

Summary
An approach to the generation of ultra-large chemical libraries of readily accessible (“REAL”) compounds is described. The strategy is based on the use of two- or three-step three-component reaction sequences and available starting materials with pre-validated chemical reactivity. After the preliminary parallel experiments, the methods with at least ∼80% synthesis success rate (such as acylation – deprotection – acylation of monoprotected diamines or amide formation – click reaction with functionalized azides) can be selected and used to generate the target chemical space. It is shown that by using only on the two aforementioned reaction sequences, a nearly 29-billion compound library is easily obtained. According to the predicted physico-chemical descriptor values, the generated chemical space contains large fractions of both drug-like and “beyond rule-of-five” members, whereas the strictest lead-likeness criteria (the so-called Churcher's rules) are met by the lesser part, which still exceeds 22 million.
Subject Areas: Chemical Compound, Cheminformatics, Computational Chemistry by Subject
Graphical Abstract
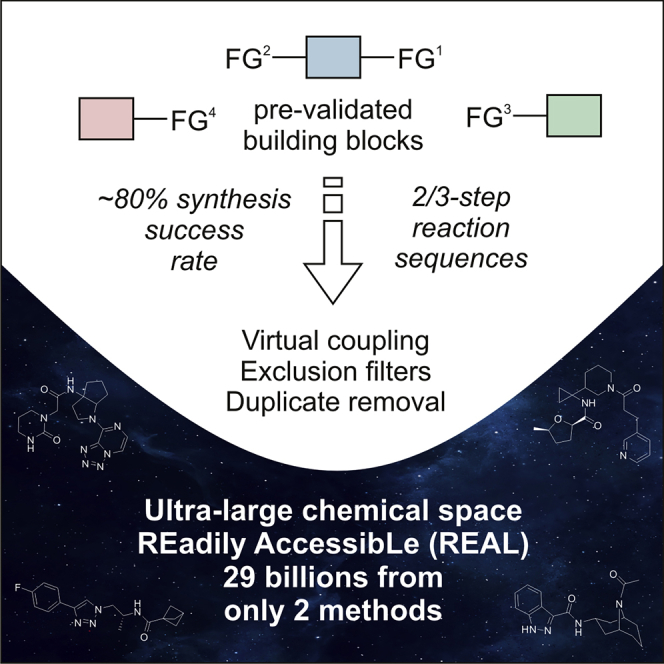
Highlights
-
•
A strategy for ultra-large readily accessible (REAL) compound libraries is described
-
•
Pre-validated two- or three-step three-component reaction sequences are used
-
•
A 29-billion chemical space with ∼80% synthesis success rate has been easily obtained
Chemical Compound; Cheminformatics; Computational Chemistry by Subject
Introduction
Modern drug discovery relies heavily on efficient mining of the chemical space, which is a descriptor space of all possible compounds (Dobson, 2004). This task is difficult owing to the enormous size of the accessible chemical space, which is estimated to include at least 1060 “observable” molecules; such a huge number makes its comprehensive enumeration and synthetic exploration impossible (at least currently). Nevertheless, significant advances in computational techniques allowed virtual exploration of reasonably large portions of chemical space efficiently (Hoffmann and Gastreich, 2019; Walters, 2019). Many recent works addressed enumeration of compounds relevant to drug discovery; a prominent example is given by works of Reymond and co-workers who described the generation of all stable molecules with up to a certain number of heavy atoms (GDB) (Reymond, 2015). In combination with virtual screening as a powerful tool for prioritizing compounds before in vitro biological tests, such databases provide a promising tool to discover chemotypes for further optimization into drug candidates.
The major drawback of many virtually enumerated compound libraries is the unpredictable synthetic feasibility of their particular members, which hampers their experimental validation against the biological targets of interest. A possible approach to address this issue is based on the so-called forward synthetic analysis (Schreiber, 2000) and includes an enumeration of virtual libraries by the combination of synthons representing readily available building blocks. This strategy typically requires a reasonably large pool of reagents with established chemical behavior; it is not surprising therefore that it has been mostly used by the big pharma companies internally (e.g., Merck's MASSIV [Walters, 2019], Boehringer Ingelheim's BIClaim [Lessel et al., 2009], Eli Lilly's Proximal Collection [Nicolaou et al., 2016] or Pfizer Global Virtual Library [PGVL] [Hu et al., 2012]).
More than a decade ago, we have launched a similar project on the generation of a virtual compound database based on the experimentally validated synthetic accessibility (the so-called REAL database, where REAL stands for REadily AccessibLe) (Shivanyuk et al., 2007). The main idea of this project follows the “forward synthetic analysis” concept described above: the available building blocks with validated reactivity are transformed into synthons with denoted reactivity features, which are then subjected to virtual coupling according to the well-established reactions and exclusion rules based on the reactivity features (Figure 1). The database was growing through the years and has reached 1.2 billion compounds with a 3- to 4-week synthesis time and ca. 85% synthesis success rate (i.e., a fraction of experiments that could produce the target compound among all the experiments performed) (Enamine REAL compounds, 2020). Recently, its utility in combination with virtual screening was confirmed by discovery of highly potent AmpC β-lactamase (AmpC) inhibitors, D4 dopamine receptor ligands (Lyu et al., 2019), and Kelch-like ECH-associated protein 1 (KEAP1) inhibitors (Gorgulla et al., 2020).
Figure 1.

A General Principle of the REAL Database Generation Using One-Step Two-Component Reactions
Further extension of this concept led to the development of the REAL Space, a searchable chemical space that is not typically stored as an enumerated database but generated upon query through a chemoinformatics software (Klingler et al., 2019). This feature tree-based (Rarey and Stahl, 2001; Boehm et al., 2008) engine allowed processing very large datasets currently reaching 13 billion molecules. In addition to that, it allowed considering more complex reaction sequences as compared with those shown in Figure 1. In this work, we describe our approach to the generation of ultra-large, multibillion chemical space of the readily accessible compounds, which is based on the one-pot parallel reactions involving at least three building blocks (Figure 2).
Figure 2.

An Approach to the Generation of Ultra-large Chemical Space Described in this Work
Results and Discussion
Validation of Parallel Reactions
To demonstrate the principles of the chemical space generation, we have selected five two- or three-step three-component reactions shown in Scheme 1. In most cases, modification of N-Boc-monoprotected diamines (building blocks 1) was envisaged, i.e., acylation – deprotection – acylation (reaction A), acylation – deprotection – arylation (B), acylation – deprotection – alkylation (C), and arylation – deprotection – acylation (D) sequences. In addition to that, the acylation – copper-catalyzed azide-alkyne click reaction sequence involving either amino azides (2) or azido acids (3) was studied. Starting from the available set of bifunctional building blocks 1–3 and capping reagents 4–10 (typically with validated reactivity in the corresponding one-step transformations), 5 × 400 members of the libraries 11–15 were generated by random selection and virtual coupling of the corresponding synthons and then subjected to parallel synthesis.
Scheme 1.

Parallel Reaction Sequences Studied in This Work.
See also Tables S1 and S2, Figures S1 and S2.
The results of these validation experiments are shown in Table 1. Thus, methods A and D worked well and gave the target products with 77% and 81% synthesis success rate, as well as 44% and 38% average yield. Two-step reaction sequence E was even more efficient (80% synthesis success rate, 51% average yield). On the contrary, methods B and C (acylation – deprotection – arylation/alkylation) showed lower success rate (60% and 53%, respectively); the corresponding library members 12 and 13 were obtained with 43% and 31% average yield, respectively. Analysis of the crude reaction mixture showed that competitive arylation/alkylation of N-hydroxybenzotriazole (formed from HATU, a coupling reagent from the acylation step) might be a major problem lowering the efficiency of the latter reaction sequences.
Table 1.
Validation Experiments for the Parallel Synthesis of Libraries 11–15
| # | Method | Conditions | Library | Success Rate, %a | Average Yield, % |
|
|---|---|---|---|---|---|---|
| All Experiments | Successful Experiments | |||||
| 1 | A | 1. HATU, i-Pr2NEt, DMSO, rt, 16 h 2. CF3COOH, i-Pr3SiH, H2O, rt, 6 h 3. HATU, i-Pr2NEt, DMSO, rt, 16 h |
11 | 77 | 34 | 44 |
| 2 | B | 1-2. Same as for A 3. i-Pr2NEt, NMP,b 100°C, 16 h |
12 | 60 | 26 | 43 |
| 3 | C | 1-2. Same as for A 3. i-Pr2NEt, DMF, 80°C, 16 h |
13 | 53 | 16 | 31 |
| 4 | D | 1. i-Pr2NEt, NMP,b 100°C, 16 h 2-3. Same as for A |
14 | 81 | 30 | 38 |
| 5 | E | 1. HATU, i-Pr2NEt, DMF, rt, 16 h 2. i-Pr2NEt, Cu(OAc)2, 80°C, 16 h |
15 | 80 | 41 | 51 |
See Also Tables S1 and S2, Figures S1 and S2.
Fraction of 400 experiments that allowed for the preparation of the target product.
NMP, N-methyl-2-pyrrolidone.
Taking into account the results described above, as well as the acceptable synthesis success rate for the REAL Database and the REAL Space (around 80%), only methods A, D, and E can be used to generate the ultra-large chemical space in further steps of this work. Methods B and C require further optimization before their incorporation into the toolbox of the studied strategy is possible; they might still be applicable for the library synthesis but with lower confidence.
In addition to that, success rates were analyzed for each of the reagents 1–3 to identify those demonstrating poor efficiency. Owing to the limited size of the dataset, only the building blocks for which at least 10 experiments were performed were taken into account. Figure 3 shows examples of the reagents showing both excellent and low reactivity in the reaction sequence studied. An obvious reason for the poor efficiency observed for compounds 1{256} and 3{5} is related to steric hindrance. Therefore, building blocks 1{256} and 3{5} were excluded from the further generation of the REAL chemical space.
Figure 3.

Examples of Reagents 1–3 Showing Excellent and Poor Efficiency in the Methods Studied (Relative Configurations are Shown)
Generation of the Chemical Space
First of all, building blocks 1–5 and 8–10 necessary for reaction sequences A and E were transformed into the synthons ready for the virtual coupling (see Figure 4 and Scheme 2). Of building blocks 4, 5, and 8–10, only those having validated reactivity in the corresponding one-step parallel syntheses were taken into consideration. In addition to that, cut-offs by molecular weight were applied for 4 and 5. For the bifunctional building blocks 1–3, visual inspection was also applied in addition to the results obtained from the preliminary tests described above. Apart from the SMILES representation (Weininger, 1988), reaction ID, and the role in the reaction sequence, reactivity features for the exclusion rules were recorded for each synthon, denoting steric hindrance around the corresponding functional groups. Again, for building blocks 4 and 5, these reactivity features were taken from the available statistical data for the one-step parallel reactions, whereas for monoprotected diamines 1, they were assigned manually for each of the functional groups after the visual inspection. It should be pointed out that the methodology does not involve quantitative reactivity measures; instead, binary (“yes/no”) qualitative reactivity features are introduced for each synthon. For reaction sequence E, the reactivity features related to the steric factor were not taken into account. Although method D is fully suitable for the REAL Space generation, it was not included in the study at this point; the corresponding synthons are currently under development.
Figure 4.

Examples of Synthons Generated from Reagents 1, 2, 4, 5, 8, and 10 (in the Corresponding SMILES Representations, Uncommon [“Dummy”] Atoms are Used Instead of the Colored Asterisks [∗] to Denote Different Types of the Variation Points)
Scheme 2.

Virtual Coupling of the Synthons Shown in Figure 4 (the Variation Points [∗] Are Connected according to Their Types)
As a result, a total of 15,153 and 46,474 synthons were generated for the reaction sequences A and E, respectively (Table 2). Further processing of these synthons followed the workflow shown in Figure 5. The workflow included virtual coupling, application of the exclusion filters (addressing the reactivity features), and duplicate removal (performed with the InChI key representations [Heller et al., 2015] to increase the performance). The synthons with negative overall reactivity feature were excluded from the process prior the coupling. As for the steric factor, combinations of synthons with both negative features were excluded at the corresponding step. Table 3 summarizes the generation of the multibillion parts of the chemical space according to the methods A and E, as well as numbers of the readily accessible compounds that could be achieved.
Table 2.
Number of Various Synthons Types Generated for Reaction Sequences A and E
| # | Method | Reagents | Number of Synthons |
||
|---|---|---|---|---|---|
| No Reactivity Features | With Steric Features | Total | |||
| 1 | A | 1 | 467 | 196a | 663 |
| 2 | 4 | 6,706 | 1,271 | 7,977 | |
| 3 | 5 | 5,451 | 1,063 | 6,514 | |
| 4 | E | 2 | 41 | 0 | 41 |
| 5 | 3 | 52 | 0 | 52 | |
| 6 | 8 | 17,944 | 550 | 18,494 | |
| 7 | 9 | 26,434 | 646 | 27,080 | |
| 8 | 10 | 807 | 0 | 807 | |
With steric hindrance at the free amino group (103), the protected amino group (82), or both (11).
Figure 5.

The Workflow of the Multibillion Chemical Space Generation
Table 3.
Results of the Multibillion Chemical Space Generation
| # | Method | No. of Synthons | No. of Library Members after |
||
|---|---|---|---|---|---|
| Virtual Coupling | Exclusion Filters | Duplicate Removal | |||
| 1 | A | 15,154 | 34,450,924,014 | 32,733,348,058 | 27,297,397,644 |
| 2 | E | 46,474 | 1,748,296,098 | 1,748,296,098 | 1,563,752,616 |
| 3 | Total | 60,431 | 36,199,220,112 | 34,481,644,156 | 28,861,150,260 |
As it is obvious from Table 3, the number of the “core” (bifunctional) building blocks, as well as sufficient and comparable accessibility of both “capping” reagents types are the key parameters affecting the size of the generated chemical space. Even with reasonably decreased sets of the “capping reagents,” multibillion numbers are easily achieved (as in the case of method A). For method E, both limited availabilities of the azide-containing bifunctional building blocks 2 and 3 and very different accessibility of the reagents 8/9 and 10 are responsible for the fact that the size of the resulting chemical space could only exceed a billion of structures. Nevertheless, even with all these limitations, we could generate a chemical space containing nearly 29 billion of readily accessible compounds using only two reaction sequences. As it was mentioned in the Introduction, this chemical space can be accessed either directly as a pre-enumerated database or through a feature-tree based search engine that performs generation of the corresponding structures upon a query. The current versions of the REAL Database and REAL Space include 0.27 and 9.9 billion members obtained according to methods A or E (since additional cut-offs on the physico-chemical and structural properties, as well as reagent availability, were applied).
Predicted Physico-Chemical Descriptors
Over the last decades, it was stressed out that physico-chemical properties of the compounds are important to drug discovery since they have a critical impact on the attrition rate of drug candidates (Grygorenko et al., 2020). It is therefore important to understand the capabilities of the generated chemical space in terms of providing the so-called drug-like or lead-like compounds (Nadin et al., 2012). To address this point, we have calculated physico-chemical descriptors of common interest to medicinal chemistry, i.e., molecular weight (MW), the logarithm of octanol-water partition coefficient (sLogP) (Wildman and Crippen, 1999), hydrogen bond acceptor/donor counts (HAcc/HDon), topologic polar surface area (TPSA), rotatable bond count (RotB), and sp3-hybrid carbon atom fraction (Fsp3).
As it follows from Figure 6 and Tables 4 and 5, the part of the chemical space generated by method A complies well with the classical drug-likeness criteria (i.e., Lipinski and Veber rules), whereas method E tends to provide heavier, more lipophilic compounds with higher hydrogen bond acceptor count, polar surface area, and rotatable bond count, an obvious consequence of the less stringent pre-selection of starting building blocks 8–10. Of course, the percentage of the fitting chemical space members goes down rapidly when more stringent lead-likeness criteria are applied. Nevertheless, a considerable number of the compounds remains even after application of the most rigorous Churcher's rules (21.2, 0.95, and 22.1 Mln members by method A, E, and in total, respectively). Moreover, the significant fraction of the readily accessible “beyond-of-Ro5” members can be sometimes considered even advantageous taking into account the recently increased interest to such compounds in medicinal chemistry (DeGoey et al., 2018).
Figure 6.

Distribution of Physico-Chemical Descriptors Predicted for the Generated Chemical Space and Approved Drugs
See also Table S3.
Table 4.
Average Values of Physico-Chemical Descriptors Predicted for the Generated Chemical Space and Approved Drugs
| # | Method | MW | sLogP | HAcc | HDon | TPSA, Å2 | RotB | Fsp3 |
|---|---|---|---|---|---|---|---|---|
| 1 | A | 440 | 2.61 | 5.3 | 1.5 | 93.7 | 6.4 | 0.56 |
| 2 | E | 502 | 3.15 | 7.8 | 1.3 | 112.1 | 8.3 | 0.51 |
| 3 | Total | 444 | 2.64 | 5.4 | 1.5 | 94.6 | 6.5 | 0.55 |
| 4 | DrugBanka | 395 | 2.05 | 5.1 | 2.4 | 96.9 | 6.4 | 0.47 |
Data for 2,470 drugs deposited in DrugBank (as of September 2020).
Table 5.
Fractions of the Generated Chemical Space (%) Compliant with the Drug- and Lead-likeness Rules
| # | Method | Rule of 5a | + Veber's Rulesb | Rule of 4.5c | Rule of 4d | Churcher's Rulese,f |
|---|---|---|---|---|---|---|
| 1 | A | 89.1 | 82.6 | 56.9 | 16.9 | 0.08 (21,167,934) |
| 2 | E | 48.4 | 40.4 | 24.3 | 7.4 | 0.06 (952,402) |
| 3 | Total | 86.9 | 80.3 | 55.1 | 16.5 | 0.08 (22,120,336) |
MW < 500, LogP<5, HAcc≤10, HDon≤5 (Lipinski et al., 1997).
RotB ≤10, TPSA <140 (Veber et al., 2002).
MW < 450, LogP<4.5 (Oprea et al., 2001).
MW < 400, LogP<4 (Hann and Oprea, 2004).
MW 200 … 350, LogP −1 … 3 (Nadin et al., 2012).
Absolute numbers of the library members are given in brackets.
Comparison of the obtained results with the physico-chemical properties of 2,470 approved drugs deposited in DrugBank database (Wishart et al., 2018) showed that compounds produced by our approach tend to be heavier and slightly more lipophilic and have somewhat lower hydrogen bond donor count, which is an obvious consequence of the chemical methodology used (Table 4 and Figure 6). They are also more sp3 enriched. All these features are in line with recent trends in drug discovery (good or bad) related to increased molecular complexity of new drug molecules (Grygorenko et al., 2020). On the contrary, the average values of total polar surface area, rotatable bond and hydrogen bond acceptor counts for the library members are more or less in line with those of the known drugs.
A short study was also performed to assess the relationship between the distribution of the synthons and the space covered by the generated databases. In particular, random selections were made from the sets of the synthons used for method A containing from 5% to 95% (with 5% step) of the initial structures, and the corresponding library members were selected from the final database. As it might be expected, the database size followed a cube function of the synthon subset size (Figure 7).
Figure 7.

Relationship between the Size of the Generated Databases and the Size of the Synthon Subsets
Obtained by random selections from the initial synthon set for Method A; average from three independent selections; see also Table S4.
In addition to that, other synthon subsets were prepared by applying molecular weight cut-offs of 100–275 (with 25 MW step) to the initial synthon set used for method A, and the corresponding databases were generated. Owing to the properties of the initial synthon set, the size of the resulting databases increased dramatically for the cut-off range 100–200 (the so-called rule-of-two for building blocks [Goldberg et al., 2015]) and reached a maximum value after MW = 275 (a general cut-off used in the design of the initial set) (Figure 8A). Expectedly, distribution of physico-chemical properties (i.e., MW and sLogP) within the resulting virtual libraries correlated with increase in the corresponding values for the synthons (Figure 8B).
Figure 8.

Properties of the Generated Chemical Space as a Function of the Molecular Weight Cut-offs Applied to the Initial Synthon Sets for Method A
(A and B) (A) The size of the generated databases. (B) Distribution of physico-chemical descriptors (MW and sLogP) for the generated chemical space.
See also Table S5.
One might argue that the technology described in the current work is mostly based on very simple chemical transformations; therefore, its capability of producing novel, complex, and diverse molecules might be questionable. Nevertheless, a recent analysis by AstraZeneca scientists shows that this is not the case: even using only the amide formation reaction, very good results can be obtained in the early drug discovery provided that sufficient access to the corresponding building blocks is possible (Tomberg and Boström, 2020).
The physico-chemical features of the chemical space generated in this work are similar to those of DNA-encoded libraries (Kunig et al., 2018). In both cases, this is related to the fact that final library members are constructed from at least three building blocks, which increases the lower MW limit. In our opinion, the huge size of both DNA-encoded libraries and multibillion chemical spaces like the one described herein can be considered as compensation for the increased molecular complexity (provided that efficient in vitro or in silico screening technologies are available to mine these ultra-large libraries). The success stories available in the literature for both technologies (Goodnow et al., 2017; Kunig et al., 2018; Lyu et al., 2019; Gorgulla et al., 2020) can serve as a justification for the above hypothesis.
Conclusions
Combined with the modern virtual screening tools, ultra-large libraries of readily accessible (“REAL”) compounds have proven their utility for the identification of highly potent hits against various biological targets. Herein, it is shown that a nearly 29-billion chemical space covering such synthetically feasible representatives can be easily generated using two- or three-step three-component reaction sequences and available starting materials with the chemical reactivity validated in one-step parallel reactions. Only the methods with at least ∼80% synthesis success rate (e.g., acylation – deprotection – acylation of monoprotected diamines, as well as amide formation – click reaction with amino azides or azido acids) are acceptable to generate the target chemical space with sufficient synthetic confidence. It is shown that diversity of the “core” (bifunctional) building blocks, as well as nearly equal (but sufficient) accessibility of the “capping” reagents are essential to obtain the largest numbers of the library members. Analysis of physico-chemical descriptors reveals that the generated chemical space contains large fractions of both drug-like and “beyond rule-of-five” members, whereas the strictest lead-likeness criteria (i.e., Churcher's rules) are met for the lesser part (which still exceeds 22 million compounds). In our opinion, a combination of ultra-large REAL libraries and modern virtual screening tools is similar to DNA-encoded libraries (that have gained momentum in recent years) in terms of physico-chemical properties and chemical space coverage.
The approach proposed in this work is a substantial extension of the previous methodology that was based mainly on the two-component parallel reactions. It is also distinct from recent approaches relying heavily on artificial intelligence (Hoffmann and Gastreich, 2019) since it relies on the very robust and straightforward algorithm (Table 6).
Table 6.
Selected Approaches to Generate (Ultra-)large Virtual Chemical Space
| Feature | Approach Described in This Work | Previous Feasibility-Based approachesa | Recent AI-Based approachesb |
|---|---|---|---|
| Virtual chemical space | Multibillion (over 3 × 1010) | Large (~109) | Varied but typically less than 109 |
| Synthetic methods | Experimentally validated three-component two- or three-step reaction sequences | Experimentally validated two-component one-step reactions (mostly) | Various; typically based on the literature data (not always validated experimentally) |
| Algorithm | Very straightforward | Sophisticated | |
| Synthetic feasibility | Average value for each method or synthon, described as average synthesis success rate | Varied; from unknown to predicted for each particular member | |
| Building block reactivity assessment | Semi-qualitative; by a chemical expert aided by a computer | Typically quantitative; by AI | |
Previous version of our REAL methodology is referred here; much larger datasets were also generated internally within big pharma companies (Hoffmann and Gastreich, 2019).
The subject was reviewed and critically accessed in a number of recent publications (Schneider, 2018; Schwaller and Laino, 2019; Brown et al., 2020; Lemonick, 2020).
Limitations of the Study
Possible limitations of the study include: (1) difficulties with handling of the full generated chemical space owing to the current hardware capabilities; this can be overcome by pre-selection of its part according to some criteria (like molecular weight) or by using special search engines like those mentioned in the Introduction; (2) a ca. 20% probability for the particular library member to be not produced according to the proposed synthetic methodology; a possible solution is to make a larger selection of the library members of interest (e.g., at least 100–200 representatives) to be synthesized with ca. 80% confidence; (3) impossibility to provide more or less precise synthetic feasibility for a particular compound—only an average value can be predicted for the method as a whole; (4) dynamic nature of the generated space due to the changes in the availability of the starting materials or information on their reactivity; this can be addressed by its regular periodic updates, as well as by applying cut-offs for the amounts of the stock reagents.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Yurii S. Moroz, ysmoroz@gmail.com.
Materials Availability
Compound library members generated in this study will be made available on request, but we shall require payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Data and Code Availability
The complete lists of reagents used to construct the chemical space supporting the current study have not been deposited in a public repository owing to the company's policy but are available from the corresponding author on request. There are restrictions on the availability of the in-house code and the synthon lists with the reactivity features that have been used to generate the chemical space owing to commercial confidentiality reasons.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
Dedicated to the memory of Dr. Andrey V. Bogolubsky. The work was funded by the NIH Grant GM133836 (to Prof. John J. Irwin and Y.S.M.). O.O.G. was also funded by the Ministry of Education and Science of Ukraine (Grant No. 19BF037-03). The authors thank Prof. Andrey A. Tolmachev for his encouragement and support, Mr. Bohdan V. Vashchenko and Mr. Dmytro Krivoruchko for their help with the Transparent Methods section preparation.
Author Contributions
Conceptualization, O.O.G. and D.S.R.; Methodology, D.S.R. and A.C.; Software, I.D. and A.C.; Validation, D.S.R., I.D., and A.C.; Formal Analysis, O.O.G. and D.S.R.; Investigation, D.S.R., I.D., and K.E.G.; Data Curation, D.S.R., Y.S.M., and K.E.G; Writing – Original Draft, O.O.G.; Writing – Review & Editing, O.O.G., D.S.R., and Y.S.M.; Visualization, O.O.G.; Supervision, O.O.G. and Y.S.M.; Project Administration, D.S.R. and Y.S.M.; Funding Acquisition, Y.S.M.
Declaration of Interests
The authors declare no competing interests.
Published: November 20, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101681.
Contributor Information
Oleksandr O. Grygorenko, Email: gregor@univ.kiev.ua.
Yurii S. Moroz, Email: yurii.moroz@chem-space.com.
Supplemental Information
References
- Boehm M., Wu T.Y., Haussen H., Lemmen C. Similarity searching and scaffold hopping in synthetically accessible combinatorial chemistry spaces. J. Med. Chem. 2008;51:2468–2480. doi: 10.1021/jm0707727. [DOI] [PubMed] [Google Scholar]
- Brown N., Ertl P., Lewis R., Luksch T., Reker D., Schneider N. Artificial intelligence in chemistry and drug design. J. Comput. Aided. Mol. Des. 2020;34:709–715. doi: 10.1007/s10822-020-00317-x. [DOI] [PubMed] [Google Scholar]
- DeGoey D.A., Chen H.-J.J., Cox P.B., Wendt M.D. Beyond the rule of 5: lessons learned from AbbVie’s drugs and compound collection. J. Med. Chem. 2018;61:2636–2651. doi: 10.1021/acs.jmedchem.7b00717. [DOI] [PubMed] [Google Scholar]
- Dobson C.M. Chemical space and biology. Nature. 2004;432:824–828. doi: 10.1038/nature03192. [DOI] [PubMed] [Google Scholar]
- Enamine REAL compounds (2020). Available at: https://enamine.net/library-synthesis/real-compounds.
- Goldberg F.W., Kettle J.G., Kogej T., Perry M.W.D., Tomkinson N.P. Designing novel building blocks is an overlooked strategy to improve compound quality. Drug Discov. Today. 2015;20:11–17. doi: 10.1016/j.drudis.2014.09.023. [DOI] [PubMed] [Google Scholar]
- Goodnow R.A., Dumelin C.E., Keefe A.D. DNA-encoded chemistry: enabling the deeper sampling of chemical space. Nat. Rev. Drug Discov. 2017;16:131–147. doi: 10.1038/nrd.2016.213. [DOI] [PubMed] [Google Scholar]
- Gorgulla C., Boeszoermenyi A., Wang Z.-F., Fischer P.D., Coote P.W., Padmanabha Das K.M., Malets Y.S., Radchenko D.S., Moroz Y.S., Scott D.A. An open-source drug discovery platform enables ultra-large virtual screens. Nature. 2020;580:663–668. doi: 10.1038/s41586-020-2117-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grygorenko O.O., Volochnyuk D.M., Ryabukhin S.V., Judd D.B. The symbiotic relationship between drug discovery and organic chemistry. Chem. Eur. J. 2020;26:1196–1237. doi: 10.1002/chem.201903232. [DOI] [PubMed] [Google Scholar]
- Hann M.M., Oprea T.I. Pursuing the leadlikeness concept in pharmaceutical research. Curr. Opin. Chem. Biol. 2004;8:255–263. doi: 10.1016/j.cbpa.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Heller S.R., McNaught A., Pletnev I., Stein S., Tchekhovskoi D. InChI, the IUPAC international chemical identifier. J. Cheminform. 2015;7:23. doi: 10.1186/s13321-015-0068-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann T., Gastreich M. The next level in chemical space navigation: going far beyond enumerable compound libraries. Drug Discov. Today. 2019;24:1148–1156. doi: 10.1016/j.drudis.2019.02.013. [DOI] [PubMed] [Google Scholar]
- Hu Q., Peng Z., Sutton S.C., Na J., Kostrowicki J., Yang B., Thacher T., Kong X., Mattaparti S., Zhou J.Z. Pfizer Global Virtual Library (PGVL): a chemistry design tool powered by experimentally validated parallel synthesis information. ACS Comb. Sci. 2012;14:579–589. doi: 10.1021/co300096q. [DOI] [PubMed] [Google Scholar]
- Klingler F.-M., Gastreich M., Grygorenko O., Savych O., Borysko P., Griniukova A., Gubina K., Lemmen C., Moroz Y. SAR by Space: enriching hit sets from the chemical space. Molecules. 2019;24:3096. doi: 10.3390/molecules24173096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunig V., Potowski M., Gohla A., Brunschweiger A. DNA-encoded libraries-an efficient small molecule discovery technology for the biomedical sciences. Biol. Chem. 2018;399:691–710. doi: 10.1515/hsz-2018-0119. [DOI] [PubMed] [Google Scholar]
- Lemonick S. Exploring chemical space: can AI take us where no human has gone before? Chem./Eng. News. 2020;98:30–35. [Google Scholar]
- Lessel U., Wellenzohn B., Lilienthal M., Claussen H. Searching fragment spaces with feature trees. J. Chem. Inf. Model. 2009;49:270–279. doi: 10.1021/ci800272a. [DOI] [PubMed] [Google Scholar]
- Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Lyu J., Irwin J.J., Roth B.L., Shoichet B.K., Levit A., Wang S., Tolmachova K., Singh I., Tolmachev A.A., Che T. Ultra-large library docking for discovering new chemotypes. Nature. 2019;566:224–229. doi: 10.1038/s41586-019-0917-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadin A., Hattotuwagama C., Churcher I. Lead-oriented synthesis: a new opportunity for synthetic chemistry. Angew. Chem. Int. Ed. 2012;51:1114–1122. doi: 10.1002/anie.201105840. [DOI] [PubMed] [Google Scholar]
- Nicolaou C.A., Watson I.A., Hu H., Wang J. The Proximal Lilly collection: mapping, exploring and exploiting feasible chemical space. J. Chem. Inf. Model. 2016;56:1253–1266. doi: 10.1021/acs.jcim.6b00173. [DOI] [PubMed] [Google Scholar]
- Oprea T.I., Davis A.M., Teague S.J., Leeson P.D. Is there a difference between leads and drugs? A historical perspective. J. Chem. Inf. Comput. Sci. 2001;41:1308–1315. doi: 10.1021/ci010366a. [DOI] [PubMed] [Google Scholar]
- Rarey M., Stahl M. Similarity searching in large combinatorial chemistry spaces. J. Comput. Aided. Mol. Des. 2001;15:497–520. doi: 10.1023/a:1011144622059. [DOI] [PubMed] [Google Scholar]
- Reymond J.-L. The chemical space project. Acc. Chem. Res. 2015;48:722–730. doi: 10.1021/ar500432k. [DOI] [PubMed] [Google Scholar]
- Schneider G. Automating drug discovery. Nat. Rev. Drug Discov. 2018;17:97–113. doi: 10.1038/nrd.2017.232. [DOI] [PubMed] [Google Scholar]
- Schreiber S.L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- Schwaller P., Laino T. Data-driven learning systems for chemical reaction prediction: an analysis of recent approaches. ACS Symp. Ser. 2019:61–79. [Google Scholar]
- Shivanyuk A., Ryabukhin S.V., Bogolubsky A.V., Tolmachev A. Enamine REAL database: making chemical diversity real. Chem. Today. 2007;25:58–59. [Google Scholar]
- Tomberg A., Boström J. Can ‘easy’ chemistry produce complex, diverse and novel molecules? ChemRxiv. 2020 doi: 10.26434/CHEMRXIV.12563231.V1. [DOI] [PubMed] [Google Scholar]
- Veber D.F., Johnson S.R., Cheng H.Y., Smith B.R., Ward K.W., Kopple K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- Walters W.P. Virtual chemical libraries. J. Med. Chem. 2019;62:1116–1124. doi: 10.1021/acs.jmedchem.8b01048. [DOI] [PubMed] [Google Scholar]
- Weininger D. SMILES, a Chemical language and information system: 1: introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988;28:31–36. [Google Scholar]
- Wildman S.A., Crippen G.M. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 1999;39:868–873. [Google Scholar]
- Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The complete lists of reagents used to construct the chemical space supporting the current study have not been deposited in a public repository owing to the company's policy but are available from the corresponding author on request. There are restrictions on the availability of the in-house code and the synthon lists with the reactivity features that have been used to generate the chemical space owing to commercial confidentiality reasons.