

Abstract
Introduction
Tau, a natively unfolded soluble protein, forms abnormal oligomers and insoluble filaments in several neurodegenerative diseases, including Alzheimer disease (AD). Tau‐induced toxicity is mainly due to oligomers rather than monomers or fibrils.
Methods
We have developed monoclonal antibodies against purified low‐n tau oligomers of the tau repeat domain as a tool to neutralize tau aggregation and toxicity. In vitro aggregation inhibition was tested by thioflavin S, dynamic light scattering (DLS), and atomic force microscopy (AFM). Using a split‐luciferase complementation assay and fluorescence‐activated cell sorting (FACS), the inhibition of aggregation was analyzed in an N2a cell model of tauopathy.
Results
Antibodies inhibited tau aggregation in vitro up to ~90% by blocking tau at an oligomeric state. Some antibodies were able to block tau dimerization/oligomerization in cells, as measured by a split‐luciferase complementation assay. Antibodies applied extracellularly were internalized and led to sequestration of tau into lysosomes for degradation.
Discussion
Novel low‐n tau oligomer specific monoclonal antibody inhibits Tau oligomerization in cells and promotes toxic tau clearance.
Keywords: aggregation, antibody, N2a cells, screening, tau
1. INTRODUCTION
Tau, a neuronal microtubule‐associated protein, aggregates to form insoluble, fibrillary deposits in a wide range of neurodegenerative diseases called tauopathies. 1 Alzheimer disease (AD) is characterized by the presence of extracellular plaques composed of amyloid beta (Aβ) and intracellular tangles of tau. Several therapies have been tried to target Aβ pathology, 2 , 3 but so far, they have failed to show significant benefits in clinical trials. 4 , 5 Therefore, therapies targeting tau pathology have gained importance, especially as cognitive decline in AD correlates better with tau pathology than with amyloid burden. 6 , 7
Mutations in the tau gene are sufficient to trigger neurodegeneration. 8 Tau undergoes multiple post‐translational modifications such as phosphorylation, acetylation, cleavage, glycation, etc. 9 Although post‐translational modifications may contribute to tau aggregation, the mechanisms involved in tau‐induced neurodegeneration are still poorly understood. Various studies of transgenic mice suggest a correlation between intracellular tau aggregation and neuronal dysfunction. 10 , 11 , 12 More specifically, tau‐induced toxicity is mainly due to tau oligomers rather than monomers or fibrillar aggregates. 13 , 14 , 15 Tau oligomers can induce toxicity by operating both on intracellular (cytosolic) and the extracellular (released) level. Extracellular tau may cause synaptic damage and also act as seeds for further aggregation in recipient neurons. 15 , 16 Attempts to scavenge the extracellular tau (with antibodies) may intercept the cell‐to‐cell transmission of tau. 17 , 18 , 19 On the other hand, this would not address the pool of cytosolic toxic tau. Thus it may be more appropriate to target the intracellular pathological tau oligomers to ameliorate tau pathology. 20 , 21
Several treatments based on small molecules aiming at reducing tau aggregation appeared to be promising in animal models 22 , 23 , 24 but failed in clinical trials. 25 , 26 , 27 As a result, tau‐based immunotherapies gained importance. Both passive and active immunization studies on tau are in progress. For example, passive immunization studies with anti‐monomeric tau antibodies injected into tau transgenic animals showed a decrease in hyperphosphorylated tau and reversal of behavior deficits. 17 Anti‐phospho tau antibodies 4E6 and 77E9 injected in 3XTg AD mice showed reduced levels of hyperphosphorylated tau and amyloid plaques with improved cognitive performance. 28 , 29 Transgenic mice treated with tau oligomer monoclonal antibodies (raised against Aβ‐cross‐seeded tau oligomers) resulted in lower cognitive and behavioral deficits. 30 , 31 , 32 Furthermore, active immunization studies with the vaccine AADvac1 (based on a peptide from the tau repeat domain) and phospho‐tau peptides in transgenic mice showed reduced tau oligomerization, phosphorylation, and improved sensorimotor functions. 19 , 33
The majority of these antibodies are directed against the tau monomers or phospho‐tau monomers. However, these might not be an ideal target as the toxicity is associated with the soluble oligomers of tau. 15 Therefore, we raised antibodies against highly purified low‐n oligomers of tau, which detected primarily assembled forms of tau. Some antibodies detected specifically low‐n (atomic force microscopy [AFM] height 2 to 3 nm) or high‐n (AFM height >10 nm) oligomers. We found that two oligomer‐specific antibodies inhibited the aggregation of tau by >90% in vitro. Further testing in a tauopathy cell model confirmed that such antibodies could enter cells and recruit the toxic oligomeric tau to lysosomes for degradation.
2. METHODS
2.1. Cell models
N2a wild type and inducible cell line (N2a‐TauRDΔK) 34 were grown in minimal essential media (Sigma, Darmstadt, Germany) supplemented with 10% fetal bovine serum (FBS), 5 mL non‐essential amino acids (PAA, Pasching, Austria), and 1X penicillin and streptomycin antibiotic. The inducible N2a cell lines expressing tau require antibiotics geneticin (G418) (300 µg/mL) and hygromycin (100 µg/mL). The TauRDΔK protein expression was induced by incubating cells with 1 µg/mL doxycycline. Minimal essential media (MEM) with complete serum deprivation (no FBS) for 72 hours were used to differentiate N2a cells in to neurons.
RESEARCH IN CONTEXT
Systematic review: Low‐n tau oligomers represent the most synaptotoxic form of tau. Such tau oligomers are in dynamic equilibrium with monomers and polymers and short lived, so that few treatment strategies have been developed so far.
Interpretation: Using well characterized purified low‐n oligomers of tau, we developed several specific antibodies inhibiting aggregation of tau up to 90% in vitro. Antibodies are internalized into cells, inhibit the oligomerization of tau, and recruit tau to lysosomes for degradation.
Future direction: Preclinical studies on animals will be used to investigate the therapeutic potential of antibodies. Antibodies may be developed as diagnostic tools for tau‐dependent toxicity.
HIGHLIGHTS
Development of antibodies specific for toxic low‐n oligomers of tau are targeted.
Low‐n tau oligomer specific antibodies can inhibit tau aggregation in vitro.
Antibodies can inhibit oligomerization of tau in cells.
Antibodies recruit tau to lysosomes for clearance.
2.2. Antibodies
Monoclonal antibodies were generated against the purified oligomers of the mutant tau repeat domain with the deletion of lysine 280 (TauRDΔK). TauRDΔK oligomers were prepared and purified as described previously, making use of hydrophobic interaction chromatography. 35 Briefly, 50 µM recombinant TauRDΔK (Mr∼13.5 kDa) was diluted in tris buffered saline (TBS) pH 9.0 and incubated at 37°C for 30 minutes for aggregation. The aggregated sample was lightly cross‐linked using 0.01% glutaraldehyde (GA) for 10 minutes at 37°C to stabilize the oligomers. The cross‐linked sample was purified using a HiPrep Butyl FF 16/10 hydrophobic column. These purified oligomers of TauRDΔK were used as an antigen for generating the monoclonal antibodies. C57BL/6 mice and Lou/C rats were injected subcutaneously and intraperitoneally with a mixture of 60 µg purified oligomers, 5 nmol CpG (Tib Molbiol, Berlin, Germany) and an equal volume of incomplete Freund's adjuvant. Six weeks later a booster injection was performed without Freund's adjuvant. Three days later, spleen cells were fused with P3 × 63Ag8.653 myeloma cells using standard procedures. Hybridoma supernatants were screened in a solid‐phase enzyme‐linked immunosorbent assay (ELISA) for binding to TauRDΔK oligomers and monomers. Positive supernatants were further assayed for their potential in capture ELISA, immunoblot, and dot blot analysis. Hybridoma cells from selected supernatants were subcloned at least twice by limiting dilution to obtain stable monoclonal cell lines. Experiments in this work were performed with antibody TAU clone 6B5 (mouse IgG2b), 8E7 (mouse IgG2b), 2H9 (mouse IgG2b), 29E2 (mouse IgG2a), 26G1 (mouse IgG2a), 28D4 (mouse IgG2b), 32E7 (mouse IgG2b), 2G9 (rat IgG1), 11D10 (rat IgG2c), 2B10 (rat IgG1) and 6H1 (rat IgG2c), 6H3 (rat IgG2a). In house generated Anti‐FLAG 6F7 (rat IgG1) was used as an immunoglobulin G (IgG) isotype control.
Commercial antibodies used in the current study include K9JA (DAKO #A0024), anti‐rabbit AMCA (Dianova #711‐155‐152), anti‐rat CF488 (VWR #20027), anti‐rabbit Alexa 555 (abcam #ab150066).
2.3. Dot blot
Activated Immobilon‐P membrane (Millipore) was loaded with 50 ng of different tau constructs (dissolved in phosphate buffered saline [PBS] buffer) and imbibed on the membrane using a vacuum pump. The membrane was washed thrice with PBS buffer. Then the membrane was blocked in 5% fat dry milk powder in1X TBST (containing 0.01% Tween 20) for 1 hour at room temperature on an orbital shaker. Later the membrane was incubated with primary antibody (in 1X TBST) at 4°C overnight followed by horseradish peroxidase (HRP) labeled secondary antibody (1X TBST) incubation at 37°C for 1 hour. The membrane was developed using enhanced chemiluminescent (ECL) reagent, and images were acquired using a LAS ImageQuant instrument (GE Healthcare).
2.4. Immunolabeling of antibody
Fluorophore labeling of the antibodies was done by maleimide coupling method (SH). Antibodies were reduced by tris(2carboxyethyl)phosphine (TCEP), which splits the molecule at the hinge region but the heavy and light chain remains intact. The SH groups of the reduced antibodies react with a maleimide‐activated fluorophore (A647) forming a stable bond. For labeling, the monoclonal antibody sample was concentrated using 3000 MWCO Amicon filters (Millipore) in a centrifuge (Eppendorf 5810R, Rotor A‐4‐62) at 2770 rpm, 4°C to achieve final concentrations of 50 to 100 µM. 10X excess molar concentrations of TCEP (Molecular Probes #T2556) to antibody was added and incubated on ice for 30 minutes, after which 11.85X excess molar concentrations of Alexa 647 dye (ThermoFisher, Waltham, MA, USA) was dissolved in dimethyl sulfoxide (DMSO), added and incubated on ice for 30 minutes. A NAP‐5 column (GE Healthcare, Chicago, IL, USA) was equilibrated with 10 mL PBS, and ∼500 µL of the antibody mixture was transferred on the column bed and eluted with 1 mL PBS buffer. Protein yield (280 nm), dye concentration (650 nm), and labeling efficiency were determined using a NanoDrop spectrophotometer (Peqlab ND‐1000). The degree of labeling (DOL) was calculated using the following equation.
Є IgG protein−203000; Є A647 dye −265000; CF280 (A647)–0.03, where Є = extinction coefficient.
DOL of antibody 2B10 = 5.4 (5.4 moles dye per mole of IgG), DOL of control antibody = 8.5 (8.5 moles dye per mole of IgG). The quality of the protein was checked using 10% sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) followed by Coomassie staining (Brilliant blue) and western blot. The antibody was intact after the labeling and a slight shift in the labeled antibody confirmed the degree of labelling (Figure S8 in supporting information).
2.5. Thioflavin S (ThS) assay
10 µM of tau monomer in buffer BES pH 7.0 with or without 2.5 µM heparin (MW 16,000), with or without different concentrations of antibody in the presence of 40 µM of ThS was prepared for a maximum of 40 µL and loaded in 384‐well plates (black microtiter 384‐plate round well, Thermo LabSystems, Dreieich, Germany), and measurements were carried out using Magellan software in a TECAN spectrofluorimeter (Ascent, Lab Systems, Frankfurt, Germany). Kinetics was carried out at 37°C for 24 hours with measurement intervals of 15 minutes using an excitation wavelength of 440 nm, an emission wavelength of 521 nm, with spectral bandwidths of 2.5 nm for excitation and emission. Samples were prepared in duplicates or triplicates, and after the measurements, the background fluorescence from ThS alone was subtracted.
2.6. Dynamic light scattering (DLS)
20 µL of the aggregated sample (with or without antibody) was placed in a quartz batch cuvette (ZEN2112) and thermally equilibrated at 25°C for 2 minutes in a Zetasizer Nano S (Malvern, Herrenberg, Germany) instrument fitted with 5‐milliwatt helium‐neon 633 nm laser at 173° measurement angle. The scattering intensity of the particles, their size, and numbers was obtained as an average of three measurements with 20 runs each. The results are expressed as a volume graph.
2.7. Atomic force microscopy (AFM)
AFM sample preparation and imaging was performed as described earlier. 36 In brief, mica discs pasted on a glass slide were freshly cleaved using a sticky tape. On this freshly cleaved mica, 1 to 2 µM of protein sample diluted in adsorption buffer (PBS, pH 7.4) was incubated for 10 minutes followed by washing with PBS, pH 7.4 (3 to 5 times) to remove loosely bound protein on the surface.
AFM imaging was done in oscillation mode for all tau samples in liquid using an MSNL10 cantilever. The oscillation of frequency and drive amplitude for oscillation mode imaging was chosen using the in‐built auto‐tuning procedure. The images were acquired at a scan rate of 1 Hz with a resolution of 512 by 512 pixels. During the scanning, the gains and amplitude set points were altered manually, often to keep a minimal force between the cantilever and the sample. The images were acquired using the JPK Nano Wizard ultra‐speed AFM microscope facility at CAESAR. The acquired images were processed by the JPK data processing software.
2.8. Bio‐layer interferometry (BLI)
Kinetics assays were performed by capturing the monoclonal antibodies using anti‐mouse IgG Fc capture (AMC) biosensors in a label‐free kinetic assay BLItz System (FortéBio, Fremont, CA, USA) followed by buffer baseline for 30 seconds. Monoclonal antibody captured sensors were submerged in a sample with different tau constructs for 5 minutes, followed by dissociation. The binding sensograms were obtained, and the binding parameters (on and off rates ka, kd, dissociation constant KD) were calculated by local fitting (1:1 Langmuir interaction model) by BLItz Pro software.
2.9. Transfection (DNA) for luciferase complementation assay
Luciferase complementation fragments were generated using plasmid pCBG99 (click beetle green). 37 The amino acid sequence of the luciferase complementation fragments 2‐413 for N‐terminus (Luc‐N), and 395‐542 for C‐terminus (Luc‐C) were amplified by polymerase chain reaction (PCR). The PCR fragments were cloned into the pTauRDΔK‐EGFPN1 (enhanced green fluorescent protein) vector by replacing the green fluorescent protein with Luc‐N or Luc‐C. N2a wild‐type cells were plated in a T25 flask to achieve uniform transfection efficiency. TauRDΔK‐Luc‐N (3.24 µg) and TauRDΔK‐Luc‐C (2.7 µg) plasmids were mixed in reduced‐serum opti‐MEM medium (1150 µL). Lipofectamine2000 (25 µL) (Invitrogen, Carlsbad, CA, USA) was also diluted in opti‐MEM medium (1150 µL). Both the DNA and lipofectamine were mixed and incubated at RT for 5 minutes. The cells were washed with warm PBS, and the reaction mixture was added to the cells and incubated for 2 hours at 37°C in 5% CO2 incubator. After 2 hours, 4 mL of complete medium was added and further incubated for another 2 hours. Later the cells were trypsinized, and all the cells were plated on a 96‐well plate equally in all wells. After 15 hours of expression in N2a cells, 5 µg/mL of D‐luciferin was added and incubated for 10 to 20 minutes before the measurement. Bioluminescence images were acquired using an IVIS imager (PerkinElmer, Waltham, MA, USA) according to the manufacturer's protocol.
2.10. Antibody transfection into cells
Xfect protein transfection reagent kit (Clontech, Mountain View, CA, USA) was used to deliver the protein of interest into the cells. N2a cells were plated in 6‐well plate with a density of 3 × 105 cells per well. After 24 hours, 15 µL of Xfect transfection reagent was mixed with 85 µL of deionized water. In another tube, the protein of interest (antibody‐ 100 µg/mL [≈666 nM]) and Xfect transfection buffer was made to the final volume of 100 µL. Both reaction mixtures were pooled, and the solution was pipetted. Afterward, the solution was incubated at RT for 30 minutes; 400 µL of serum‐free media was added to the mixture, and the whole content of 600 µL was added to the cells and incubated for 2 hours at 37°C in a CO2 incubator. After 2 hours, 1.4 mL of complete media was added on the cells and incubated for 15 hours.
2.11. Flow cytometry
2.11.1. Annexin V staining
Inducible N2a cells expressing TauRDΔK were treated with desired antibodies and grown for 4 days. Cells were trypsinized and centrifuged, the supernatant was removed, and the cell pellet was suspended in 100 µL of 1X Annexin binding buffer and 5 µL of Annexin V dye was added and incubated in the dark for 30 minutes at room temperature (RT). Cells were washed in 1X binding buffer and collected in PBS buffer. Cell counting was performed in Gallios Beckman‐Coulter Flow Cytometer. Twenty thousand cells per sample were counted to check Annexin V positive cells.
2.11.2. Thioflavin S staining of N2a cells
Inducible N2a cells expressing TauRDΔK were treated with desired antibodies and 0.0005% of ThS dye and grown for 4 days at 37°C in a CO2 incubator. Cells were trypsinized and centrifuged, the supernatant was removed, and the cell pellet was resuspended in PBS buffer. Twenty thousand cells per sample were counted to check for ThS positive cells using the Gallios Beckman‐Coulter Flow Cytometer.
2.11.3. Settings
Blue laser (488 nm) was used to excite ThS (excitation‐440 nm; emission 521 nm), and red laser (683 nm) was used to excite Annexin V (excitation‐633 nm; emission 665 nm) with a flow‐rate of 30 µL/minute. Counting was carried out at RT.
2.12. Reactive oxygen species (ROS) measurements
Toxic tau oligomers can induce the production of superoxides and peroxy radicals in cells which can be measured with fluorescent dye DCF. N2a‐wt cells were plated in 96‐well plates with density 25000 cells/well. At 70 to 80% confluence, the cells were washed once with warm PBS and then incubated with 20 µM of 2´, 7´‐dichlorodihydrofluorescein diacetate (Abcam #ab113851) diluted in 1X dilution buffer for 30 minutes at 37°C. After 30 minutes the cells were washed once with 1X PBS. After washing, the cells were incubated with desired concentrations of different samples (oligomers ± treated with antibodies or controls) for 30 minutes. The fluorescence intensity was measured using a TECAN spectrofluorometer (excitation at 485 nm and emission at 535 nm).
2.13. Calcium measurements
25000 cells/well of N2a‐wt cells were plated in a 96‐well format cell culture plates. At 70% to 80% confluence, the cells were washed with warm PBS once and then incubated with 100 µL of Fluo 4‐AM NW dye (Molecular Probes #F36206) loading solution for 30 minutes at 37°C. After 30 minutes the cells were washed once with 1X Hanks’ balanced salt solution. After washing, the cells were incubated in different samples (oligomers ± treated with antibodies or controls) for 30 minutes. The fluorescence intensity was measured using a TECAN spectrofluorometer (excitation at 494 nm and emission at 516 nm).
2.13.1. Staining of lysosomes
N2a cells were grown to maximum confluence. Different concentrations of antibodies were incubated extracellularly for 24 hours. After 22 hours, 75 nM Lysotracker red DND 99 (L7528, Molecular Probes, Eugene, OR, USA) dye dissolved in fresh complete warm medium was added to the cells and incubated for 2 hours at 37°C in 5% CO2 incubator. After 2 hours the medium was removed, and the cells were fixed using 4% sucrose and 3.7% formaldehyde in PBS for 30 minutes at 37°C, and then cells were washed thrice with PBS buffer at RT. If required, co‐staining of other antibodies can be performed using immunocytochemistry protocol.
2.14. Immunocytochemistry
N2a cells were treated with antibodies at different concentrations for 24 hours. Afterward, the cells were fixed in solution with 4% sucrose and 3.7% formaldehyde in PBS for 30 minutes at 37°C and then washed thrice with PBS buffer. Cells were permeabilized and blocked using 5% BSA and 0.5% TritonX‐100 in PBS for 6 minutes at RT and then washed thrice with PBS buffer. Primary antibody was diluted in blocking solution and incubated on cells at 4°C overnight on a shaker. Unbound or loosely bound antibodies were washed off thrice using PBS. The secondary antibody was diluted in blocking solution and incubated on cells for 1 hour at 37°C. Cells were washed thrice with PBS. 10 µg/mL of 4´, 6‐diamidino‐2‐phenylindole (DAPI) in PBS buffer was incubated for 10 minutes at RT, followed by washing with PBS. Finally, cells were mounted on a glass slide using a poly mount solution and dried at 4°C.
2.15. Immunohistochemistry (IHC)
Animals were sacrificed by cervical dislocation, and the whole brain was removed. Half of the brain (hemisphere) was fixed using 3.7% formaldehyde in PBS for 24 hours at 4°C, followed by sucrose gradient (10%, 20%, and 30%) fixation for 24 hours in each gradient at 4°C. The brain was sliced (free‐floating sections) into 50 µm equal sections from top to bottom using a vibratome (Leica VT 1200S, Wetzlar, Germany), and the sections were collected and stored in PBS. Antigen epitope retrieval was carried out by adding 10 mM sodium citrate buffer (hot [80°C]) to the sections and incubating the sections for 30 minutes at 80°C. Sections were washed with PBS (three times, 10 minutes each). For permeabilization, sections were incubated in 0.1% Triton‐X 100 for 1 hour at RT on an orbital shaker. Sections were washed with PBS (three times, 10 minutes each). Sections were incubated in a blocking solution (0.3% Triton‐X 100, 2% horse serum in PBS) for 2 hours at RT on a shaker. Primary antibody was diluted in blocking solution and incubated at 4°C overnight on a shaker. Sections were washed with 0.1% Triton‐X100 in PBS (three times, 10 minutes each) on a shaker. The secondary antibody was diluted in blocking solution and incubated on sections for 2 hours at 37°C (shaker). Sections were washed with 0.1% Triton‐X100 in PBS (three times, 10 minutes each) on a shaker. 10 µg/mL of DAPI in PBS buffer was incubated for 10 minutes at RT followed by washing with PBS. Finally, sections were mounted using the poly‐mount solution, and the coverslips were dried at 4°C.
2.16. Confocal microscopy
Image acquisition of cells and tissue sections was carried out using a Zeiss LSM 700 confocal microscope. Images were captured at 20X, 40X, and 60X magnification. Image quantification was carried out using the software Zen blue edition (2012, Zeiss). For image acquisition, gain values of positive control were considered a reference to minimize the overexposure of the samples.
2.17. Statistics
All experimental data were normalized with respective controls. The statistics were done using Graph Pad (Prism) software. The significance of differences was analyzed by one‐way analysis of variance test with Tukey's post hoc test. In all the experiments, the confidence interval was set at 95%. Hence a p value <0.05 was considered to be statistically significant.
3. RESULTS
3.1. Specificity and affinity analysis of anti‐tau low‐n oligomeric antibodies
Monoclonal antibodies raised against tau oligomers were extensively characterized in in vitro and in cellular models to select potential candidates for in vivo studies. Various biochemical methods like dot blot, western blot, immunocytochemistry (IC), and immunohistochemistry (IHC) were used to test the specificity of antibodies. To determine the specificity of the monoclonal antibodies, different mutants of tau and different preparations of tau were used (monomers, oligomers, and aggregates, etc., Figure 1A). Polyclonal pan‐tau antibody K9JA (detecting all constructs and assembly forms of tau) was used as an internal control to demonstrate that the protein was loaded in all lanes (Figure 1, row 1a–j). Different monoclonal antibodies showed varied affinities to different constructs of tau (Figure S1 in supporting information). Antibody 2B10 bound selectively only for TauRDΔK purified oligomers (containing 1% monomers, 99% low‐n oligomers; Figure 1A, row 2c, green box). Antibody 6H1 mainly bound to crude oligomers (containing 35% monomers, 63% low‐n oligomers, 2% high‐n oligomers) and high molecular weight aggregates of TauRDΔK (Figure 1A, row 3b, and 3d, pink box), but did not detect purified low‐n tau oligomers. The antibodies yielded different results when tested under denaturing conditions in western blots; in this case, antibodies 2B10 and 6H1 detected all forms of tau in western blots with broad specificity (data not shown).
FIGURE 1.
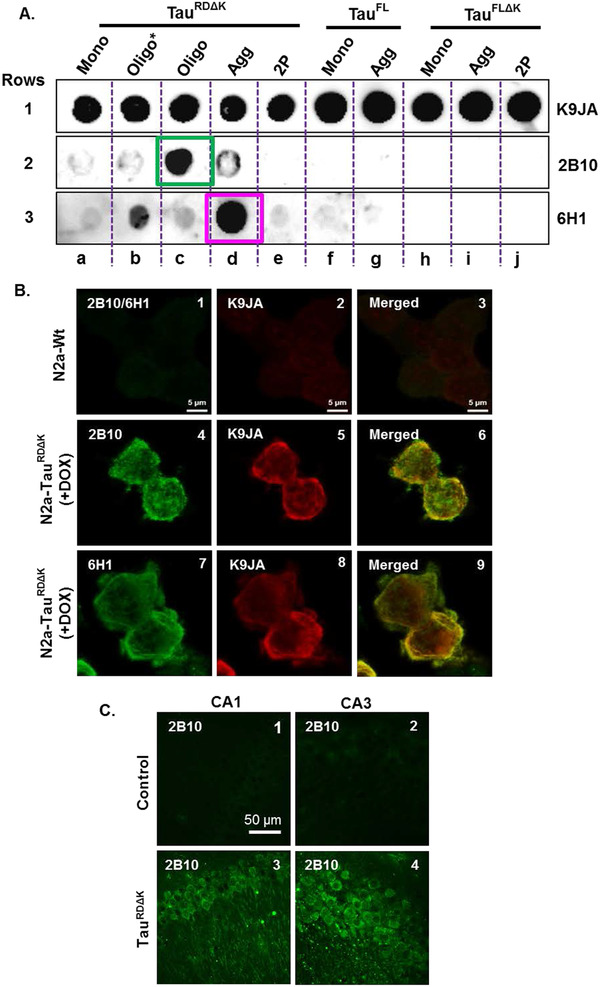
Analysis of the specificity of anti‐tau oligomer antibodies. Anti‐tau oligomer monoclonal antibodies exhibit differential abilities to bind to different tau species or constructs. A, Representative dot blot images show the affinity of antibodies to tau monomers, oligomers, and fibrils. 50 ng/well of protein was loaded on the polyvinylidene difluoride membrane, and 3 µg of antibody was used to detect the protein. Row 1a–j: A representative blot (pan‐tau polyclonal antibody K9JA) acts as a positive control that detects all tau constructs and assembly forms. By contrast, different monoclonal antibodies have different specificities and affinities to different constructs and assembly forms of tau. Row 2: Antibody 2B10 shows high specificity for the oligomer‐enriched sample (containing 1% monomers, 99% low‐n oligomers; blot 2c, green box) whereas antibody 6H1 (row 3) show high affinity to crude oligomers (containing 35% monomers, 63% low‐n oligomers, 2% high‐n oligomers; blot 3b) and aggregates (blot 3d, pink box) of the repeat domain (RD) of tau with the ΔK280 mutation. B, Immunocytochemistry of monoclonal antibodies detects tau with high specificity in cell models. 2B10 (1:100) and 6H1 (1:100) antibodies (green) detected tau in N2a cells expressing TauRDΔK (image 4, 7) but with minimal or no staining in N2a‐wt cells (image 1; no signal for either 2B10 [shown] or 6H1 [not shown]; anti‐rat CF488). Pan tau K9JA antibody (1:1000; anti‐rabbit Alexa 555) was used as a total tau antibody (images 2, 5, 6) co‐localizing with anti‐tau oligomer antibodies (image 6, 9). C, Fluorescence imaging of free‐floating mouse brain sections probed with 2B10 antibody (anti‐rat CF488) showing tau staining mostly in the soma and some dendritic processes of CA1 and CA3 neurons in the transgenic mouse expressing TauRDΔK (image 3, 4) versus non‐transgenic controls (image 1, 2). 2B10 antibody (1:100) does not show any staining in the control brain (image 1, 2) owing to its high specificity to TauRDΔK. Abbreviations: Mono, monomer; Oligo, cross‐linked and purified oligomers; Oligo*, not cross‐linked and crude oligomers; Agg, Aggregates; RD, repeat domain; FL, full length
Fixed N2a‐wt cells and N2a cells expressing TauRDΔK were probed with primary antibodies 2B10 and 6H1. Antibody K9JA (recognizing the repeats+C‐terminal tail) was used as a total anti‐tau antibody. Antibodies 2B10 and 6H1 detected human tau with high specificity in N2a‐TauRDΔK cells after induction of tau (Figure 1B, images 4, 7). Positive control antibody K9JA detected tau strongly in N2a‐TauRDΔK cells (Figure 1B, images 5, 8), co‐localizing with antibodies 2B10 and 6H1 (yellow; Figure 1B, images 6, 9) and thus confirming the specificity of 2B10 and 6H1 for human tau in cell models. All antibodies showed little or no background fluorescence in N2a‐wt cells lacking human tau and containing only low amounts of mouse tau (Figure 1B, images 1, 2, 3). Figure 1C shows the binding of 2B10 antibody to fixed free‐floating brain slices of control and TauRDΔK mice. Antibody 2B10 detected human tau in these animals with high specificity in the soma and dendritic processes of the CA1 and CA3 regions of the hippocampus (Figure 1C, images 3, 4) without showing any affinity to mouse tau expressed in control brains (Figure 1C, images 1, 2). The immunofluorescence data suggest that antibody 2B10 specifically binds to human tau in cell and animal models.
The binding affinities of the antibodies with different tau constructs were determined by biolayer interferometry (BLItz; see Methods). Sensograms in Figure 2A showed an association of two antibodies (2B10, 6H1) with human tau (TauFL, TauRDΔK) and mouse tau (mTau). Antibody 2B10 showed a higher association rate constant (ka) for TauRDΔK compared to TauFL and mouse tau, but similar dissociation rate constants (kd; Figure 2B) to TauFL and mouse tau, which resulted in a high dissociation constant (KD = 1.21 µM) for TauRDΔK (≈10 times higher than TauFL and mouse tau. By contrast, antibody 6H1 showed nanomolar (nM) range affinities to all three antigens with similar ka to human tau and low ka for mouse tau. The kd value for TauRDΔK was higher compared to the other two antigens (human tau and mouse tau). Therefore, the affinity of the 6H1 toward TauRDΔK (KD = 19.19 nM) was higher than to the other two antigens (TauFL–51.34 nM and mouse tau–133.2 nM). We also performed affinity analysis for the antibodies 6B5 and 8E7, which showed strong affinities (KD = 4.92 nM, 0.59 nM, respectively) to TauRDΔK (Figure S2 in supporting information). Of all the tested antibodies, the 2B10 antibody showed the weakest affinities (micromolar [µM] range). The other antibodies showed binding affinities in the nM range to human and mouse tau.
FIGURE 2.
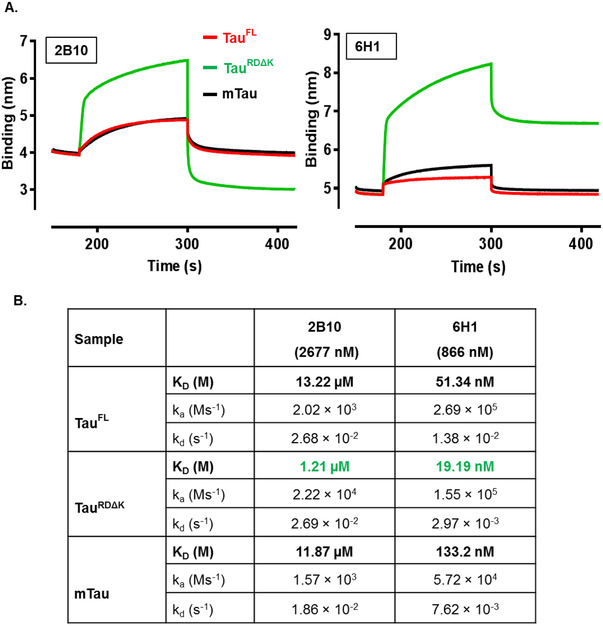
Analysis of affinity of antibodies by biolayer interferometry (BLITz). BLItz shows the binding affinities of immobilized monoclonal antibodies toward different constructs of recombinant human tau (full length or repeat domain) and mouse tau. A, Graphical BLItz sensogram (raw data) shows the binding affinities of immobilized antibodies 2B10 (2677 nM) and 6H1 (866.7 nM) toward TauRDΔK (green), TauFL (red), and mTau (black). The sensograms show ka and kd values of different tau construct toward antibodies. KD values are calculated from the obtained ka and kd values. B, Table showing affinities (ka, kd, and KD) of antibodies to tau protein based on analyzed sensogram. Abbreviations; RD, repeat domain tau; ka, association rate constant; kd, dissociation rate constant; KD, dissociation constant/binding affinity
3.2. Anti‐tau oligomer antibodies block the aggregation of tau in vitro
Several studies reported the close association of the neurofibrillary tangle (NFT) burden and the degree of cognitive impairment in AD. 38 , 39 , 40 As the process begins at least 20 years before any clinical manifestations of AD, targeting tau aggregation offers a rational approach to treatment and prevention. 4 The use of monoclonal antibodies is one such approach for inhibiting tau aggregation. We therefore tested the abilities of our antibodies to inhibit tau aggregation using ThS assay. Equimolar concentrations of tau (aggregation prone mutants TauRDΔK and TauFL‐P301L) and antibody were deployed in an aggregation inhibition assay. In the presence of heparin tau aggregated and reached saturation in 3 hours for TauRDΔK (blue curves in Figure 3A) and 15 hours in TauFL‐P301L (blue curve in Figure 3B) compared to tau monomers in the absence of heparin (red curves in Figure 3A, B), which did not show any increase in ThS fluorescence even after 24 hours. Antibodies 2B10 (green curve) and 6H1 (pink curve) inhibited the formation of ThS positive aggregates of tau dramatically (~90%).
FIGURE 3.

Anti‐tau oligomer monoclonal antibodies block tau aggregation in vitro. Monoclonal antibodies block the aggregation of tau into thioflavin S (ThS) positive aggregates.10 µM of TauRDΔK or TauFL‐P301L (1N4R) was incubated in the presence of heparin (2.5 µM) and 10 µM of different monoclonal antibodies in BES pH 7.0. Aggregation was monitored for 24 hours at 37°C by fluorescence of ThS (40 µM). A and B, TauRDΔK and TauFL‐P301L aggregate into ThS positive aggregates in the presence of heparin (blue curve; A and B). Antibodies 2B10 (green curve) 6H1 (pink curve) block the aggregation of TauRDΔK and TauFL‐P301L compared to their controls (blue curve). In the presence of antibodies, the ThS signal is at baseline similar to monomeric conditions. C, Quantification of the ability of antibodies to block the aggregation of TauFL‐P301L (% aggregation inhibition). Antibodies 2B10 and 6H1 (green) block tau aggregation by ~90%
Quantification of the aggregation inhibition of TauFL‐P301L (tau:antibodies = 1:1 molar ratio) of 10 µM concentration showed that the two antibodies 2B10 and 6H1 were able to inhibit the aggregation of tau up to 90%, whereas antibodies 8E7, 26G1, 28D4, and 29E2 could inhibit aggregation up to >50% (Figure 3C). Antibodies 2H9, 6B5 inhibited aggregation poorly (<20%). This result confirms that antibodies specific for oligomers can have inhibitory efficiencies that do not correlate with their affinity constants (Figure 2B).
3.3. Antibodies reduce tau aggregation by blocking the low‐n oligomeric state
Fluorescence‐based aggregation assays allow high throughput screening, which makes them well suited for primary screens. In contrast, DLS and AFM approaches provide detailed insights regarding the quantity, composition, and morphology of aggregation species. 36 The size of the tau species in the presence of antibodies after 24 hours of aggregation was measured by DLS. Figure 4A shows the average of TauFL‐P301L aggregates (blue curve) as ~10 to 200 nm in diameter (diameter = 2x hydrodynamic radius RH), whereas the monomer (black curve) shows diameters of ~4 to 10 nm. Thus the size of aggregates was >20‐fold larger than monomers. This value reflects the presence of high molecular weight aggregates or fibrils in the TauFL‐P301L +heparin sample (blue curve). In the presence of antibody 2B10 (green curve) and 6H1 (red curve), the average diameter was ~10 nm and 20 nm, respectively. Thus, the average diameter values of antibody treated samples correspond to the low‐n and high‐n oligomers of tau. From the above observation, we conclude that antibody 2B10 binds to low‐n oligomers (~2–3 mers) and prevents the tau aggregation after that. Antibody 6H1 binds to both low‐n and high‐n oligomers and prevents further aggregation.
FIGURE 4.
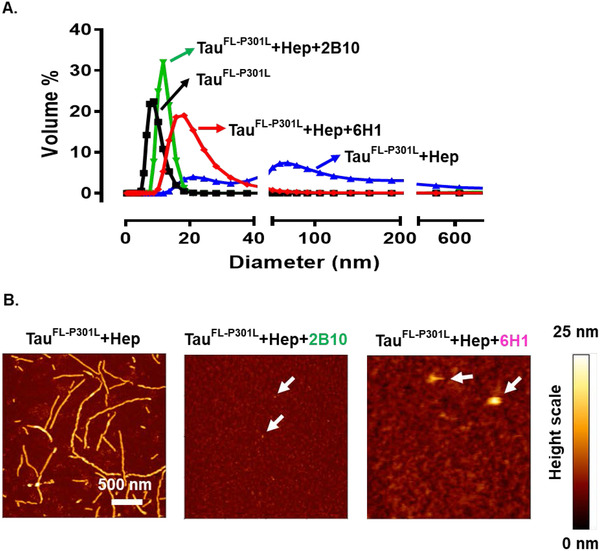
Structure of tau assemblies in the presence of monoclonal antibodies. 10 µM of TauFL‐P301L incubated with or without heparin (2.5 µM) or different antibodies (10 µM) in BES pH 7.0 for 24 hours at 37°C. A, The size (measured by dynamic light scattering) of TauFL‐P301L in the presence of antibodies 2B10 (green) and 6H1 (pink) was in the range of low‐n oligomers, ie, < 30 nm compared to that of the control (blue) in the range of 10 to 200 nm. B, Height images of atomic force microscopy reveal that antibody (2B10 or 6H1) treated TauFL‐P301L was mostly in the form of oligomers (height ~2–3 nm) whereas aggregated paired helical filaments are in the height range of 10 to 18 nm. (Note that atomic force microscopy height is a more precise measure of molecular size than the apparent extend in the x‐y plane)
We performed AFM analysis to reveal the morphology of the tau in the presence and absence of the antibodies. Figure 4B shows the height images of the tau. In the absence of antibodies, TauFL‐P301L formed lengthy paired helical filaments, which are visible in AFM height images, with heights in the range of 10 to 18 nm. In the presence of antibody 2B10, tau aggregation was limited to the level of low‐n oligomers with globular shapes and average heights of 2 to 3 nm (white arrowheads), corresponding to dimers‐tetramers of tau. In the presence of the antibody 6H1 tau was restricted to low‐n oligomers and some bigger globular aggregates (>10 nm, white arrows heads) (Figure 4B). In both cases, no filaments were observed. Antibodies 6B5 and 8E7 poorly inhibited aggregation of TauFL‐P301L evident from AFM (Figure S3 in supporting information). Taken together, we conclude that anti‐tau oligomer‐specific monoclonal antibodies 2B10 and 6H1 are active in inhibiting the aggregation of tau in vitro and halt the growth at the oligomer state.
3.4. Interaction of oligomer‐specific antibodies with TauRDΔK in N2a cells
Inhibition of tau aggregation by antibodies is one of the primary targets in the field of tauopathies. Antibodies could be used to scavenge extracellular tau to reduce transcellular spreading, 16 , 17 or to neutralize toxic forms of tau (eg, oligomers) inside cells. We, therefore, tested the oligomer‐specific antibodies in cell culture models.
N2a cells with inducible expression of TauRDΔK forms fibrous aggregates, 41 and the population of cells with aggregates can be measured by the dye ThS. ThS is cell permeable when added to the cell culture medium, and its fluorescence can be recorded in live cells using flow cytometry. 34 The extracellular application of antibodies (100 µg/mL, ~660 nM) did not inhibit the intracellular tau aggregation (data not shown). We also applied pre‐incubated (24 hours at 37°C) tau oligomer with or without antibody complex extracellularly to inhibit the tau oligomer induced ROS and Ca+2 elevation. Antibody 2B10 pre‐incubated with TauRDΔK oligomers inhibited only the oligomer induced intracellular calcium elevation and not the ROS production (Figure S5 in supporting information). However, the effect was minimal. Therefore, the antibodies had to be delivered into the cells to combat tau aggregation, which was achieved using Xfect protein transfection reagent. Cells treated with antibodies 2B10, 6H1, 6B5, and 8E7 at 100 µg/mL concentration did not show any significant differences in the ThS+ cell population compared to the control (3.38%; Figure 5A). This result indicates that the antibodies were inefficient at blocking the aggregation of tau in the cells.
FIGURE 5.
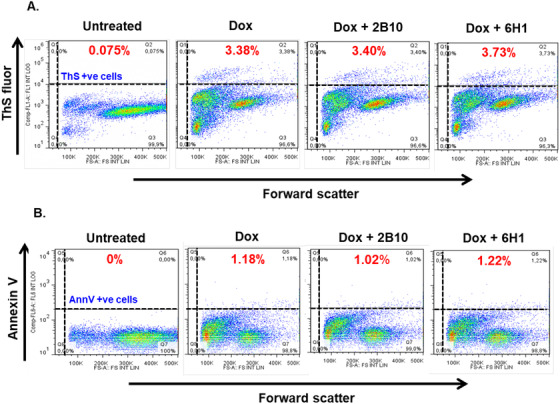
Intracellular delivery of antibodies has no effect on markers of aggregation (ThS) and apoptosis (Ann V) in N2a cells expressing TauRDΔK. 100 µg/mL (~666 nM) of anti‐tau oligomer antibodies were delivered into the cells (N2a‐TauRDΔK) using Xfect reagent to combat the aggregation of tau in cells (as judged by the decrease in ThS+ cells) and rescue the cells from apoptosis (by decreasing APC‐Annexin V staining—a pre‐apoptotic marker). A, Forward scatter plot (reporting on cell volume, x‐axis) versus “green” channel (ThS fluorescence, y‐axis). Area II (Q2) represents the ThS positive cell population. Doxycycline induced cells showed 3.38% ThS positive cells. Neither of the two antibodies reduces the ThS positive cell population. B, Forward scatter plot (cell volume, x‐axis) versus “red” channel (APC Annexin V fluorescence, y‐axis). Area II (Q6) represents the Ann V positive cell population. Doxycycline induced cells showed 1.18% Ann V positive cells. Neither of the two antibodies rescues the cells from pathological effects caused by TauRDΔK. Abbreviations: Ann V, Annexin V; ThS, Thioflavin S; Q2 or 6, Quadrant 2 or 6
N2a cells overexpressing TauRDΔK in the presence of doxycycline showed enhanced cell death due to the expression and aggregation of TauRDΔK, as seen by staining methods, eg, Annexin V assay. Therefore, we tested whether the antibodies can reduce toxic effects induced by TauRDΔK. Annexin V positive cells were quantified by fluorescence‐activated cell sorting after Xfect‐mediated antibody treatment (96 hours in cells). However, none of the tested antibodies (2B10, 6H1, 6B5, and 8E7) showed any significant reductions in Annexin V positive N2a cells (Figure 5B). Thus, the intracellular delivery of anti‐tau oligomer antibodies had no significant effects on blocking tau aggregation or decreasing apoptotic signals caused by the pro‐aggregant tau. One reason for the inefficiency of these antibodies may be the large excess of intracellular tau after induction (~95 µM), compared to the antibodies (~660 nM) in N2a cells.
3.5. Antibodies inhibit tau dimerization in tau‐luciferase‐fragment complementation assay
We further tested whether the antibodies can inhibit the initial stages of tau aggregation, that is, dimerization or oligomerization. For this purpose, we adopted the split‐luciferase complementation assay. 37 TauRDΔK was fused C‐terminally with either N‐terminal (Luc‐N) or C‐terminal (Luc‐C) fragments of click beetle green luciferase (Figure S6A in supporting information). After 15 hours of co‐transfection of both tau constructs (TauRDΔK‐Luc‐N + TauRDΔK‐Luc‐C) in N2a wildtype cells, the dimerization of tau was observed by the bioluminescence of luciferin (Figure S6B, well 1 and S6C bar 1), whereas either construct alone did not generate a signal. This indicates that the interaction of TauRDΔK moieties enforces the combination of Luc‐N and Luc‐C into a functional bioluminescent unit (Figure S6B, wells 2, 3, and S6C bars 2, 3). To rule out the possibility of non‐specific interactions (bioluminescence signal without tau), we co‐transfected the N2a cells with Luc‐N and Luc‐C constructs without tau, which failed to generate a bioluminescence signal (Figure S6B, well 4 and S6C bar 4). These results confirm that the tau‐split luciferase complementation assay is an ideal tool to detect early steps in tau aggregation.
Monoclonal antibodies were further tested to explore their ability to attenuate the tau aggregation process at the stage of dimerization. N2a cells were co‐transfected with tau‐luciferase constructs (TauRDΔK‐Luc‐N + TauRDΔK‐Luc‐C). After 5 to 8 hours of DNA‐transfection, antibodies were incubated for 15 hours in the extracellular medium (without any transfection reagent) followed by imaging. Figure 6A shows that treatment with antibodies caused significant differences in the bioluminescence signal, which relates to changes in tau dimerization in cells. 2B10 antibody (Figure 6A, well 2 and bar 2) decreased (~90%) tau dimerization, compared to the controls (untreated and non‐specific IgG control antibody; bars 1, 4, and wells 1, 4). By contrast, antibodies 6H1 (Figure 6A, bar 3, well 3), 11D10, 2H9, 29E2, and 26G1 did not show any significant difference compared to controls (Figure S7 in supporting information). Further testing of the antibody 2B10 with concentrations ranging from 6.25 to 100 µg/mL (41–666 nM) showed a decrease in bioluminescence (Figure 6B). From these results we conclude that antibody 2B10 efficiently blocks the dimerization/oligomerization of tau in a concentration‐dependent manner.
FIGURE 6.
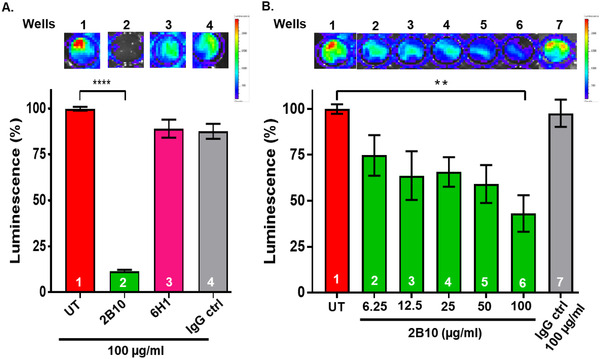
Antibodies inhibit tau dimerization in tau‐luciferase protein‐fragment complementation assay. N2a cells were co‐transfected with TauRDΔK‐Luc‐N and TauRDΔK‐Luc‐C. Assembly of tau molecules (dimerization or oligomerization) leads to complementation of both split luciferase fragments resulting in a bioluminescence signal. A, N2a cells expressing the two constructs treated with 100 µg/mL of 2B10 or 6H1 antibodies extracellularly for 15 hours. Representative bioluminescence images of 96‐well plate showing the ability of antibodies to block tau aggregation. The histogram below compares bioluminescence intensity, normalized to untreated cells = 100%. Antibody 2B10 (bar 2) reduces intensity by ~90%. The non‐specific antibody IgG control (bar 4) has no significant effects. Likewise, antibody 6H1 has no effect (bar 3), presumably because it interferes with aggregation at the stage of high‐n oligomers and is not sensitive to this assay. B, N2a cells co‐transfected with both constructs and treated with various concentrations of 2B10 (6.25, 12.5, 25, 50, 100 µg/mL) or control IgG control antibodies for 15 hours. Representative images illustrating the decrease in bioluminescence intensity are shown on top and quantified in the histogram. 100 µg/mL (bar 6) of 2B10 antibody show a decrease in bioluminescence to ~40% of controls (bar 1). By contrast, 100 µg/mL of the IgG control antibody have no effect (bar 7)
3.6. Uptake and lysosomal localization of antibodies in N2a cells
From the results described above, it is evident that antibodies applied extracellularly (without using Xfect) can inhibit the intracellular oligomerization of tau (split‐luciferase complementation assay). However, the question remains how these extracellular antibodies are acting on intracellular tau aggregation? We, therefore, performed antibody uptake assay with a A647 fluorophore tagged 2B10 antibody and a non‐specific isotype control antibody (anti‐Flag). Different antibody concentrations ranging from 10 to 60 µg/mL (66 to 400 nM) were added to N2a wildtype cells and N2a cells expressing TauRDΔK (differentiated and undifferentiated) in the extracellular medium (without using Xfect). After 24 hours, uptake of 2B10 antibody was observed in both cells types (N2a‐wt, N2a‐TauRDΔK) irrespective of the presence or absence of the tau inside the cell (Figure 7A) and at all antibody concentrations (10‐60 µg/mL of 2B10; N2a‐wt: Figure S9 in supporting information, images 1, 4, 7; N2a‐TauRDΔK: Figure S9, images 2, 5, 8). Surprisingly, the non‐specific isotype control antibody was also taken up by N2a‐ TauRDΔK cells (Figure S9, images 3, 6). Thus, both differentiated and undifferentiated cells take up the antibody with similar efficiency (Figure S9, A, B).
FIGURE 7.

Uptake and lysosomal localization of antibodies in N2a cells. A, Uptake of antibodies by N2a cells monitored by immunofluorescence. N2a‐wt cells or N2a cells expressing TauRDΔK treated extracellularly (without using Xfect) with 60 µg/mL of Alexa647 tagged 2B10 (red) antibody for 24 hours, then fixed for immunocytochemistry. Alexa tagged 2B10 antibody showed uptake both in N2a‐wt (images 1) and N2a‐TauRDΔK (images 2) cells, and the antibody was localized in punctate structures (white arrows) in the cells. B, 60 µg/mL of A647 labeled 2B10 (red, top row) or immunoglobulin G (IgG) control (red, bottom row) antibodies were applied extracellularly for 24 hours with 75 nM of Lysotracker dye (green) on N2a‐TauRDΔK cells induced with doxycycline. Lysotracker dye stained the lysosomes (image 1, 4). 2B10 and IgG control antibodies were taken up by cells and localized as punctate structures (images 2, 5). Some of these antibodies showed co‐localization (yellow) with lysosomes (images 3, 6). C, Quantification of the lysosomes and co‐localized puncta per cell shows increased localization of antibody 2B10 in lysosomes (Lys). n = 3; one‐way analysis of variance with Tukey's multiple comparison; ****P = < .0001; ***P = .0002
The extracellular antibodies are internalized by N2a cells and are localized in punctate structures (Figure 7A. white arrow). To understand the specific nature of these punctate antibody‐containing structures, we stained the cells with fluorescent dyes that label acidic organelles (lysosomes) in living cells. 24 hours’ incubation of fluorophore‐labeled 2B10 (red) and non‐specific IgG control (red) antibodies (60 µg/mL) on N2a‐TauRDΔK cells showed co‐localization (yellow) with the lysotracker dye (Figure 7B, images 3, 6), which confirms that the tau specific antibody 2B10 and the non‐specific isotype control antibody were sorted to the lysosomes. Although both antibodies are sorted to lysosomes, a much larger fraction of 2B10 antibody co‐localized with lysosomes compared to the isotype control antibody (Figure 7C, compare red and gray bars).
3.7. Antibodies promote tau entry to lysosomes for clearance
The antibodies added extracellularly were internalized by the cells and are sorted to lysosomes. However, it is unknown whether antibodies in lysosomes are bound to tau or whether they promote the sorting of tau to lysosomes. To test this, we incubated N2a‐TauRDΔK cells (doxycycline, 48 hours) with lysotracker dye (green) and fluorophore‐labeled 2B10 (red) or IgG control (red) antibodies (extracellularly) for 24 hours followed by cell fixation and stained with unlabeled K9JA antibody (blue) followed by fluorophore labeled secondary antibody. Lysotracker dye stained the lysosomes (green; Figure 8A, images 1, 5). Antibodies 2B10 (red) and IgG control (red) were taken up by the cells and compartmentalized as punctate structures (Figure 8A, images 2, 6). K9JA antibody (blue) detected TauRDΔK in N2a cells (Figure 8A, images 3, 7). N2a TauRDΔK cells treated with 2B10 antibody showed clear co‐localization (white) with lysotracker dye and pan tau antibody K9JA (Figure 8A, image 4, white arrows). In contrast, cells treated with IgG control antibody cells did not show co‐localization with the pan tau antibody (Figure 8A, image 8). It is evident that tau was compartmentalized in cells treated with 2B10 antibody (Figure 8A, image 3), whereas in cells treated with IgG control antibody tau was distributed all over (Figure 8A, image 7). This result shows that the 2B10 antibody recruits the tau to lysosomes presumably for its clearance, unlike the control antibody (Figure 8B).
FIGURE 8.

Antibodies promote tau entry to lysosomes for clearance. 60 µg/mL of Alexa647 labeled 2B10 (red) or immunoglobulin G (IgG) control (red) antibodies applied extracellularly for 24 hours with 75 nM of Lysotracker dye (green) on N2a‐TauRDΔK cells induced with doxycycline. A, Cells fixed and probed with antibody K9JA (blue) (anti‐rabbit AMCA) to detect TauRDΔK (image 3, 7). Lysotracker dye stained lysosomes (images 1, 5). 2B10 and IgG control antibodies were taken up by cells and compartmentalized into puncta (images 2, 6). 2B10 antibody shows co‐localization with tau and lysosomes (image 4, white arrows), whereas the IgG control antibody shows co‐localization only with lysosomes but not with tau (image 8). B, Quantification of the triple co‐localized (tau+antibody in lysosomes) puncta per cell shows increased localization of tau and 2B10 in lysosomes. n = 3; Unpaired t test; ****P = < .0001
In conclusion, the monoclonal antibody 2B10 binds specifically to low‐n oligomers of tau (dot blot, IC, and IHC) with weak affinity (BLItz assay). Antibody 2B10 blocks the aggregation of tau in vitro (ThS, DLS, and AFM) but fails to do so in N2a cells expressing TauRDΔK (flow cytometry‐ThS). Interestingly, 2B10 antibody added extracellularly can block the dimerization of tau in a concentration‐dependent manner (split luciferase complement assay). Moreover, extracellularly added antibody enters the cells, binds to cytosolic tau (TauRDΔK), and recruits tau (presumably toxic oligomers) to lysosomes for degradation.
4. DISCUSSION
Tauopathies, including AD, are characterized by abnormal changes of tau, notably aggregation and posttranslational modifications (eg, phosphorylation). 1 , 42 Traditional attempts to develop treatments were aimed at drugs to inhibit aggregation, or to reduce posttranslational modifications, eg, kinase inhibitors. However, these approaches were not successful. 27 More recent attempts focused on targeting either intracellular or extracellular tau by immunotherapies (active or passive). The majority of these approaches targeted non‐toxic tau monomers or tau aggregates by antibodies. 17 , 28 , 32 , 33 On the other hand, we and others demonstrated that oligomers are the predominant toxic tau species. 13 , 43 , 44 In fact, low‐n oligomers of tau are highly toxic to synapses even before neurons show signs of degeneration. 15 Thus it would be rational to target these species of tau for therapeutic approaches, and hence we generated antibodies against selectively purified low‐n oligomers.
Monoclonal antibodies raised against these low‐n tau oligomers were analyzed by biophysical, biochemical, and cell biological methods. In dot blot analysis, the antibodies showed reactivity for low‐n oligomers (2B10), high‐n oligomers (6H1), and aggregates (2G9, 26G1, 28D4, 29E2, and 32E7) of tau (Figure 1A and Figure S1). The reactivity of the antibodies was changed in western blots presumably due to denaturation of the proteins by SDS, whereas dot blots retain the protein in their native conformation. The results confirmed that some of the antibodies were conformation‐dependent and showed specificity under native conditions, reminiscent of observations with monoclonal antibodies specific for Aβ aggregates. 45
Aggregation of tau is the primary hallmark for the disease pathology in AD and other tauopathies. We tested our antibodies in assays to inhibit tau aggregation in vitro, using different tau constructs K19, K18 (3‐repeat, 4‐repeat domain, respectively, data not shown), TauRDΔK (pro‐aggregant 4 repeats with ΔK280 mutation) and TauFL‐P301L (full‐length tau with pro‐aggregant P301L mutation). Several antibodies inhibit tau aggregation dramatically at ~1:1 (antibody:tau) equimolar ratio. More specifically, antibodies 2B10 and 6H1 strongly (~90%) inhibited the aggregation (Figure 3) even at low concentrations (1:0.5 and 0.25 = tau:antibody ratio; Figure S4 in supporting information). This is similar to the case of Aβ in which aggregation was inhibited by Aβ‐specific antibodies either only at a 4:1 (antibody:Aβ) molar ratio 46 or at 20µM equimolar concentrations. 47 2B10 and 6H1 antibodies bind to low‐n and high‐n oligomers of tau and prevent further aggregation, as observed by DLS and AFM (Figure 4).
Tau is an intracellular protein that forms fibrous aggregates inside the neurons. Aggregated tau inside cells can cause microtubule disruption, transport inhibition, and dysregulation of signaling or degradation pathways. 1 , 48 A majority of the immunotherapeutic approaches aimed to scavenge extracellular tau to block the transcellular spreading of tau. However, this approach does not address tau packed in extracellular vesicles (eg, exosomes) and the pool of intracellular pathological tau. 16 , 17 , 19 , 49 Because cytosolic tau is the source of extracellular tau and tau pathology, it would be more beneficial to target the cytosolic tau with the antibodies to clear intracellular pathological tau. 21 , 50 Using the N2a cell model of tauopathy expressing TauRDΔK, we tested the ability of our antibodies to inhibit aggregation of intracellular tau (ThS positive) and the associated toxic effects (early apoptosis‐Annexin V). Despite the uptake of our antibodies (added extracellularly) by N2a cells, the number of ThS positive cells and Annexin V positive cells remained unchanged (data not shown). Even intracellular delivery of antibodies (Xfect transfection—to increase the intracellular antibody concentration) at high concentration did not affect ThS and Annexin V staining in this cell model (Figure 5).
There are several possible explanations for these observations.
Excess of tau over antibodies: The concentration of antibodies applied (extra and intracellularly) was very low (~660 nM) compared to the high concentration (~95 µM) of the tau expressed in N2a cells. 34 The in vitro data clearly show that antibody 2B10 or 6H1 inhibits tau aggregation at equimolar concentrations. However, such a high concentration (~95 µM) of antibody could not be reached in the cell culture system.
Degradation of antibodies: The antibody was present in the cells for 96 hours. During this time, the antibodies could have been degraded by the cell. 51
Detection of tau oligomers: In N2a cells, the ThS dye robustly labels filamentous tau aggregates assembled with β‐structure, but incipient forms of aggregation (oligomers) are not labeled reliably. Therefore, reduction of oligomers by antibodies might not have been detectable. This would require a more sensitive assay where the initial stages of tau aggregation (oligomerization) can be monitored.
To achieve this, we developed a tau‐split luciferase complementation assay 37 to monitor the oligomerization of TauRDΔK. This assay monitors the early changes that occur during tau dimerization 52 (Figure S6). Interestingly, antibody 2B10 (specific for low‐n oligomers) inhibited the tau dimerization completely in a concentration dependent manner (Figure 6A, B), whereas all others tested antibodies had no effect (Figure S7). Even the 6H1 antibody, despite inhibiting tau aggregation in vitro, did not have any effect in the bioluminescence assay. The possible explanation could be that antibody 6H1 binds to high‐n oligomers and blocks tau aggregation, which cannot be detected by this assay. The results were intriguing, considering that the antibody with the lowest affinity when applied extracellularly inhibited the tau‐dimerization better than antibodies with higher affinity. Analogous observations were made by others. 53
Because antibody 2B10 inhibited intracellular tau oligomerization when added extracellularly, we hypothesized that antibody 2B10 must have been internalized by the N2a cells. There is contradictory evidence about the internalization of antibodies by neurons and the mechanism of the entry. 17 , 18 , 20 However, different modes of antibody entry into neuronal cells (receptor‐dependent and ‐independent) to combat intracellular tau pathology had been reported previously. 5 , 50 Alexa 647‐tagged 2B10 antibody enters both N2a wild type cells and N2a‐TauRDΔK cells in a concentration dependent manner (Figure S9 in supporting information). Even the control antibodies (secondary antibody, IgG control non‐specific antibody) are taken up by these cells. Unlike previously reported, uptake of antibody 2B10 by N2a cells was not dependent either on the concentration of tau or the pathological tau inside the neurons. 20 , 54 Interestingly, the internalized antibodies were detected as puncta in the cytoplasm (Figure 7A). Consistent with previous observations, 54 2B10 antibody co‐localized with lysosomal markers (Figure 7B). In our studies, all other internalized antibodies also co‐localized with the lysosomes (Figure 7B). A likely reason is that the internalized antibodies are degraded in the lysosomes, whether they are specific to tau protein or not. We observed that only the 2B10 antibody was able to bind to tau (TauRDΔK) in the cytosol and the lysosomes, but the control antibody did not bind to tau (Figure 8A). The internalized antibody 2B10 appears to be released into the cytosol, possibly by potocytosis, 55 where it binds to tau and presumably diverts them to lysosomes for degradation. These data are consistent with the previous findings that the endosomal–lysosomal pathway is involved in antibody‐mediated clearance of tau aggregates. 20 , 21 , 56 The exact mechanism of entry of our antibodies into N2a cells is not known yet. In our hands, the blockers used to inhibit different modes of endocytosis were highly toxic to our N2a cell model (not shown). However, it has been reported that antibody uptake is through bulk endocytosis as well as receptor‐mediated endocytosis. 50
In spite of the different effects of the antibody actions, it is crucial to understand the mechanism of antibody‐mediated reduction and clearance of tau aggregates. In general, tau antibodies could use different mechanisms to eliminate or reduce tau protein and hence aggregation. One possibility is that antibodies bind extracellular tau, which are then internalized by microglia via cell surface receptors (FcγRII and FcγRIII). This could inhibit the spreading of tau pathology via trans‐neuronal spreading of tau, as illustrated in Figure 9A. 18 Another possibility is that antibodies are internalized by neurons, bind to tau, and guide it to lysosomes for degradation. This could reduce intracellular tau and, therefore, tau aggregation (see Figure 9B). 20
FIGURE 9.
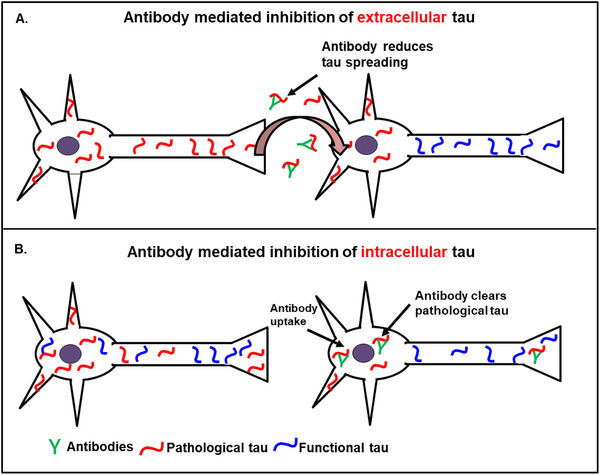
Modes of activities of anti‐tau antibodies. A, Antibody mediated inhibition of extracellular tau: Pathological tau (red) released into the extracellular space is taken up by the neighboring cell and induces the aggregation of endogenous tau (violet) in healthy neurons (prion‐like spreading model). Anti‐tau antibodies bind to the extracellular tau and prevent the uptake and spreading of toxic tau to the neighboring neurons. B, Antibody mediated inhibition of intracellular tau: Extracellular antibodies are taken up by neurons, where they interact with their antigen (eg, pathological tau oligomers), inhibit tau aggregation, and promote their entry to lysosomes for degradation
In conclusion, this study focused on the generation of tau oligomer‐specific antibodies and their uptake into neurons. Different antibodies can be taken up by cells, but only oligomer‐specific antibodies bind to toxic tau and form clusters in lysosomes. This would be the basis for reducing cytoplasmic tau and thus reducing aggregation. The mechanistic details of antibody‐mediated inhibition of tau aggregation and clearance still have to be elucidated.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
Supporting information
Supplementary information
Supplementary information
ACKNOWLEDGMENTS
We are greatly indebted to Dr. Elisabeth Kremmer (Helmholtz Center Munich) for her excellent support in antibody production. We thank Dr. Anja Schneider and Dr. Eckhard Mandelkow (DZNE, Bonn) for their constructive criticism. We thank AFM facility at CAESAR and the light microscope facility at DZNE. This project was supported by DZNE, MPG, and Katharina‐Hardt Foundation.
Chandupatla RR, Flatley A, Feederle R, Mandelkow E‐M, Kaniyappan S. Novel antibody against low‐n oligomers of tau protein promotes clearance of tau in cells via lysosomes. Alzheimer's Dement. 2020;6:e12097 10.1002/trc2.12097
REFERENCES
- 1. Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17:5‐21. [DOI] [PubMed] [Google Scholar]
- 2. Liu E, Wang D, Sperling R, et al, GROUP, ELNI. . Biomarker pattern of ARIA‐E participants in phase 3 randomized clinical trials with bapineuzumab. Neurology. 2018;90:e877‐e886. [DOI] [PubMed] [Google Scholar]
- 3. Honig LS, Vellas B, Woodward M, et al. Trial of solanezumab for mild dementia due to Alzheimer's disease. N Engl J Med. 2018;378:321‐330. [DOI] [PubMed] [Google Scholar]
- 4. Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer's disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71:362‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Elmaleh DR, Farlow MR, Conti PS, Tompkins RG, Kundakovic L, Tanzi RE. Developing effective Alzheimer's disease therapies: clinical experience and future directions. J Alzheimers Dis. 2019;71:715‐732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reas ET. Amyloid and tau pathology in normal cognitive aging. J Neurosci. 2017;37:7561‐7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huber CM, Yee C, May T, Dhanala A, Mitchell CS. Cognitive decline in preclinical Alzheimer's disease: amyloid‐Beta versus tauopathy. J Alzheimers Dis. 2018;61:265‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spillantini MG, Van Swieten JC, Goedert M. Tau gene mutations in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP‐17). Neurogenetics. 2000;2:193‐205. [DOI] [PubMed] [Google Scholar]
- 9. Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med. 2012;2:a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ramsden M, Kotilinek L, Forster C, et al. Age‐dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J Neurosci. 2005;25:10637‐10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337‐351. [DOI] [PubMed] [Google Scholar]
- 12. Mocanu MM, Nissen A, Eckermann K, et al. The potential for beta‐structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci. 2008;28:737‐748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lasagna‐Reeves CA, Castillo‐Carranza DL, Guerrero‐Muoz MJ, Jackson GR, Kayed R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry. 2010;49:10039‐10041. [DOI] [PubMed] [Google Scholar]
- 14. Flach K, Hilbrich I, Schiffmann A, et al. Tau oligomers impair artificial membrane integrity and cellular viability. J Biol Chem. 2012;287:43223‐43233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kaniyappan S, Chandupatla RR, Mandelkow EM, Mandelkow E. Extracellular low‐n oligomers of Tau cause selective synaptotoxicity without affecting cell viability. Alzheimers Dement. 2017;13:1270‐1291. [DOI] [PubMed] [Google Scholar]
- 16. Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans‐cellular propagation of Tau aggregation by fibrillar species. J Biol Chem. 2012;287:19440‐19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yanamandra K, Kfoury N, Jiang H, et al. Anti‐tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron. 2013;80:402‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Funk KE, Mirbaha H, Jiang H, Holtzman DM, Diamond MI. Distinct therapeutic mechanisms of Tau antibodies: promoting microglial clearance versus blocking neuronal uptake. J Biol Chem. 2015;290:21652‐21662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weisova P, Cehlar O, Skrabana R, et al. Therapeutic antibody targeting microtubule‐binding domain prevents neuronal internalization of extracellular tau via masking neuron surface proteoglycans. Acta Neuropathol Commun. 2019;7:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gu J, Congdon EE, Sigurdsson EM. Two novel Tau antibodies targeting the 396/404 region are primarily taken up by neurons and reduce Tau protein pathology. J Biol Chem. 2013;288:33081‐33095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gallardo G, Wong CH, Ricardez SM, et al. Targeting tauopathy with engineered tau‐degrading intrabodies. Mol Neurodegener. 2019;14:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hosokawa M, Arai T, Masuda‐Suzukake M, et al. Methylene blue reduced abnormal tau accumulation in P301L tau transgenic mice. PLoS One. 2012;7:e52389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lai AY, Mclaurin J. Inhibition of amyloid‐beta peptide aggregation rescues the autophagic deficits in the TgCRND8 mouse model of Alzheimer's disease. Biochim Biophys Acta. 2012;1822:1629‐1637. [DOI] [PubMed] [Google Scholar]
- 24. Hochgrafe K, Sydow A, Matenia D, et al. Preventive methylene blue treatment preserves cognition in mice expressing full‐length pro‐aggregant human Tau. Acta Neuropathol Commun. 2015;3:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Panza F, Solfrizzi V, Imbimbo BP, Logroscino G. Amyloid‐directed monoclonal antibodies for the treatment of Alzheimer's disease: the point of no return? Expert Opin Biol Ther. 2014;14:1465‐1476. [DOI] [PubMed] [Google Scholar]
- 26. Gauthier S, Feldman HH, Schneider LS, et al. Efficacy and safety of tau‐aggregation inhibitor therapy in patients with mild or moderate Alzheimer's disease: a randomised, controlled, double‐blind, parallel‐arm, phase 3 trial. Lancet. 2016;388:2873‐2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Medina M. An overview on the clinical development of Tau‐based therapeutics. Int J Mol Sci. 2018;19:1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dai CL, Chen X, Kazim SF, et al. Passive immunization targeting the N‐terminal projection domain of tau decreases tau pathology and improves cognition in a transgenic mouse model of Alzheimer disease and tauopathies. J Neural Transm (Vienna). 2015;122:607‐617. [DOI] [PubMed] [Google Scholar]
- 29. Dai CL, Tung YC, Liu F, Gong CX, Iqbal K. Tau passive immunization inhibits not only tau but also Abeta pathology. Alzheimers Res Ther. 2017;9:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Castillo‐Carranza DL, Gerson JE, Sengupta U, Guerrero‐Munoz MJ, Lasagna‐Reeves CA, Kayed R. Specific targeting of tau oligomers in Htau mice prevents cognitive impairment and tau toxicity following injection with brain‐derived tau oligomeric seeds. J Alzheimers Dis. 2014a;40( Suppl 1):S97‐S111. [DOI] [PubMed] [Google Scholar]
- 31. Castillo‐Carranza DL, Sengupta U, Guerrero‐Munoz MJ, et al. Passive immunization with Tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J Neurosci. 2014b;34:4260‐4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ward SM, Himmelstein DS, Lancia JK, Fu Y, Patterson KR, Binder LI. TOC1: characterization of a selective oligomeric tau antibody. J Alzheimers Dis. 2013;37:593‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kontsekova E, Zilka N, Kovacech B, Novak P, Novak M. First‐in‐man tau vaccine targeting structural determinants essential for pathological tau‐tau interaction reduces tau oligomerisation and neurofibrillary degeneration in an Alzheimer's disease model. Alzheimers Res Ther. 2014;6:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pickhardt M, Biernat J, Hubschmann S, et al. Time course of Tau toxicity and pharmacologic prevention in a cell model of Tauopathy. Neurobiol Aging. 2017;57:47‐63. [DOI] [PubMed] [Google Scholar]
- 35. Kaniyappan S, Chandupatla RR, Mandelkow E. Purification and characterization of low‐n tau oligomers. Methods Mol Biol. 2018;1779:99‐111. [DOI] [PubMed] [Google Scholar]
- 36. Wegmann S, Muller DJ, Mandelkow E. Investigating fibrillar aggregates of Tau protein by atomic force microscopy. Methods Mol Biol. 2012;849:169‐183. [DOI] [PubMed] [Google Scholar]
- 37. Villalobos V, Naik S, Bruinsma M, et al. Dual‐color click beetle luciferase heteroprotein fragment complementation assays. Chem Biol. 2010;17:1018‐1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Arriagada PV, Growdon JH, Hedley‐Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631‐639. [DOI] [PubMed] [Google Scholar]
- 39. Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013;12:609‐622. [DOI] [PubMed] [Google Scholar]
- 40. Braak H, Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991;1:213‐216. [DOI] [PubMed] [Google Scholar]
- 41. Khlistunova I, Biernat J, Wang Y, et al. Inducible expression of Tau repeat domain in cell models of tauopathy: aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem. 2006;281:1205‐1214. [DOI] [PubMed] [Google Scholar]
- 42. Holtzman DM, Carrillo MC, Hendrix JA, et al. Tau: from research to clinical development. Alzheimers Dement. 2016;12:1033‐1039. [DOI] [PubMed] [Google Scholar]
- 43. Patterson KR, Remmers C, Fu Y, et al. Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer's disease. J Biol Chem. 2011;286:23063‐23076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kumar S, Tepper K, Kaniyappan S, et al. Stages and conformations of the Tau repeat domain during aggregation and its effect on neuronal toxicity. J Biol Chem. 2014;289:20318‐20332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hatami A, Albay R 3RD, Monjazeb S, Milton S, Glabe C. Monoclonal antibodies against Abeta42 fibrils distinguish multiple aggregation state polymorphisms in vitro and in Alzheimer's disease brain. J Biol Chem. 2014;289:32131‐32143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Solomon B, Koppel R, Hanan E, Katzav T. Monoclonal antibodies inhibit in vitro fibrillar aggregation of the Alzheimer's beta‐amyloid peptide. Proc Natl Acad Sci U S A. 1996;93:452‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu R, Yuan B, Emadi S, et al. Single chain variable fragments against beta‐amyloid (Abeta) can inhibit abeta aggregation and prevent abeta‐induced neurotoxicity. Biochemistry. 2004;43:6959‐6967. [DOI] [PubMed] [Google Scholar]
- 48. Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156:1051‐1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lasagna‐Reeves CA, Castillo‐Carranza DL, Sengupta U, et al. Alzheimer brain‐derived tau oligomers propagate pathology from endogenous tau. Sci Rep. 2012;2:700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Congdon EE, Gu J, Sait HB, Sigurdsson EM. Antibody uptake into neurons occurs primarily via clathrin‐dependent Fcgamma receptor endocytosis and is a prerequisite for acute tau protein clearance. J Biol Chem. 2013;288:35452‐35465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Press OW, Farr AG, Borroz KI, Anderson SK, Martin PJ. Endocytosis and degradation of monoclonal antibodies targeting human B‐cell malignancies. Cancer Res. 1989;49:4906‐4912. [PubMed] [Google Scholar]
- 52. Wegmann S, Nicholls S, Takeda S, Fan Z, Hyman BT. Formation, release, and internalization of stable tau oligomers in cells. J Neurochem. 2016;139:1163‐1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Congdon EE, Lin Y, Rajamohamedsait HB, et al. Affinity of Tau antibodies for solubilized pathological Tau species but not their immunogen or insoluble Tau aggregates predicts in vivo and ex vivo efficacy. Mol Neurodegener. 2016;11:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shamir DB, Rosenqvist N, Rasool S, Pedersen JT, Sigurdsson EM. Internalization of tau antibody and pathological tau protein detected with a flow cytometry multiplexing approach. Alzheimers Dement. 2016;12:1098‐1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mineo C, Anderson RG. Potocytosis. Robert Feulgen Lecture. Histochem Cell Biol. 2001;116:109‐118. [DOI] [PubMed] [Google Scholar]
- 56. Krishnamurthy PK, Deng Y, Sigurdsson EM. Mechanistic studies of antibody‐mediated clearance of tau aggregates using an ex vivo brain slice model. Front Psychiatry. 2011;2:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information
Supplementary information