

This observation study characterizes regions and rates of atrophy in the 3 primary familial frontotemporal lobar degeneration genes (MAPT, GRN, and C9orf72) across all disease stages from asymptomatic to dementia.
Key Points
Question
How does the trajectory of atrophy differ between the 3 primary genetic groups (MAPT, GRN, and C9orf72) associated with familial frontotemporal lobar degeneration?
Findings
Among 160 members of families affected by familial frontotemporal lobar degeneration in this case-control study, MAPT and GRN pathogenic variants were associated with increases in the rate of volume loss as a function of disease stage, whereas C9orf72 expansion carriers showed minimal increases in the rate of volume loss with disease progression.
Meaning
This study advances the knowledge of between-gene differences in atrophy rates as a function of disease severity; treatment studies enrolling familial frontotemporal dementia cases should consider the heterogeneity conferred by both the altered gene and the disease stage.
Abstract
Importance
Several clinical trials are planned for familial forms of frontotemporal lobar degeneration (f-FTLD). Precise modeling of brain atrophy in f-FTLD could improve the power to detect a treatment effect.
Objective
To characterize regions and rates of atrophy in the 3 primary f-FTLD genetic groups (MAPT, GRN, and C9orf72) across all disease stages from asymptomatic to dementia.
Design, Setting, and Participants
This investigation was a case-control study of participants enrolled in the Advancing Research and Treatment for Frontotemporal Lobar Degeneration or Longitudinal Evaluation of Familial Frontotemporal Dementia studies. The study took place at 18 North American academic medical centers between January 2009 and September 2018. Participants with f-FTLD (n = 100) with a known pathogenic variant (MAPT [n = 28], GRN [n = 33], or C9orf72 [n = 39]) were grouped according to disease stage (ie, Clinical Dementia Rating [CDR] plus National Alzheimer’s Coordinating Center [NACC] FTLD module). Included were participants with at least 2 structural magnetic resonance images at presymptomatic (CDR + NACC FTLD = 0 [n = 57]), mild or questionable (CDR + NACC FTLD = 0.5 [n = 15]), or symptomatic (CDR + NACC FTLD = ≥1 [n = 28]) disease stages. The control group included family members of known pathogenic variant carriers who did not carry the pathogenic variant (n = 60).
Main Outcomes and Measures
This study fitted bayesian linear mixed-effects models in each voxel of the brain to quantify the rate of atrophy in each of the 3 genes, at each of the 3 disease stages, compared with controls. The study also analyzed rates of clinical decline in each of these groups, as measured by the CDR + NACC FTLD box score.
Results
The sample included 100 participants with f-FTLD with a known pathogenic variant (mean [SD] age, 50.48 [13.78] years; 53 [53%] female) and 60 family members of known pathogenic variant carriers who did not carry the pathogenic variant (mean [SD] age, 47.51 [12.43] years; 36 [60%] female). MAPT and GRN pathogenic variants were associated with increased rates of volume loss compared with controls at all stages of disease. In MAPT pathogenic variant carriers, statistically significant regions of accelerated volume loss compared with controls were identified in temporal regions bilaterally in the presymptomatic stage, with global spread in the symptomatic stage. For example, mean [SD] rates of atrophy in the left temporal were −231 [47] mm3 per year during the presymptomatic stage, −381 [208] mm3 per year during the mild stage, and −1485 [1025] mm3 per year during the symptomatic stage (P < .05). GRN pathogenic variant carriers generally had minimal increases in atrophy rates between the presymptomatic and mild stages, with rapid increases in atrophy rates in the symptomatic stages. For example, in the right frontal lobes, annualized volume loss was −267 [81] mm3 per year in the presymptomatic stage and −182 [90] mm3 per year in the mild stage, but −1169 [555] mm3 per year in the symptomatic stage. Compared with the other groups, C9orf72 expansion carriers showed minimal increases in rate of volume loss with disease progression. For example, the mean (SD) annualized rates of atrophy in the right frontal lobe in C9orf72 expansion carriers was −272 (118) mm3 per year in presymptomatic stages, −310 (189) mm3 per year in mildly symptomatic stages, and −251 (145) mm3 per year in symptomatic stages.
Conclusions and Relevance
These findings are relevant to clinical trial planning and suggest that the mechanism by which C9orf72 pathogenic variants lead to symptoms may be fundamentally different from the mechanisms associated with other pathogenic variants.
Introduction
Frontotemporal lobar degeneration (FTLD) is a neurodegenerative disorder associated with a variety of pathological mechanisms. As many as 30% of FTLD cases are associated with pathogenic gene variants that are autosomal dominant (familial forms of FTLD [f-FTLD]), and over half of these are associated with pathogenic variants in 1 of the following 3 genes: microtubule-associated protein tau (MAPT [OMIM 157140]), progranulin (GRN [OMIM 138945]), and a repeat expansion in the chromosome 9 open reading frame 72 (C9orf72 [OMIM 614260]) gene. Pathogenic variants in each of these genes are associated with overlapping but unique clinical and neuroimaging manifestations.1,2,3,4,5,6,7,8
Accurate characterization of the natural history of each genetic group is important for clinical care and clinical trials because precise modeling of the disease course can improve the ability to detect a treatment effect.9,10 Furthermore, there is a need for a working model of disease and biomarker progression in f-FTLD to inform hypotheses about when biomarker changes develop in the course of disease and how biomarkers change over time.11 In addition, natural history data can help clinicians prognosticate and assist family planning.
Many studies have used brain atrophy to describe the evolution of neurodegeneration in f-FTLD, yielding the following observations: (1) cross-sectional atrophy can be detected in the presymptomatic stages, and each genetic group has different regional predilection for atrophy3,6,8,12,13,14; (2) atrophy rates in the presymptomatic stages may exceed those of age-matched control cases5,15,16; (3) the rate of volume loss may accelerate near the transition from asymptomatic to symptomatic5,13; and (4) volume loss in symptomatic cases is usually well in excess of that in control cases.16,17,18 However, conclusions from these observations are tempered because many analyses focused only on 1 genetic group or disease stage, limiting comparisons across genes and stages. Moreover, many prior estimates of change over time were derived from cross-sectional rather than longitudinal data.
The emergence of large, comprehensive studies3,19,20 of f-FTLD that include presymptomatic and symptomatic pathogenic variant carriers allows direct study of the natural history of disease using longitudinal observations. The present analysis, based on data from 2 of these large natural history studies,19,21 addresses limitations in previous work by incorporating longitudinal data across the disease course in participants carrying the 3 most common f-FTLD–associated pathogenic variants. Based on theoretical models11 and previous observational studies of Alzheimer disease22,23 and FTLD,13,24 our hypothesis was that pathogenic variants in all 3 genes would produce a nonlinear pattern of neurodegeneration, with acceleration of volume loss as patients develop symptoms.25 We investigated this question using longitudinal voxelwise analyses of gray matter volume and assessed whether comparable results were observed for a clinical measure of daily functioning, the Clinical Dementia Rating (CDR) plus behavioral and language domains from the National Alzheimer’s Coordinating Center (NACC) FTLD module (CDR + NACC FTLD).
Methods
Participants
In this longitudinal case-control study, we included 160 members of families affected by f-FTLD, most of whom were enrolled in the Advancing Research and Treatment for Frontotemporal Lobar Degeneration (ARTFL) or Longitudinal Evaluation of Familial Frontotemporal Dementia (LEFFTDS) studies, which were conducted through a consortium of 18 academic medical centers across the United States and Canada between May 2015 and September 2018. For LEFFTDS,19 at least 1 family member must have a pathogenic variant in the MAPT, GRN, or C9orf72 genes. For ARTFL,21 families with any f-FTLD pathogenic variant or without a known pathogenic variant can enroll, but only carriers of MAPT, GRN, or C9orf72 pathogenic variants were included in this analysis. The ARTFL and LEFFTDS protocols include annual follow-up with clinical reassessment. Additional f-FTLD cases included those enrolled in another study26 of FTLD at the University of California, San Francisco, and who had undergone a similar brain imaging protocol (grants AG032306 and AG019724 from the National Institutes of Health) from January 2009 to October 2016. Exclusion and inclusion criteria are provided in the eMethods in the Supplement. Local ethics committees at each of the sites approved the study, and participants provided written informed consent. This study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline.
The sample included 100 participants with f-FTLD with a known pathogenic variant (MAPT+ [28 individuals with MAPT pathogenic variants], GRN+ [33 individuals with GRN pathogenic variants], and C9orf72+ [39 individuals with C9orf72 repeat expansions]) and 60 family members of known pathogenic variant carriers who did not carry the pathogenic variant (demographic characteristics are listed in the Table and eTable 1 in the Supplement). Participants with f-FTLD were grouped into the following 3 disease stages using CDR + NACC FTLD27: presymptomatic (CDR + NACC FTLD = 0 [n = 57]), mild or questionable (CDR + NACC FTLD = 0.5 [n = 15]), or symptomatic (CDR + NACC FTLD = ≥1 [n = 28]). Included were participants who had at least 2 structural magnetic resonance images within 1 of these stages (Table and eTable 1 in the Supplement); all available scans within that disease stage for each participant were used for the study. Each participant was only included in a single disease stage.
Table. Sample Characteristics.
| Characteristic | Control cases | All pathogenic variant carriers | MAPT | GRN | C9orf72 | Group comparisona | P value | Post hocb |
|---|---|---|---|---|---|---|---|---|
| No. of individuals (No. of visits) | 60 (138) | 100 (250) | 28 (68) | 33 (81) | 39 (101) | NA | NA | NA |
| Age, mean (SD), y | 47.51 (12.43) | 50.48 (13.78) | 43.97 (11.49) | 56.89 (13.52) | 50.50 (12.94) | F2,97 = 7.21 | .001 | MAPT < GRN and C9orf72 |
| Educational level, mean (SD), y | 15.61 (2.61) | 15.35 (2.48) | 15.56 (2.17) | 15.40 (2.55) | 15.17 (2.63) | F2,97 = 0.69 | .50 | NA |
| Sex, No./total No. (%) | ||||||||
| Female | 36/60 (60) | 53/100 (53) | 23/39 (59) | 19/33 (58) | 11/28 (39) | NA | NA | NA |
| Male | 24/60 (40) | 47/100 (47) | 16/39 (41) | 14/33 (42) | 17/28 (61) | χ2 = 4.24 | .12 | NA |
| Race/ethnicity, No./total No. (%) | ||||||||
| White | 57/60 (95) | 93/100 (93) | 38/39 (97) | 28/33 (85) | 27/28 (96) | NA | NA | NA |
| Otherc | 3/60 (5) | 7/100 (7) | 1/39 (3) | 5/33 (15) | 1/28 (4) | NA | NA | NA |
| Functional severity, No. of individuals (No. of visits) | ||||||||
| CDR + NACC FTLD = 0 | 60 (138) | 57 (142) | 19 (47) | 18 (42) | 20 (53) | NA | NA | NA |
| CDR + NACC FTLD = 0.5 | NA | 15 (35) | 4 (9) | 6 (14) | 5 (12) | NA | NA | NA |
| CDR + NACC FTLD = ≥1d | NA | 28 (73) | 5 (12) | 9 (25) | 14 (36) | NA | NA | NA |
| CDR + NACC FTLD = 1 | NA | 12 (31) | 2 (5) | 7 (18) | 3 (8) | NA | NA | NA |
| CDR + NACC FTLD = 2 | NA | 14 (37) | 2 (4) | 2 (7) | 10 (26) | NA | NA | NA |
| CDR + NACC FTLD = 3 | NA | 2 (5) | 1 (3) | 0 | 1 (2) | NA | NA | NA |
Abbreviations: CDR + NACC FTLD, Clinical Dementia Rating plus National Alzheimer’s Coordinating Center Frontotemporal Lobar Degeneration; NA, not applicable.
MAPT, GRN, and C9orf72 groups were compared on age and educational level using regression and on sex using χ2 test.
Post hoc comparisons reported if P < .05 for group difference.
Other includes Native American, Asian, Asian Indian, Mixed, and not reported. These groups were combined to protect confidentiality.
The 3 rows below are the detailed breakdown for CDR + NACC FTLD = ≥1.
Clinical Assessment
The multidisciplinary assessment included neurological history and examination and collateral interview. Neuropsychological tests included the Uniform Data Set (version 3.0) neuropsychological battery.28 Functional status was quantified using CDR + NACC FTLD27,29,30 (details are provided in the Supplement). Brain imaging was not used for diagnosis or severity rating. Clinical diagnoses are listed in eTable 2 in the Supplement. All participants had genetic testing at the University of California, Los Angeles, using published methods31 (specific pathogenic variants are provided in the eAppendix in the Supplement).
Neuroimaging
Image Acquisition
Participants underwent 3 tesla (3-T) imaging on MRI scanners (scanner types are listed in eTable 3 in the Supplement). A standard imaging protocol was used across all centers and was managed and reviewed for quality by a core group (including K.K.) at the Mayo Clinic, Rochester, Minnesota. Details of image acquisition, processing, and harmonization are provided in the eMethods in the Supplement and have been published elsewhere.8 All participants except 3 were scanned on the same scanner at all visits (for 2 participants, the scanner was upgraded; the third changed sites).
Bayesian Voxelwise Mixed-Effects Modeling
Group-level and participant-level rates of atrophy at each brain voxel were longitudinally modeled as a function of age using a bayesian hierarchical mixed-effects framework32 introduced by Friston and colleagues33 and reproduced in our in-house software suite at the Memory and Aging Center, University of California, San Francisco. The model consists of the following 2 hierarchical levels: (1) a single-participant level for individual structural trajectories and (2) a group level for an ensemble of trajectories (eMethods in the Supplement). Researchers interested in the code for the bayesian mixed-effects models can find information in the publication by Zeigler and colleagues32 or may contact the corresponding author of this study.
Statistical Analysis
Details of the analytic approach are provided in the eMethods in the Supplement. To address the main hypothesis that f-FTLD pathogenic variants are associated with high rates of volume loss that increase with disease stage, we examined voxelwise maps of rates of annualized brain volume loss at each disease stage in each genetic group and compared these with rates in the control group. We fit a 3-way interaction model at each voxel as the rate of atrophy by disease stage by gene. Statistically significant voxels indicated that the association of increasing disease stage with volume loss is moderated by gene. Voxelwise maps showing where rates of volume loss were statistically significantly increased in the pathogenic variant carrier groups compared with the control group were produced using the FMRIB Software Library.34,35 To understand the cumulative associations of volume loss, we analyzed cross-sectional volume using the last observation for all participants in their disease stage.34 P < .05 was considered statistically significant, and all tests were 2 tailed.
To summarize rates of volume loss in various brain regions, we analyzed data for several large regions of interest (ROIs),36 including bilateral frontal, temporal, parietal, and occipital lobes and the thalamus and cerebellum. Thalamic and cerebellar ROIs were chosen because of their involvement in f-FTLD.3,6 For each ROI, we extracted the specific slope of each participant.
To examine patterns of change in clinical measures, we created linear mixed-effects regression models using participant-specific rates of change in CDR + NACC FTLD box score as the dependent variable. Higher box scores indicate more severe functional impairment. Analysis of clinical data was performed using Stata, version 14.2 (StataCorp LLC).
Results
The sample included 100 participants with f-FTLD; the mean (SD) age was 50.48 (13.78) years, 53 (53%) were female, and 47 (47%) were male. Noncarriers made up a control group with otherwise similar genetic and environmental backgrounds compared with the carriers. The control group included 60 family members; the mean (SD) age was 47.51 (12.43) years, 36 (60%) were female, and 24 (40%) were male.
Longitudinal Atrophy Rates
Maps of annualized rates of atrophy (Figures 1, 2, and 3 and eFigure 1 in the Supplement) revealed statistically significant increases in the rate of volume loss for pathogenic variant carriers compared with control cases for all genes at all stages. The mean (SD) regional rates of atrophy for control participants (eTable 4 in the Supplement) were as follows for 6 lobes of interest: −170 (12) mm3 per year for left frontal, −160 (15) mm3 per year for right frontal, −77 (13) mm3 per year for left frontal, −73 (17) mm3 per year for right temporal, −105 (14) mm3 per year for left parietal, and −102 (16) mm3 per year for right parietal.
Figure 1. Maps of Voxelwise Atrophy Rate in MAPT Pathogenic Variant Carriers at 3 Levels of Disease Severity.

A, More positive values represent faster rates of atrophy. Based on our hypothesis, only those voxels that show rates of atrophy are presented; extending the color scale to voxels that were estimated to show volume growth would decrease interpretability by compressing the color scale in voxels of interest (those showing volume loss). B, Green voxels are statistically significant at P < .05 after familywise error correction for multiple comparison at each voxel. Statistically significant increased rates of volume loss compared with controls were observed at all stages. Statistically significant regions of accelerated volume loss were identified in temporal regions bilaterally in the presymptomatic stage and mild or questionable stage, with global spread in the symptomatic stage; the largest effect sizes were observed in the frontal and temporal lobes. CDR + NACC FTLD indicates Clinical Dementia Rating plus National Alzheimer’s Coordinating Center Frontotemporal Lobar Degeneration.
Figure 2. Maps of Voxelwise Atrophy Rate in GRN Pathogenic Variant Carriers at 3 Levels of Disease Severity.

A, More positive values represent faster rates of atrophy. Based on our hypothesis, only those voxels that show rates of atrophy are presented; extending the color scale to voxels that were estimated to show volume growth would decrease interpretability by compressing the color scale in voxels of interest (those showing volume loss). B, Green voxels are statistically significant at P < .05 after familywise error correction for multiple comparison at each voxel. Statistically significant increased rates of volume loss compared with controls were observed at all stages. In GRN+, the rate of volume loss was fairly uniform across the brain, with little evidence of acceleration between the presymptomatic stage and mild or questionable stage except for a possible area of accelerated atrophy in the putamen. With development of dementia, GRN+ showed accelerated loss of volume in portions of the frontal, temporal, and parietal lobes bilaterally. CDR + NACC FTLD indicates Clinical Dementia Rating plus National Alzheimer’s Coordinating Center Frontotemporal Lobar Degeneration.
Figure 3. Maps of Voxelwise Atrophy Rate in C9orf72 Repeat Expansion Carriers at 3 Levels of Disease Severity.

A, More positive values represent faster rates of atrophy. Based on our hypothesis, only those voxels that show rates of atrophy are presented; extending the color scale to voxels that were estimated to show volume growth would decrease interpretability by compressing the color scale in voxels of interest (those showing volume loss). B, Green voxels are statistically significant at P < .05 after familywise error correction for multiple comparison at each voxel. Statistically significant increased rates of volume loss compared with controls were observed at all stages. In contrast to carriers of pathogenic variants in the other 2 genes, C9orf72 repeat expansion carriers showed little acceleration across disease stages, even with transition to dementia. Regions with the largest effect sizes were distributed among frontal, temporal, and parietal regions in C9orf72+. CDR + NACC FTLD indicates Clinical Dementia Rating plus National Alzheimer’s Coordinating Center Frontotemporal Lobar Degeneration.
In MAPT+ carriers, statistically significant regions of accelerated volume loss compared with controls (P < .05 for all) were identified in temporal regions bilaterally in the presymptomatic stage. In the ROI analysis, mean (SD) values were −231 (47) mm3 per year for left temporal and −150 (36) mm3 per year for right temporal lobe. For the mild or questionable stage, the mean (SD) values were −381 (208) mm3 per year for left temporal and −315 (201) mm3 per year for right temporal lobe.There was global spread in the symptomatic stage. The largest effect sizes were observed in the frontal and temporal lobes (Figure 1). The mean (SD) values were −2269 (1574) mm3 per year for the left frontal lobe, −2053 (2006) mm3 per year for right frontal lobe, −1485 (1025) mm3 per year for left temporal, and −1164 (882) mm3 per year for right temporal lobe.
In GRN+ carriers, the rate of volume loss was fairly uniform across the brain in the presymptomatic and mild stages, with little evidence of acceleration between stages. For example, in the ROI analysis, annualized right frontal volume loss was −267 (81) mm3 per year in the presymptomatic stage and −182 (90) mm3 per year in the mild stage. The exception was a possible area of accelerated atrophy in the putamen (Figure 2). With development of dementia, GRN+ carriers showed accelerated loss of volume in portions of the frontal (mean [SD], −1530 [388] mm3 per year for left frontal and −1169 [555] mm3 per year for right frontal ROIs), temporal (mean [SD], −867 [308] mm3 per year for left temporal and −433 [119] mm3 per year for right temporal ROIs), and parietal (mean [SD], −896 [217] mm3 per year for left parietal and −484 [108] mm3 per year for right parietal ROIs) lobes bilaterally.
In contrast to pathogenic variants in the other 2 genes, C9orf72+ carriers showed minimal increase in atrophy rates across disease stages (Figure 3). For example, in the ROI analysis, the mean (SD) annualized right frontal lobe volume loss was −272 (118) mm3 per year in the presymptomatic stage, −310 (189) mm3 per year in the mild or questionable stage, and −251 (145) mm3 per year in the symptomatic stage. Regions with the largest effect sizes were distributed among frontal (mean [SD], −285 [199] mm3 per year for left frontal and −251 [145] mm3 per year for right frontal ROIs), temporal (mean [SD], −77 [44] mm3 per year for left temporal and −64 [46] mm3 per year for right temporal ROIs), and parietal (mean [SD], −122 [157] mm3 per year for left parietal and −124 [160] mm3 per year for right parietal ROIs) regions in C9orf72+.
Because the maps of volume loss indicated differences in rates of stage-dependent volume loss across groups, we fit an omnibus, disease stage by gene interaction model for rates of volume loss at each voxel. Almost every voxel in the brain (91% [247 910 of 273 039 voxels]) showed a statistically significant interaction (eFigure 2 in the Supplement), indicating that the association of disease severity with atrophy rates differs across genetic groups.
The ROI analysis highlighted the increases in the rate of volume loss for MAPT+ between the presymptomatic and mild or questionable stages in the right (mean [SD], −277 [119] mm3 per year for presymptomatic and −576 [276] for mild or questionable) and left (mean [SD], −259 [99] mm3 per year for presymptomatic and −544 [301] mm3 per year for mild or questionable) frontal, temporal (eg, mean [SD], −231 [47] mm3 per year for presymptomatic and −381 [208] for mild or questionable for left temporal), and parietal (eg, mean [SD], −139 [27] mm3 per year for presymptomatic and −303 [151] for mild or questionable for left parietal) regions (Figure 4 and eFigure 3 and eTable 4 in the Supplement). Smaller increases in the rate of volume loss with increasing disease severity were observed in the occipital lobes (mean [SD], −38 [10] for presymptomatic and −110 [68] for mild or questionable) and thalamus (mean [SD], −13 [13] for presymptomatic and −78 [50] for mild or questionable). The ROI analysis also underscored how the genetic groups differed in the degree of increased atrophy when transitioning from the mild to symptomatic stage. Even in regions where the rate of volume loss increased between the mild and symptomatic stages in C9orf72+ carriers, the magnitude of acceleration of atrophy between these 2 stages was much higher in MAPT+ and GRN+ carriers. For example, in the right frontal lobe, the increase in atrophy rate between the 2 stages was about 6 to 9 times higher in GRN+ (mean [SD], −182 [90] for mild or questionable and −1169 [555] for symptomatic) and MAPT+ (mean [SD], −576 [276] for mild or questionable and −2053 [2006] for symptomatic) carriers, respectively, compared with C9orf72+ (mean [SD], −310 [189] for mild or questionable and −251 [145] for symptomatic) carriers (eTable 4 in the Supplement). Overall, these data supported the voxelwise pattern of results.
Figure 4. Mean Rates of Volume Loss for Frontal and Temporal Regions of Interest.
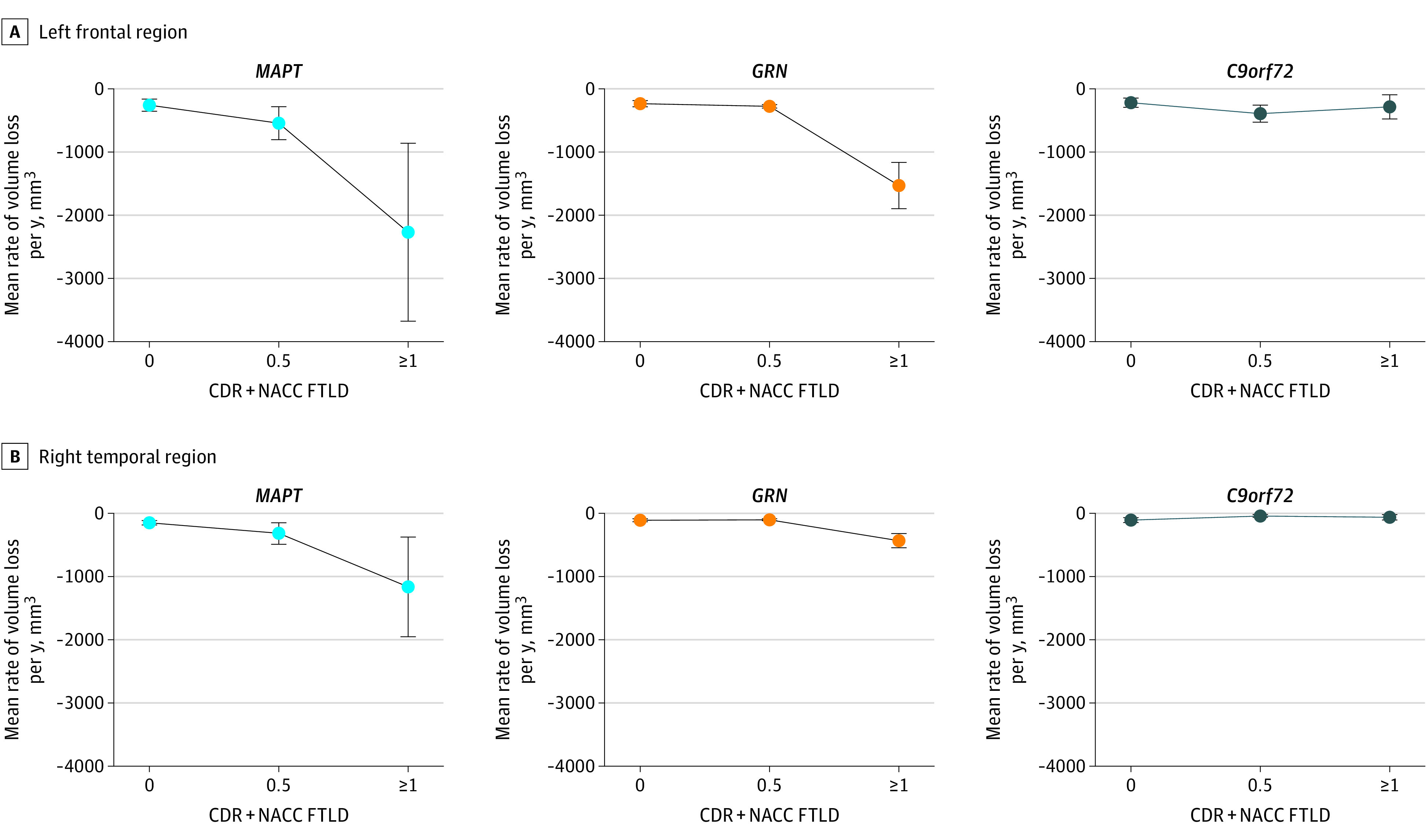
Examination of mean rates of volume loss in several regions of interest highlights how the consequences of disease stage vary by genetic group, with C9orf72 repeat expansion carriers showing the least increase in the rate of atrophy as disease severity increases. GRN pathogenic variant carriers showed almost no differences in the rate of volume loss between CDR + NACC FTLD 0 and 0.5 stages, whereas a large increase in the rate of volume loss was observed between the 0.5 and 1 or greater stages. CDR + NACC FTLD indicates Clinical Dementia Rating plus National Alzheimer’s Coordinating Center Frontotemporal Lobar Degeneration. Error bars indicate SDs.
One potential reason why the rate of volume loss may appear erroneously low in the C9orf72+ group is that the spatial location of atrophy may vary across C9orf72+ carriers, such that mean rates of change in any single region might be low at the group level. We examined this question by creating voxelwise maps of variance in rates of change (eFigure 4 in the Supplement) and by plotting mean lobar rates of change for each pathogenic variant carrier (eFigure 5 in the Supplement) for each genetic group at each stage. We also created maps of annualized volume loss for each individual in the CDR + NACC FTLD = 1 or greater stage (eFigure 6 in the Supplement). These maps and plots revealed that variability was highest in MAPT+ and lowest in C9orf72+ carriers, suggesting that excessive variability across C9orf72+ carriers, either in the spatial location of atrophy or the rate of atrophy, does not account for the group-level findings.
Clinical Decline
Rates of functional decline, as measured by the CDR + NACC FTLD box score, showed a disease stage by gene interaction, similar to the rates of atrophy (bottom of eTable 4 in the Supplement). In contrast to the imaging results, the C9orf72+ and GRN+ groups showed similar differences in the rate of change from 0 (mean [SD], 0.1 [0] box score units per year for C9orf72+ and 0.1 [0] box score units per year for GRN+) to 0.5 (mean [SD], 0.4 [0.1] box score units per year for C9orf72+ and 0.3 [0.2] box score units per year for GRN+) and from 0.5 to 1 (mean [SD], 1.5 [0.3] box score units per year for C9orf72+ and 1.4 [0.5] box score units per year for GRN+). The estimated difference in rate of clinical decline from 0.5 (mean [SD], 0.3 [0.1] for MAPT+) to 1 (mean [SD], 2.2 [1.0] for MAPT+) was almost twice as large in the MAPT+ group as it was for GRN+ or C9orf72+ groups, consistent with neuroimaging.
Cross-sectional Atrophy
The small increases in the rate of volume loss in the C9orf72+ group prompted the question of whether expansions in this gene are associated with accumulation of brain atrophy to a similar degree as pathogenic variants in the other genes. Voxelwise maps depicting cross-sectional atrophy at each stage in each gene are shown in eFigure 7A, C, and E in the Supplement, with maps of statistical significance shown in eFigure 7B, D, and F in the Supplement. At CDR + NACC FTLD = 1 or greater, all groups showed more atrophy in all ROIs compared with the control group (eFigure 8 and eTable 5 in the Supplement). The MAPT+ group showed the greatest degree of frontal (mean [SD] volume, left frontal: 12 683 [2345] mm3; right frontal: 13 235 [2015] mm3) and temporal (mean [SD] volume, left temporal: 8652 [1090] mm3; right temporal: 8628 [1237] mm3) atrophy at this stage, followed by GRN+ (eg, mean [SD] volume, right frontal: 13 679 [2448] mm3; right temporal: 9271 [1530] mm3) and C9orf72+ groups (eg, mean [SD] volume, right frontal: 14 012 [1485] mm3; right temporal: 9336 [734] mm3), with similar degrees of atrophy in GRN+ and C9orf72+ groups.
Discussion
The objective of this study was to characterize the evolution of neurodegeneration in FTLD associated with pathogenic variants in 3 different genes. Consistent with our hypothesis, we found evidence for acceleration of neurodegeneration, as measured by loss of brain volume, in MAPT+ and GRN+. Compared with these 2 genetic groups, C9orf72+ was associated with attenuated increases in the rate of volume loss, even with transition to dementia. The differences in mean rates of change across groups were not accounted for by differences in interparticipant variability. Despite differences in patterns of acceleration, cross-sectional maps of atrophy indicated that pathogenic variants in all 3 genes were associated with substantial accumulation of atrophy by the time patients developed dementia. Progression in a clinical measure of disease severity diverged from this pattern, with the rate of functional decline in C9orf72+ being similar to that in GRN+. Together, these findings suggest that, although the destiny for the brain in C9orf72+ is similar to that of other pathogenic variants, the path to this point is different, being slower and more constant over time. This finding has implications for models that would target prediction of symptom onset or tracking of disease progression. In addition, it raises important questions about the unique pathophysiology associated with C9orf72 repeat expansions and how this finding relates to symptoms.
These results have several implications for work predicated on accurate prognostication. Treatment studies enrolling groups with f-FTLD must consider the heterogeneity conferred by both the disease stage and the altered gene. In addition, a critical goal for trials in f-FTLD is to identify predictors for when symptoms will develop so that participants who are close to symptom onset and demonstrate delay in this transition can be enrolled.37,38,39 Recent publications from studies of f-FTLD and other familial neurodegenerative diseases indicate that cross-sectional40 and longitudinal5 measurements of imaging and fluid biomarkers can predict development of symptoms. Our results suggest that models assuming rapid change in biomarkers preceding or accompanying development of symptoms may apply well to MAPT+ and GRN+ carriers, but not as well to C9orf72+ carriers. However, whereas the nonlinear nature of change in MAPT+ and GRN+ may make it difficult to predict onset of symptoms using measures collected in the stable or asymptomatic phase, such measurements may be more useful in C9orf72+, where decline is more linear.
Different dynamics of change across these 3 genetic groups may be associated with the unique pathophysiology of pathogenic variants in each gene. We observed regions of accelerated volume loss in the medial temporal regions relative to the rest of the brain in MAPT+ early in the course of illness. This finding indicates fairly consistent associations in this region across participants, consistent with prior literature,3,4,13 indicating that the medial temporal lobes are particularly vulnerable to MAPT pathogenic variants. The MAPT pathogenic variants lead to accumulation of modified tau molecules that damage neurons, although the mechanisms are not completely understood,41 and our model suggests that the associations of interventions in the early stages of disease might be measurable in reduced rates of volume loss in medial temporal regions or reduced spread of atrophy to other regions. Compared with the other genetic groups, MAPT+ tends to exhibit atrophy more focally and symmetrically, which could improve the power to detect atrophy at the group level.
In contrast to MAPT+, little acceleration of volume loss occurred in any region until symptom onset in GRN+. This observation is consistent with studies showing minimal cross-sectional13,42 or longitudinal2,15,42 atrophy in presymptomatic GRN+. Moreover, other studies have shown that rapid neuroimaging changes13 and 3-fold to 4-fold increases in cerebrospinal fluid neurofilament light chain levels43 occur around the time of symptom onset in GRN+. The primary consequence of the GRN pathogenic variant is reduced production of the progranulin protein. This reduction is detectable early in life, and levels of progranulin are similar in the presymptomatic and symptomatic stages, indicating that progranulin reduction may not be directly responsible for symptoms.44,45,46 These observations could be consistent with the theory that a secondary biological process (“hit”) occurring in the context of low progranulin sets off a rapid cascade of neurodegeneration13 or that there is a tipping point in the accumulation of cellular or tissue damage. If this 2-hit model does indeed apply to GRN+, progranulin-raising medications administered in the presymptomatic stage may delay onset of symptoms but might have only a minimal impact on measurable imaging changes in this phase.
The observation that C9orf72+ showed only a small degree of acceleration yet the degree of volume loss accumulated was close to the amount seen with GRN+ in the symptomatic phase might suggest that atrophy starts at a younger age, which is supported by previous studies.6,12 Furthermore, studies15,47,48,49 of small cohorts of C9orf72+ have highlighted slow progression with insidious transition from presymptomatic to symptomatic phases. Our findings indicate that this insidious transition may be a common feature of disease associated with C9orf72 repeat expansions, although rapid deterioration may still occur in some cases or later in the illness.50 Divergence in rates of volume loss and clinical decline in C9orf72+ is consistent with prior findings suggesting that neuronal dysfunction (particularly salience network and medial pulvinar dysfunction quantified with task-free functional magnetic resonance imaging) rather than global neuronal loss may best predict clinical severity in C9orf72+.49
Limitations
These results should be interpreted in the context of several limitations. First, because of the rarity of this disease, the sample sizes are small. Although the longitudinal nature of this study improves our ability to directly quantify changes, replication will be important given the small sample sizes in some of the groups. A second consequence of the small sample size is that, although we separated participants into 3 genetic groups, we were unable to look at the association of specific pathogenic variants, which produce overlapping but distinct atrophy patterns51,52 and different disease durations.20 We addressed this limitation in part by producing variability maps to understand the consequences of within-group heterogeneity. Third, the small sample size required careful consideration of covariates, and we were unable to fully explore all potential factors, such as sex. This limitation is a topic that will be the focus of future investigation.
Conclusions
To our knowledge, this investigation is the first study to analyze the natural history of longitudinal volumetric changes in pathogenic variant carriers in 3 genes, across the entire disease spectrum. This study advances the knowledge of between-gene differences in atrophy rates as a function of disease severity, and the results have implications for clinical trial design. These findings suggest that the mechanism by which C9orf72 pathogenic variants engender symptoms may be fundamentally different from the mechanisms associated with MAPT and GRN pathogenic variants.
eMethods. Supplemental Methods
eAppendix. Specific MAPT and GRN Mutations Included in This Study
eFigure 1. Maps of Voxel-Wise Atrophy Rate in Each Genetic Group at Three Levels of Disease Severity
eFigure 2. Voxel-Wise Mutation Type by Disease Severity Interaction
eFigure 3. Mean Rates of Volume Loss for Several Regions of Interest
eFigure 4. SD of Velocity Maps in Each Genetic Group at Three Levels of Disease Severity
eFigure 5. Individual Variability in Mean Rates of Atrophy for Several Regions of Interest
eFigure 6. Individual Maps of Voxel-Wise Atrophy Rates for All Symptomatic Mutation Carriers
eFigure 7. Cross-Sectional Atrophy Maps in Each Genetic Group at Three Levels of Disease Severity
eFigure 8. Cross-Sectional Atrophy by Region of Interest
eTable 1. Demographic Information for Each Subgroup
eTable 2. Diagnostic Composition
eTable 3. Distribution of Scanner Vendors by Genetic Group
eTable 4. Mean and SD of Annualized Rate of Volume Loss
eTable 5. Cross-sectional Volume by Region of Interest
References
- 1.Rohrer JD, Rosen HJ. Neuroimaging in frontotemporal dementia. Int Rev Psychiatry. 2013;25(2):221-229. doi: 10.3109/09540261.2013.778822 [DOI] [PubMed] [Google Scholar]
- 2.Chen Q, Boeve BF, Senjem M, et al. . Trajectory of lobar atrophy in asymptomatic and symptomatic GRN mutation carriers: a longitudinal MRI study. Neurobiol Aging. 2020;88:42-50. doi: 10.1016/j.neurobiolaging.2019.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rohrer JD, Nicholas JM, Cash DM, et al. . Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal Dementia Initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol. 2015;14(3):253-262. doi: 10.1016/S1474-4422(14)70324-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Q, Boeve BF, Senjem M, et al. ; LEFFTDS Consortium . Rates of lobar atrophy in asymptomatic MAPT mutation carriers. Alzheimers Dement (N Y). 2019;5:338-346. doi: 10.1016/j.trci.2019.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiskoot LC, Panman JL, Meeter LH, et al. . Longitudinal multimodal MRI as prognostic and diagnostic biomarker in presymptomatic familial frontotemporal dementia. Brain. 2019;142(1):193-208. doi: 10.1093/brain/awy288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee SE, Sias AC, Mandelli ML, et al. . Network degeneration and dysfunction in presymptomatic C9ORF72 expansion carriers. Neuroimage Clin. 2016;14:286-297. doi: 10.1016/j.nicl.2016.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitwell JL, Weigand SD, Boeve BF, et al. . Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain. 2012;135(pt 3):794-806. doi: 10.1093/brain/aws001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olney NT, Ong E, Goh SM, et al. . Clinical and volumetric changes with increasing functional impairment in familial frontotemporal lobar degeneration. Alzheimer’s Dement. 2020;16(1):49-59. doi: 10.1016/j.jalz.2019.08.196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boxer AL, Gold M, Huey E, et al. . The advantages of frontotemporal degeneration drug development (part 2 of frontotemporal degeneration: the next therapeutic frontier). Alzheimers Dement. 2013;9(2):189-198. doi: 10.1016/j.jalz.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romero K, Ito K, Rogers JA, et al. ; Alzheimer’s Disease Neuroimaging Initiative; Coalition Against Major Diseases . The future is now: model-based clinical trial design for Alzheimer’s disease. Clin Pharmacol Ther. 2015;97(3):210-214. doi: 10.1002/cpt.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jack CR Jr, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12(2):207-216. doi: 10.1016/S1474-4422(12)70291-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bertrand A, Wen J, Rinaldi D, et al. ; Predict to Prevent Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis (PREV-DEMALS) Study Group . Early cognitive, structural, and microstructural changes in presymptomatic C9orf72 carriers younger than 40 years. JAMA Neurol. 2018;75(2):236-245. doi: 10.1001/JAMANEUROL.2017.4266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cash DM, Bocchetta M, Thomas DL, et al. ; Genetic FTD Initiative, GENFI . Patterns of gray matter atrophy in genetic frontotemporal dementia: results from the GENFI study. Neurobiol Aging. 2018;62:191-196. doi: 10.1016/j.neurobiolaging.2017.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Papma JM, Jiskoot LC, Panman JL, et al. . Cognition and gray and white matter characteristics of presymptomatic C9orf72 repeat expansion. Neurology. 2017;89(12):1256-1264. doi: 10.1212/WNL.0000000000004393 [DOI] [PubMed] [Google Scholar]
- 15.Panman JL, Jiskoot LC, Bouts MJRJ, et al. . Gray and white matter changes in presymptomatic genetic frontotemporal dementia: a longitudinal MRI study. Neurobiol Aging. 2019;76:115-124. doi: 10.1016/j.neurobiolaging.2018.12.017 [DOI] [PubMed] [Google Scholar]
- 16.Floeter MK, Bageac D, Danielian LE, Braun LE, Traynor BJ, Kwan JY. Longitudinal imaging in C9orf72 mutation carriers: relationship to phenotype. Neuroimage Clin. 2016;12:1035-1043. doi: 10.1016/j.nicl.2016.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whitwell JL, Boeve BF, Weigand SD, et al. . Brain atrophy over time in genetic and sporadic frontotemporal dementia: a study of 198 serial magnetic resonance images. Eur J Neurol. 2015;22(5):745-752. doi: 10.1111/ene.12675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whitwell JL, Weigand SD, Gunter JL, et al. . Trajectories of brain and hippocampal atrophy in FTD with mutations in MAPT or GRN. Neurology. 2011;77(4):393-398. doi: 10.1212/WNL.0b013e318227047f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boeve B, Bove J, Brannelly P, et al. . The Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects protocol: framework and methodology. Alzheimers Dement. 2020;16(1):22-36. doi: 10.1016/j.jalz.2019.06.4947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore KM, Nicholas J, Grossman M, et al. . Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an international retrospective cohort study. Lancet Neurol. 2020;19(2):145-156. doi: 10.1016/S1474-4422(19)30394-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosen HJ, Boeve BF, Boxer AL. Tracking disease progression in familial and sporadic frontotemporal lobar degeneration: recent findings from ARTFL and LEFFTDS. Alzheimers Dement. 2020;16(1):71-78. doi: 10.1002/alz.12004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jack CR Jr, Lowe VJ, Weigand SD, et al. ; Alzheimer’s Disease Neuroimaging Initiative . Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: implications for sequence of pathological events in Alzheimer’s disease. Brain. 2009;132(pt 5):1355-1365. doi: 10.1093/brain/awp062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li D, Iddi S, Thompson WK, Rafii MS, Aisen PS, Donohue MC. Bayesian latent time joint mixed-effects model of progression in the Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement (Amst). 2018;10:657-668. doi: 10.1016/j.dadm.2018.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiskoot LC, Panman JL, van Asseldonk L, et al. . Longitudinal cognitive biomarkers predicting symptom onset in presymptomatic frontotemporal dementia. J Neurol. 2018;265(6):1381-1392. doi: 10.1007/s00415-018-8850-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jack CR Jr, Vemuri P, Wiste HJ, et al. ; Alzheimer’s Disease Neuroimaging Initiative . Shapes of the trajectories of 5 major biomarkers of Alzheimer disease. Arch Neurol. 2012;69(7):856-867. doi: 10.1001/archneurol.2011.3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Staffaroni AM, Ljubenkov PA, Kornak J, et al. . Longitudinal multimodal imaging and clinical endpoints for frontotemporal dementia clinical trials. Brain. 2019;142(2):443-459. doi: 10.1093/brain/awy319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyagawa T, Brushaber D, Syrjanen J, et al. ; ARTFL/LEFFTDS Consortium . Use of the CDR plus NACC FTLD in mild FTLD: data from the ARTFL/LEFFTDS Consortium. Alzheimers Dement. 2020;16(1):79-90. doi: 10.1016/j.jalz.2019.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weintraub S, Besser L, Dodge HH, et al. . Version 3 of the Alzheimer Disease Centers’ neuropsychological test battery in the Uniform Data Set (UDS). Alzheimer Dis Assoc Disord. 2018;32(1):10-17. doi: 10.1097/WAD.0000000000000223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knopman DS, Kramer JH, Boeve BF, et al. . Development of methodology for conducting clinical trials in frontotemporal lobar degeneration. Brain. 2008;131(pt 11):2957-2968. doi: 10.1093/brain/awn234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyagawa T, Brushaber D, Syrjanen J, et al. . Utility of the global CDR plus NACC FTLD rating and development of scoring rules: data from the ARTFL/LEFFTDS Consortium. Alzheimers Dement. 2020;16(1):106-117. doi: 10.1002/alz.12033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramos EM, Dokuru DR, Van Berlo V, et al. ; ARTFL/LEFFTDS Consortium . Genetic screening of a large series of North American sporadic and familial frontotemporal dementia cases. Alzheimers Dement. 2020;16(1):118-130. doi: 10.1002/alz.12011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ziegler G, Penny WD, Ridgway GR, Ourselin S, Friston KJ; Alzheimer’s Disease Neuroimaging Initiative . Estimating anatomical trajectories with Bayesian mixed-effects modeling. Neuroimage. 2015;121:51-68. doi: 10.1016/j.neuroimage.2015.06.094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friston KJ, Penny W, Phillips C, Kiebel S, Hinton G, Ashburner J. Classical and Bayesian inference in neuroimaging: theory. Neuroimage. 2002;16(2):465-483. doi: 10.1006/nimg.2002.1090 [DOI] [PubMed] [Google Scholar]
- 34.Winkler AM, Ridgway GR, Webster MA, Smith SM, Nichols TE. Permutation inference for the general linear model. Neuroimage. 2014;92:381-397. doi: 10.1016/j.neuroimage.2014.01.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith SM, Nichols TE. Threshold-free cluster enhancement: addressing problems of smoothing, threshold dependence and localisation in cluster inference. Neuroimage. 2009;44(1):83-98. doi: 10.1016/j.neuroimage.2008.03.061 [DOI] [PubMed] [Google Scholar]
- 36.Desikan RS, Ségonne F, Fischl B, et al. . An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31(3):968-980. doi: 10.1016/j.neuroimage.2006.01.021 [DOI] [PubMed] [Google Scholar]
- 37.Galimberti D, Fenoglio C, Scarpini E. Progranulin as a therapeutic target for dementia. Expert Opin Ther Targets. 2018;22(7):579-585. doi: 10.1080/14728222.2018.1487951 [DOI] [PubMed] [Google Scholar]
- 38.Boeve BF, Rosen HJ. Multimodal imaging in familial FTLD: phenoconversion and planning for the future. Brain. 2019;142(1):8-11. doi: 10.1093/brain/awy314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sperling RA, Rentz DM, Johnson KA, et al. . The A4 study: stopping AD before symptoms begin? Sci Transl Med. 2014;6(228):228fs13. doi: 10.1126/scitranslmed.3007941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Staffaroni AM, Cobigo Y, Goh SM, et al. ; ARTFL/LEFFTDS Consortium . Individualized atrophy scores predict dementia onset in familial frontotemporal lobar degeneration. Alzheimers Dement. 2020;16(1):37-48. doi: 10.1016/j.jalz.2019.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17(1):5-21. doi: 10.1038/nrn.2015.1 [DOI] [PubMed] [Google Scholar]
- 42.Caroppo P, Habert MO, Durrleman S, et al. ; Predict-PGRN study group . Lateral temporal lobe: an early imaging marker of the presymptomatic GRN disease? J Alzheimers Dis. 2015;47(3):751-759. doi: 10.3233/JAD-150270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meeter LH, Dopper EG, Jiskoot LC, et al. . Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol. 2016;3(8):623-636. doi: 10.1002/acn3.325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meeter LHH, Patzke H, Loewen G, et al. . Progranulin levels in plasma and cerebrospinal fluid in granulin mutation carriers. Dement Geriatr Cogn Dis Extra. 2016;6(2):330-340. doi: 10.1159/000447738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghidoni R, Benussi L, Glionna M, Franzoni M, Binetti G. Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology. 2008;71(16):1235-1239. doi: 10.1212/01.wnl.0000325058.10218.fc [DOI] [PubMed] [Google Scholar]
- 46.Galimberti D, Fumagalli GG, Fenoglio C, et al. ; Genetic FTD Initiative (GENFI) . Progranulin plasma levels predict the presence of GRN mutations in asymptomatic subjects and do not correlate with brain atrophy: results from the GENFI study. Neurobiol Aging. 2018;62:245.e9-245.e12. doi: 10.1016/j.neurobiolaging.2017.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Devenney E, Hornberger M, Irish M, et al. . Frontotemporal dementia associated with the C9ORF72 mutation: a unique clinical profile. JAMA Neurol. 2014;71(3):331-339. doi: 10.1001/jamaneurol.2013.6002 [DOI] [PubMed] [Google Scholar]
- 48.Khan BK, Yokoyama JS, Takada LT, et al. . Atypical, slowly progressive behavioural variant frontotemporal dementia associated with C9ORF72 hexanucleotide expansion. J Neurol Neurosurg Psychiatry. 2012;83(4):358-364. doi: 10.1136/jnnp-2011-301883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee SE, Khazenzon AM, Trujillo AJ, et al. . Altered network connectivity in frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. Brain. 2014;137(pt 11):3047-3060. doi: 10.1093/brain/awu248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chester C, de Carvalho M, Miltenberger G, et al. . Rapidly progressive frontotemporal dementia and bulbar amyotrophic lateral sclerosis in Portuguese patients with C9orf72 mutation. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(1):70-72. doi: 10.3109/17482968.2012.690418 [DOI] [PubMed] [Google Scholar]
- 51.Whitwell JL, Jack CR Jr, Boeve BF, et al. . Atrophy patterns in IVS10+16, IVS10+3, N279K, S305N, P301L, and V337M MAPT mutations. Neurology. 2009;73(13):1058-1065. doi: 10.1212/WNL.0b013e3181b9c8b9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cordes M, Wszolek ZK, Calne DB, Rodnitzky RL, Pfeiffer RF. Magnetic resonance imaging studies in rapidly progressive autosomal dominant parkinsonism and dementia with pallido-ponto-nigral degeneration. Neurodegeneration. 1992;1:217-224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods. Supplemental Methods
eAppendix. Specific MAPT and GRN Mutations Included in This Study
eFigure 1. Maps of Voxel-Wise Atrophy Rate in Each Genetic Group at Three Levels of Disease Severity
eFigure 2. Voxel-Wise Mutation Type by Disease Severity Interaction
eFigure 3. Mean Rates of Volume Loss for Several Regions of Interest
eFigure 4. SD of Velocity Maps in Each Genetic Group at Three Levels of Disease Severity
eFigure 5. Individual Variability in Mean Rates of Atrophy for Several Regions of Interest
eFigure 6. Individual Maps of Voxel-Wise Atrophy Rates for All Symptomatic Mutation Carriers
eFigure 7. Cross-Sectional Atrophy Maps in Each Genetic Group at Three Levels of Disease Severity
eFigure 8. Cross-Sectional Atrophy by Region of Interest
eTable 1. Demographic Information for Each Subgroup
eTable 2. Diagnostic Composition
eTable 3. Distribution of Scanner Vendors by Genetic Group
eTable 4. Mean and SD of Annualized Rate of Volume Loss
eTable 5. Cross-sectional Volume by Region of Interest