcoxon matched pairs test (GraphPad Prism 5.0). (C) The residual virus replication at maximum IFN-I doses (Vres) was calculated for each donor based on the plateau of the best-fit equation. The differences were not significant based on a one-way ANOVA using Friedman’s test (p>0.05). For panels (B) and (C), the central line corresponds to the median values.
(JPG)
{kind=link}
(A) (Left panel) Multidimensional scaling plot of distances between gene expression profiles of different LPMC donors, using mock and IFNβ-treated data as an example. Donor 4 is highlighted in red. (Right panels) Inclusion of donor 4 increased the biological coefficient of variation (BCV) when comparing mock to IFN-I treatment. As an example, BCV plots with or without donor 4 are shown for mock vs IFNβ treatment. The red line corresponds to the common dispersion for all genes; the blue curve is the dispersion trend; each black dot is the genewise dispersion rate. Higher dispersion trends for mock versus IFNα1, IFNα2, IFNα5, IFNα8 and IFNα14 were also observed if donor 4 was included (not shown). These were used to justify exclusion of donor 4 from subsequent analysis. (B) Confirmation of transcriptome data via qPCR. Fold-induction relative to mock values were compared for 4 genes between the RNASeq data (based on TPM values) and qPCR (based on ΔΔCt method). Note that one outlier in the LAT qPCR for IFNα14 (orange arrow) drove a positive fold induction with a large error bar. The individual data points are 7.03, -1.00 and -0.44.
(JPG)
{kind=link}
(Left panels) IFNα5-specific genes (n = 201) were classified based on significant induction/suppression in IFNα5-treated cells relative to mock at a 20% FDR cut-off. However, when these genes were evaluated in transcriptome datasets for IFNα1, IFNα2, IFNα8 and IFNα14, most had FDR values >90%, suggesting that these IFNα5-specific genes were not even close to being statistically-significant in these other IFNα subtypes. (Right panels) Evaluation of IFNα14-specific genes against gene datasets for IFNα1, IFNα2, IFNα8 and IFNα5. Bars correspond to the frequency of genes that fell within the FDR values noted in the x-axis.
(TIF)
IFN regulated genes (IRGs) in primary gut CD4+ T cells (n = 3 donors) were determined via RNAseq. Euler diagrams are used to show the overlap between IFNα subtype interferomes at 1.5, 2.0, 2.5 and 3.0 fold-change (FC) cut-offs. The number of IFN-regulated genes (IRGs) decrease with higher FC cut-offs, but genes unique to each IFNα subtype remained significant.
(TIF)
Heat maps of genes that were differentially regulated by various IFN-Is in primary gut CD4+ T cells (n = 3 donors) at various fold-change cut-offs. Red bars correspond to upregulated genes relative to mock, whereas blue bars correspond to genes that were downregulated. Brackets indicate genes that were weakly downregulated by IFNα5, but upregulated by IFNα1, 2, 8 and 14. These IFNα5-differentially regulated genes were absent at a fold-change cut-off of 3.0.
(TIF)
Visualization of unwanted variation of filtered RNAseq data in clinical biopsy samples from HIV uninfected (n = 13) and PWH (n = 19) samples using Relative Log Expression (RLE) Plots. (A) Without normalization; (B) Normalization via Transcripts per Million; (C) DESeq2; and (D) Trimmed Mean of M-values (TMM) using edgeR. The RLE plot, which is a boxplot of deviations from gene medians, shows the TMM normalization is the best in our case, based on the position at 0 of medians and narrow widths.
(TIF)
Linear regression plots are shown for (A) IFNβ mRNA levels, IL10, IL10RA and LAG3; and (B) IFNβ mRNA levels and cytokines IFNG, TNFA, TGFB1 and IL10. IFNβ mRNA levels were obtained previously [38]; the remaining genes were TMM values from the RNAseq experiment described here. R2 and p-values are shown for these relationships.
(JPG)
{kind=link}
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(PDF)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Acknowledgments
We thank Eric Lee and Christine Purba for expert technical assistance with the Lamina Propria Aggregate Culture model, and Cheyret Wood for helpful statistical advice. We also thank the patients who agreed to use discarded tissue for research purposes and the clinical study participants.
Data Availability
Raw next generation sequencing data were deposited in the Sequence Read Archive. PRJNA558974 (interferome dataset) and PRJNA558500 (colon pinch biopsies). All pertinent data are found in the manuscript and Supporting Information files.
Funding Statement
This work was supported by the National Institutes of Health R01 AI134220 (MLS and CCW), The RNA Bioscience Initiative (MLS, CCW and TG), the Biostatistics, Epidemiology and Research Design program of the Colorado Clinical and Translational Sciences Institute (GS, MK and KK), the Deutsche Forschungsgemeinschaft SPP 1923/1/2 Program (KS and UD), the Intramural Research Program of the National Institute of Allergy and Infectious Diseases Grant 1ZIAAI001141, National Institute of Health (KJH). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Meager A. The Interferons: characterization and application. Germany: Wiley-VCH; 2006. [Google Scholar]
- 2.Sutter K, Dickow J, Dittmer U. Interferon alpha subtypes in HIV infection. Cytokine Growth Factor Rev. 2018;40:13–8. Epub 2018/02/25. 10.1016/j.cytogfr.2018.02.002 [DOI] [PubMed] [Google Scholar]
- 3.Wang B, Kang W, Zuo J, Kang W, Sun Y. The Significance of Type-I Interferons in the Pathogenesis and Therapy of Human Immunodeficiency Virus 1 Infection. Front Immunol. 2017;8:1431 Epub 2017/11/23. 10.3389/fimmu.2017.01431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stark GR, Darnell JE Jr. The JAK-STAT pathway at twenty. Immunity. 2012;36(4):503–14. Epub 2012/04/24. 10.1016/j.immuni.2012.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Samarajiwa SA, Forster S, Auchettl K, Hertzog PJ. INTERFEROME: the database of interferon regulated genes. Nucleic acids research. 2009;37(Database issue):D852–7. 10.1093/nar/gkn732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Azzoni L, Foulkes AS, Papasavvas E, Mexas AM, Lynn KM, Mounzer K, et al. Pegylated Interferon alfa-2a monotherapy results in suppression of HIV type 1 replication and decreased cell-associated HIV DNA integration. The Journal of infectious diseases. 2013;207(2):213–22. 10.1093/infdis/jis663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun H, Buzon MJ, Shaw A, Berg RK, Yu XG, Ferrando-Martinez S, et al. Hepatitis C therapy with interferon-alpha and ribavirin reduces CD4 T-cell-associated HIV-1 DNA in HIV-1/hepatitis C virus-coinfected patients. The Journal of infectious diseases. 2014;209(9):1315–20. 10.1093/infdis/jit628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.George J, Mattapallil JJ. Interferon-alpha Subtypes As an Adjunct Therapeutic Approach for Human Immunodeficiency Virus Functional Cure. Front Immunol. 2018;9:299 10.3389/fimmu.2018.00299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moron-Lopez S, Gomez-Mora E, Salgado M, Ouchi D, Puertas MC, Urrea V, et al. Short-term Treatment With Interferon Alfa Diminishes Expression of HIV-1 and Reduces CD4+ T-Cell Activation in Patients Coinfected With HIV and Hepatitis C Virus and Receiving Antiretroviral Therapy. The Journal of infectious diseases. 2016;213(6):1008–12. 10.1093/infdis/jiv521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van der Sluis RM, Zerbato JM, Rhodes JW, Pascoe RD, Solomon A, Kumar NA, et al. Diverse effects of interferon alpha on the establishment and reversal of HIV latency. PLoS pathogens. 2020;16(2):e1008151 10.1371/journal.ppat.1008151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palesch D, Bosinger SE, Mavigner M, Billingsley JM, Mattingly C, Carnathan DG, et al. Short-Term Pegylated Interferon alpha2a Treatment Does Not Significantly Reduce the Viral Reservoir of Simian Immunodeficiency Virus-Infected, Antiretroviral Therapy-Treated Rhesus Macaques. Journal of virology. 2018;92(14):e00279–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harper MS, Guo K, Gibbert K, Lee EJ, Dillon SM, Barrett BS, et al. Interferon-alpha Subtypes in an Ex Vivo Model of Acute HIV-1 Infection: Expression, Potency and Effector Mechanisms. PLoS pathogens. 2015;11(11):e1005254 10.1371/journal.ppat.1005254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lavender KJ, Gibbert K, Peterson KE, Van Dis E, Francois S, Woods T, et al. Interferon Alpha Subtype-Specific Suppression of HIV-1 Infection In Vivo. Journal of virology. 2016;90(13):6001–13. 10.1128/JVI.00451-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abraham S, Choi JG, Ortega NM, Zhang J, Shankar P, Swamy NM. Gene therapy with plasmids encoding IFN-beta or IFN-alpha14 confers long-term resistance to HIV-1 in humanized mice. Oncotarget. 2016;7(48):78412–20. Epub 2016/10/13. 10.18632/oncotarget.12512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schlaepfer E, Fahrny A, Gruenbach M, Kuster SP, Simon V, Schreiber G, et al. Dose-Dependent Differences in HIV Inhibition by Different Interferon Alpha Subtypes While Having Overall Similar Biologic Effects. mSphere. 2019;4(1):e00637–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lavoie TB, Kalie E, Crisafulli-Cabatu S, Abramovich R, DiGioia G, Moolchan K, et al. Binding and activity of all human alpha interferon subtypes. Cytokine. 2011;56(2):282–9. 10.1016/j.cyto.2011.07.019 [DOI] [PubMed] [Google Scholar]
- 17.Manry J, Laval G, Patin E, Fornarino S, Itan Y, Fumagalli M, et al. Evolutionary genetic dissection of human interferons. The Journal of experimental medicine. 2011;208(13):2747–59. 10.1084/jem.20111680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gibbert K, Joedicke JJ, Meryk A, Trilling M, Francois S, Duppach J, et al. Interferon-alpha subtype 11 activates NK cells and enables control of retroviral infection. PLoS pathogens. 2012;8(8):e1002868 Epub 2012/08/23. 10.1371/journal.ppat.1002868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cull VS, Tilbrook PA, Bartlett EJ, Brekalo NL, James CM. Type I interferon differential therapy for erythroleukemia: specificity of STAT activation. Blood. 2003;101(7):2727–35. 10.1182/blood-2002-05-1521 [DOI] [PubMed] [Google Scholar]
- 20.Hasenkrug KJ, Lavender KJ, Santiago ML, Sutter K, Dittmer U. Different Biological Activities of Specific Interferon Alpha Subtypes. mSphere. 2019;4(4):e00127–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schlaepfer E, Fahrny A, Gruenbach M, Kuster SP, Simon V, Schreiber G, et al. Reply to Hasenkrug et al., "Different Biological Activities of Specific Interferon Alpha Subtypes". mSphere. 2019;4(4):e00275–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerlach N, Gibbert K, Alter C, Nair S, Zelinskyy G, James CM, et al. Anti-retroviral effects of type I IFN subtypes in vivo. Eur J Immunol. 2009;39(1):136–46. Epub 2009/01/09. 10.1002/eji.200838311 [DOI] [PubMed] [Google Scholar]
- 23.Sandler NG, Bosinger SE, Estes JD, Zhu RT, Tharp GK, Boritz E, et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature. 2014;511(7511):601–5. 10.1038/nature13554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carnathan D, Lawson B, Yu J, Patel K, Billingsley JM, Tharp GK, et al. Reduced Chronic Lymphocyte Activation following Interferon Alpha Blockade during the Acute Phase of Simian Immunodeficiency Virus Infection in Rhesus Macaques. Journal of virology. 2018;92(9):e01760–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science. 2013;340(6129):202–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science. 2013;340(6129):207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stylianou E, Aukrust P, Bendtzen K, Muller F, Froland SS. Interferons and interferon (IFN)-inducible protein 10 during highly active anti-retroviral therapy (HAART)-possible immunosuppressive role of IFN-alpha in HIV infection. Clin Exp Immunol. 2000;119(3):479–85. Epub 2000/02/26. 10.1046/j.1365-2249.2000.01144.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sedaghat AR, German J, Teslovich TM, Cofrancesco J Jr., Jie CC, Talbot CC Jr., et al. Chronic CD4+ T-cell activation and depletion in human immunodeficiency virus type 1 infection: type I interferon-mediated disruption of T-cell dynamics. Journal of virology. 2008;82(4):1870–83. Epub 2007/12/14. 10.1128/JVI.02228-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harris LD, Tabb B, Sodora DL, Paiardini M, Klatt NR, Douek DC, et al. Downregulation of robust acute type I interferon responses distinguishes nonpathogenic simian immunodeficiency virus (SIV) infection of natural hosts from pathogenic SIV infection of rhesus macaques. Journal of virology. 2010;84(15):7886–91. 10.1128/JVI.02612-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview. The Journal of experimental medicine. 2016;213(12):2527–38. 10.1084/jem.20161596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhen A, Rezek V, Youn C, Lam B, Chang N, Rick J, et al. Targeting type I interferon-mediated activation restores immune function in chronic HIV infection. J Clin Invest. 2017; 127(1):260–268. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng L, Ma J, Li J, Li D, Li G, Li F, et al. Blocking type I interferon signaling enhances T cell recovery and reduces HIV-1 reservoirs. J Clin Invest. 2017; 127(1):269–279. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nganou-Makamdop K, Billingsley JM, Yaffe Z, O'Connor G, Tharp GK, Ransier A, et al. Type I IFN signaling blockade by a PASylated antagonist during chronic SIV infection suppresses specific inflammatory pathways but does not alter T cell activation or virus replication. PLoS pathogens. 2018;14(8):e1007246 10.1371/journal.ppat.1007246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dillon SM, Santiago ML, Wilson CC. HIV-1 pathogenesis in the gut In: Hope TJ, Stevenson M, Richman D, editors. Encyclopedia of AIDS. New York: Springer; 2016. p. 1–8. [Google Scholar]
- 35.Ericsen AJ, Lauck M, Mohns MS, DiNapoli SR, Mutschler JP, Greene JM, et al. Microbial Translocation and Inflammation Occur in Hyperacute Immunodeficiency Virus Infection and Compromise Host Control of Virus Replication. PLoS pathogens. 2016;12(12):e1006048 10.1371/journal.ppat.1006048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klatt NR, Funderburg NT, Brenchley JM. Microbial translocation, immune activation, and HIV disease. Trends in microbiology. 2013;21(1):6–13. 10.1016/j.tim.2012.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nature medicine. 2006;12(12):1365–71. Epub 2006/11/23. 10.1038/nm1511 [DOI] [PubMed] [Google Scholar]
- 38.Dillon SM, Guo K, Austin GL, Gianella S, Engen PA, Mutlu EA, et al. A compartmentalized type I interferon response in the gut during chronic HIV-1 infection is associated with immunopathogenesis. AIDS (London, England). 2018;32(12):1599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mahlakoiv T, Hernandez P, Gronke K, Diefenbach A, Staeheli P. Leukocyte-derived IFN-alpha/beta and epithelial IFN-lambda constitute a compartmentalized mucosal defense system that restricts enteric virus infections. PLoS pathogens. 2015;11(4):e1004782 Epub 2015/04/08. 10.1371/journal.ppat.1004782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iyer SS, Bibollet-Ruche F, Sherrill-Mix S, Learn GH, Plenderleith L, Smith AG, et al. Resistance to type 1 interferons is a major determinant of HIV-1 transmission fitness. Proceedings of the National Academy of Sciences of the United States of America. 2017;114(4):E590–E9. Epub 2017/01/11. 10.1073/pnas.1620144114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fenton-May AE, Dibben O, Emmerich T, Ding H, Pfafferott K, Aasa-Chapman MM, et al. Relative resistance of HIV-1 founder viruses to control by interferon-alpha. Retrovirology. 2013;10:146 10.1186/1742-4690-10-146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jaks E, Gavutis M, Uze G, Martal J, Piehler J. Differential receptor subunit affinities of type I interferons govern differential signal activation. J Mol Biol. 2007;366(2):525–39. 10.1016/j.jmb.2006.11.053 [DOI] [PubMed] [Google Scholar]
- 43.Jaitin DA, Roisman LC, Jaks E, Gavutis M, Piehler J, Van der Heyden J, et al. Inquiring into the differential action of interferons (IFNs): an IFN-alpha2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-beta. Mol Cell Biol. 2006;26(5):1888–97. 10.1128/MCB.26.5.1888-1897.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dillon SM, Guo K, Castleman MJ, Santiago ML, Wilson CC. Quantifying HIV-1-Mediated Gut CD4+ T Cell Death in the Lamina Propria Aggregate Culture (LPAC) Model Bio-Protocol. 2020;10(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holmes M, Zhang F, Bieniasz PD. Single-Cell and Single-Cycle Analysis of HIV-1 Replication. PLoS pathogens. 2015;11(6):e1004961 10.1371/journal.ppat.1004961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mohammadi P, Desfarges S, Bartha I, Joos B, Zangger N, Munoz M, et al. 24 hours in the life of HIV-1 in a T cell line. PLoS pathogens. 2013;9(1):e1003161 10.1371/journal.ppat.1003161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mariotti B, Servaas NH, Rossato M, Tamassia N, Cassatella MA, Cossu M, et al. The Long Non-coding RNA NRIR Drives IFN-Response in Monocytes: Implication for Systemic Sclerosis. Front Immunol. 2019;10:100 Epub 2019/02/26. 10.3389/fimmu.2019.00100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barriocanal M, Carnero E, Segura V, Fortes P. Long Non-Coding RNA BST2/BISPR is Induced by IFN and Regulates the Expression of the Antiviral Factor Tetherin. Front Immunol. 2014;5:655 Epub 2015/01/27. 10.3389/fimmu.2014.00655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kramer A, Green J, Pollard J Jr., Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics (Oxford, England). 2014;30(4):523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dillon SM, Lee EJ, Kotter CV, Austin GL, Dong Z, Hecht DK, et al. An altered intestinal mucosal microbiome in HIV-1 infection is associated with mucosal and systemic immune activation and endotoxemia. Mucosal immunology. 2014; 7(4):983–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics (Oxford, England). 2010;26(1):139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic acids research. 2012;40(10):4288–97. Epub 2012/01/31. 10.1093/nar/gks042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550 Epub 2014/12/18. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gandolfo LC, Speed TP. RLE plots: Visualizing unwanted variation in high dimensional data. PloS one. 2018;13(2):e0191629 Epub 2018/02/06. 10.1371/journal.pone.0191629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107(1):55–65. Epub 2001/10/12. 10.1016/s0092-8674(01)00506-2 [DOI] [PubMed] [Google Scholar]
- 56.Popov S, Rexach M, Ratner L, Blobel G, Bukrinsky M. Viral protein R regulates docking of the HIV-1 preintegration complex to the nuclear pore complex. The Journal of biological chemistry. 1998;273(21):13347–52. Epub 1998/05/28. 10.1074/jbc.273.21.13347 [DOI] [PubMed] [Google Scholar]
- 57.Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, et al. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302(5647):1056–60. [DOI] [PubMed] [Google Scholar]
- 58.Francois-Newton V, Magno de Freitas Almeida G, Payelle-Brogard B, Monneron D, Pichard-Garcia L, Piehler J, et al. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon alpha response. PloS one. 2011;6(7):e22200 10.1371/journal.pone.0022200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fukui R, Saitoh S, Kanno A, Onji M, Shibata T, Ito A, et al. Unc93B1 restricts systemic lethal inflammation by orchestrating Toll-like receptor 7 and 9 trafficking. Immunity. 2011;35(1):69–81. 10.1016/j.immuni.2011.05.010 [DOI] [PubMed] [Google Scholar]
- 60.Dillon SM, Lee EJ, Kotter CV, Austin GL, Gianella S, Siewe B, et al. Gut dendritic cell activation links an altered colonic microbiome to mucosal and systemic T-cell activation in untreated HIV-1 infection. Mucosal immunology. 2016;9(1):24–37. 10.1038/mi.2015.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dillon SM, Castleman MJ, Frank DN, Austin GL, Gianella S, Cogswell AC, et al. Brief Report: Inflammatory Colonic Innate Lymphoid Cells Are Increased During Untreated HIV-1 Infection and Associated With Markers of Gut Dysbiosis and Mucosal Immune Activation. J Acquir Immune Defic Syndr. 2017;76(4):431–7. 10.1097/QAI.0000000000001523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dillon SM, Kibbie J, Lee EJ, Guo K, Santiago ML, Austin GL, et al. Low abundance of colonic butyrate-producing bacteria in HIV infection is associated with microbial translocation and immune activation. AIDS (London, England). 2017;31(4):511–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dillon SM, Lee EJ, Donovan AM, Guo K, Harper MS, Frank DN, et al. Enhancement of HIV-1 infection and intestinal CD4+ T cell depletion ex vivo by gut microbes altered during chronic HIV-1 infection. Retrovirology. 2016;13:5 10.1186/s12977-016-0237-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu Z, Cumberland WG, Hultin LE, Prince HE, Detels R, Giorgi JV. Elevated CD38 antigen expression on CD8+ T cells is a stronger marker for the risk of chronic HIV disease progression to AIDS and death in the Multicenter AIDS Cohort Study than CD4+ cell count, soluble immune activation markers, or combinations of HLA-DR and CD38 expression. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;16(2):83–92. Epub 1997/11/14. 10.1097/00042560-199710010-00003 [DOI] [PubMed] [Google Scholar]
- 65.Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, et al. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(31):13794–9. Epub 2010/07/20. 10.1073/pnas.1008684107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Groettrup M, Standera S, Stohwasser R, Kloetzel PM. The subunits MECL-1 and LMP2 are mutually required for incorporation into the 20S proteasome. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(17):8970–5. Epub 1997/08/19. 10.1073/pnas.94.17.8970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maruhashi T, Okazaki IM, Sugiura D, Takahashi S, Maeda TK, Shimizu K, et al. LAG-3 inhibits the activation of CD4(+) T cells that recognize stable pMHCII through its conformation-dependent recognition of pMHCII. Nat Immunol. 2018;19(12):1415–26. Epub 2018/10/24. 10.1038/s41590-018-0217-9 [DOI] [PubMed] [Google Scholar]
- 68.Graydon CG, Balasko AL, Fowke KR. Roles, function and relevance of LAG3 in HIV infection. PLoS pathogens. 2019;15(1):e1007429 Epub 2019/01/18. 10.1371/journal.ppat.1007429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rogers GW Jr., Richter NJ, Lima WF, Merrick WC. Modulation of the helicase activity of eIF4A by eIF4B, eIF4H, and eIF4F. The Journal of biological chemistry. 2001;276(33):30914–22. Epub 2001/06/22. 10.1074/jbc.M100157200 [DOI] [PubMed] [Google Scholar]
- 70.Hata A, Chen YG. TGF-beta Signaling from Receptors to Smads. Cold Spring Harb Perspect Biol. 2016;8(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gladyshev VN, Arner ES, Berry MJ, Brigelius-Flohe R, Bruford EA, Burk RF, et al. Selenoprotein Gene Nomenclature. The Journal of biological chemistry. 2016;291(46):24036–40. Epub 2016/09/21. 10.1074/jbc.M116.756155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Labunskyy VM, Hatfield DL, Gladyshev VN. Selenoproteins: molecular pathways and physiological roles. Physiol Rev. 2014;94(3):739–77. Epub 2014/07/06. 10.1152/physrev.00039.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Laguette N, Bregnard C, Hue P, Basbous J, Yatim A, Larroque M, et al. Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell. 2014;156(1–2):134–45. Epub 2014/01/15. 10.1016/j.cell.2013.12.011 [DOI] [PubMed] [Google Scholar]
- 74.Rodriguez-Berriguete G, Granata G, Puliyadi R, Tiwana G, Prevo R, Wilson RS, et al. Nucleoporin 54 contributes to homologous recombination repair and post-replicative DNA integrity. Nucleic acids research. 2018;46(15):7731–46. Epub 2018/07/10. 10.1093/nar/gky569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fu H, Martin MM, Regairaz M, Huang L, You Y, Lin CM, et al. The DNA repair endonuclease Mus81 facilitates fast DNA replication in the absence of exogenous damage. Nat Commun. 2015;6:6746 Epub 2015/04/17. 10.1038/ncomms7746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen J, Li Y, Lai F, Wang Y, Sutter K, Dittmer U, et al. Functional Comparison of IFN-alpha Subtypes Reveals Potent HBV Suppression by a Concerted Action of IFN-alpha and -gamma Signaling. Hepatology. 2020; 10.1002/hep.31282 [DOI] [PubMed] [Google Scholar]
- 77.Der SD, Zhou A, Williams BR, Silverman RH. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(26):15623–8. 10.1073/pnas.95.26.15623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dierckx T, Khouri R, Menezes SM, Decanine D, Farre L, Bittencourt A, et al. IFN-beta induces greater antiproliferative and proapoptotic effects and increased p53 signaling compared with IFN-alpha in PBMCs of Adult T-cell Leukemia/Lymphoma patients. Blood Cancer J. 2017;7(1):e519 10.1038/bcj.2016.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pestka S. The interferons: 50 years after their discovery, there is much more to learn. The Journal of biological chemistry. 2007;282(28):20047–51. 10.1074/jbc.R700004200 [DOI] [PubMed] [Google Scholar]
- 80.Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity. 2012;36(3):491–501. 10.1016/j.immuni.2012.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ickler J, Francois S, Widera M, Santiago ML, Dittmer U, Sutter K. HIV infection does not alter interferon alpha/beta receptor 2 expression on mucosal immune cells. PloS one. 2020;15(1):e0218905 10.1371/journal.pone.0218905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yoder AC, Guo K, Dillon SM, Phang T, Lee EJ, Harper MS, et al. The transcriptome of HIV-1 infected intestinal CD4+ T cells exposed to enteric bacteria. PLoS pathogens. 2017;13(2):e1006226 10.1371/journal.ppat.1006226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Burdo TH, Lentz MR, Autissier P, Krishnan A, Halpern E, Letendre S, et al. Soluble CD163 made by monocyte/macrophages is a novel marker of HIV activity in early and chronic infection prior to and after anti-retroviral therapy. The Journal of infectious diseases. 2011;204(1):154–63. 10.1093/infdis/jir214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Knudsen TB, Ertner G, Petersen J, Moller HJ, Moestrup SK, Eugen-Olsen J, et al. Plasma Soluble CD163 Level Independently Predicts All-Cause Mortality in HIV-1-Infected Individuals. The Journal of infectious diseases. 2016;214(8):1198–204. 10.1093/infdis/jiw263 [DOI] [PubMed] [Google Scholar]
- 85.Kaplan A, Lee MW, Wolf AJ, Limon JJ, Becker CA, Ding M, et al. Direct Antimicrobial Activity of IFN-beta. J Immunol. 2017;198(10):4036–45. Epub 2017/04/16. 10.4049/jimmunol.1601226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ng CT, Sullivan BM, Teijaro JR, Lee AM, Welch M, Rice S, et al. Blockade of interferon Beta, but not interferon alpha, signaling controls persistent viral infection. Cell host & microbe. 2015;17(5):653–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nan J, Wang Y, Yang J, Stark GR. IRF9 and unphosphorylated STAT2 cooperate with NF-kappaB to drive IL6 expression. Proceedings of the National Academy of Sciences of the United States of America. 2018;115(15):3906–11. Epub 2018/03/28. 10.1073/pnas.1714102115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pelka K, Bertheloot D, Reimer E, Phulphagar K, Schmidt SV, Christ A, et al. The Chaperone UNC93B1 Regulates Toll-like Receptor Stability Independently of Endosomal TLR Transport. Immunity. 2018;48(5):911–22 e7. 10.1016/j.immuni.2018.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang W, Yin Y, Xu L, Su J, Huang F, Wang Y, et al. Unphosphorylated ISGF3 drives constitutive expression of interferon-stimulated genes to protect against viral infections. Sci Signal. 2017;10(476):eaah4248 [DOI] [PubMed] [Google Scholar]
- 90.Cheon H, Holvey-Bates EG, Schoggins JW, Forster S, Hertzog P, Imanaka N, et al. IFNbeta-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 2013;32(20):2751–63. Epub 2013/09/26. 10.1038/emboj.2013.203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Herbeuval JP, Grivel JC, Boasso A, Hardy AW, Chougnet C, Dolan MJ, et al. CD4+ T-cell death induced by infectious and noninfectious HIV-1: role of type 1 interferon-dependent, TRAIL/DR5-mediated apoptosis. Blood. 2005;106(10):3524–31. 10.1182/blood-2005-03-1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sivaraman V, Zhang L, Su L. Type I interferon contributes to CD4+ T cell depletion induced by infection with HIV-1 in the human thymus. Journal of virology. 2011;85(17):9243–6. 10.1128/JVI.00457-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tsuge M, Uchida T, Hiraga N, Kan H, Makokha GN, Abe-Chayama H, et al. Development of a Novel Site-Specific Pegylated Interferon Beta for Antiviral Therapy of Chronic Hepatitis B Virus. Antimicrob Agents Chemother. 2017;61(6):e00183–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Makowska Z, Duong FH, Trincucci G, Tough DF, Heim MH. Interferon-beta and interferon-lambda signaling is not affected by interferon-induced refractoriness to interferon-alpha in vivo. Hepatology. 2011;53(4):1154–63. 10.1002/hep.24189 [DOI] [PubMed] [Google Scholar]
- 95.Hung IF, Lung KC, Tso EY, Liu R, Chung TW, Chu MY, et al. Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet. 2020; 395(10238):1695–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Channappanavar R, Fehr AR, Zheng J, Wohlford-Lenane C, Abrahante JE, Mack M, et al. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J Clin Invest. 2019;130:3625–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell host & microbe. 2016;19(2):181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cheon H, Stark GR. Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(23):9373–8. Epub 2009/05/30. 10.1073/pnas.0903487106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Steele AK, Lee EJ, Manuzak JA, Dillon SM, Beckham JD, McCarter MD, et al. Microbial exposure alters HIV-1-induced mucosal CD4+ T cell death pathways Ex vivo. Retrovirology. 2014;11:14 10.1186/1742-4690-11-14 [DOI] [PMC free article] [PubMed] [Google Scholar]