

SUMMARY
Hallucinogens like lysergic acid diethylamide (LSD), psilocybin, and substituted N-benzyl phenylalkylamines are widely used recreationally with psilocybin being considered as a therapeutic for many neuropsychiatric disorders including depression, anxiety, and substance abuse. How psychedelics mediate their actions—both therapeutic and hallucinogenic—are not understood, although activation of the 5-HT2A serotonin receptor (HTR2A) is key. To gain molecular insights into psychedelic actions, we determined the active-state structure of HTR2A bound to 25-CN-NBOH—a prototypical hallucinogen—in complex with an engineered Gαq heterotrimer by cryoelectron microscopy (cryo-EM). We also obtained the X-ray crystal structures of HTR2A complexed with the arrestin-biased ligand LSD or the inverse agonist methiothepin. Comparisons of these structures reveal determinants responsible for HTR2A-Gαq protein interactions as well as the conformational rearrangements involved in active-state transitions. Given the potential therapeutic actions of hallucinogens, these findings could accelerate the discovery of more selective drugs for the treatment of a variety of neuropsychiatric disorders.
In Brief
Roth et al. reveal structurally how psychedelics, including LSD, psilocin, mescaline, and various N-BOH analogs, mediate their therapeutic and hallucinogenic effects by binding to and activating their molecular target, the serotonin (5-HT) 2A receptor coupled with G-protein Gαq.
Graphical Abstract
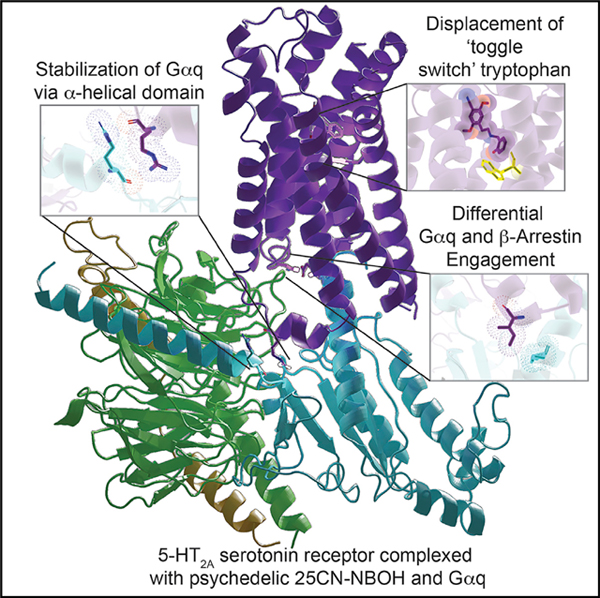
INTRODUCTION
Naturally occurring psychedelics from plants as diverse as the mescaline-containing cactus Lophophora williamsii, the psilocy-bin-containing mushrooms Psilocybe sp., and the lysergamide-containing seeds from “Morning Glories” (Ipomoea sp.) have been reportedly used for millennia for religious purposes and shamanism (Nichols, 2016). Beginning in the 1950s and 1960s, semi-synthetic hallucinogens like lysergic acid diethylamide (LSD) and synthetic hallucinogens related to mescaline, including substituted amphetamines such as 2,5-dimethoxy-4-methylamphetamine (Snyder et al., 1968), were available and used recreationally. More recently so-called “designer hallucinogens” with a scaffold related to N-benzyl-2,5-dimethoxy-phene-thylamine (NBOMe) (Figure 1A) have become popular (Poulie et al., 2019) albeit with scattered reports of toxicity (Halberstadt, 2017). Of the NBOMe series, 25CN-NBOH is reported to be among the most potent and selective in vitro and in vivo (Fantegrossi et al., 2015; Hansen et al., 2014).
Figure 1. The Overall Cryo-EM Structure of the HTR2A-Gαq Bound to 25CN-NBOH and Crystal Structures of the HTR2A Bound to LSD or Methiothepin.

(A) Cartoon view of the cryo-EM structure of HTR2A (green color) with agonist, 25CN-NBOH (yellow stick and 2D chemical structure, left), mini-Gαq (olive color), Gβ (red color), and Gγ (yellow color) with scFv16 omitted. See Figures S1 and Table S1.
(B) Cartoon view of the crystal structure of HTR2A (pink color) with the partial agonist LSD (light blue stick and 2D chemical structure, left).
(C) Cartoon view of the crystal structure of HTR2A (dark blue color) with the antagonist methiothepin (orange stick and 2D chemical structure, left). See Figure S2 and Table S1.
(D) Schematic diagram of the bioluminescence resonance energy transfer (BRET) assays, BRET 1 and BRET 2.
(E) The comparison of mini-Gαq or Gαq wild-type interactions with HTR2A upon the agonist 25CN-NBOH stimulation; data represent mean ± SEM of three biological replicates. See Table S4 for fitted parameter values.
(F) Ligand-mediated mini-Gαq recruitment to HTR2A. Several hallucinogen agonists were compared in BRET 1 assays. See Table S4 for fitted parameter values.
Hallucinogens like psilocybin and LSD have been described to have potential therapeutic actions for many neuropsychiatric diseases (Nutt et al., 2020). Psilocybin, for instance, has shown efficacy in two phase II clinical trials of depression and anxiety (Carhart-Harris et al., 2018; Ross et al., 2016). Remarkably, psilocybin’s effects appear to be both rapid and enduring (Carhart-Harris et al., 2018; Ross et al., 2016; Griffiths et al., 2016). Likewise, LSD has been reported in pilot studies to have efficacy in cluster headaches (Sewell et al., 2006), to alleviate anxiety in terminal illness (Gasser et al., 2015), and a number of uncontrolled and anecdotal reports have suggested its therapeutic actions in other neuropsychiatric disorders (Dos Santos et al., 2018; Nichols, 2016; Nutt et al., 2020).
Although the precise mechanisms of action of hallucinogens remain largely unclear, 5-HT2A serotonin (5-hydroxytryptamine [5-HT]) receptor agonism is essential for their psychedelic effects in humans (Barrett et al., 2018; Kometer et al., 2013; Preller et al., 2018; Nutt et al., 2020). The key to an understanding of psychedelic actions, then, is an appreciation of how they interact with and activate 5-HT2A serotonin receptors (HTR2A). To elucidate hallucinogen actions at the molecular level, we report here the active-state structure of 5-HT2A bound to 25CN-NBOH in complex with a mini-Gαq-βγ heterotrimer stabilized by a single-chain variable fragment (scFv16) obtained by cryoelectron microscopy (cryo-EM). For comparison, we also report X-ray structures of HTR2A complexed with the prototypical hallucinogen LSD and the inverse agonist methiothepin. Collectively, these results provide insights into how a model hallucinogen stabilizes a specific HTR2A transducer-coupled state. These findings should accelerate structure-guided efforts for discovering more selective HTR2A agonists with potential therapeutic activity for many neuropsychiatric diseases.
RESULTS
A Gαq-Coupled HTR2A Structure Bound to the Hallucinogen 25CN-NBOH
For structural studies with a transducer-coupled HTR2A, we initially evaluated the HTR2A non-selective hallucinogen LSD (Kroeze et al., 2015) and the HTR2A-selective agonist 25CN-NBOH (Fantegrossi et al., 2015; Hansen et al., 2014). In preliminary experiments, we found that 25CN-NBOH provided more effective stabilization of the final complex than LSD (not shown). We used an engineered Gαq that was previously demonstrated to faithfully recapitulate wild-type Gαq-coupled G-protein-coupled receptor (GPCR) activation for several GPCRs, including HTR2A (Nehmé et al., 2017; Wan et al., 2018). Here, 35 amino acids at the N terminus of engineered mini-Gαq were replaced by the corresponding sequence in Gαi2 to facilitate binding of a single-chain variable fragment (scFv16) (Maeda et al., 2018), thereby further stabilizing the HTR2A-Gαq protein complex for structural studies (Figure S1). An analogous approach was recently used to obtain a structure of the M1-muscarinic receptor complexed with Gα11 (Maeda et al., 2019). To verify the functionality of the complex we performed bioluminescence resonance energy transfer (BRET 1 and 2) (see Olsen et al., 2020 for details) (Figure 1D) studies similar to those previously reported (Wan et al., 2018), which revealed that the functional activity of this engineered mini-Gαq construct is comparable to the wild-type Gαq (Figure 1E; Table S4). The studies also showed that 25CN-NBOH was the most efficacious agonist tested in our Gαq BRET recruitment assays (Figure 1F; Table S4).
To improve the expression of HTR2A, parts of the N- and C-terminal of HTR2A were truncated and then cloned into a pFASTBac 1 vector (Figure S1A). To obtain a stable complex of HTR2A-Gαq, the sequences of the engineered GαqiN, Gβ1, and Gγ2 were integrated into a pFast dual expression vector to afford simultaneous expression of all three components and pre-formation of the GαqiN-Gβ1-Gγ2 heterotrimer (Figure S1B). We expressed HTR2A and G protein complex (GαqiN-Gβ1-Gγ2) in sf9 insect cells and purified them individually. The full complex of HTR2A-GαqiN-Gβ1-Gγ2 was then assembled in the presence of the agonist 25CN-NBOH and further stabilized by the single-chain antibody scFv16 (Figures S1C and S1D). The structure of G protein-coupled HTR2A complexed with 25CN-NBOH was determined by single-particle cryo-EM at a global nominal resolution of 3.27 Å, enabling near-atomic resolution modeling of the complex (Figures 1A and S1; Table S1). The 25CN-NBOH pose was further confirmed and validated through the GemSpot pipeline (Robertson et al., 2020) and was virtually identical to the pose predicted by independent molecular docking studies (Figures S6E and S6F). The overall structure of 25CN-NBOH-stabilized HTR2A-Gαq/β1/γ2 structure is consistent with the fully active-state receptor conformation that has been observed in several other GPCRs bound to a G-protein ternary complex (Weis and Kobilka, 2018). We found that scFv16 binds to the crevice between the αN helix of mini-GαqiN and the β-propeller of Gβ1 similar to that observed in the MOR-Gαi complex (Koehl et al., 2018), thereby providing additional stabilization of the complex (Figure S1E).
Structures of LSD- and Methiothepin-Bound HTR2A
To obtain a more thorough understanding of how hallucinogens might interact with HTR2A, we determined crystal structures of two HTR2A-ligand complexes. LSD is the prototypical hallucinogen that exerts effects its psychedelic primarily via HTR2A (Nichols, 2016), and we previously reported the structure of the 5-HT2B (HTR2B) receptor complexed with LSD (Wacker et al., 2017b) and related ergolines (McCorvy et al., 2018). These structures provided important insights into mechanisms of biased agonism at HTR2B (McCorvy et al., 2018; Roth, 2019) and hints regarding how LSD might interact with HTR2A (Wacker et al., 2017b). To obtain a better understanding of the actions of LSD on HTR2A, we solved the X-ray crystal structures of HTR2A complexed with LSD (Figure 1B), and, for comparison, the potent inverse agonist methiothepin (Figure 1C; Table S2). Both structures were obtained at 3.4 Å resolution using an identical HTR2A construct with apocytochrome b562RIL (BRIL) fused to the third intracellular loop 3 (ICL3) of the receptor and two thermostabilizing HTR2A mutations (L247A5.51 and L371A7.44) (Figures S2A–S2D). Binding assays showed that this engineered construct maintained WT ligand-binding affinities (Figure S2E). The asymmetric unit (ASU) contains three molecules, and crystal contacts are mediated by TM1, TM4, and H8 (Figure S2F). The superposition of these three molecules shows that the orientation of the receptors is nearly identical, although slight differences are observed in the BRIL fusion protein. We focused on HTR2A molecule “chain A” for subsequent analysis (Figure S2G).
The electron density maps for LSD and methiothepin were well resolved (Figure S2H). The overall differences between LSD bound to HTR2A and the previous LSD-HTR2B structure are relatively subtle, as predicted (Wacker et al., 2017b). In the HTR2B/LSD structure, ECL2 forms a “lid” over LSD, prolonging LSD’s residence time (Wacker et al., 2017b). In confirmation of our prior mutagenesis, kinetic, and molecular modeling studies (Wacker et al., 2017b), we find that ECL2 in the LSD-HTR2A complex is located in a similar position, occluding LSD (Figure 2A). Using a previously described constitutively active HTR2A mutant (Shapiro et al., 2002), we found that methiothepin (a non-selective serotonin, dopamine, and adrenergic receptor antagonist) was a potent and efficacious HTR2A inverse agonist (Figure S2I). Methiothepin’s binding pose in HTR2A displays unique features that are detailed below.
Figure 2. Overall Conformational Differences in the Ligand-Binding Pockets of 25CN-NBOH and LSD-Bound HTR2A.

(A) View of the HTR2A ligand-binding pocket from the extracellular side. Conformational changes in cylindric helix and loop positions due to receptor activation are highlighted by red arrows. Distances were measured between the Cα atoms of I2104.60, L229ECL2, V2355.39, N3436.55, and E3557.38.
(B and C) Electrostatic surface representation from the extracellular view of HTR2A/LSD and HTR2A/25CN-NBOH, respectively. Electrostatic potential surfaces were calculated using the APBS plugin in PyMOL (Baker et al., 2001).
(D) Expansion of binding pocket of HTR2A bound to full agonist 25CN-NBOH compared to partial agonist LSD. See Table S3 for volume estimates.
Structural Comparisons of HTR2A Inactive- and Active-State Structures
With these available structures in hand, we first assessed the overall conformational rearrangements between the inactive and active state structures. A surface view from the extracellular side also shows that the Gαq-coupled state is considerably more open than the LSD-bound state (Figures 2A–2C). We calculated the size of the ligand-binding pockets for all three structures using the CASTp 3.0 server (Tian et al., 2018) and found that the solvent-accessible volumes of the ligand-binding pockets were 188.1 A3 for methiothepin, 153.1 A3 for LSD, and 287.0 A3 for 25CN-NBOH (Figure 2D; Table S3). Typically, based on analysis of other class A active-state GPCR/G protein complex structures (e.g., β2-adrenergic/Gαs [Rasmussen et al., 2011], M1-muscarininc/Gα11 and M2-muscarinic/Go [Maeda et al., 2019], CB1/Gαi1 [Krishna Kumar et al., 2019], A1-adenosine/Gαi [Draper-Joyce et al., 2018], and the κ-opioid-nanobody [Che et al., 2018]), receptor activation leads to a contraction of the extracellular binding pocket and expansion of the intracellular end, thereby providing space for the engagement of transducers (e.g., G proteins or arrestins). Conceivably, the observed expansion of the ligand-binding pocket in the 25CN-NBOH-bound complex is ligand-specific, coinciding with its unique binding pose compared to LSD and methiothepin, although additional Gαq-coupled structures will be needed to test this hypothesis.
In the cryo-EM map, the density of the ligand 25CN-NBOH, the transmembrane domains, and the G protein binding interface are all well resolved, thus allowing us to examine conformational changes upon agonist binding and transducer coupling (Figures S3A–S3D). Consistent with previous G protein- and arrestin-complexed GPCR structures (Lee et al., 2020; Huang et al., 2020; Staus et al., 2020), the intracellular ends of TM5 and TM6 in HTR2A-Gαq complex undergo the largest displacement and are tilted outward by 4.3 Å (Cα of A2655.69) and 7.1 Å (Ca of I3156.27), respectively, while TM7 in the HTR2A-Gαq complex is shifted inward by 2.0 Å (Cα of F3837.56) (Figure S4A). It is noteworthy that the structure of the muscarinic receptor M1 bound to Gα11, which is also a Gαq family subtype, shows an outward movement of TM6 by 8 Å (Maeda et al., 2019) that displays a slightly different tilt of the α5 helix of G11.
Because of the stabilization provided by Gαq coupling, the overall structure of HTR2A resides in a rigid conformation, and ICL2 now displays helical turns reminiscent of that predicted by computational studies (Perez-Aguilar et al., 2014) that are not resolved in the inactive structures (Figure S4B). Other features related to receptor activation were observed in conserved motifs including (1) an inward shift of residues in NPXXY motif, (2) breaking of the ionic lock between R1733.50 and E3186.30 (Shapiro et al., 2002) due to side chain rearrangement of R1733.50 in the E/DRY motif, and (3) rotation of the side chain of the toggle switch residue, W3366.48, and subsequent movement of the side chain of F3326.44 in the P-I-F motif (Figure S4C). Although these residues or motifs have been implicated in playing important roles in receptor activation and signaling transduction, the initiation and sequence of these events are still understudied.
25CN-NBOH Displays a Distinct Binding Mode at HTR2A
We next examined the ligand-binding pocket of the three structures as they represent unique subfamilies (25CN-NBOH as an N-benzyl phenethylamine full-agonist, LSD is an ergoline arrestin-biased partial agonist, and methiothepin is an inverse agonist). Conserved interactions observed in all three structures reveal a shared mechanism for HTR2A binding (Figures 3A–3C). A salt bridge is observed between D1553.32 and a positively charged nitrogen in each ligand, which is a critical interaction for ligand binding in serotonin and other monoamine receptors (Kristiansen et al., 2000). Not surprisingly, mutation of D1553.32 leads to the loss of function for almost all HTR2A ligands (Figure 3D). Other hydrophobic interactions have also been observed in all three liganded structures, such as V1563.33, V2355.39, and, as predicted previously (Roth et al., 1997), W3366.48, F3396.51, and F3406.52 (Figures 3A–3C). Mutagenesis and functional experiments suggested that many of these residues are essential for ligand binding and subsequent receptor activation (Figures 3D, S5A, and S5B; Table S6).
Figure 3. Ligand-Specific Interactions with HTR2A.

(A–C) Specific residues in the binding pockets that interact with 25CN-NBOH (yellow) (A), LSD (light blue) (B), and methiothepin (brown) (C), respectively. Alternative 2D diagrams showing direct interactions with each ligand are also provided at the bottom of each panel. The salt bridge interaction, as well as hydrogen bond interactions, is shown as red dashed lines.
(D) Mutagenesis studies showing the effects of orthosteric-site residues on ligand-binding affinity and functional activity. Heatmap of ΔpEC50 (EC50WT-EC50mt) (by BRET 2, HTR2A/Gαq) and ΔpKi (Kiwt-Kimt) (by binding assay, [3H]-LSD) shows differences between HTR2A wild-type and mutants. See Figure S6 and Tables S5 and S6 for fitted parameter values that represent mean ± SEM of n = 3 biological replicates.
See also Figure S5.
Quite recently, inactive-state structures of HTR2A bound to risperidone or zotepine were reported (Kimura et al., 2019). Thus, a total of five ligand-HTR2A complexes (risperidone and zotepine [Kimura et al., 2019], LSD, 25CN-NBOH, and methiothepin here) thereby providing the opportunity to examine ligand-specific features related to binding. Alignment of all five ligand-complexed HTR2A structures shows that the HTR2A-se-lective agonist 25CN-NBOH displays a unique binding pose compared with the non-selective partial agonist LSD and the non-selective inverse agonists methiothepin, risperidone, and zotepine (Figure 4A). We note that these following comparisons represent a single active state with four mainly inactively state structures. It will be important going forward to obtain more active-state HTR2A structures to determine if the binding pose of 25CN-NBOH is unique. The 2-hydroxyphenyl moiety of 25CN-NBOH dives deep down into a previously undescribed pocket between TM3 and TM6, forming hydrophobic interactions with the indole ring of W3366.48 (Figure 4B). For many years, this highly conserved W6.48 in class A GPCRs has been proposed as a “toggle-switch” to control GPCRs transitioning between “on” and “off” states during signaling transduction (Shi et al., 2002). The close interaction between 25CN-NBOH and W3366.48 coincides with a large displacement of the side chain of W3366.48, acting as a pivot for the outward movement of TM6 (Figure 4C). This displacement of W3366.48 is accompanied by a movement of F3326.44 that is part of the conserved P5.50-I3.40-F6.44 motif (Figure 4D) (Wacker et al., 2017a). Both W6.48 and the P5.50-I3.40-F6.44 motif have been shown to undergo conformational changes upon receptor activation (Wacker et al., 2013). Here, the dynamic coupling between W6.48 and the P5.50-I3.40-F6.44 motif may partially explain how allosteric changes are transmitted in the active state similar to what was observed in the cryo-EM structure of the CB1 cannabinoid-Gi in complex with fubinaca (Krishna Kumar et al., 2019). A comparison of all available serotonin-receptor structures, including active and inactive, reveals that the displacement of W3366.48 of HTR2A is also greater than those observed in other structures (Figure S5C) (García-Nafría et al., 2018; McCorvy et al., 2018; Peng et al., 2018; Yin et al., 2018). In addition to an edge-to-face π–π interaction with W3366.48, the 2-OH of 25-CN-NBOH is the only observed ligand to form a hydrogen bond with S1593.36, and this results in a 2.9 Å side-chain movement. As anticipated, the S159A3.36 mutation dramatically attenuated 25CN-NBOH’s agonist potency (Figure S5B). The 2-hydroxyphenyl moiety is also accommodated by the conserved G3697.42 which moves “in” upon agonist binding to form hydrophobic interactions. Slightly increasing the size of the side chain through mutation G369A7.42 dramatically attenuated 25CN-NBOH’s agonist potency (Figure S5B; Table S5 and Table S6) However, the binding affinity of G369A7.42 does not significantly change compared to the wild-type Figure S5A and Table S6), indicating that this residue may play a role in signal transduction in HTR2A. These interactions, as will be shown below, are not observed in either the LSD- or methiothepin-bound structures.
Figure 4. Differential 25-CN-NBOH and LSD Binding Modes.

(A) The superimposed structures of HTR2A with 25CN-NBOH (yellow), LSD (light blue), methiothepin (orange), risperidone (pink, PDB: 6A93), and zotepine (green, PDB: 6A94).
(B) S159, W336, and G369 form a binding pocket that is important for 25CN-NBOH’s agonist activity.
(C) W3366.48 acts as a pivot for the outward movement of TM6.
(D) Conformational displacement of side-chain of W3366.48, followed by F3326.44 in the P-I-F motif. See Figure S4.
(E)The sequence alignment of the serotonin receptor family with an HTR2A specific residue S2425.46 is highlighted.
(F)The S242A5.46 mutation accelerates LSD dissociation from HTR2A; data represent mean ± SEM of n = 3 biological replicates.
(G)The overall structural comparison of HTR2A/LSD (pink/magenta color) and HTR2B/LSD (olive/lime color) and inset shows side view of LSD (magenta)-bound HTR2A (pink) crystal structure overlaid with the LSD (lime)-bound HTR2B (olive) structure. Hydrogen-bond interactions are highlighted by red dash lines.
Given that LSD and methiothepin are non-selective HTR ligands, the observed binding pose between 25CN-NBOH and HTR2A could contribute to the selectivity of 25CN-NBOH at 5HTR2A over many other biogenic amine receptors (Figures S5D and S6). As reported (Halberstadt et al., 2016) and summarized (Figure S6A), 25CN-NBOH has negligible affinity for all other human biogenic amine receptors while preferring HTR2A over HTR2C and HTR2B (Figure S6A). An alignment of binding site residues among biogenic amine receptors discloses that G2385.42 is replaced by Ser5.42 or Thr5.42 in most other biogenic amine receptors (Figure S6A). As predicted from this alignment, a G2385.42S mutation diminished 25CN-NBOH’s agonist potency (Figure S6B), and this likely represents one of the structural determinants essential for 25CN-NBOH’s selective pharmacology.
Although the phenethylamine group of 25CN-NBOH is located in the orthosteric pocket similar to LSD and other antagonists, 25CN-NBOH does not, like LSD, have substantial interactions with the conserved serines S2395.43 and S2425.46. In particular, S2425.46 is unique to HTR2A among the serotonin family (Figure 4E). A comparison of LSD-bound HTR2A with HTR2B structures shows that the overall orientation of LSD is similar (Figure 4G). However, the steric extrusion of L229ECL2 in HTR2A and the H-bond interaction of S2425.46 together may cause a slight rotation and leftward shift of the ergoline ring. (Figure 4G). In the LSD-HTR2A structure, LSD forms a hydrogen bond between the indole-NH and side chain of S2425.46, and we next preformed additional experiments to gain insight into this interaction. We found that mutation of S2425.46 to an alanine does not significantly change the binding affinity of LSD, although this mutation accelerates its dissociation rate, implying that this unique residue may contribute to LSD’s unusually long binding kinetics (Figure 4F). As expected, because the S2425.46 does not directly interact with 25CN-NBOH, the S242A5.46 mutation has no effect on 25CN-NBOH’s potency or efficacy in BRET (Figure S6B; Table S5).
Guided by this structure, we also evaluated potential binding modes of the endogenous agonist 5-HT using molecular docking (Coleman et al., 2013). In the docked conformation (Figure S6D), the positively charged primary amine of serotonin forms a salt-bridge with the anionic D1553.32 (distance 2.8 Å ) as predicted by mutagenesis studies (Kristiansen et al., 2000). In further agreement with many prior mutagenesis and molecular modeling studies (Kristiansen et al., 2000; Roth et al., 1997; Braden and Nichols, 2007) 5-HT is also predicted to be stabilized by aromatic interactions with F3406.52 and F3396.51 and hydrogen bonds with N3436.55 and S2425.46. N3436.55 was previously implicated from molecular modeling studies (Kristiansen et al., 2000) and was here found to be essential for 5-HT’s agonist potency (Figure S5B; Table S5). As predicted from the HTR2A-LSD structure, the N3436.55A mutation did not affect LSD’s agonist potency, efficacy, or binding affinity (Figures S5A and S5B).
The Agonist-Gαq Complex Binding Interface
A major interface between HTR2A and the Gαq subunit is mediated by the C-terminal helix (α5 helix) of Gαq as suggested by G protein-bound structures (Kato et al., 2019; Krishna Kumar et al., 2019; Maeda et al., 2019). In detail, HTR2A residues N1072.37, D1723.49, N3176.29, and N3848.47 form H-bonds with Gαq residues E242H5.22, Y243H5.23, Q237H5.17, and N244H5.24, respectively (superscription is CGN numbering system) (Flock et al., 2015). Additionally, the residues A3216.33, L2615.65, I1773.54, L3256.37, and V3246.36 of HTR2A form a hydrophobic core with L236H5.16, L240H5.20, and L245H5.25 of Gαq (Figure 5A). Considering these residues form the major interactions between HTR2A and Gαq, alanine mutagenesis studies were conducted, and BRET2 assays were performed to determine which might be key for Gαq recognition. Measurement of receptor expression levels showed that these mutants, for both receptors and Gαq proteins, generally maintain robust whole cell (Figure S5E) and receptor cell surface (Figure S5F) expression, although some mutations slightly decreased receptor expression levels compared to wild-type (Figures S5E and S5F). Among the HTR2A mutations, N384A8.47 and L325A6.37 dramatically reduced receptor-Gαq coupling compared to wild-type, whereas R185AICL2 and N107A2.37 decreased the efficacy of 25CN-NBOH by 50%, indicating that these residues probably play a crucial role in G protein signaling (Figure 5B). Among the Gαq mutations, the agonist activity of 25CN-NBOH was completely abolished by Q237AH5.17 (Figure 5C). Q237H5.17 is a highly conserved residue in the Gαq family (Lys in Gαi/o family and Arg in Gαs family), indicating that this residue may be involved in Gαq specificity (Figure S7D). N244H5.24 is also a conserved residue existing only in the Gαq family (Gly in Gαi/o, Met in G12/13, and Glu in Gαs family), and expectedly, the N244AH5.24 mutant significantly decreases both the potency and efficacy of 25CN-NBOH (Figures 5A and 5C), indicating that it may also contribute to receptor-Gαq coupling.
Figure 5. The Interface between the HTR2A and Gαq Protein and Confirmation of Its Functional Relevance.

(A) The close up view of interaction of HTR2A (green color) and H5 helix (olive color) of Gαq protein. All residues involved in interaction show stick model. Hydrogen bond interactions are highlighted by the red dashed line.
(B) The detailed view of interaction of ICL2 of HTR2A and Gαq and showing I181ICL2 of HTR2A and its surrounded hydrophobic residues of Gαq.
(C) BRET validation of residues in the HTR2A interface. See Table S7 for fitted parameter values.
(D) BRET validation of residues in the Gαq interface. See Table S7 for fitted parameter values where data represent mean ± SEM of n = 3 biological replicates.
See also Table S5.
An unexpected interaction was exemplified by R185ICL2 that undergoes a dramatic ~13 Å rearrangement from being displaced upward toward the helical core to a close engagement via backbone interactions with R32 near the bottom of the α5 helix (Figures 5B and 7H). The essential nature of this interaction was demonstrated by the R185AICL2 mutation that significantly attenuated agonist potency (Figure 5C; Table S7). In a similar manner, N3848,47 transitions toward the α5 helix with an ~3 Å movement where it engages N244H5.24 near the tip of the α5 helix (Figure 7G).
Figure 7. Mutations of the ICL2 Residue I188 Differentially Modulate Gαq and Arrestin Interactions at HTR2A.

(A) BRET2 HTR2A-Gαq assays reveal that I881A/E ICL2/34.51 mutations abolish 25CN-NBOH-potentiated Gαq activation; data represent mean ± SEM of n = 3 biological replicates. See Table S5 for fitted parameters.
(B) BRET2 HTR2A-Gαq assays reveal that I881A/E ICL2/34.51 mutations abolished LSD-potentiated Gαq activation; data represent mean ± SEM of n = 3 biological replicates. See Table S5 for fitted parameters.
(C) BRET2 HTR2A-Gαq assays reveal that I881A/E ICL2/34.51 mutations attenuate 5-HT-potentiated Gαq activation; data represent mean ± SEM of n = 3 biological replicates. See Table S5 for fitted parameters.
(D–F) BRET1 HTR2A-βArr2 translocation assays show that I881A/E ICL2,34.51 mutations enhance the efficacy of 25CN-NBOH (D), LSD (E), and 5-HT (F); data represent mean ± SEM of n = 3 biological replicates. See Table S5 for fitted parameters.
(G) The structural comparison in TM7 of active- (HTR2A-Gq, green and olive color, respectively) and inactive- (pink color) structures of HTR2A.
(H) The structural comparison in ICL2 of active- (HTR2A-Gq, green and olive color, respectively) and inactive- (pink color) structures of HTR2A.
(I) The structural comparison in ICL234.51 residue of β2AR-Gs (light green and gray, respectively, PDB: 3SN6) and β1AR-βarr1 (brown and lime, respectively, PDB: 6TKO).
(J) The structural comparison in ICL234.51 residue of NT1- βarr1 (light blue and sky blue color, respectively, PDB: 6UP7) and M2R-βarr1 (magenta and yellow, respectively, PDB: 6U1N).
(K) A diagram showing how ICL2E34.51 differentially affects HTR2A-mediated G protein signaling and arrestin translocation.
We next compared the Gq interface with the recent M1-muscarinic G11 interface to explore features responsible for Gαq subunit specificity. We found that the terminal hydroxyl of Y243, which is comparable to Y356 on the extreme terminus of the α5 helix and that has been implicated in receptor-G protein selectivity, was shifted 2.9 Å to create additional interactions with HTR2A that are not present in the M1 muscarinic receptor (Figure 6A). Additionally the terminal N-L-V motif, which is conserved among all Gq-like Gα subunits, was displaced downward to achieve a previously unreported interacting surface with the terminus of TM7 of M1 (Figure 5A). By contrast, in the Gq-HTR2A structure, the terminal V246 (that corresponds to V359 in M1-G11) is shifted upward 4.8 Å to avoid a potential clash with TM7 (Figure 5A). Overall, this illustrates a striking rearrangement of the key terminal residues in HTR2A-Gq versus M1-G11 to afford productive interactions with the core of the respective Gq/11-coupled GPCRs (Figure 5A). These results indicate that although the motifs used by different Gq-family subunits to afford receptor selectivity might be conserved, the interactions to mediate this selectivity are apparently distinct.
Figure 6. HTR2A Couples Efficiently to Gq-Family Members In Vitro.

(A)Shown is an alignment of the HTR2A-Gq (green and olive, respectively) and the M1-G11 (lavender and pink, respectively) interface at the tip of the α5 helix of the respective Gα subunits. As depicted, V359H5.26 unde rgoes a 4.8 Å shift in HTR2A compared with M1 while Y356H5.23 is shifted 2.9 Å relative to M1.
(B) Shows a comparison of HTR2A-Gq with the HTR1B-Go (yellow and gray, respectively) showing that the HTR2A Y354H5.26 cognate residue V246H5.26 is shifted up 10.9 Å.
(C) Shows a heatmap of the relative efficacy for selected agonists at HTR2A versus a reference agonist for 14 distinct Gα subunits.
(D)Shows concentration-response curves (N = 3 biological replicates each) for the 14 Gα subunits in 7C along with controls for the Gz study.
See also Table S7.
Recent studies performed with chimeric G proteins have indicated that GPCRs may interact with a much larger diversity of Gα subunits than previously anticipated (Inoue et al., 2019). These authors (Inoue et al., 2019) provided data indicating that HTR2A can activate not only Gq-family Gα subunits but also several members of the Gi (Gαi1, Gαi3, GαO, and Gαz) and, minimally, Gαs family members (albeit with low relative intrinsic activity). Because these results are surprising, given the specific and non-conserved interactions we visualized in our 5-HT2A-Gq structure, we compared the structures of another 5-HT receptor in complex with a Gi-family member—5-HT1B-Gαo (García-Nafría et al., 2018). A comparison of the two structures revealed that the terminal Y354 of Gαo is displaced 10 Å down from the terminal V246 of Gαq where it cannot interact with any known residues of 5-HT2A (Figure 6B). As well, the interaction surface formed by the final 5 residues of the α5 helix of Gαo are not expected to form productive interactions with any of the closest residues in either TM7 or TM6 (Figure 5B) in HTR2A.
These results suggested to us that it was unlikely, based on an examination of these two structures, that HTR2A would productively interact with Gαo or related Gαi proteins. To test this hypothesis, we quantified the ability of HTR2A to productively interact with 14 distinct Gα subunits using our recently described BRET-based technology (Olsen et al., 2020). As shown in Figures 6C and 6D, HTR2A coupled robustly to Gq-family members and minimally to conventional Gi- or Gs-family members. A diminished but reproducible response was seen for the pertussis-toxin-insensitive Gz. We verified the Gz interactions by showing a lack of response to the agonists in the presence of the HTR2A-selective antagonist M100907 (10 μM) and a lack of response in cells not transfected with HTR2A plasmid (Figure 6D).
Residue I181(ICL2)34.51 is highly conserved among the GPCR family and has been reported to play a crucial role in Gαs or Gαq coupling, but not Gαi/o (Moro et al., 1993). In the HTR2A/Gαq structure, I18134.51 interacts with L34S1.02, V79S3.01, F228H5.08, and I235H5.15 of Gαq via hydrophobic interactions (Figure 5B). We found that the I181A34.51 and I181E34.51 mutations completely abolished the Gαq activation by 25CN-NBOH, while potentiating its activity in arrestin recruitment (Figures 7A and 7D). As shown in Figures 7B and 7C, the I181A34.51 and I181E34.51 mutations similarly either attenuated (Figure 7B) or abolished (Figure 7C) the ability of LSD and 5-HT respectively, to activate Gαq (see Table S5 for fitted parameters).
Finally, we observed that when most Family A active state structures are superimposed, the phenolic hydroxyl group of Y5.58 is located in the inner core of TM6. It has been reported that Y5.58 forms a water mediated-hydrogen bond network with R3.50 (the middle part of the E/DRY motif) and Y7.53 (the latter part of the NPXXY motif) as previously suggested (Weis and Kobilka, 2018). By contrast, in the case of HTR2A, HTR2B, and HTR2C, the phenolic hydroxyl of Y5.58 is displaced outward from TM6 (Figure S7A). Y5.58 apparently interacts with F6.41 by π-stacking interactions (Figure S7B). The sequence alignment of all aminergic receptors shows that F6.41 in the HTR2s is highly conserved (Figure S7C), and this represents one of the possible reasons for the observed outward orientation of Y5.58 in TM6.
DISCUSSION
In this paper, we determined the agonist-activated structure of the 5-HT2A serotonin receptor coupled to its canonical transducer Gαq, compared it with inactive state structures stabilized by both the partial agonist LSD and the inverse agonist methiothepin, and identified key determinants essential for agonist actions and receptor-Gq coupling. Because the 5-HT2A receptor is essential for the actions of classical psychedelics including LSD, psilocin, mescaline, and various N-BOH analogs, these studies provide structural insights into the actions of psychedelics at their molecular target. Importantly, we also discovered that the specific interactions the 5-HT2A receptor achieves with Gq residues are essential for the apparently selective engagement of this receptor with Gq-family proteins in vitro.
Some of these findings contrast with predictions from a recent study utilizing chimeric Gα subunits that indicated that HTR2A interact efficiently with all of the 11 tested Gα subunits, albeit with low efficacy at Gas (Inoue et al., 2019). However, the Gα subtypes in that study (Inoue et al., 2019) were created by replacing the terminal seven amino acids of Gq with the corresponding sequences from several other Gα subunits using the motif LXXXLX (where X = subtype selective sequences) (Inoue et al., 2019). Pertinent to this, we note that immediately prior to L353 is Q352 that is conserved among all of the chimeric G proteins employed in the study by Inoue et al. (2019). Here, we discovered that the Q352 cognate residue directly interacts with HTR2A TM6 residue N3176.29, and mutation of this residue abolishes the ability of HTR2A to activate Gq. We also point out that in the only other Gq-GPCR structure available, the same Q352 interacts directly with the TM5 residue R5.46 (Maeda et al., 2019). Importantly, using assays in which full-length heterotrimeric G proteins were used, we find that HTR2A productively couples weakly with only one Gi-family member, Gαz.
Relevant to these findings, we also found important interactions outside the α5 helix that are essential for HTR2A-Gq interactions. Thus, R132ICL2 interacts via an extended backbone interaction with N32 of the α-helical domain, and we further demonstrated via mutagenesis and functional assays that this interaction is essential for agonist-induced activation of Gq. This GPCR-Gq interaction is not seen in the M1-G11 structure, although R134ICL2 in M1 could weakly interact with the backbone carbonyl of R32 in Gq based on an examination of that prior structure. These results underscore suggestions made decades ago that the selectivity determinants for Gα subunits may not be entirely specified by reside in their terminal 5–7 amino acids (Wess, 1997).
We also discovered that a key hydrophobic residue essential for G protein coupling among various GPCRs—I181ICL2— when mutated, abolishes Gαq coupling while potentiating arrestin binding at HTR2A. This result was unexpected because there was no prior data with sufficiently high resolution of GPCR-arrestin complexes that could illuminate how the mutation of a single amino acid at the GPCR-Gα interface would switch the coupling of HTR2A from an unbiased to an arrestin-exclusive state. While this paper was in revision, however, a 3.3 Å resolution structure of the turkey β1-adrenergic receptor complexed with human β-arrestin1 was reported (Lee et al., 2020). A comparison of this structure with the Gs-coupled β2-adrenergic receptor (Rasmussen et al., 2011) and our HTR2A-Gq structure reveals that the cognate hydrophobic residue F139ICL2 in β2-AR interacts with the Gs α5-helix in a manner similar to HTR2A-Gq. In the β1 adrenergic receptor-Arrestin structure, by contrast, F147ICL2 is displaced upward where it would potentially clash with residues in the α5 helix of Gs (Figure 7I) and where it is engaged in no productive interactions. A similar relative lack of extensive engagement of this cognate residue with arrestins can be seen in the lower resolution muscarinic- and neurotensin-receptor arrestin complexes (Huang et al., 2020; Staus et al., 2020) (Figure 7J) This analysis provides a potential structural explanation for how loss of this hydrophobic interaction impairs Gq subunit coupling while preserving arrestin interaction—at least for HTR2A.
In addition to these fundamental insights into GPCR-Gq interactions, our findings have relevance for neuropsychiatric drug discovery. Psilocybin and LSD have emerged as potential therapeutics for a number of neuropsychiatric conditions including depression, anxiety, substance abuse, and cluster headaches (Carhart-Harris et al., 2016; Bogenschutz, 2013; Gasser et al., 2015; Sewell et al., 2006; Nutt et al., 2020). Because the 5-HT2A receptor is essential for the actions of psilocybin in humans (Kometer et al., 2013; Nutt et al., 2020), insights into the molecular details of both HTR2A ligand recognition and HTR2A-effector coupling are key to arriving at a molecular understanding of hallucinogen actions. Additionally, these studies will provide a framework for a structure-guided search to identify more selective and efficacious HTR2A agonists as potential innovative neuropsychiatric therapeutics as recently exemplified by us for other GPCRs (Lyu et al., 2019; Wang et al., 2017; Stein et al., 2020).
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact,Bryan Roth (bryan_roth@med.unc.edu)
Materials Availability
Plasmids generated from this study may be obtained directly from the Lead Contact.
Data and Code Availability
The following structure files are available from the Protein Databank: 6WHA, 6WGT and 6WH4.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The following eukaryotic cell lines were used: Spodoptera frugiperda (Sf9) cells and HEK293T cells. The HEK293T cells were obtained directly from the American Type Culture Collection (CRL-11268) and have been validated by analysis of short tandem repeat (STR) DNA profiles and these profiles showed 100% match at the STR database from ATCC. The Sf9 cells were obtained as Sf-900 II SFM cells from Invitrogen and were not validated further.
HEKT cells were grown in a humidified 37°C incubator with 5% CO2 using media supplemented with 100 I.U./mL penicillin and 100 μg/mL streptomycin (Invitrogen, Cat#15140-122). The human cell lines HEK293T were maintained in DMEM (VWR, #45000) containing 10% fetal bovine serum (FBS, VWR, #89510-186). At 24–48 hr prior to studies, HEKT cells were cultured in the above media containing 1% dialyzed fetal bovine serum (Omega Scientific, #FB-03) instead of 10% fetal bovine serum to remove serotonin. For expression studies, Sf9 cells were grown at 27 C at a cell density of 2 × 106 cells/ml in ESF921 medium (Expression systems) with P1 virus at a multiplicity of infection (MOI) of 3.
METHOD DETAILS
Generation of HTR2A constructs for X-ray crystallography and cryoEM
For crystallization, the modified thermostabilized apocytochrome b562RIL (BRIL) as a fusion partner was inserted into the receptor’s third intracellular loop (ICL3) at A265 and T311 of the human 5-HT2A gene. The construct was further optimized by truncation of N-terminal residues 1–65 and C-terminal residues 405–471. The ΔN65–5HT2A-BRIL-ΔC405 DNA was subcloned into a modified pFASTBac1 vector for expression in Spodoptera frugiperda (Sf9) cells. All constructs also contained a hemagglutinin (HA) signal sequence followed by a FLAG tag at the N terminus and a PresCission protease site followed by a 10X histidine tag (His tag) at the C terminus to enable purification by immobilized metal affinity chromatography. To increase the thermostability and homogeneity of the 5-HT2A, two point mutations, Leu247Ala and Leu371Ala, were introduced in by standard Quick Change PCR (Figure S2A). For the cryoEM study, wild-type human HTR2A was truncated in N-terminal residues 1–65 and C-terminal residues 405–471 to enhance expression levels, and a haemagglutinin (HA) signal sequence followed by FLAG-, His10-tag, TEV protease site, BRIL, HRV3C and linker “GSGSG” at the N terminus were indroduced (Figure S1A). This construct also was subcloned into a modified pFASTBac1 vector.
Expression for HTR2A-XTAL and -cryoEM constructs
The Bac-to-Bac Baculovirus Expression System (Invitrogen) was used to generate high-titer recombinant baculovirus (> 109 viral particles per ml). Recombinant baculovirus was obtained by transfecting ~5 μg of recombinant bacmid into 5×105 per well settled Spodoptera frugiperda (Sf9) in a 12-well plate (Corning) using 3 μL of Cellfectin II reagent (Invitrogen). After 5–12h, medium was exchanged for 1 mL of Sf-900 II SFM medium (Invitrogen), and the plates were incubated for 4–6d at 27°C. P0 viral stock was harvested as the supernatant and used to generate high-titer baculovirus stock by infection of 40 mL of 3×106 Sf9 cells/ml and incubation for 3d. Viral titers were determined by flow cytometry analysis of cells that were immobilized with phycoerythrin (PE)-conjugated gp64 antibody (Expression Systems) (Hanson et al., 2007). Expression of 5-HT2A was carried out by infection of sf9 cells at a cell density of 2 × 106 cells/ml in ESF921 medium (Expression systems) with P1 virus at a multiplicity of infection (MOI) of 3. Cells were harvested by centrifugation at 48 h after infection, washed in TN buffer (20 mM Tris-Cl, 100 mM NaCl, pH7.5) and stored at −80°C until use.
Purification for HTR2A-XTAL and -cryoEM
Thawed insect cell membranes were disrupted in a hypotonic buffer containing 10 mM HEPES (pH7.5), 10 mM MgCl2, and 20 mM KCl and protease inhibitors containing 500 μM AEBSF, 1 μM E-64, 1 μM Leupeptin and 0.15 μM Aprotinin. Subsequently, soluble and membrane associated proteins were removed in a high osmotic buffer containing 10 mM HEPES (pH7.5), 1,000 mM NaCl, 10 mM MgCl2, and 20 mM KCl. Purified membranes were incubated in the presence of desired ligands (50 μM 25CNNBOH, LSD or methiothepin) and protease inhibitor cocktail at 4°C for 2 h. The membranes were incubated with 2.0 mg/ml iodoacetamide (Sigma) for 30 min and were solubilized in the buffer containing 50 mM HEPES (pH 7.5), 1% (w/v) n-dodecyl-beta-D-maltopyranoside (DDM, Anatrace), 0.2% (w/v) cholesterol hemisuccnate (CHS, Sigma) and 150 mM NaCl, at 4°C for 2h. The solubilized HTR2A proteins in the supernatants were isolated by ultra-centrifugation in 40,000 rpm at 4°C for 50 min, and then incubated at 4°C overnight with TALON IMAC resin (Clontech), 800 mM NaCl and 20 mM imidazole as the final buffer concentration. The resin was washed with 10 column volumes of washing buffer I containing 50 mM HEPES (pH 7.5), 0.1% (w/v) DDM, 0.02% (w/v) CHS, 800 mM NaCl, 10% (v/v) glycerol, 20 mM imidazole and 50 μM of desired ligands (25CN-NBOH, LSD or methiothepin) and 10 column volumes of washing buffer II 50 mM HEPES (pH 7.5), 0.05% (w/v) DDM, 0.01% (w/v) CHS, 500 mM NaCl, 10% (v/v) glycerol and 50 μM of desired ligands (50 μM 25CN-NBOH, LSD or methiothepin) without imidazole. The protein was eluted using 3 column volumes of elution buffer containing 50 mM HEPES (pH 7.5), 0.05% (w/v) DDM, 0.01% (w/v) CHS), 500 mM NaCl, 10% (v/v) glycerol, 250 mM imidazole and 50 μM of desired ligands (25CN-NBOH, LSD or methiothepin) and concentrated in a Vivaspin 20 concentrator with a molecular weight cutoff of 100 kDa (Satorius Stedim) to 500 μl. The followed methods differed with crystal and CryoEM construct purifications. For the crystal construct purification, the 500 μL of 5HT2A protein sample was applied to PD MiniTrap G-25 columns (GE Healthcare) to remove imidazole. The C-terminal 10 × His tag was removed by addition of His-tagged PreScission protease (GeneScript) and incubation overnight at 4°C. Protease, cleaved His tag and uncleaved protein were trapped by equilibrated TALON IMAC resin (Clontech) and collecting the flow-through. 5-HT2A/LSD or methiothepin complexes were then concentrated to ~30 mg/ml using a Vivaspin 500 centrifuge concentrator with a molecular weight cutoff of 100 kDa (Sartorius Stedim). Protein purity and monodispersity were tested by analytical size-exclusion chromatography column, SRT-300 (Sepax scientific) and Ultimate 3000 UHPLC systems (Thermo Scientific). For the CryoEM construct purification, the 500 μL of 5HT2A protein sample was applied to PD MiniTrap G-25 columns (GE Healthcare) to remove imidazole with buffer containing 20 mM HEPES (pH 7.5), 100 mM NaCl, 0.5% (w/v) LMNG, 0.05% (w/v) CHS, 0.00025% (w/v) GDN, 100 uM TCEP, and 50 μM 25CN-NBOH. The N-terminal BRIL was removed by addition of His-tagged PreScission protease (GeneScript) and incubation overnight at 4°C. Protease, cleaved BRIL and uncleaved protein were trapped by equilibrated TALON IMAC resin (Clontech) and the flow-through was collected. The BRIL removed HTR2A was further purified by size exclusion chromatography on Superdex 200 10/300 gel filtration column (GE) with SEC buffer containing 20 mM HEPES (pH7.5), 100 mM NaCl, 0.001% (w/v) LMNG, 0.0001% (w/v) CHS, 0.00025% (w/v) GDN, 100 uM TCEP, 1 mM MgCl2 and 10 uM GDP, and collected until use.
Generation of heterotrimeric mini-Gαq protein complex
The mini-GαqiN chimeric construct was designed for the binding of scFv16 and sub-cloned into a designed vector that help form heterotrimeric G complex in situ (Figure S1). For the expression, Sf9 insect cells were infected with one virus, encoding three subunits including the mini-GαqiN subunit and Gβ1/γ2 subunits with histidine tag inserted at the amino terminus of the β subunit. Cells expressing the heterotrimeric G-protein were harvested 72 hours post infection. Cells were lysed in lysis buffer containing 20 mM HEPES (pH 7.5), 100 mM NaCl, 30 mM imidazole, 5 mM β-mercaptoethanol, 0.1 mM GDP, 1 mM MgCl2, 0.2 % (v/v) Triton X-100 and protease inhibitors, and the soluble fraction was isolated by ultra-centrifugation at 40,000 rpm at 4°C for 50 min. The heterotrimer containing soluble fraction was purified using Ni-NTA chromatography. Human Rhinovirus 3C protease (HRV3C protease) was added and the histidine tag was cleaved at 4° C for overnight. The histidine tag removed heterotrimeric G protein was further purified by size exclusion chromatography on a Superdex 200 10/300 gel filtration column (GE) with SEC buffer containing 20 mM HEPES (pH 7.5), 100 mM NaCl, 0.001% (w/v) LMNG, 0.0001% (w/v) CHS, 0.00025% (w/v) GDN, 100 uM TCEP, 1 mM MgCl2, and 10 uM GDP, and collected and concentrated to ~25 mg/ml, and stored at −80°C until use.
Formation of HTR2A/mini-Gαq heterotrimer and scFv16
ScF16 was expressed from Spodoptera frugiperda (Sf9) insect cells as a secreted protein using baculovirus infection system and the purification process exactly as previously reported (Maeda et al., 2018). Media expressing scFv16 from Spodoptera frugiperda (Sf9) was pH balanced to pH 8.0 by addition of Tris powder. Chelating agents were quenched by addition of 1 mM nickel and 5 mM calcium chloride and incubation with stirring for 1 hour at 25°C. The precipitants were removed by centrifugation at 16,263 g for 30 min and supernatant was incubated with His60 Ni Superflow Resin (Takara) for 5 hours. The resin was loaded over Poly-Prep Chromatography column (Bio-Rad) and washed with the washing buffer (20 mM HEPES pH7.5, 100 mM NaCl and 20 mM Imidazole). The protein was eluted with the elution buffer (20 mM HEPES pH 7.5, 100 mM NaCl and 300 mM Imidazole) and treated the HRC-3C protease to cleave the carboxy-terminal octa-histidine tag. Cleaved protein was further purified by size exclusion chromatography using a Superdex 200 16/60 column (GE healthcare). Monomeric fractions were pooled, concentrated, flash frozen in liquid nitrogen and stored at −80°C freezer until use. Purified scFv16 was concentrated, and flash frozen in liquid nitrogen until further use. Purified HTR2A/25CN-NBOH was mixed with a 1.2 molar excess of mini-GαqiN heterotrimer. The coupling reaction was allowed to proceed at 24°C for 1 hr and was followed by addition of 0.2 U/ml (final concentration) of apyrase to catalyze hydrolysis of unbound GDP. After one hour, a 1.5 molar excess of scFv16 was added to the HTR2A /mini-Gαq mixture and incubated overnight at 4°C. HTR2A/mini-GαqiN heterotrimer/scFv16 complex was further purified by size exclusion chromatography on a Superdex 200 10/300 column in 20 mM HEPES (pH 7.5), 100 mM NaCl, 0.001% (w/v) LMNG, 0.0001% (w/v) CHS, 0.00025% (w/v) GDN, 100 uM TCEP, 1 mM MgCl2, and 10 μM GDP. Peak fractions were concentrated to ~17 mg/ml for electron microscopy studies.
CryoEM Data Collection and 3D Reconstruction
The sample (3.5 μL) was applied at a concentration of 17 mg/mL to glow-discharged holey carbon grids (Quantifoil R1.2/1.3) and vitrified using a Vitrobot Mark IV (FEI) at 22°C and 100% humidity, and plunged frozen into liquid ethane. CryoEM imaging was performed on a Titan Krios (ThermoFisher) electron microscope operated at 300 kV with a K3 Summit direct electron detector (Gatan) at a magnification of 57,050x in counting mode. 2368 movies of 50 frames each (4 s exposure) were obtained at a total dose of 82 electrons/Å2 and defocus ranging from −0.8 to −2.1 μm. Dose-fractionated image stacks were subjected to beam-induced motion correction and dose-weighting using MotionCor2. Contrast transfer function parameters for corrected micrographs were determined by Gctf (Zhang, 2016). A total of 2.7 million particles were extracted from 2368 micrographs using semi-automated particle selection. 2D and 3D classification rounds were performed on a binned dataset (pixel size 1.704Å ) using Relion 3.0 (Scheres, 2012). A subset of 168,570 particles were selected for the final map. The unbinned (with pixel size of 0.8421Å ) particle set was subjected to Ctf Refinement and two rounds of Bayesian Polishing before the final refinement and sharpening was applied.
The final map was postprocessed in Relion with a temperature factor of −90 Å 2. The resolution of the final map at the 0.143 FSC threshold was estimated to be 3.27 Å by Relion and 3.23 Å using Mtriage in PHENIX (Table S1).
Model building and refinement
Homology models of active-state HTR2A were built by SWISS-MODEL (Guex and Peitsch, 1997) using HTR2C (PDB code 6BQG) (Peng et al., 2018) for the receptor and M1R/G11 (PDB code 6OIJ) (Maeda et al., 2019) for the G-protein as template models, respectively. All models were docked into the EM density map using Chimera (Pettersen et al., 2004) followed by iterative manual adjustment in COOT (Emsley and Cowtan, 2004) and phenix.real_space_refine in Phenix (Adams et al., 2010). The model statistics was validated using Molprobity (Chen et al., 2010). Structural figures were prepared by Chimera or Pymol (https://pymol.org/2). The final refinement statistics were provided in Table S1.
Lipidic Cubic Phase Crystallization
The purified 5-HT2A protein in complex with LSD and methiothepin was screened for crystallization in lipidic cubic phase (LCP) with mixed molten lipid (90% (w/v) monoolein and 10% (w/v) cholesterol) at a protein:lipid ratio of 1:1.5 (v/v) using a mechanical syringe mixer (Caffrey and Cherezov, 2009). Crystallization was done on 96-well glass sandwich plates (Marienfeld) in 50 nL LCP drops that were dispersed from a 10 μL gas-tight pipette (Hamilton) using a handheld dispenser (Art Robbins Instruments) and overlaid with 1 μL of precipitant solution. After optimization, crystals were obtained in 100 mM Tris-HCl (pH 7.0), 380–430 mM potassium phosphate monobasic, 27%–32% (v/v) polyethylene glycol 400 (PEG400), 100 mM guanidine hydrochloride and 300 mM NDSB-195. Crystals grew to a maximum size of ~30 μm × 20 μm × 20 μm within 2 weeks and were harvested directly from the LCP plates using MiTeGen micromounts and storage in liquid nitrogen.
Data Collection and Structure Determination
X-ray diffraction data of 5-HT2A/LSD and methiothepin crystals were collected at beam line 23ID-B (GM/CA CAT) of the Advanced Photon Source (Argonne, IL, USA) using a 10-μm minibeam at a wavelength of 1.0330 Å and an Eiger-16 m detector (Dectris) and beam line 17ID-2 (FMX) of the NSLS-II (Brookhaven, NY, USA). The data collection strategy was set up as an exposing the crystals for 0.2 s to an unattenuated beam using 0.2° oscillation. Diffraction images were indexed, integrated and scaled, and merged using XDS (Kabsch, 2010a, 2010b) and further scaled using AIMLESS (Evans, 2006). Initial phase was obtained by molecular replacement (MR) method with Phaser (McCoy, 2007; McCoy et al., 2007) using the receptor and BRIL portion of 5-HT2B/LSD (PDB: 5TVN) as independent search models (Wacker et al., 2017b). Refinement was carried out with Phenix (Adams et al., 2010) and REFMAC (Murshudov et al., 1997) followed by manual examination and adjustments of the refined structures in the program COOT (Emsley and Cowtan, 2004) with both 2|Fo|-|Fc| and |Fo|-|Fc| maps. After refinement, we did not observe any Ramachandran outliers in any of the structure: 95.98% and 4.02% for LSD, and 93.73% and 6.27% for methiothepin of favored and allowed regions, respectively, as defined by Ramachandran statistics. We further observed Molprobity scores of 2.04 for LSD, and 2.1 for methiothepin (Table S2).
Radioligand binding assays
Competitive binding assays were performed using membrane preparations from HEK293 T cells transiently expressing HTR2A wt or mutants. Binding assays were set up in 96-well plates in the standard binding buffer (50 μM Tris, 0.1 mM EDTA, 10 mM MgCl2, 0.1% BSA, 0.01% ascorbic acid, pH 7.40). Saturation binding assays with 0.1–20 nM [3H]-LSD in standard binding buffer were performed to determine equilibrium dissociation constant (Kd) and Bmax, whereas 10 μM final concentration of LSD was used to define nonspecific binding. For the competition binding, 50 μL each of 3H-LSD (final 0.6 nM), drug solution (3X) and homogeneous HTR2A membrane solution was incubated in 96-well plates in the standard binding buffer. Reactions (either saturation or competition binding) were incubated for 2 h at room temperature in the dark and terminated by rapid vacuum filtration onto chilled 0.3% PEI-soaked GF/A filters followed by three quick washes with cold washing buffer (50 mM Tris HCl, pH 7.40) and read. Results were analyzed using the equation ‘one-site fit Ki’ in GraphPad Prism 8.0.
Molecular docking of 5-HT
The agonist 5-HT was docked into the active HTR2A structure bound to 25-CN-NBOH (chain A, current work) using DOCK3.7 (Coleman et al., 2013). During the docking, 30280 complexes were sampled at the orthosteric binding site. Each pose was scored against three pre-calculated enegy grids which represent AMBER van der Waals potential (Meng et al., 1992), Poisson–Boltzmann electrostatic potential by QNIFFTT (Sharp et al., 1995) (Gallagher and Sharp, 1998), and ligand desolvation (Mysinger and Shoichet, 2010), respectively. The best scoring 5-HT conformation received a docking score of −37.5 kcal/mol. The 3D dockable db2 file of 5-HT was downloaded from ZINC15 (http://zinc15.docking.org/substances/ZINC000000057058/).
Molecular docking of 25-CN-NBOH
The 3D dockable db2 file of 25-CN-NBOH was generated using our standard protocol (http://wiki.docking.org/index.php/Ligand_preparation_-_2017-04). The protonation states at pH 7.4 (Jchem version 15.11.23.0, ChemAxon, 2015; https://chemaxon.com), 3D structures (Corina 4.2 https://www.mn-am.com/products/corina), conformer ensembles (omega v.2.5.1.4, https://www.eyesopen.com/omega), partial charges and desolvation energies (AMSOL version 7.1 https://comp.chem.umn.edu/amsol/) were calculated as previously described (Sterling and Irwin, 2015). The docking grids used were identical to those used for the 5-HT docking as follows. All steps in the standard protocol are identical to the steps in the ZINC15 pipeline (Sterling and Irwin, 2015): Jchem (version 15.11.23.0, ChemAxon, 2015; https://chemaxon.com) is used to enumerate the protonation states at pH 7.4 from an input SMILES. Each protonated SMILES is used to produce an initial 3D structure by Corina (version 4.2 https://www.mn-am.com/products/corina). Based on the 3D conformer, AMSOL (version 7.1 https://comp.chem.umn.edu/amsol/) is used to compute partial charges and desolvation energies. OMEGA (version 2.5.1.4, https://www.eyesopen.com/omega) is used to generate conformer ensembles. At the final step, mol2db2 compresses all the information calculated above into a dockable db2 file. Over 7,500 orientations were explored in the orthosteric site, resulting 2,723,040 complexes sampled during the docking (orientations × conformations). The best scoring pose received a favorable docking score of −44.2 kcal/mol. The ligand symmetry accounted r.m.s.d (0.16Å) between the docked pose and the cryo-EM pose is calculated by the Hungarian algorithm in DOCK6 (Allen and Rizzo, 2014).
Ligand Dissociation Radionligand Binding Assay
Radioligand dissociation assays were performed in parallel with the competitive binding assays utilizing the same concentrations of radioligand, membrane preparations, and binding buffer (50 mM Tris, 10 mM MgCl2, 0.1 mM EDTA, 0.1% BSA, 0.01% ascorbic acid, pH 7.4). All assays utilized at least two concentrations of radioligand ([3H]-LSD = 0.4 and 1.2 nM) (PerkinElmer). For dissociation assays, membranes were incubated with radioligand for at least two hours at room temperature before the addition of 10 μL of 10 μM excess cold ligand LSD to the 200 μL membrane suspension at designated time points. Time points spanned 2 minutes to 7 hours. Immediately at time = 0 min, plates were harvested by vacuum filtration onto 0.3% polyethyleneimine pre-soaked 96-well filter mats (Perkin Elmer) using a 96-well Filtermate harvester, followed by three washes of cold wash buffer (50 mM Tris pH 7.4). Scintillation (Meltilex) cocktail (Perkin Elmer) was melted onto dried filters and radioactivity was counted using a Wallac Trilux MicroBeta counter (PerkinElmer). Data were analyzed using “Dissociation – One phase exponential decay” in Graphpad Prism 8.0.
Bioluminescence resonance energy transfer assays (BRET)
To measure HTR2A-mediated β-arrestin2 recruitment, HEK293T cells were co-transfected in a 1:5 ratio with human 5-HT2AR containing C-terminal Renilla luciferase (RLuc8), and Venus-tagged N-terminal β-arrestin2. After at least 16 hours, transfected cells were plated in poly-lysine coated 96-well white clear bottom cell culture plates in plating media (DMEM + 1% dialyzed FBS) at a density of 25–50,000 cells in 200 μl per well and incubated overnight. The next day, media was decanted and cells were washed twice with 60 μL of drug buffer (1 × HBSS, 20 mM HEPES, 0.1% BSA, 0.01% ascorbic acid, pH 7.4), then 60 mL of drug buffer was added per well. For kinetic experiments, plates were incubated at 37°C at least 20 minutes prior to receiving drug stimulation. Afterward, 30 μL of drug (3X) was added per well and incubated for designated time points. Before reading, 10 μL of the RLuc substrate, coelenterazine h (Promega, 5 μM final concentration) was added per well, incubated an additional 5 minutes to allow for substrate diffusion, and plates were immediately read for both luminescence at 485 nm and fluorescent eYFP emission at 530 nm for 1 s per well using a Mithras LB940 multimode microplate reader. The ratio of eYFP/RLuc was calculated per well and the net BRET ratio was calculated by subtracting the eYFP/RLuc per well from the eYFP/RLuc ratio in wells without Venus-β-Arrestin present. The net BRET ratio was plotted as a function of drug concentration using Graphpad Prism 5 (Graphpad Software Inc., San Diego, CA). Data were normalized to % 5-HT stimulation and analyzed using nonlinear regression “log(agonist) vs. response” in GraphPad Prism 8.0. For measurement of arrestin translocation at other GPCRs an identical approach was used.
For HTR2A-mediated G protein activation, HEK293T cells were plated either in six-well dishes containing 700–800,000 cells per well, or 10-cm dishes at approximately 7–8 million cells/dish. Cells were transfected 2–4 hours later, using a 1:1:1:1 ratio of the receptor:GαrLuc8:Gβ:GγGFP DNA (Olsen et al., 2020). Transit 2020 (Mirus biosciences) was used to complex the DNA at a ratio of 3 μL Transit/mg DNA, in OptiMEM (GIBCO) at a concentration of 10 ng DNA/μL OptiMEM. The next day, cells were harvested from the plate using Versene (0.1M PBS + 0.5 mM EDTA, pH 7.4), and plated in poly-D-lysine-coated 96-well white assay plates (Greiner) at a density of 25–50,000 cells per well.
One day after plating in 96-well assay plates, white backings (Perkin Elmer) were applied to the clear bottoms of the plate, and media was carefully aspirated and replaced with 50 μM coelenterazine 400a (nanolight technology) in 60 μL of drug buffer (1 × HBSS, 20 mM HEPES, 0.1% BSA, 0.01% ascorbic acid, pH 7.4). After a five-minute equilibration period, cells were treated with 30 μL of the drug for an additional 5 minutes. Plates were then read in an LB940 Mithras plate reader (Berthold Technologies) with a 395 nm (RLuc8-coelenterazine 400a) and 510 nm (GFP2) emission filters, at 1 s integration times. Plates were read six times, and measurements from the sixth read were used in all analyses. BRET ratio was computed as the ratio of the GFP2 emission to rLuc8 emission. Data were normalized to % 5-HT stimulation and analyzed using nonlinear regression “log(agonist) vs. response” in GraphPad Prism 8.0. For measurement of G protein activation with reference ligands the approach above was used identically as described except that neurotensin was used as a control for NTSR1-neurotensin.
Surface expression enzyme-linked immunosorbent assay
To confirm cell surface expression of HTR2A and its mutants, after 48hr transfected, immunocytochemistry was done using cells plated on 384-white plates at 10,000 cells/well. Cells were fixed with 20 μl/well of 4% paraformaldehyde (Fisher, #AAJ19943K2) for 10 minutes at room temperature. After fixation, cells were washed twice with 40 μl/well of PBS. Blocking was performed with 20 μl/well of 5% BSA (Akron, #AK8909) in PBS for 30 minutes at room temperature. After blocking, 20 μl/well of monoclonal ANTI-FLAG M2-Peroxidase (HRP) antibody (Sigma-Aldrich, A8592) diluted 1/10,000 in PBS was added and incubated for 1 hour at room temperature. This was followed by two washes with 80 μl/well of PBS. Then, 20 ml/well of SuperSignal enzyme-linked immunosorbent assay (ELISA) Pico Chemiluminescent Substrate (Thermo Fisher, #37070) was added, and luminescence was counted using a PHERAstar FSX (BMG Labtech). Data were plotted as relative luminescent units (RLU) in GraphPad Prism 8.0.
QUANTIFICATION AND STATISTICAL ANALYSIS
For radioligand binding assays data were analyzed using “Dissociation – One phase exponential decay” in Graphpad Prism 8.0 for the kinetic studies and the the equation ‘one-site fit Ki’ in GraphPad Prism 8.0 for the inhibition studies. For the BRET studies, the net BRET ratio was plotted as a function of drug concentration using Graphpad Prism 8 (Graphpad Software Inc., San Diego, CA) and data were normalized to % 5-HT or the reference agonist stimulation and analyzed using nonlinear regression “log(agonist) vs. response” in GraphPad Prism 8.0. For the cell surface expression studies Data were plotted as relative luminescent units (RLU) in GraphPad Prism 8.0. For the analysis of all fitted curves the goodness-of-fit was calculated by GraphPad Prism 8.0 and the data analyzed for parameter estimates was not evaluated for hetereskedasticity.
Data in figures and tables are reported as mean ± standard error of the mean (SEM) with the number of biological and technical replicates indicated in the figure and table legends where “n” represents the number of biological replicates performed. EC50 and Emax were analyzed by analysis of variance (ANOVA), and then a Dunnett’s multiple comparison test using comparing each mutant to the wild-type receptor. The family wise-significance and confidence level was set at 0.05. For the depiction of the heatmap of agonist efficacies, the on-line program MORPHEUS (https://software.broadinstitute.org/morpheus/) was used with the map paramters listed in the figure.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| gp64-PE antibody | Expression Systems | Cat#97-201 |
| monoclonal ANTI-FLAG M2-Peroxidase (HRP) antibody | Sigma-Aldrich | Cat#A8592 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| AEBSF | GoldBio | Cat#A-540-5 |
| Leupeptin | Sigma | Cat#L2884 |
| Aprotinin | GoldBio | Cat#A-655-100 |
| Iodoacetamide | Sigma | Cat#I1149 |
| n-dodecyl-beta-D-Maltopyranoside (DDM) | Anatrace | Cat#D310 |
| Lauryl maltose neopentyl glycol (LMNG) | Anatrace | Cat#NG310 |
| Glyco-Diosgenin (GDN) | Anatrace | Cat#GDN101 |
| GDP | Sigma Aldrich | Cat#M0398L |
| His-tagged PreScission protease | GenScript | Cat#Z0392-500 |
| Tris HCL | VWR | Cat#0497 |
| HEPES | Fisher Scientific | Cat#BP310 |
| Glycerol | Fisher Scientific | Cat#BP229 |
| EDTA | Sigma Aldrich | Cat#E6758 |
| Imidazole | Sigma | Cat#I0250 |
| Sodium Chloride | Fisher | Cat#BP358 |
| Potassium Chloride | Sigma | Cat#793590 |
| Magnesium Chloride | Sigma | Cat#M0250 |
| BSA, Free Fatty Acid | Akron | Cat#AK8909 |
| Ascorbic Acid | Sigma | Cat#A5960 |
| Cholesteryl heisucinate (CHS) | Sigma | Cat#C6512 |
| TALON IMAC resin | Clontech | Cat#635507 |
| 1-Oleoyl-rac-glycerol (monoolein) | Sigma | Cat#M7765 |
| Cholesterol | Sigma | Cat#C8667 |
| Coelenterazine-h | Promega | Cat#S2011 |
| Coelenterazine 400a (Deep Blue C) | Nanolight | Cat#340-1 |
| Poly-L-lysine | Sigma | Cat#P2636 |
| Penicillin/Streptomycin | Invitrogen | Cat#15140-122 |
| Zeocin | Invitrogen | Cat#R25005 |
| Sf-900 II SFM | Invitrogen | Cat#10902096 |
| ESF921 | Expression Systems | Cat#96-001-01 |
| CCM3 media | GE healthcare | Cat#SH3061.04 |
| DMEM | VWR | Cat#45000-306 |
| FBS | VWR | Cat#97068-085 |
| Dialyzed FBS | Omega Scientific | Cat#FB-03 |
| 10xHBSS | Invitrogen | Cat#14065-056 |
| Cellfectin II Reagent | Invitrogen | Cat#10362-100 |
| TransIT-2020 | Mirus | Cat#MIR5400 |
| Polyethylenimine (PEI) | Polysciences, Inc | Cat#23966 |
| [3H]-LSD | Perkin Elmer | Cat#NET638 |
| Methiothepin mesylate salt | Sigma | Cat#M149 |
| Polyethylene glycol 400 | Hampton Research | HR2-603 |
| StockOptions Salt Kit | Hampton Research | HR2-245 |
| Additive Screen HT | Hampton Research | HR2-138 |
| Paraformaldehyde Solution, 4% in PBS | Fisher | Cat#AAJ 19943K2 |
| Critical Commercial Assays | ||
| Bac-to-Bac Baculovirus Expressiokn system | Invitrogen | Cat#A11100 |
| pcDNA 3.1 | Thermo Fisher | Cat#V79020 |
| SuperSignal ELISA Pico Chemiluminescent Substrate | Thermo Fisher | Cat#37070 |
| Deposited Data | ||
| HTR2A/25CN-NBOH/mGq/Gβ/G | This paper | PDB: 6WHA |
| HTR2A/LSD complex structure | This paper | PDB: 6WGT |
| HTR2A/methiothepin complex structure | This paper | PDB: 6WH4 |
| Experimental Models: Cell Lines | ||
| Spodoptera frugiperda Sf9 cells | Expression Systems | Cat#94-001S |
| HEK293T | ATCC | Cat# |
| Recombinant DNA | ||
| Human 5HT2A gene | Integrated DNA Technologies (IDT) | N/A |
| miniGqiN chimera | Integrated DNA Technologies (IDT) | N/A |
| Gβ1 subunit | Integrated DNA Technologies (IDT) | N/A |
| Gγ2 subunit | Integrated DNA Technologies (IDT) | N/A |
| pFastBac dual expression vector | Invitrogen | Cat#10712024 |
| pFastBac 1 expression vector | Invitrogen | Cat#10359016 |
| Software and Algorithms | ||
| COOT | COOT | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| XDS | Kabsch, 2010a, 2010b | http://xds.mpimf-heidelberg.mpg.de/ |
| IUPHAR | Southan et al., 2016 | https://www.guidetopharmacology.org/ |
| Marvin v.15.11.23.0 | ChemAxon | https://chemaxon.com |
| Phaser | McCoy et al., 2007 | https://www.ccp4.ac.uk |
| Phenix | Adams et al., 2010 | https://www.phenix-online.org |
| PyMOL | Schrödinger | https://pymol.org/2/ |
| Prism v8.0 | GraphPad Software | N/A |
| Relion | Scheres, 2012 | https://www3.mrc-lmb.cam.ac.uk/relion/index.php?title=Main_Page |
| SerialEM | Mastronarde, 2003 | https://bio3d.colorado.edu/SerialEM/ |
| REFMAC | Murshudov et al., 1997 | https://www.ccp4.ac.uk |
| MORPHEUS | N/A | https://software.broadinstitute.org/morpheus/ |
| Adobe Illustrator CC | Adobe | https://www.adobe.com |
| Adobe Photoshop CC | Adobe | https://www.adobe.com |
| Other | ||
| PD MiniTrap G-25 columns | GE Healthcare | Cat#28-9180-07 |
| 100 kDa molecular weight cut-off Vivaspin 20 concentrator | Sartorius Stedim | Cat#VS2042 |
| 100 kDa molecular weight cut-off Vivaspin 500 centrifuge concentrator | Sartorius Stedim | Cat#VS0142 |
| 96-well LCP glass sandwich set | Marienfeld GmbH | Cat#0890003 |
| Meltilex | Perkin Elmer | Cat#1450-441 |
| Filtermat A | Perkin Elmer | Cat#1450-421 |
| 96-well black plates | Greiner Bio-one GmbH | Cat#655090 |
| Superdex 200 Increase column | GE healthcare | Cat#289909944 |
| 96-well white plates | Greiner Bio-one GmbH | Cat#655098 |
| Amicon Ultra –4 10K | Merck Millipore | Cat#UFC801024 |
| Amicon Ultra –4 50K | Merck Millipore | Cat#UFC805024 |
Highlights.
Cryo-EM 5-HT2A serotonin receptor structure complexed with hallucinogen and Gαq
The hallucinogen 25CN-NBOH displaces “toggle switch” tryptophan
Interactions essential for Gαq-specific signaling identified
X-ray crystal structure of LSD complexed with 5-HT2A elucidated
ACKNOWLEDGMENTS
We thank M.J. Miley and the UNC macromolecular crystallization core for advice and use of their equipment for crystal harvesting and transport, which is supported by the National Cancer Institute (P30CA016086), and the staff of GM/CA@APS, which has been funded by the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). This research used resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science user facility operated for the DOE Office of Science by Argonne National Laboratory (DE-AC02-06CH11357). K.K. was partially supported by Kortney Rose Foundation (KRF) postdoctoral fellowship (2017R1A6A3A03012547).This work was supported by NIH (R37DA04567 and R01MH112205), a cooperative agreement from DARPA (HR0011-20-2-0029 to B.L.R.), and by NIH (R35GM122481 to B.K.S.). The views, opinions, and/or findings contained in this material are those of the authors and should not be interpreted as representing the official views, policies, or endorsement of the Department of Defense or the US Government.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.08.024.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N,Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen WJ, and Rizzo RC (2014). Implementation of the Hungarian algorithm to account for ligand symmetry and similarity in structure-based design. J. Chem. Inf. Model. 54, 518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, and McCammon JA (2001). Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett FS, Preller KH, Herdener M, Janata P, and Vollenweider FX (2018). Serotonin 2A Receptor Signaling Underlies LSD-induced Alteration of the Neural Response to Dynamic Changes in Music. Cereb. Cortex 28, 3939–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogenschutz MP (2013). Studying the effects of classic hallucinogens in the treatment of alcoholism: rationale, methodology, and current research with psilocybin. Curr. Drug Abuse Rev. 6, 17–29. [DOI] [PubMed] [Google Scholar]
- Braden MR, and Nichols DE (2007). Assessment of the roles of serines 5.43(239) and 5.46(242) for binding and potency of agonist ligands at the human serotonin 5-HT2A receptor. Mol. Pharmacol. 72, 1200–1209. [DOI] [PubMed] [Google Scholar]
- Caffrey M, and Cherezov V (2009). Crystallizing membrane proteins using lipidic mesophases. Nat. Protoc. 4, 706–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carhart-Harris RL, Bolstridge M, Rucker J, Day CM, Erritzoe D, Kaelen M, Bloomfield M, Rickard JA, Forbes B, Feilding A, et al. (2016). Psilocybin with psychological support for treatment-resistant depression: an open-label feasibility study. Lancet Psychiatry 3, 619–627. [DOI] [PubMed] [Google Scholar]
- Carhart-Harris RL, Bolstridge M, Day CMJ, Rucker J, Watts R, Erritzoe DE, Kaelen M, Giribaldi B, Bloomfield M, Pilling S, et al. (2018). Psilocybin with psychological support for treatment-resistant depression: six-month follow-up. Psychopharmacology (Berl.) 235, 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che T, Majumdar S, Zaidi SA, Ondachi P, McCorvy JD, Wang S, Mosier PD, Uprety R, Vardy E, Krumm BE, et al. (2018). Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell 172, 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RG, Carchia M, Sterling T, Irwin JJ, and Shoichet BK (2013). Ligand pose and orientational sampling in molecular docking. PLoS ONE 8, e75992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Santos RG, Bouso JC, Alcázar-Córcoles MA, and Hallak JEC (2018). Efficacy, tolerability, and safety of serotonergic psychedelics for the management of mood, anxiety, and substance-use disorders: a systematic review of systematic reviews. Expert Rev. Clin. Pharmacol. 11, 889–902. [DOI] [PubMed] [Google Scholar]
- Draper-Joyce CJ, Khoshouei M, Thal DM, Liang YL, Nguyen ATN, Furness SGB, Venugopal H, Baltos JA, Plitzko JM, Danev R, et al. (2018). Structure of the adenosine-bound human adenosine A1 receptor-Gi complex. Nature 558, 559–563. [DOI] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Evans P (2006). Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82. [DOI] [PubMed] [Google Scholar]
- Fantegrossi WE, Gray BW, Bailey JM, Smith DA, Hansen M, and Kristensen JL (2015). Hallucinogen-like effects of 2-([2-(4-cyano-2,5-dimethoxyphenyl) ethylamino]methyl)phenol (25CN-NBOH), a novel N-benzylphe-nethylamine with 100-fold selectivity for 5-HT2A receptors, in mice. Psychopharmacology (Berl.) 232, 1039–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flock T, Ravarani CNJ, Sun D, Venkatakrishnan AJ, Kayikci M, Tate CG, Veprintsev DB, and Babu MM (2015). Universal allosteric mechanism for Gα activation by GPCRs. Nature 524, 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher K, and Sharp K (1998). Electrostatic contributions to heat capacity changes of DNA-ligand binding. Biophys. J. 75, 769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Nafría J, Nehmé R, Edwards PC, and Tate CG (2018). Cryo-EM structure of the serotonin 5-HT1B receptor coupled to heterotrimeric Go. Nature 558, 620–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser P, Kirchner K, and Passie T (2015). LSD-assisted psychotherapy for anxiety associated with a life-threatening disease: a qualitative study of acute and sustained subjective effects. J. Psychopharmacol. (Oxford) 29, 57–68. [DOI] [PubMed] [Google Scholar]
- Griffiths RR, Johnson MW, Carducci MA, Umbricht A, Richards WA, Richards BD, Cosimano MP, and Klinedinst MA (2016). Psilocybin produces substantial and sustained decreases in depression and anxiety in patients with life-threatening cancer: A randomized double-blind trial. J. Psychopharmacol. (Oxford) 30, 1181–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N, and Peitsch MC (1997). SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723. [DOI] [PubMed] [Google Scholar]
- Halberstadt AL (2017). Pharmacology and Toxicology of N-Benzylphenethylamine (“NBOMe”) Hallucinogens. Curr. Top. Behav. Neurosci. 32, 283–311. [DOI] [PubMed] [Google Scholar]
- Halberstadt AL, Sindhunata IS, Scheffers K, Flynn AD, Sharp RF, Geyer MA, and Young JW (2016). Effect of 5-HT2A and 5-HT2C receptors on temporal discrimination by mice. Neuropharmacology 107, 364–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Phonekeo K, Paine JS, Leth-Petersen S, Begtrup M, Bräuner-Osborne H, and Kristensen JL (2014). Synthesis and structure-activity relationships of N-benzyl phenethylamines as 5-HT2A/2C agonists. ACS Chem. Neurosci. 5, 243–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MA, Brooun A, Baker KA, Jaakola VP, Roth C, Chien EY, Alexandrov A, Velasquez J, Davis L, Griffith M, et al. (2007). Profiling of membrane protein variants in a baculovirus system by coupling cell-surface detection with small-scale parallel expression. Protein Expr. Purif. 56, 85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Masureel M, Qu Q, Janetzko J, Inoue A, Kato HE, Robertson MJ, Nguyen KC, Glenn JS, Skiniotis G, and Kobilka BK (2020). Structure of the neurotensin receptor 1 in complex with beta-arrestin 1. Nature 579, 303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Raimondi F, Kadji FMN, Singh G, Kishi T, Uwamizu A, Ono Y, Shinjo Y, Ishida S, Arang N, et al. (2019). Illuminating G-Protein-Coupling Selectivity of GPCRs. Cell 177, 1933–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010a). Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010b). Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato HE, Zhang Y, Hu H, Suomivuori CM, Kadji FMN, Aoki J, Krishna Kumar K, Fonseca R, Hilger D, Huang W, et al. (2019). Conformational transitions of a neurotensin receptor 1-Gi1 complex. Nature 572, 80–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura KT, Asada H, Inoue A, Kadji FMN, Im D, Mori C, Arakawa T, Hirata K, Nomura Y, Nomura N, et al. (2019). Structures of the 5-HT2A receptor in complex with the antipsychotics risperidone and zotepine. Nat. Struct. Mol. Biol. 26, 121–128. [DOI] [PubMed] [Google Scholar]
- Kometer M, Schmidt A, Jäncke L, and Vollenweider FX (2013). Activation of serotonin 2A receptors underlies the psilocybin-induced effects on α oscillations, N170 visual-evoked potentials, and visual hallucinations. J. Neurosci. 33, 10544–10551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna Kumar K, Shalev-Benami M, Robertson MJ, Hu H, Banister SD, Hollingsworth SA, Latorraca NR, Kato HE, Hilger D, Maeda S, et al. (2019). Structure of a Signaling Cannabinoid Receptor 1-G Protein Complex. Cell 176, 448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen K, Kroeze WK, Willins DL, Gelber EI, Savage JE, Glennon RA, and Roth BL (2000). A highly conserved aspartic acid (Asp-155) anchors the terminal amine moiety of tryptamines and is involved in membrane targeting of the 5-HT(2A) serotonin receptor but does not participate in activation via a “salt-bridge disruption” mechanism. J. Pharmacol. Exp. Ther. 293, 735–746. [PubMed] [Google Scholar]
- Kroeze WK, Sassano MF, Huang XP, Lansu K, McCorvy JD, Giguère PM, Sciaky N, and Roth BL (2015). PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 22, 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Warne T, Nehme R, Pandey S, Dwivedi-Agnihorti H, Chaturvedi M, Edwards PC, Garcia-Nafria J, Leslie AG, Shukla AK, and Tate CG (2020). Molecular basis of β-arrestin coupling to formoterol-bound β1-adrenoceptor. Nature 583, 862–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu J, Wang S, Balius TE, Singh I, Levit A, Moroz YS, O’Meara MJ, Che T, Algaa E, Tolmachova K, et al. (2019). Ultra-large library docking for discovering new chemotypes. Nature 566, 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Koehl A, Matile H, Hu H, Hilger D, Schertler GFX, Manglik A, Skiniotis G, Dawson RJP, and Kobilka BK (2018). Development of an antibody fragment that stabilizes GPCR/G-protein complexes. Nat. Commun. 9, 3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Qu Q, Robertson MJ, Skiniotis G, and Kobilka BK (2019). Structures of the M1 and M2 muscarinic acetylcholine receptor/G-protein complexes. Science 364, 552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCorvy JD, Wacker D, Wang S, Agegnehu B, Liu J, Lansu K, Tribo AR, Olsen RHJ, Che T, Jin J, and Roth BL (2018). Structural determinants of 5-HT2B receptor activation and biased agonism. Nat. Struct. Mol. Biol. 25, 787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ (2007). Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng EC, Shoichet B, and Kuntz ID (1992). Automated docking with grid-based energy evaluation. J. Comput. Chem. 13, 505–524. [Google Scholar]
- Moro O, Lameh J, and Sadée W (1993). Serine- and threonine-rich domain regulates internalization of muscarinic cholinergic receptors. J. Biol. Chem. 268, 6862–6865. [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, and Dodson EJ (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Mysinger MM, and Shoichet BK (2010). Rapid context-dependent ligand desolvation in molecular docking. J. Chem. Inf. Model. 50, 1561–1573. [DOI] [PubMed] [Google Scholar]
- Nehmé R, Carpenter B, Singhal A, Strege A, Edwards PC, White CF, Du H, Grisshammer R, and Tate CG (2017). Mini-G proteins: Novel tools for studying GPCRs in their active conformation. PLoS ONE 12, e0175642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols DE (2016). Psychedelics. Pharmacol. Rev. 68, 264–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt D, Erritzoe D, and Carhart-Harris R (2020). Psychedelic Psychiatry’s Brave New World. Cell 181, 24–28. [DOI] [PubMed] [Google Scholar]
- Olsen RHJ, DiBerto JF, English JG, Glaudin AM, Krumm BE, Slocum ST, Che T, Gavin AC, McCorvy JD, Roth BL, and Strachan RT (2020). TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat. Chem. Biol. 16, 841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, McCorvy JD, Harpsoe K, Lansu K, Yuan S, Popov P, Qu L, Pu M, Che T, Nikolajsen LF, et al. (2018). 5-HT2C Receptor Structures Reveal the Structural Basis of GPCR Polypharmacology. Cell 172, 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Aguilar JM, Shan J, LeVine MV, Khelashvili G, and Weinstein H (2014). A functional selectivity mechanism at the serotonin-2A GPCR involves ligand-dependent conformations of intracellular loop 2. J. Am. Chem. Soc. 136, 16044–16054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Poulie CBM, Jensen AA, Halberstadt AL, and Kristensen JL (2019). DARK Classics in Chemical Neuroscience: NBOMes. ACS Chem. Neurosci. Published online November 12, 2019. 10.1021/acschem-neuro.9b00528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preller KH, Burt JB, Ji JL, Schleifer CH, Adkinson BD, Stämpfli P, Seifritz E, Repovs G, Krystal JH, Murray JD, et al. (2018). Changes in global and thalamic brain connectivity in LSD-induced altered states of consciousness are attributable to the 5-HT2A receptor. eLife 7, e35082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. (2011). Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson MJ, van Zundert GCP, Borrelli K, and Skiniotis G (2020). GemSpot: A Pipeline for Robust Modeling of Ligands into Cryo-EM Maps. Structure 28, 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross S, Bossis A, Guss J, Agin-Liebes G, Malone T, Cohen B, Mennenga SE, Belser A, Kalliontzi K, Babb J, et al. (2016). Rapid and sustained symptom reduction following psilocybin treatment for anxiety and depression in patients with life-threatening cancer: a randomized controlled trial. J. Psychopharmacol. (Oxford) 30, 1165–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL (2019). Molecular pharmacology of metabotropic receptors targeted by neuropsychiatric drugs. Nat. Struct. Mol. Biol. 26, 535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL, Shoham M, Choudhary MS, and Khan N (1997). Identification of conserved aromatic residues essential for agonist binding and second messenger production at 5-hydroxytryptamine2A receptors. Mol. Pharmacol. 52, 259–266. [DOI] [PubMed] [Google Scholar]
- Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewell RA, Halpern JH, and Pope HG Jr. (2006). Response of cluster headache to psilocybin and LSD. Neurology 66, 1920–1922. [DOI] [PubMed] [Google Scholar]
- Shapiro DA, Kristiansen K, Weiner DM, Kroeze WK, and Roth BL (2002). Evidence for a model of agonist-induced activation of 5-HT2A serotonin receptors which involves the disruption of a strong ionic interaction between helices 3 and 6. J. Biol. Chem. 277, 11441–11449. [DOI] [PubMed] [Google Scholar]
- Sharp KA, Friedman RA, Misra V, Hecht J, and Honig B (1995). Salt effects on polyelectrolyte-ligand binding: comparison of Poisson-Boltzmann, and limiting law/counterion binding models. Biopolymers 36, 245–262. [DOI] [PubMed] [Google Scholar]
- Shi L, Liapakis G, Xu R, Guarnieri F, Ballesteros JA, and Javitch JA (2002). Beta2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J. Biol. Chem. 277, 40989–40996. [DOI] [PubMed] [Google Scholar]
- Snyder SH, Faillace LA, and Weingartner H (1968). DOM (STP), a new hallucinogenic drug, and DOET: effects in normal subjects. Am. J. Psychiatry 125, 113–120. [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH, Buneman OP, Davenport AP, McGrath JC, Peters JA, et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res. 44, D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staus DP, Hu H, Robertson MJ, Kleinhenz ALW, Wingler LM, Capel WD, Latorraca NR, Lefkowitz RJ, and Skiniotis G (2020). Structure of the M2 muscarinic receptor-β-arrestin complex in a lipid nanodisc. Nature 579, 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein RM, Kang HJ, McCorvy JD, Glatfelter GC, Jones AJ, Che T, Slocum S, Huang XP, Savych O, Moroz YS, et al. (2020). Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nature 579, 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling T, and Irwin JJ (2015). ZINC 15–Ligand Discovery for Everyone. J. Chem. Inf. Model. 55, 2324–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W, Chen C, Lei X, Zhao J, and Liang J (2018). CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 46 (W1), W363–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, et al. (2013). Structural features for functional selectivity at serotonin receptors. Science 340, 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D, Stevens RC, and Roth BL (2017a). How Ligands Illuminate GPCR Molecular Pharmacology. Cell 170, 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D, Wang S, McCorvy JD, Betz RM, Venkatakrishnan AJ, Levit A, Lansu K, Schools ZL, Che T, Nichols DE, et al. (2017b). Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell 168, 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q, Okashah N, Inoue A,Nehmé R, Carpenter B, Tate CG, and Lambert NA (2018). Mini G protein probes for active G protein-coupled receptors (GPCRs) in live cells. J. Biol. Chem. 293, 7466–7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wacker D, Levit A, Che T, Betz RM, McCorvy JD, Venkatakrishnan AJ, Huang XP, Dror RO, Shoichet BK, and Roth BL (2017). D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 358, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis WI, and Kobilka BK (2018). The Molecular Basis of G Protein-Coupled Receptor Activation. Annu. Rev. Biochem. 87, 897–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wess J (1997). G-protein-coupled receptors: molecular mechanisms involved in receptor activation and selectivity of G-protein recognition. FASEB J. 11, 346–354. [PubMed] [Google Scholar]
- Yin W, Zhou XE, Yang D, de Waal PW, Wang M, Dai A, Cai X, Huang CY, Liu P, Wang X, et al. (2018). Crystal structure of the human 5-HT1B serotonin receptor bound to an inverse agonist. Cell Discov. 4, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The following structure files are available from the Protein Databank: 6WHA, 6WGT and 6WH4.