

Abstract
Chronic short sleep or extended wake periods are commonly observed in most industrialized countries. Previously neurobehavioral impairment following sleep loss was considered to be a readily reversible occurrence, normalized upon recovery sleep. Recent clinical studies suggest that chronic short sleep and sleep disruption may be risk factors for neurodegeneration. Animal models have been instrumental in determining whether disturbed sleep can injure the brain. We now understand that repeated periods of extended wakefulness across the typical sleep period and/or sleep fragmentation can have lasting effects on neurogenesis and select populations of neurons and glia. Here we provide a comprehensive overview of the advancements made using animal models of sleep loss to understand the extent and mechanisms of chronic short sleep induced neural injury.
Keywords: Sleep loss, neurodegeneration, neural injury, neurogenesis, amyloid-beta, tau
1. Introduction
With chronic short sleep in humans defined as obtaining less than 7 h of sleep per 24 h period, approximately one third of individuals in most industrialized countries report chronic short sleep (Centers for Disease Control and Prevention, n.d.; Eaton et al., 2010; Khubchandani & Price, 2019). Notably, the percentage of individuals reporting short sleep has increased over the past few decades (Hisler, Muranovic, & Krizan, 2019; Knutson, Van Cauter, Rathouz, DeLeire, & Lauderdale, 2010; Norell-Clarke & Hagquist, 2017). In addition to chronic short sleep, common disorders and conditions, for example obstructive sleep apnea and chronic pain, may result in significant disruption of sleep despite a normal sleep time. While the immediate neurobehavioral consequences of short-term sleep loss have been well-described and are considered reversible in humans, much less is known of the consequences of chronic short sleep and sleep disruption on brain function.
Epidemiologic studies in humans suggest that sleep disruption and chronic short sleep may be risk factors for neurodegenerative disorders, including Alzheimer’s disease (AD), the most common neurodegenerative disorder (D. W. Kang, Lee, & Lim, 2017; Lutsey et al., 2018; Pase et al., 2017). However, there are substantial challenges in critically testing whether chronic short sleep in humans results in neurodegeneration. Specifically, the reported poor sleep in individuals with AD and even early AD may be a consequence of AD related neural injury rather than a primary source of degeneration. Second, because AD occurs typically after the 7th decade of life, researchers exploring sleep and dementia typically look only at sleep in elderly subjects at the time of cognitive assessment. Ideally, sleep would be measured over many decades before the earliest of AD findings, as sleep times vary across the lifespan with changes in jobs, commutes, families, and illnesses, and presently, we do not know when across the lifespan sleep disruption is most likely to impart neural injury. In light of the high prevalence of sleep disorders worldwide and increasing prevalence of AD and other neurodegenerative processes, it is imperative to understand what is and is not known regarding the role of sleep disruption and neural injury and neurodegeneration and then critically test unknown effects of sleep loss on brain health in animal models. At the same time, it is important to understand the limitations and confounds of various animal models and the sleep disruption paradigms used and how with respect to neurodegeneration animal models differ. The purpose of this review is to provide a comprehensive examination of the effects of sleep loss and sleep disruption on the different facets of neural injury and neurodegeneration in animal models.
Paradigms for sleep loss in animal models vary widely, yet are important to understand as these variations may impact neural injury findings. Here we have grouped sleep loss into three exposure durations: acute (<24 h), intermediate (1-4 days) and chronic (>4 days). Protocols using acute and intermediate durations typically implement either total or rapid-eye-movement (REM) sleep deprivation, while chronic sleep loss has been performed as REM sleep deprivation and as chronic short sleep, where wakefulness is enforced for 6-8 h of the sleep-predominant period or in extreme paradigms for up to 20 h per 24 h period. Modes of sleep disruption vary from rotating drums, discs over water, running wheels or treadmills that force the animal to move periodically or when it falls asleep, platform techniques (where animals are housed on small platforms over water, and when an animal falls asleep it will fall into the water, wake up and climb back up to the platform), gentle handling (where an animal is kept in its home cage and brushed when the animal appears sleepy/asleep, or the cage is shaken) and exploratory wakefulness, which involves an enriched environment of constantly changing novel climbing toys. Each paradigm has its own advantages and disadvantages. Many of these techniques allow the animal to fall asleep and then quickly awaken the animal resulting in sleep fragmentation/disruption, while others such as the platform method can be designed to prevent primarily REM sleep when the platform is small enough that the animal will fall from the platform only during REM sleep atonia. The exploratory wakefulness technique results in constant attentive wakefulness but adds the confound of increased motor activity. Sleep loss paradigms may variably influence the hypothalamic-pituitary-adrenal (HPA) stress axis. Corticosterone levels (as one indicator of stress) have been shown elevated in platform paradigms but not in exploratory wakefulness models (Suchecki, Lobo, Hipolide, & Tufik, 1998; J. Zhang et al., 2014). Absence of change in a one-time corticosterone measurement does not exclude increased stress as the circadian rhythm may have shifted, and there are several studies to support the concept that sleep deprivation, even short-term influences circadian regulators in humans and animal models (Foo et al., 2019; Lo et al., 2012; Orozco-Solis et al., 2017; Wisor et al., 2008). Thus, we must acknowledge that sleep deprivation studies, in animal models and in humans influence not only sleep but also HPA responses and circadian rhythmicity.
A host of markers have been used to characterize neural injury. Some of these markers indicate metabolic stress in the cell, mitochondria or nucleus. Perhaps the clearest indicator of neural injury is neurodegeneration. Neurodegeneration is defined as the loss of neurons in association with behavioural impairments. Regional brain volume is often used as a surrogate for neuronal loss indicative of neurodegeneration. In general, reductions in brain region volume signify injury, but volume loss can occur in response to changes in vasculature, myelin, glial cells and neurites in the absence of neuron loss. Both neuronal loss and regional brain volume have been studied in the context of sleep loss and will be discussed below. Here we will summarize effects of sleep loss on neuronal counts and volumes of specific brain regions. Other aspects of neural injury that are observed in sleep loss and in neurodegeneration will also be described, including markers of neural injury and death, neuroinflammation, changes in myelin and protein aggregation. Overall, increasing durations of sleep loss lead to cumulative brain injury, and while early injury may be reversible chronic sleep loss leads to irreversible neurodegeneration (Figure 1).
Figure 1. Time dependent effects of sleep disruption.
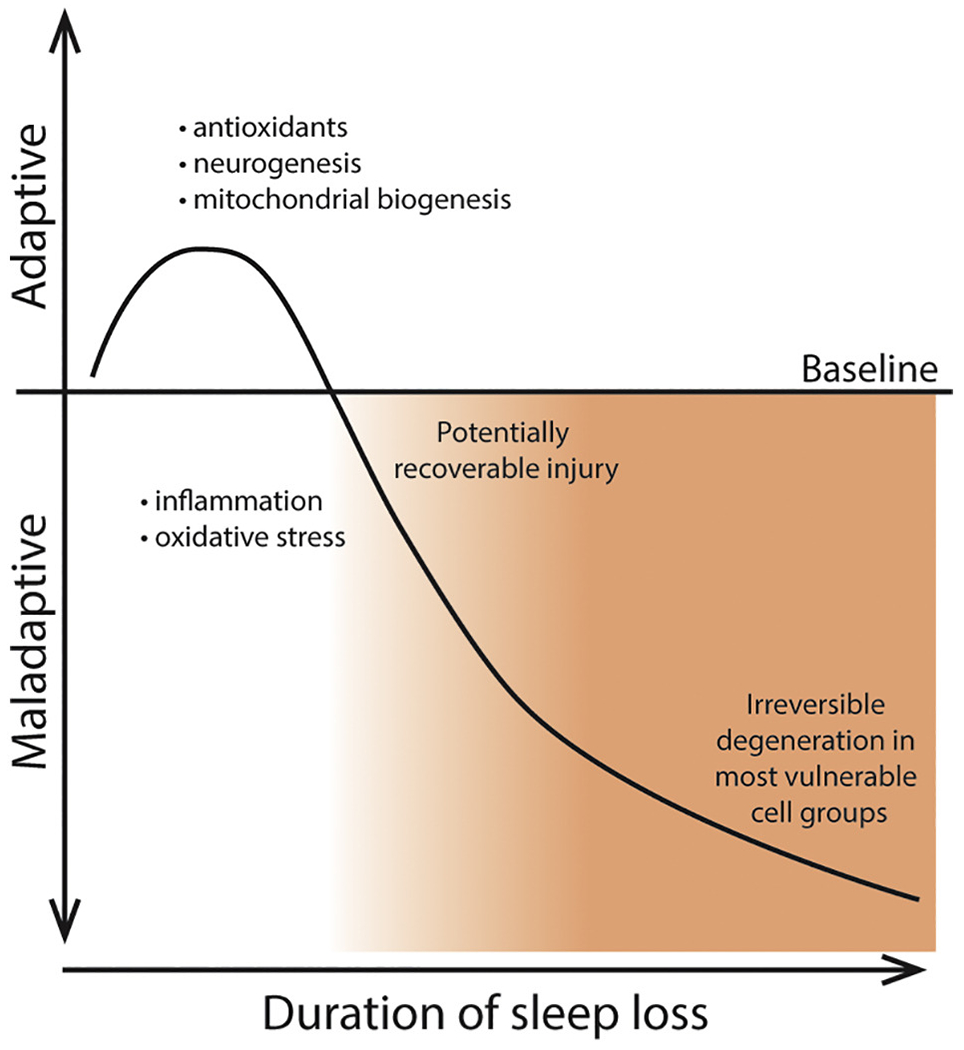
Very brief (≤ 3 h) wakefulness extension may be adaptive by increasing antioxidants, hippocampal neurogenesis and mitochondrial biogenesis, longer durations are maladaptive with increased inflammation and oxidative stress which may be reversible. Even longer durations of sleep disruption ultimately result in degeneration, irreversible injury and impairment.
2. Regional volume and neuron loss
In human studies, brain imaging techniques allow regional brain volume to be assessed as a proxy in vivo for cell loss and therefore neurodegeneration. In animal models the best way to determine the volume of a brain region or accurately estimate the number of cells is via stereological analysis (West & Gundersen, 1990).
Brain volume changes have been estimated after chronic sleep deprivation using the multiple platform method (Kamali, Noorafshan, Karimi, Karbalay-Doust, & Nami, 2017; Noorafshan, Karimi, Kamali, Karbalay-Doust, & Nami, 2017; Noorafshan, Karimi, Karbalay-Doust, & Kamali, 2017) and the rotating drum apparatus (Novati, Hulshof, Koolhaas, Lucassen, & Meerlo, 2011). The authors reports that chronic sleep restriction for 3 or 4 weeks in rats resulted in decreased volume of two brainstem respiratory nuclei (nucleus of the solitary tract and the parabrachial nucleus)(Kamali et al., 2017), in addition to medial prefrontal cortex (Noorafshan, Karimi, Karbalay-Doust, et al., 2017), and the hippocampus (both dorsal and CA2/CA3 pyramidal cell layers) (Novati et al., 2011). In a separate study, volume loss was observed for hippocampal regions, CA1 and the dentate gyrus (Noorafshan, Karimi, Kamali, et al., 2017). Volume reductions in the medial prefrontal cortex were examined after a 3-4 week recovery period (Noorafshan, Karimi, Karbalay-Doust, et al., 2017), indicating that at least in this region the effect of sleep restriction does not fully reverse. The other brain regions were examined within a few days of sleep loss; so it is unclear whether the volume losses observed represent permanent or readily reversible changes. Of note the methodologies for volume and cell count measurements are not provided in great detail to understand the robustness of the findings.
Cell loss has also been found in a number of regions after sleep disruption. As little as three days of sleep restriction via exploratory wakefulness for 8 h/day plus 4 days of recovery sleep resulted in an approximately 30% decrease in the number of locus ceruleus (LC) neurons (J. Zhang et al., 2014; Zhu et al., 2016). Four weeks of this paradigm resulted in a similar (30%) loss of orexinergic neurons in young adult mice and a 4 week recovery opportunity of normal sleeping conditions did not improve cell counts in either region (Zhu et al., 2016). However, not all neuronal populations are vulnerable. A group of sleep-active neurons, melanin-concentrating hormone neurons, adjacent to orexinergic neurons are not lost in response to chronic short sleep (Zhu et al., 2016). Using the same paradigm for a longer duration (12 weeks followed by a 6 month recovery) did not result in a further loss of LC neurons (Zhu et al., 2018), suggesting that vulnerable neurons are only lost early on in sleep loss. Additionally this study found a similar, 30%, decrease in neurons of the basolateral amygdala. Other brain regions that succumb to cell loss after a different sleep restriction paradigm (platform technique in rats) include the nucleus of the solitary tract and the medial, lateral and sub- parabrachial nuclei each of which had approximately 10% fewer cells after chronic sleep restriction (18 h/day for 21 days, no recovery) (Kamali et al., 2017). The same paradigm decreased medial prefrontal cortex neuronal numbers by 36% and interestingly found a 31% decrease in the number of glial cells in the same region (Noorafshan, Karimi, Karbalay-Doust, et al., 2017). Neuronal subtypes susceptible to sleep loss in the cortices have not yet been identified. Collectively, this body of work strongly supports loss of select neurons in response to chronic sleep loss.
A more significant form of chronic sleep restriction (18 h/day for 21 days) in adult rats using the platform technique resulted in a 48% decrease in the CA1 pyramidal neurons and a 25% loss of dentate gyrus neurons compared to controls (Noorafshan, Karimi, Kamali, et al., 2017). Additionally, REM sleep deprivation for 2 or 4 days found significantly less viable pyramidal neurons in the CA1 of mice (Hou et al., 2019; Olonode, Aderibigbe, Adeoluwa, Eduviere, & Ben-Azu, 2019). However a large caveat is that neither of these studies used stereology to estimate cell numbers; thus, these results should be confirmed in future studies in order to be confident that sleep loss can lead to cell loss in the hippocampus. In support of significant neural injury hippocampal dependent memory was impaired after sleep loss (Hou et al., 2019; Noorafshan, Karimi, Kamali, et al., 2017).
Collectively, these results suggest that there is significant volumetric and neuronal loss in multiple brain regions after sleep loss and the reductions do not readily reverse with recovery sleep. Elucidation of all susceptible regions should now be pursued. Neuronal and volume loss could be occurring via a number of different mechanisms including changes in vasculature, changes in the extracellular space, increased cell death, decreased neurogenesis, changes to dendrites and/or myelination or any combination of these factors. The relationship between some of these mechanisms and sleep loss is discussed below.
2.1. Neurogenesis
Adult neurogenesis is known to occur throughout life, although slowing progressively across advanced aging and declining substantially in AD (Moreno-Jiménez et al., 2019). This process occurs in both the subventricular zone of the forebrain and the subgranular zone of the hippocampus. A decrease in the rate of neurogenesis is a possible explanation for the volume/cell loss observed as a consequence of sleep disruption. Studies using a variety of sleep deprivation methods and durations have investigated neurogenesis almost exclusively in the subgranular zone. Cell proliferation may be measured by injection of bromodeoxyuridine (BrdU), which is incorporated into newly synthesised DNA effectively labeling cells in the S-phase of the cell cycle that will shortly divide. For example, if BrdU is injected 2h prior to the end of deprivation, BrdU will indicate the effect of sleep loss on cell proliferation at the end of deprivation. Ki-67 is a complementary stain that labels cells in all active phases of the cell cycle (G1, S, G2 and M) and not cells in the resting phase (G0) and would indicate only cell phase at the time of perfusion. Doublecortin (DCX) is a marker of immature, post-mitotic neurons that is sometimes double labeled with BrdU.
Using these markers, most studies report no change in the number of proliferating cells with acute sleep deprivation (≤24 h) (Guzman-Marin, Bashir, Suntsova, Szymusiak, & McGinty, 2007; Mirescu, Peters, Noiman, & Gould, 2006; Murata et al., 2018; van der Borght et al., 2006). An absence of effect on proliferation has been found using a variety of sleep disruption methods including gentle handling (total sleep deprivation)(van der Borght et al., 2006), intermittent forced activity on a treadmill (sleep fragmentation) (Guzman-Marin et al., 2007) and the small platform method (REM sleep deprivation) (Mirescu et al., 2006; Murata et al., 2018). Of the acute sleep deprivation studies showing changes effects have been divergent. For example, while finding no evidence for proliferation in the subgranular zone after 24 h sleep disruption, a decrease in ki-67+ cell numbers in the hilus was observed after 24 h, without a change in BrdU+ cells (Roman, Van der Borght, Leemburg, Van der Zee, & Meerlo, 2005). The hilus is not a known location of neurogenesis, but has been suggested to be a location of gliogenesis (Steiner et al., 2004), suggesting that sleep disruption may effect glial cell proliferation and not neuronal cell proliferation within the first 24 h. Two studies have found significantly increased numbers of BrdU+ cells after 12 h of sleep loss by gentle handling (Grassi Zucconi, Cipriani, Balgkouranidou, & Scattoni, 2006) and forced activity (Junek, Rusak, & Semba, 2010). Interestingly, the number of BrdU+ cells remained elevated 15 and 30 days after the one sleep loss exposure (Grassi Zucconi et al., 2006). The only other study to investigate this sleep loss time point (van der Borght et al., 2006) used the gentle handling method but found no change in the number of proliferating cells. Differences between the studies include species used and the BrdU injection timing; mice (van der Borght et al., 2006) or rats injected only at 2 h prior to the end of sleep disruption (Junek et al., 2010), or rats injected twice at 4 and 2 h prior to the end of sleep disruption (Grassi Zucconi et al., 2006). Differences in species and/or study protocols may contribute to varying results.
Changes in neurogenesis after longer periods of sleep interruption (2-4 days) have been investigated in both rats and mice using a variety of paradigms including REM sleep disruption (small/multiple platforms) (Cui et al., 2019; Mirescu et al., 2006; Mueller et al., 2008; Murata et al., 2018), near total sleep loss (gentle handling or disk over water) (García-García et al., 2011; Tung, Takase, Fornal, & Jacobs, 2005), intermittent sleep restriction (6h/day for 4 days with a enriched environment) (Hairston et al., 2005) and forced activity whether on a timed schedule (eg. 3 s on; 30 s off) (Guzman-Marin et al., 2007; Guzmán-Marín et al., 2003; Wadhwa, Prabhakar, et al., 2017) or inactivity/REM sleep detected (Guzman-Marin et al., 2008; Sahu et al., 2013). The consensus is that sleep interruption for 2-4 days leads to a decrease in cell proliferation (ki-67+, BrdU+ and DCX+ cells) (Cui et al., 2019; García-García et al., 2011; Guzman-Marin et al., 2007, 2008; Guzmán-Marín et al., 2003; Hairston et al., 2005; Mirescu et al., 2006; Mueller et al., 2008; Murata et al., 2018; Sahu et al., 2013; Tung et al., 2005; Wadhwa, Prabhakar, et al., 2017). In support of a differential effect between acute and intermediate sleep loss, it has been shown that 3 h of sleep disruption results in an increase in neuroprotective factors including multiple antioxidants in the brain which is no longer apparent after 3 days of sleep loss for 8 h each day (J. Zhang et al., 2014). However, one study found no change in the number of BrdU labeled cells in BALBc mice after 72 h of REM sleep deprivation by the multiple platforms method (Soto-Rodriguez et al., 2016). Another study used exactly the same sleep loss procedure in a different strain of mice, C57B6, and found a decrease in the number of BrdU labeled cells (Cui et al., 2019). It is possible that vulnerability to sleep deprivation varies with genetics of mouse strains. In support of this, chronic sleep restriction increased markers of inflammation and the permeability of the blood brain barrier in C57B6 mice and not BALBc mice when compared directly (G. Hurtado-Alvarado et al., 2018).
Studies investigating neurogenesis have also been implemented using sleep disruption paradigms longer than 4 days, 2 using a slow rotating drum for 20h/day (Novati et al., 2011; Roman et al., 2005) and 1 using forced treadmill activity every 30s (Guzman-Marin et al., 2007). These studies have induced sleep interruption for 1-4 weeks in duration. Guzman and colleagues (2007) investigated the effect of chronic sleep interruption (7 days) on cell proliferation by injecting BrdU 2 h prior to the end of the sleep disruption period as was done in studies using intermittent sleep loss paradigms. They found that the number of BrdU+ cells decreased compared to control animals not sleep deprived but also exposed to forced activity. Other studies investigated cell survival not proliferation by injecting BrdU 5 days prior to the onset of sleep disruption (Novati et al., 2011; Roman et al., 2005), and found no change in the number of BrdU+ cells. Novati et al also found no change in the number of DCX+ cells. Taken together these findings suggest that cells dividing prior to sleep interruption are not affected by subsequent sleep loss, while cells proliferating during sleep disruption are susceptible.
2.2. Neuronal injury markers
The most extreme measure of neural injury is considered irreversible cell loss; however, numerous studies have used biomarkers of neuronal demise, including markers of apoptosis, mitochondrial injury, and DNA changes consistent with dying cells. It is critical to understand that many of these markers are not specific for “dying or dead” neurons; some are evident simply upon neuronal activation, in senescent cells, or even non-specific labeling.
Overall, evidence supports the conclusion that neuronal injury/cell death does not occur after sleep loss of less than 24 h in young adult rodents (Artamokhina, Belova, & Romanova, 2011; Cirelli, Shaw, Rechtschaffen, & Tononi, 1999; Naidoo, Ferber, Master, Zhu, & Pack, 2008). One of these studies found an increase in the pro-apoptotic marker p53 in two hypothalamic nuclei, but also found an increase in the anti-apoptotic marker Bcl2 in the same nuclei and no morphological evidence of apoptosis (Artamokhina et al., 2011).
Sleep loss (REM sleep disruption or intermittent exploratory wakefulness) occurring for 2-4 days results in some signs of neuronal injury. Caspase-3, a marker of apoptotic and necrotic cell death, was increased in the LC after 3 days of sleep loss for 8 h/day (J. Zhang et al., 2014) and in the hippocampus after 48 h of REM sleep loss using the multiple platforms method (Cao et al., 2019). Cao (2019) also found a decreased ratio of Bcl2 (anti-apoptotic) to Bax (pro-apoptotic) proteins suggesting that mitochondrial stress occurred, however another study found no change in Bcl2 in any region examined, including the hippocampus after 96 h of REM sleep deprivation (Hipolide, D’Almeida, Raymond, Tufik, & Nobrega, 2002). Additionally, no evidence of damaged, degenerating or degenerated cells (amino-cupric-silver staining) was found after 4 days of REM sleep deprivation (Biswas, Mishra, & Mallick, 2006). Conversely, 72 h of REM sleep deprivation increased the number of TUNEL+ cells (cells with substantial DNA fragmentation, suggestive of apoptosis) in the CA1 (Soto-Rodriguez et al., 2016). Interestingly, this study also found that after 2 weeks of recovery mice still had increased TUNEL+ cells in the CA1 and also had increased TUNEL+ cells in the dentate gyrus and CA3, 3 weeks of recovery resulted in increased TUNEL+ cells in the CA1 and the dentate gyrus suggesting that injury may be progressive. Studies are now needed to determine ultimate cell loss by stereology in TUNEL+ brains to determine if this signal truly represents apoptosis. While the results of these studies seem contradictory an important factor to note is that the studies that did not find evidence of cell death/injury were performed in rats (Biswas et al., 2006; Hipolide et al., 2002) and the studies that did find evidence of cell death/injury after sleep loss were performed in mice (Cao et al., 2019; Soto-Rodriguez et al., 2016; J. Zhang et al., 2014). This may be indicative of an earlier vulnerability of mice to sleep loss that is not seen in rats, the two species should be compared in the same study to investigate this.
In support of significant neural injury, chronic sleep restriction (>4 days) in mice leads to increased cleaved caspase 3, TUNEL+ cells, Bax and decreased anti-apoptotic marker Bcl2 and abnormal mitochondria in the hippocampus (Qiu et al., 2016), specifically the dentate gyrus (Yin et al., 2017). Some of these markers remained abnormal after 3 weeks of recovery (Yin et al., 2017), and returned to normal levels after 3 months of recovery (Qiu et al., 2016).
The effects of chronic sleep restriction on cell death/injury in rats is less clear. Two studies report no evidence of cell death after up to 14 days of sleep disruption (Cirelli et al., 1999; Eiland et al., 2002), one of which reported increased amino-cupric-silver staining in one hypothalamic nucleus (Eiland et al., 2002). However, neither study examined cell counts in specific brain regions. Another study reported increased TUNEL+ cells only in aged rats, not young rats (de Souza et al., 2012). Conversely, two studies reported evidence of cell death after 6 days of REM sleep disruption including increased caspase 3, TUNEL+ cells and pro-apoptotic markers in multiple brain regions (Biswas et al., 2006; Somarajan, Khanday, & Mallick, 2016). The reason for the discrepancy here is unclear, however it is noteworthy that only one of these studies reported the age of the rats. Given that older rodents may be more susceptible to sleep loss imposed cell death/injury than their young counterparts (de Souza et al., 2012; Naidoo et al., 2008), this information could provide an important clue.
2.3. Dendrites and axons
Factors other than increased cell death and decreased neurogenesis may account for the volume loss observed after sleep loss. These include dendritic changes and decreased myelination of axons.
After short term sleep loss (<24 h) both increased and decreased dendritic spine densities have been observed in the hippocampus (Acosta-peña et al., 2015; Havekes et al., 2016; Raven, Meerlo, Van der Zee, Abel, & Havekes, 2018; Spano et al., 2019). Spano and colleagues suggest this differential effect could be accounted for by the difference in sleep loss procedures used with most studies using gentle handling and finding decreasing hippocampal spine density (Acosta-peña et al., 2015; Havekes et al., 2016; Raven et al., 2018) and their study using an enriched environment found increasing hippocampal spine density, the consequence of a method that increases synaptic plasticity (Spano et al., 2019). However, a recent study found increased spine density in the hippocampus after sleep loss by gentle handling for 5 h in adult mice (Gisabella, Scammell, Bandaru, & Saper, 2019). It is possible that the difference is due to different methods for visualising dendritic spines. The three studies that found decreased spine density all used Golgi staining which doesn’t stain all neurons and the reason a neuron might or might not stain with Golgi is unknown. The two more recent studies that found increased spine density used electron microscopy (Spano et al., 2019) and confocal microscopy with optogenetic expression of excitatory glutamatergic neurons (Gisabella et al., 2019), both of which allow more complete and detailed examination of dendritic spines. The only study examining dendrites after chronic sleep loss found decreased spine density and decreased total dendrite length in the CA1 after 21 days of chronic REM sleep restriction (18 h a day) using the multiple platform method (Noorafshan, Karimi, Kamali, Karbalay-Doust, & Nami, 2018). Additionally, acute (8h) total sleep loss by environmental enrichment and longer term (4.5 days) sleep restriction by environmental enrichment and forced locomotion resulted in decreased myelin thickness (Bellesi et al., 2018). Together these studies suggest changes in spine density and myelination could impact the volume loss observed after sleep loss, however more still needs to be done particularly in chronic sleep loss.
3. Sleep loss and glia
The effects of sleep loss on non-neuronal cell types may influence the observed volume loss as well as being a potential mechanism for neuronal loss given that glial cells provide vital metabolic support to neurons.
The key astrocyte marker GFAP increased after 12 h of sleep deprivation by gentle handling in the hippocampus (Mishra et al., 2016) and piriform cortex (Kaur et al., 2017), interestingly both of these studies were conducted in aged female animals whereas all other studies were conducted in males. Longer sleep deprivation protocols have found both increased GFAP (Hou et al., 2019; Gabriela Hurtado-Alvarado, Domínguez-Salazar, Velázquez-Moctezuma, & Gómez-González, 2016; Manchanda, Singh, Kaur, & Kaur, 2018; Wadhwa, Prabhakar, et al., 2017) and unchanged GFAP (Wadhwa, Kumari, et al., 2017; Yin et al., 2017) in the hippocampus, piriform cortex, cortex and basal nuclei. Morphological changes indicative of reactive astrocytosis have also been reported (Wadhwa, Kumari, et al., 2017; Wadhwa, Prabhakar, et al., 2017). Astrocytes have a variety of functions within the brain and the heterogeneity in astrocyte responses may reflect this. It would be useful to examine a wider variety of astrocyte markers concurrently after sleep loss to determine which functional changes are occurring. For example Bellesi and colleagues found increased astrocytic phagocytosis of synapses after acute (8 h) and longer term (4.5 days) sleep loss which may be a neuroprotective mechanism to allow adequate synaptic functioning during extended wakefulness (Bellesi et al., 2017).
Microglia are immune cells that produce inflammatory responses to injury in the brain, several studies have investigated the inflammatory response after sleep disruption. While some microglial markers such as OX18 and OX42 increase after less than 24 h of sleep loss, many inflammatory markers remain unchanged (Kaur et al., 2017; Mishra et al., 2016). Further, microglial morphology remains unchanged in this acute stage of sleep loss (Bellesi et al., 2017). After 24-96 h of sleep loss pro-inflammatory markers typically increase (Hou et al., 2019; Wadhwa, Kumari, et al., 2017; Wadhwa, Prabhakar, et al., 2017) and anti-inflammatory markers decrease in the hippocampus (Wadhwa, Kumari, et al., 2017; Wadhwa, Prabhakar, et al., 2017). More than 4 days of sleep loss consistently results in increased pro-inflammatory markers (G. Hurtado-Alvarado et al., 2018; Gabriela Hurtado-Alvarado et al., 2016; Kincheski et al., 2017; Manchanda et al., 2018) and increased signs of reactive microgliosis including upregulation of Iba-1(G. Hurtado-Alvarado et al., 2018; Gabriela Hurtado-Alvarado et al., 2016; Manchanda et al., 2018) and decreased microglial process length (Bellesi et al., 2017). Taken together this suggests a cumulative inflammatory response with increasing sleep loss duration.
4. Sleep loss and protein aggregation
Neurodegeneration, as stated previously is defined as a progressive loss of functional neurons with neurobehavioral impairment. Specific neurodegenerative diseases are defined by stereotypical progression of regional neural injury associated with characteristic aberrant protein aggregation. The relationship between sleep abnormalities and increasing risk for developing different neurodegenerative diseases has been discussed previously and is discussed elsewhere in this special edition. Here, relevant animal models investigating the effect of chronic sleep loss on the processing of proteins associated with specific neurodegenerative mechanisms will be discussed in detail.
4.1. Sleep loss and amyloid processing
Alzheimer’s disease (AD) is the most common neurodegenerative disorder. While AD is a tauopathy, it is the presence of amyloid plaque that helps distinguish AD from other tauopathies. The amyloid hypothesis proposes that amyloid β (Aβ) deposition underlies clinical and pathologic findings in familial AD (J. A. Hardy & Higgins, 1992; J. Hardy & Selkoe, 2002). Aβ is produced by way of proteolytic processing of amyloid precursor protein (APP) typically on neuronal membranes, where most Aβ produced is secreted extracellularly. Most Aβ secreted is Aβ40, but under various genetic and metabolic conditions, a greater relative amount of Aβ42 may be produced. Aβ42 has increased proclivity to nucleate and drive fibril formation. Interestingly, upregulation of neuronal activity increases the production of Aβ42 more so than Aβ40 (Kamenetz et al., 2003), and in contrast silencing neuronal activity in vitro results in an abrupt reduction in Aβ levels. There is some controversy whether the observed increase in Aβ upon neuronal activation occurs in response to increased Aβ production or decreased Aβ clearance, or both. Work from the Holtzman lab suggests that Aβ production increases with neuronal activation while clearance is unchanged (Cirrito et al., 2005). In contrast, the Nedergaard lab has shown that Aβ movement across the interstitial space is more rapid when neuronal activity is suppressed (Xie et al., 2013). Importantly these two labs have also shown that sleep/wake can acutely influence Aβ production and clearance. Specifically, using cortical reverse microdialysis, levels of Aβ in the interstitial fluid vary across the sleep/wake cycle, increasing in wakefulness and sleep deprivation compared to sleep (J.-E. Kang et al., 2009), and with waking compared to anesthesia, Aβ clearance is slowed across the interstitial space (Xie et al., 2013).
Several studies have examined the effects of chronic sleep loss on cognitive function and/or amyloid pathology in murine models of AD. Using a triple transgenic model with mutations in APP, presenilin and tau, 6 h/day for 6 weeks using small platforms resulted in impaired contextual and cued memory, yet no change in soluble amyloid oligomers, and amyloid pathology was not examined (Rothman, Herdener, Frankola, Mughal, & Mattson, 2013). Importantly, corticosterone levels were elevated at least initially with this sleep deprivation paradigm, and, thus, stress may have contributed to the observed cognitive impairments. More recently a double transgenic (APP and presenilin mutation) mouse was examined for effects of small platform sleep deprivation for 20 h/day for 2 months (Qiu et al., 2016). Mice showed impaired spatial memory and impaired reversal of memory, and effects were still evident 3 months after exposure to sleep deprivation. Additionally both amyloid plaques and insoluble Aβ42 were elevated in response to sleep loss and remained elevated suggesting irreversible effects. A second study in the same double transgenic mouse model found that sleep fragmentation also increases amyloid plaque burden, and that the plaque burden can be predicted by the severity of sleep fragmentation (Minakawa et al., 2017). Using a model of very severe sleep deprivation in wild type mice (a rotating drum to deprive mice of sleep 20 out of 24 h/day for 2 months), Zhao et al., demonstrated increased Aβ in the cortex in wild type mice along with impaired spatial memory performance (Zhao et al., 2017). The rotating drum paradigm is confounded by single housing and potential head trauma, as the animal is tossed around within the drum when it falls asleep. Whether chronic sleep loss in the absence of these stressors and potential physical trauma can induce AD-like pathology and/or memory impairments remains to be determined.
Administration of orexin increases wakefulness. Thus, not surprisingly, levels of Aβ can be increased by administering orexin and decreased by administering an orexin antagonist (J.-E. Kang et al., 2009); although whether the orexin effects were specific to orexin or secondary to sleep/wake effects has not been discerned. A specific role for orexin in amyloid and AD has been queried. Individuals with narcolepsy lack orexin from early on in life and appear to be as likely to be diagnosed with AD (Scammell, Matheson, Honda, Thannickal, & Siegel, 2012), however this study had a very small sample size (n=12). A recent PET study found significantly lower amyloid burden in patients with N1 type narcolepsy compared to controls (Gabelle et al., 2019) supporting a role of orexin in amyloid pathology. Further, orexin knockout in mice (Roh et al., 2014) or orexin antagonist therapy administered long-term to mice (J.-E. Kang et al., 2009) with a genetic predisposition to AD-like pathology markedly reduced the pathology, supporting the concept that increasing sleep and/or reducing orexin may be an effective intervention in slowing the development of AD-like pathology. Importantly, additional measures of enhancing sleep can slow the progression of AD pathology in an AD mouse model. Specifically, by optogenetically-enhancing slow wave activity in sleep in the double transgenic (APP and presenilin mutation) mice, slows the progression of amyloid plaque in the area with enhanced local sleep (Kastanenka et al., 2017).
Amyloid deposition may also impact sleep resulting in a feedforward cycle of sleep loss amyloid injury. Specifically, mice with transgenic overexpression of various familial AD mutations in APP have disturbances in sleep (Huitrón-Reséndiz et al., 2002; Roh et al., 2012; Schneider, Baldauf, Wetzel, & Reymann, 2014; B. Zhang et al., 2005). Sleep disturbances in mouse models of AD are consistent with sleep disturbances reported in human AD, even in cognitively normal individuals with amyloid identified by imaging (Y.-E. S. Ju et al., 2013). Based on these findings and many supporting findings, a bi-directional relationship between sleep disturbance and amyloid has been proposed and recently reviewed (Y. E. S. Ju, Lucey, & Holtzman, 2014; C. Wang & Holtzman, 2019).
4.2. Sleep loss effects on tau and tauopathy
Tau is a neuronal cytoskeletal protein that when post-translationally modified at specific sites can aggregate into fibrils and neurofibrillary tangles that are characteristic of tauopathies including AD. Tau may also be cleaved resulting in direct neural injury. A characteristic feature of each specific tauopathy is a predictable spatiotemporal pattern of tau progression through specific brain regions of connectivity. This robust trait across tauopathies prompted exploration and ultimately confirmation of tau transmission from neuron to neuron at least in part by trans-synaptic spread (de Calignon et al., 2012; Liu et al., 2012). Sleep loss may influence tau modifications and propagation. Using a method of small platform sleep restriction (6 h/day for 6 weeks) mice with APP, presenilin and tau mutations were found to have increased phosphorylated tau in the cortex (Rothman et al., 2013). Because of the additional APP and presenilin mutations, it is unclear whether the effects of sleep loss on tau are primary or downstream of amyloid effects. Recently, chronic short sleep (via active exploration in a novel environment) was found to hasten the progression of tauopathy in a murine model, the P301S mouse (Zhu et al., 2018). Specifically, this paradigm of chronic short sleep (8 h/day, 3 days/week for 4 weeks) resulted in impairments in motor performance and novel object memory, while increasing phosphorylated and misfolded tau throughout brain regions susceptible to tauopathy, including the LC, entorhinal cortex and hippocampus and importantly hastens loss of susceptible neurons in the LC and in the amygdala (Zhu et al., 2018). Sleep fragmentation, in the absence of an enriched environment, also increased tauopathy in the same model (Zhu et al., 2018). Because sleep loss affected all of the examined behavioral, biochemical and pathologic features of tauopathy and because the effects were sustained (6 month recovery time), it is expected that sleep loss impacts a fundamental initiating and/or propagating feature of tauopathy. There is some evidence that chronic sleep loss can hasten tau propagation. Pathogenic tau fibrils injected into the hippocampus unilaterally in tauopathy mice will result in tau propagation. In mice chronically sleep-deprived after injection of tau fibrils into the hippocampus unilaterally, phosphorylated and misfolded tau accumulates to a greater extent in LC neurons ipsilateral to the tau injection (Holth et al., 2019). It is possible, however, that the increase in ipsilateral tau accumulation is an additive effect of injury from the injection to LC projections within the hippocampus in addition to chronic sleep loss. One of the most intriguing features of this LC response is that only a few LC neurons are densely packed with modified tau while adjacent neurons which presumably project to the same region appear unaffected.
The awake brain is highly responsive to stimuli and shows increased overall metabolic activity and neuronal activity relative to other behavioral states, including both non-REM sleep and REM sleep. Thus, prolonged awakening provokes sustained activation in many groups of neurons and could influence tau in wake-activated neurons. Importantly, tau may be released from neurons, largely into the extracellular space, in a physiological manner (independent of injury) in response to neuronal activation (Pooler, Noble, & Hanger, 2014). Intriguingly, the tau response to neuronal activation within the interstitial space shows a continued rise for several hours after stimulus and an even slower clearance rate from the interstitial space (Yamada et al., 2014). Here, too, the increase in interstitial tau in response to neuronal activation occurs independent of neuronal injury (Yamada et al., 2014). Chronic neuronal stimulation (using chemogenetic activation for 6 weeks) is sufficient to accelerate tau pathology in the entorhinal cortex and enhance tau propagation to connected regions (Wu et al., 2016). How specifically neuronal activation promotes the release of tau remains unclear. Tau has not been identified in the synaptic vesicle proteome to support vesicular transfer (Takamori et al., 2006), yet has been identified in exosomes and tau can transfer synaptically by exosomes (Y. Wang et al., 2017). Moreover, exosome-derived tau is neurotoxic (Winston et al., 2019), while inhibiting exosome synthesis can slow tau propagation (Asai et al., 2015). Thus, neuronal activation could increase exosomal transfer of tau across the interstitial space to adjacent neurons. Recently, acute sleep deprivation was shown to increase tau levels in the interstitial space, and as with the tau response to increasing neuronal activity, the increase in tau occurs gradually over several hours and is sustained after sleep loss across the next day (Holth et al., 2019). Interestingly, chemogenetic stimulation of a brain region that results in increased wakefulness (the supramammillary nucleus) causes an abrupt increase in lactate in the interstitial space supporting acute neuronal activation, yet tau rises only slowly across 10 h after chemogenetic stimulation suggesting that the increase in tau may not necessarily be a consequence of neuronal activation, as much as it is a consequence of metabolic stress related to neuronal activation.
Overall, the above studies demonstrate that neuronal activation and sleep loss at least in part via neuronal activation can increase extracellular tau, and can propagate tau and importantly can hasten the temporal progression of tauopathy.
4.3. Sleep loss effects on α-synuclein
It is important to recognize that sleep loss influences proteins beyond those found in aggregated form in AD, and also increases α-synuclein which aggregates in the brains of people with Parkinson’s disease. Intriguingly this observation was made by Holth et al, using the same samples used to show wake-induced increases in tau (Holth et al., 2019). The timing of the increase in α-synuclein as well as the normalization after recovery sleep temporally coincide with the tau responses and are of the same magnitude. Common features of these two proteins are that they are intrinsically-disordered proteins, meaning the proteins may readily change shape with various post-translational modifications or interactions; they also have prion properties and they can seed normal tau or α-synuclein protein and propagate it. The importance of sleep loss increasing release of both of these proteins is that exposure of one to the other increases oligomerization of one, which may facilitate seeding and propagation. While it is known that sleep loss increases tauopathy and tau propagation, it is less clear at present whether sleep loss influences the temporal progression of Parkinson’s or Parkinson’s with dementia.
5. Initial clues about mechanisms of differential susceptibility
While it is evident that chronic sleep loss and/or sleep disruption can impart meaningful neural injury (summarized in Figure 2), the above review of injury patterns and other responses to sleep disruption highlights the complexity of factors underlying specific responses, including the type, duration and circadian timing of sleep disruption; age at the time of sleep disruption; and genetics (strain and/or species) of animals tested. This complexity (differential susceptibility) to neural injury should now be taken advantage of to better understand molecular mechanism underlying sleep loss neural injury. Sleep homeostatic mechanism may provide one defence system against over activation of specific wake-activated neural circuits. Recently, galanin was identified as a critical sleep homeostatic neuropeptide, necessary for effective recovery sleep (Reichert, Pavón Arocas, & Rihel, 2019). It will now be important to determine whether differences in galanin across rats and mice and various strains of mice more susceptible to sleep loss have poorer recovery sleep and reduced galaninergic responses to sleep loss. Sleep homeostasis may also involve changes in connectivity and excitability of wake-activated neurons. Specifically, sleep deprivation results in pre-synaptic inhibition of excitatory inputs to orexinergic (wake-activated) neurons (Briggs, Bowes, Semba, & Hirasawa, 2019; Briggs, Hirasawa, & Semba, 2018), while inhibitory GABAergic neurons in wake-activating regions have increased activity and augmented receptor density (del Cid-Pellitero, Plavski, Mainville, & Jones, 2017; Toossi, del Cid-Pellitero, & Jones, 2017). Interestingly, while slow wave activity dissipates over a 6-12 hour period, suppressed activity of neurons in some brain regions, including the hypothalamus, can occur for at least 48 hours (Fifel, Meijer, & Deboer, 2018). Whether this delay is secondary to increased galaninergic tone or an underlying inflammatory or metabolic response to sleep disruption has not been determined.
Figure 2. Cellular response to sleep disruption over time.
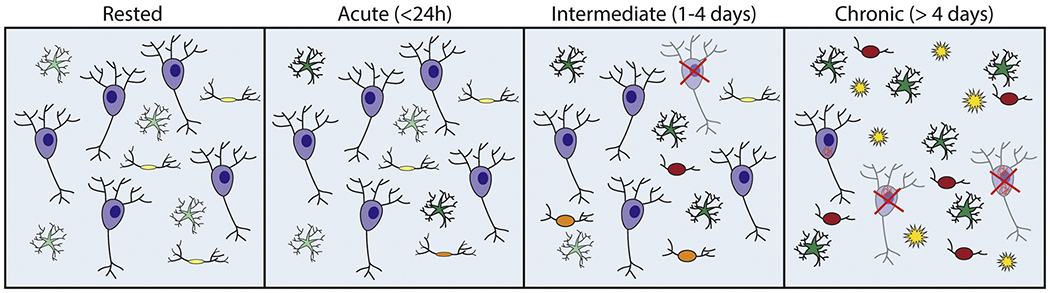
Diagrammatic representation of injury to different cell types with increasing durations of sleep disruption. The rested or baseline condition shows neurons (purple), astrocytes (green) and microglia (yellow). Acute sleep disruption shows minor changes to astrocytes (darker green) and microglia (yellow & orange) which include upregulation of some neuroinflammatory markers. Intermediate sleep disruption shows increased neuroinflammatory changes in astrocytes and microglia as well as some neuronal loss. Chronic sleep disruption shows many reactive astrocytes and microglia (red) and further neuronal loss. Additionally, increased amyloid beta plaques (yellow stars) and tauopathy (orange squiggles) can be seen in models that predispose animals to developing this pathology.
Several studies support the concept that metabolic and inflammatory changes also contribute to neural injury. Curcumin, an anti-oxidant derived from turmeric, can prevent platform sleep deprivation neuron loss in the cortices in rats when administered across the sleep deprivation period (Noorafshan, Karimi, Karbalay-Doust, et al., 2017). However, the investigators have not used stereological methods to assess cell loss, demonstrated that sleep deprivation increases oxidative stress or that curcumin lowered oxidative stress in animals protected from sleep loss neural injury. LC neurons do show increased oxidative stress in response to chronic sleep loss; thus whether curcumin or other anti-oxidant approaches could rescue these neurons would be of significant interest. Melatonin administration across platform sleep deprivation appears to reduce oxidative stress in the cortex, as evidenced by reducing oxidants: nitric oxide, malodialdehyde, and increasing superoxide dismutase activity (L. Zhang et al., 2013). Administration of melatonin across platform sleep deprivation in rats has been shown to reduce caspase-3 activation in LC neurons and to prevent neuron loss (Jameie et al., 2019). The LC is the sole source of noradrenaline to the cortices, and noradrenaline itself may serve anti-oxidant functions in the cortices (Singh, Das, Kaur, & Mallick, 2019); thus, protecting the LC across sleep deprivation may help to minimize injuries in other susceptible brain regions. It is interesting then that LC neurons projecting to select brain regions (e.g., prefrontal cortex) can show fatigue (reduced noradrenergic delivery upon extended sleep loss) (Bellesi, Tononi, Cirelli, & Serra, 2016). There is some evidence in the fly that Aβ that is increased in chronic sleep loss can counter homeostatic mechanisms and actually increase neuronal excitability (Tabuchi et al., 2015). Thus, conditions with increased Aβ production, including advanced age and oxidative stress may impair homeostasis and actually increase activity of neurons already under duress. Sleep loss induces inflammatory responses in the brain, but whether the inflammatory responses are detrimental or protective is unclear. Specifically, cytokine interleukin-1β is critical for sleep homeostasis (Davis et al., 2015), while complement activation in sleep loss suppresses neurogenesis and contributes to sleep loss memory impairments (Wadhwa et al., 2019).
6. Conclusions and Future Directions
Three decades ago, chronic sleep loss was shown to have potent effects on peripheral physiology yet have little impact on brain health. We now understand that sleep loss early in life can have lasting profound effects in animal models. Specifically, cell and volume loss occurs in some brain regions after sleep loss but not all regions are susceptible and further investigation is needed to give a more complete picture of regional vulnerability to sleep loss. Neurogenesis decreases with sleep loss of more than 24 h. Neuronal injury mechanisms are activated after sleep loss in mice more so than rats and with increasing duration of sleep loss exposure. The neuroinflammatory response seems cumulative based on duration of sleep loss also. There are indications of dendritic spine and myelin changes after acute sleep loss. It is possible that any number of these factors could be contributing to the volume/cell loss observed after sleep disruption.
Overall, animal models suggest that chronic more so than acute sleep loss can negatively impact the brain including many factors that contribute to neurodegeneration/neurodegenerative diseases (Figure 2). Given that the acute sleep loss response is seen mostly in glial cells that are known to have adaptive functions these processes could be neuroprotective (Figure 1). It would seem that the response to sleep loss of more than 24 h in rodents is neurotoxic given that after this time cell loss, reduced neurogenesis, signs of neural injury and protein aggregation begin to appear.
Many studies investigating sleep loss neural injury have not examined long-term, or residual effects. Of those that do simultaneously investigate injury immediately after sleep loss and after a recovery period suggest that the injury remains the same or deteriorates further with recovery sleep. The other unknown factor is whether longer recovery sleep periods or some kind of intervention could prevent this injury. Additionally, sex differences after sleep loss remain largely unknown with most studies being conducted only in male animals and only one (Zhu et al., 2018) to our knowledge directly comparing males and females. These are areas for future investigation.
Because of species differences in sleep patterns in humans, relative to rodents, and given species-variance in lifespan durations, it is quite difficult to relate durations of sleep loss in rodent models with duration equivalents in humans. We do know, however, that humans are not immune to residual neurobehavioral impairments days after chronic short sleep exposures to shift work-like sleep patterns (Belenky et al., 2003; Lamond et al., 2007; St Hilaire et al., 2017; Van Dongen, Maislin, Mullington, & Dinges, 2003). We still do not know whether sleep loss in humans can result in neurodegeneration, but given the wealth of detrimental findings shown in diverse animal models, we can anticipate that humans are also susceptible to chronic sleep loss. A next direction should include development of biomarkers of sleep loss-induced neural injury, verified in animal models with differential responses to injury and then assessed in humans to provide windows into sleep loss neural injury. These markers might include newer brain imaging techniques that may be performed in both animal models and humans and various plasma and cerebrospinal fluid markers. In the interim, animal models of sleep loss neural injury will be invaluable in elucidation of molecular mechanisms of sleep loss brain injury.
Highlights.
Chronic sleep loss imparts significant injury in the brain consistent with neurodegeneration.
Select populations of neurons are vulnerable to loss in response to chronic short sleep.
Effects of sleep loss on neurogenesis vary with duration of sleep loss.
Sleep loss influences the processing and/or clearance of amyloid-beta, tau and α-synuclein and may hasten progression of tauopathies, including Alzheimer’s disease in animal models.
References
- Acosta-peña E, Camacho-Abrego I, Melgarejo-Gutiérrez M, Flores G, Drucker-Colín R, & García-García F (2015). Sleep deprivation induces differential morphological changes in the hippocampus and prefrontal cortex in young and old rats. Synapse, 69(1), 15–25. 10.1002/syn.21779 [DOI] [PubMed] [Google Scholar]
- Artamokhina IV, Belova VA, & Romanova IV (2011). Immunohistochemical investigation of bcl-2 and p53 levels in rat hypothalamus after sleep deprivation. Journal of Evolutionary Biochemistry and Physiology, 47(5), 458–463. 10.1134/s0022093011050082 [DOI] [PubMed] [Google Scholar]
- Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, … Ikezu T (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nature Neuroscience, 18(11), 1584–1593. 10.1038/nn.4132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belenky G, Wesensten NJ, Thorne DR, Thomas ML, Sing HC, Redmond DP, … Balkin TJ (2003). Patterns of performance degradation and restoration during sleep restriction and subsequent recovery: a sleep dose-response study. Journal of Sleep Research, 12(1), 1–12. 10.1046/j.1365-2869.2003.00337.x [DOI] [PubMed] [Google Scholar]
- Bellesi M, de Vivo L, Chini M, Gilli F, Tononi G, & Cirelli C (2017). Sleep Loss Promotes Astrocytic Phagocytosis and Microglial Activation in Mouse Cerebral Cortex. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 37(21), 5263–5273. 10.1523/JNEUROSCI.3981-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellesi M, Haswell JD, de Vivo L, Marshall W, Roseboom PH, Tononi G, & Cirelli C (2018). Myelin modifications after chronic sleep loss in adolescent mice. Sleep, 41(5). 10.1093/sleep/zsy034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellesi M, Tononi G, Cirelli C, & Serra PA (2016). Region-Specific Dissociation between Cortical Noradrenaline Levels and the Sleep/Wake Cycle. Sleep, 39(1), 143–154. 10.5665/sleep.5336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S, Mishra P, & Mallick BN (2006). Increased apoptosis in rat brain after rapid eye movement sleep loss. Neuroscience, 142(2), 315–331. 10.1016/J.NEUROSCIENCE.2006.06.026 [DOI] [PubMed] [Google Scholar]
- Briggs C, Bowes SC, Semba K, & Hirasawa M (2019). Sleep deprivation-induced pre- and postsynaptic modulation of orexin neurons. Neuropharmacology, 154, 50–60. 10.1016/j.neuropharm.2018.12.025 [DOI] [PubMed] [Google Scholar]
- Briggs C, Hirasawa M, & Semba K (2018). Sleep deprivation distinctly alters glutamate transporter 1 apposition and excitatory transmission to orexin and MCH neurons. Journal of Neuroscience, 38(10), 2505–2518. 10.1523/JNEUROSCI.2179-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Yang Y, Wu H, Lu Y, Wu S, Liu L, … Wang Z (2019). Stem-leaf saponins from Panax notoginseng counteract aberrant autophagy and apoptosis in hippocampal neurons of mice with cognitive impairment induced by sleep deprivation. Journal of Ginseng Research. 10.1016/j.jgr.2019.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. (n.d.). Short Sleep Duration Among Workers — United States, 2010. Retrieved from https://www.cdc.gov/mmwr/preview/mmwrhtml/mm6116a2.htm
- Cirelli C, Shaw PJ, Rechtschaffen A, & Tononi G (1999). No evidence of brain cell degeneration after long-term sleep deprivation in rats. Brain Research, 840(1–2), 184–193. 10.1016/S0006-8993(99)01768-0 [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, … Holtzman DM (2005). Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron, 48(6), 913–922. 10.1016/j.neuron.2005.10.028 [DOI] [PubMed] [Google Scholar]
- Cui L, Xue R, Zhang X, Chen S, Wan Y, & Wu W (2019). Sleep deprivation inhibits proliferation of adult hippocampal neural progenitor cells by a mechanism involving IL-17 and p38 MAPK. Brain Research, 1714, 81–87. 10.1016/J.BRAINRES.2019.01.024 [DOI] [PubMed] [Google Scholar]
- Davis CJ, Dunbrasky D, Oonk M, Taishi P, Opp MR, & Krueger JM (2015). The neuron-specific interleukin-1 receptor accessory protein is required for homeostatic sleep and sleep responses to influenza viral challenge in mice. Brain, Behavior, and Immunity, 47, 35–43. 10.1016/j.bbi.2014.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, … Hyman BT (2012). Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron, 73(4), 685–697. 10.1016/j.neuron.2011.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza L, Smaili SS, Ureshino RP, Sinigaglia-Coimbra R, Andersen ML, Lopes GS, & Tufik S (2012). Effect of chronic sleep restriction and aging on calcium signaling and apoptosis in the hippocampus of young and aged animals. Progress in Neuro-Psychopharmacology and Biological Psychiatry, 39(1), 23–30. 10.1016/J.PNPBP.2012.01.018 [DOI] [PubMed] [Google Scholar]
- del Cid-Pellitero E, Plavski A, Mainville L, & Jones BE (2017). Homeostatic changes in GABA and glutamate receptors on excitatory cortical neurons during sleep deprivation and recovery. Frontiers in Systems Neuroscience, 11 10.3389/fnsys.2017.00017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton DK, McKnight-Eily LR, Lowry R, Perry GS, Presley-Cantrell L, & Croft JB (2010). Prevalence of Insufficient, Borderline, and Optimal Hours of Sleep Among High School Students - United States, 2007. Journal of Adolescent Health, 46(4), 399–401. 10.1016/j.jadohealth.2009.10.011 [DOI] [PubMed] [Google Scholar]
- Eiland MM, Ramanathan L, Gulyani S, Gilliland M, Bergmann BM, Rechtschaffen A, & Siegel JM (2002). Increases in amino-cupric-silver staining of the supraoptic nucleus after sleep deprivation. Brain Research, 945(1), 1–8. 10.1016/S0006-8993(02)02448-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fifel K, Meijer JH, & Deboer T (2018). Long-term effects of sleep deprivation on neuronal activity in four hypothalamic areas. Neurobiology of Disease, 109, 54–63. 10.1016/j.nbd.2017.10.005 [DOI] [PubMed] [Google Scholar]
- Foo JC, Trautmann N, Sticht C, Treutlein J, Frank J, Streit F, … Rietschel M (2019). Longitudinal transcriptome-wide gene expression analysis of sleep deprivation treatment shows involvement of circadian genes and immune pathways. Translational Psychiatry, 9(1). 10.1038/s41398-019-0671-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabelle A, Jaussent I, Bouallègue F. Ben, Lehmann S, Lopez R, Barateau L, … Dauvilliers Y (2019). Reduced brain amyloid burden in elderly patients with narcolepsy type 1. Annals of Neurology, 85(1), 74–83. 10.1002/ana.25373 [DOI] [PubMed] [Google Scholar]
- García-García F, De la Herrán-Arita AK, Juárez-Aguilar E, Regalado-Santiago C, Millán-Aldaco D, Blanco-Centurión C, & Drucker-Colín R (2011). Growth hormone improves hippocampal adult cell survival and counteracts the inhibitory effect of prolonged sleep deprivation on cell proliferation. Brain Research Bulletin, 84(3), 252–257. 10.1016/J.BRAINRESBULL.2011.01.003 [DOI] [PubMed] [Google Scholar]
- Gisabella B, Scammell T, Bandaru SS, & Saper CB (2019). Regulation of hippocampal dendritic spines following sleep deprivation. Journal of Comparative Neurology, (July), 1–9. 10.1002/cne.24764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi Zucconi G, Cipriani S, Balgkouranidou I, & Scattoni R (2006). ‘One night’ sleep deprivation stimulates hippocampal neurogenesis. Brain Research Bulletin, 69(4), 375–381. 10.1016/J.BRAINRESBULL.2006.01.009 [DOI] [PubMed] [Google Scholar]
- Guzman-Marin R, Bashir T, Suntsova N, Szymusiak R, & McGinty D (2007). Hippocampal neurogenesis is reduced by sleep fragmentation in the adult rat. Neuroscience, 148(1), 325–333. 10.1016/j.neuroscience.2007.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman-Marin R, Suntsova N, Bashir T, Nienhuis R, Szymusiak R, & McGinty D (2008). Rapid Eye Movement Sleep Deprivation Contributes to Reduction of Neurogenesis in the Hippocampal Dentate Gyrus of the Adult Rat. Sleep, 31(2), 167–175. 10.1093/sleep/31.2.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzmán-Marín R, Suntsova N, Steward DR, Gong H, Szymusiak R, & McGinty D (2003). Sleep deprivation reduces proliferation of cells in the dentate gyrus of the hippocampus in rats. Journal of Physiology, 549(2), 563–571. 10.1113/jphysiol.2003.041665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hairston IS, Little MTM, Scanlon MD, Barakat MT, Palmer TD, Sapolsky RM, & Heller HC (2005). Sleep Restriction Suppresses Neurogenesis Induced by Hippocampus-Dependent Learning. Journal of Neurophysiology, 94(6), 4224–4233. 10.1152/jn.00218.2005 [DOI] [PubMed] [Google Scholar]
- Hardy JA, & Higgins GA (1992). Alzheimer ’ s Disease : The Amyloid Cascade Hypothesis. Science, 256(5054), 184–185. 10.1155/2012/369808 [DOI] [PubMed] [Google Scholar]
- Hardy J, & Selkoe DJ (2002). The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science, 297, 353–356. 10.1126/science.1072994 [DOI] [PubMed] [Google Scholar]
- Havekes R, Park AJ, Tudor JC, Luczak VG, Hansen RT, Ferri SL, … Abel T (2016). Sleep deprivation causes memory deficits by negatively impacting neuronal connectivity in hippocampal area CA1. ELife, 5(AUGUST), 1–22. 10.7554/eLife.13424.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipolide DC, D’Almeida V, Raymond R, Tufik S, & Nobrega JN (2002). Sleep deprivation does not affect indicies of necrosis or apoptosis in rat brain. Intern. J. Neuroscience, (112), 155–166. [DOI] [PubMed] [Google Scholar]
- Hisler GC, Muranovic D, & Krizan Z (2019). Changes in sleep difficulties among the U.S. population from 2013 to 2017: results from the National Health Interview Survey. Sleep Health. 10.1016/j.sleh.2019.08.008 [DOI] [PubMed] [Google Scholar]
- Holth JK, Fritschi SK, Wang C, Pedersen NP, Cirrito JR, Mahan TE, … Holtzman DM (2019). The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science (New York, N.Y.), eaav2546 10.1126/science.aav2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J, Shen Q, Wan X, Zhao B, Wu Y, & Xia Z (2019). REM sleep deprivation-induced circadian clock gene abnormalities participate in hippocampal-dependent memory impairment by enhancing inflammation in rats undergoing sevoflurane inhalation. Behavioural Brain Research, 364, 167–176. 10.1016/J.BBR.2019.01.038 [DOI] [PubMed] [Google Scholar]
- Huitrón-Reséndiz S, Sánchez-Alavez M, Gallegos R, Berg G, Crawford E, Giacchino JL, … Criado JR (2002). Age-independent and age-related deficits in visuospatial learning, sleep-wake states, thermoregulation and motor activity in PDAPP mice. Brain Research, 928(1–2), 126–137. 10.1016/s0006-8993(01)03373-x [DOI] [PubMed] [Google Scholar]
- Hurtado-Alvarado G, Becerril-Villanueva E, Contis-Montes de Oca A, Domínguez-Salazar E, Salinas-Jazmín N, Pérez-Tapia SM, … Gómez-González B (2018). The yin/yang of inflammatory status: Blood-brain barrier regulation during sleep. Brain, Behavior, and Immunity, 69, 154–166. 10.1016/J.BBI.2017.11.009 [DOI] [PubMed] [Google Scholar]
- Hurtado-Alvarado Gabriela, Domínguez-Salazar E, Velázquez-Moctezuma J, & Gómez-González B (2016). A2A Adenosine Receptor Antagonism Reverts the Blood-Brain Barrier Dysfunction Induced by Sleep Restriction. PLOS ONE, 11(11), e0167236 10.1371/journal.pone.0167236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jameie SB, Mesgar S, Aliaghaei A, Raoofi A, Amini M, Khodagholi F, … Sadeghi Y (2019). Neuroprotective effect of exogenous melatonin on the noradrenergic neurons of adult male rats’ locus coeruleus nucleus following REM sleep deprivation. Journal of Chemical Neuroanatomy, 100, 101656 10.1016/J.JCHEMNEU.2019.101656 [DOI] [PubMed] [Google Scholar]
- Ju Y-ES, McLeland JS, Toedebusch CD, Xiong C, Fagan AM, Duntley SP, … Holtzman DM (2013). Sleep quality and preclinical Alzheimer disease. JAMA Neurology, 70(5), 587–593. 10.1001/jamaneurol.2013.2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju YES, Lucey BP, & Holtzman DM (2014, February). Sleep and Alzheimer disease pathology-a bidirectional relationship. Nature Reviews Neurology. 10.1038/nrneurol.2013.269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junek A, Rusak B, & Semba K (2010). Short-term sleep deprivation may alter the dynamics of hippocampal cell proliferation in adult rats. Neuroscience, 170(4), 1140–1152. 10.1016/J.NEUROSCIENCE.2010.08.018 [DOI] [PubMed] [Google Scholar]
- Kamali A-M, Noorafshan A, Karimi F, Karbalay-Doust S, & Nami M (2017). The Impact of Chronic Sleep Restriction on Neuronal Number and Volumetric Correlates of the Dorsal Respiratory Nuclei in a Rat Model. Sleep, 40(8). 10.1093/sleep/zsx072 [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, … Malinow R (2003). APP Processing and Synaptic Function. Neuron, 37(6), 925–937. 10.1016/S0896-6273(03)00124-7 [DOI] [PubMed] [Google Scholar]
- Kang DW, Lee CU, & Lim HK (2017). Role of Sleep Disturbance in the Trajectory of Alzheimer’s Disease. Clinical Psychopharmacology and Neuroscience : The Official Scientific Journal of the Korean College of Neuropsychopharmacology, 15(2), 89–99. 10.9758/cpn.2017.15.2.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J-E, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, … Holtzman DM (2009). Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science (New York, N.Y.), 326(5955), 1005–1007. 10.1126/science.1180962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastanenka KV, Hou SS, Shakerdge N, Logan R, Feng D, Wegmann S, … Bacskai BJ (2017). Optogenetic Restoration of Disrupted Slow Oscillations Halts Amyloid Deposition and Restores Calcium Homeostasis in an Animal Model of Alzheimer’s Disease. PloS One, 12(1), e0170275 10.1371/journal.pone.0170275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur T, Singh H, Mishra R, Manchanda S, Gupta M, Saini V, … Kaur G (2017). Withania somnifera as a potential anxiolytic and immunomodulatory agent in acute sleep deprived female Wistar rats. Molecular and Cellular Biochemistry, 427(1–2), 91–101. 10.1007/s11010-016-2900-1 [DOI] [PubMed] [Google Scholar]
- Khubchandani J, & Price JH (2019). Short Sleep Duration in Working American Adults, 2010–2018. Journal of Community Health. 10.1007/s10900-019-00731-9 [DOI] [PubMed] [Google Scholar]
- Kincheski GC, Valentim IS, Clarke JR, Cozachenco D, Castelo-Branco MTL, Ramos-Lobo AM, … Ferreira ST (2017). Chronic sleep restriction promotes brain inflammation and synapse loss, and potentiates memory impairment induced by amyloid-β oligomers in mice. Brain, Behavior, and Immunity, 64, 140–151. 10.1016/J.BBI.2017.04.007 [DOI] [PubMed] [Google Scholar]
- Knutson KL, Van Cauter E, Rathouz PJ, DeLeire T, & Lauderdale DS (2010). Trends in the prevalence of short sleepers in the USA: 1975-2006. Sleep, 33(1), 37–45. 10.1093/sleep/33.1.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamond N, Jay SM, Dorrian J, Ferguson SA, Jones C, & Dawson D (2007). The dynamics of neurobehavioural recovery following sleep loss. Journal of Sleep Research, 16(1), 33–41. 10.1111/j.1365-2869.2007.00574.x [DOI] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, & Duff K (2012). Trans-synaptic spread of tau pathology in vivo. PloS One, 7(2), e31302 10.1371/journal.pone.0031302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo JC, Groeger JA, Santhi N, Arbon EL, Lazar AS, Hasan S, … Dijk DJ (2012). Effects of Partial and Acute Total Sleep Deprivation on Performance across Cognitive Domains, Individuals and Circadian Phase. PLoS ONE, 7(9). 10.1371/journal.pone.0045987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutsey PL, Misialek JR, Mosley TH, Gottesman RF, Punjabi NM, Shahar E, … Alonso A (2018). Sleep characteristics and risk of dementia and Alzheimer’s disease: The Atherosclerosis Risk in Communities Study. Alzheimer’s and Dementia, 14(2), 157–166. 10.1016/j.jalz.2017.06.2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchanda S, Singh H, Kaur T, & Kaur G (2018). Low-grade neuroinflammation due to chronic sleep deprivation results in anxiety and learning and memory impairments. Molecular and Cellular Biochemistry, 449(1–2), 63–72. 10.1007/s11010-018-3343-7 [DOI] [PubMed] [Google Scholar]
- Minakawa EN, Miyazaki K, Maruo K, Yagihara H, Fujita H, Wada K, & Nagai Y (2017). Chronic sleep fragmentation exacerbates amyloid β deposition in Alzheimer’s disease model mice. Neuroscience Letters, 653, 362–369. 10.1016/J.NEULET.2017.05.054 [DOI] [PubMed] [Google Scholar]
- Mirescu C, Peters JD, Noiman L, & Gould E (2006). Sleep deprivation inhibits adult neurogenesis in the hippocampus by elevating glucocorticoids. Proceedings of the National Academy of Sciences of the United States of America, 103(50), 19170–19175. 10.1073/pnas.0608644103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra R, Manchanda S, Gupta M, Kaur T, Saini V, Sharma A, & Kaur G (2016). Tinospora cordifolia ameliorates anxiety-like behavior and improves cognitive functions in acute sleep deprived rats. Scientific Reports, 6, 25564 10.1038/srep25564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Jiménez EP, Flor-García M, Terreros-Roncal J, Rábano A, Cafini F, Pallas-Bazarra N, … Llorens-Martín M (2019). Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nature Medicine, 1 10.1038/s41591-019-0375-9 [DOI] [PubMed] [Google Scholar]
- Mueller AD, Pollock MS, Lieblich SE, Epp JR, Galea LAM, & Mistlberger RE (2008). Sleep deprivation can inhibit adult hippocampal neurogenesis independent of adrenal stress hormones. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, 294(5), R1693–R1703. 10.1152/ajpregu.00858.2007 [DOI] [PubMed] [Google Scholar]
- Murata Y, Oka A, Iseki A, Mori M, Ohe K, Mine K, & Enjoji M (2018). Prolonged sleep deprivation decreases cell proliferation and immature newborn neurons in both dorsal and ventral hippocampus of male rats. Neuroscience Research, 131, 45–51. 10.1016/J.NEURES.2017.08.008 [DOI] [PubMed] [Google Scholar]
- Naidoo N, Ferber M, Master M, Zhu Y, & Pack AI (2008). Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 28(26), 6539–6548. 10.1523/JNEUROSCI.5685-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noorafshan A, Karimi F, Kamali A-M, Karbalay-Doust S, & Nami M (2017). Restorative effects of curcumin on sleep-deprivation induced memory impairments and structural changes of the hippocampus in a rat model. Life Sciences, 189, 63–70. 10.1016/J.LFS.2017.09.018 [DOI] [PubMed] [Google Scholar]
- Noorafshan A, Karimi F, Kamali A-M, Karbalay-Doust S, & Nami M (2018). Could curcumin protect the dendritic trees of the CA1 neurons from shortening and shedding induced by chronic sleep restriction in rats? Life Sciences, 198, 65–70. 10.1016/J.LFS.2018.02.021 [DOI] [PubMed] [Google Scholar]
- Noorafshan A, Karimi F, Karbalay-Doust S, & Kamali AM (2017). Using curcumin to prevent structural and behavioral changes of medial prefrontal cortex induced by sleep deprivation in rats. EXCLI Journal, 16, 510–520. 10.17179/excli2017-139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norell-Clarke A, & Hagquist C (2017). Changes in sleep habits between 1985 and 2013 among children and adolescents in Sweden. Scandinavian Journal of Public Health, 45(8), 869–877. 10.1177/1403494817732269 [DOI] [PubMed] [Google Scholar]
- Novati A, Hulshof HJ, Koolhaas JM, Lucassen PJ, & Meerlo P (2011). Chronic sleep restriction causes a decrease in hippocampal volume in adolescent rats, which is not explained by changes in glucocorticoid levels or neurogenesis. Neuroscience, 190, 145–155. 10.1016/J.NEUROSCIENCE.2011.06.027 [DOI] [PubMed] [Google Scholar]
- Olonode ET, Aderibigbe AO, Adeoluwa OA, Eduviere AT, & Ben-Azu B (2019). Morin hydrate mitigates rapid eye movement sleep deprivation-induced neurobehavioural impairments and loss of viable neurons in the hippocampus of mice. Behavioural Brain Research, 356, 518–525. 10.1016/J.BBR.2017.12.024 [DOI] [PubMed] [Google Scholar]
- Orozco-Solis R, Montellier E, Aguilar-Arnal L, Sato S, Vawter MP, Bunney BG, … Sassone-Corsi P (2017). A Circadian Genomic Signature Common to Ketamine and Sleep Deprivation in the Anterior Cingulate Cortex. Biological Psychiatry, 82(5), 351–360. 10.1016/j.biopsych.2017.02.1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pase MP, Himali JJ, Grima NA, Beiser AS, Satizabal CL, Aparicio HJ, … Seshadri S (2017). Sleep architecture and the risk of incident dementia in the community. Neurology, 89(12), 1244–1250. 10.1212/WNL.0000000000004373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler AM, Noble W, & Hanger DP (2014). A role for tau at the synapse in Alzheimer’s disease pathogenesis. Neuropharmacology, 76 Pt A, 1–8. 10.1016/j.neuropharm.2013.09.018 [DOI] [PubMed] [Google Scholar]
- Qiu H, Zhong R, Liu H, Zhang F, Li S, & Le W (2016). Chronic Sleep Deprivation Exacerbates Learning-Memory Disability and Alzheimer’s Disease-Like Pathologies in AβPPswe/PS1ΔE9 Mice. Journal of Alzheimer’s Disease, 50(3), 669–685. 10.3233/JAD-150774 [DOI] [PubMed] [Google Scholar]
- Raven F, Meerlo P, Van der Zee EA, Abel T, & Havekes R (2018). A brief period of sleep deprivation causes spine loss in the dentate gyrus of mice. Neurobiology of Learning and Memory. 10.1016/J.NLM.2018.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert S, Pavón Arocas O, & Rihel J (2019). The Neuropeptide Galanin Is Required for Homeostatic Rebound Sleep following Increased Neuronal Activity. Neuron, 104(2), 370–384. e5 10.1016/j.neuron.2019.08.010 [DOI] [PubMed] [Google Scholar]
- Roh JH, Huang Y, Bero AW, Kasten T, Stewart FR, Bateman RJ, & Holtzman DM (2012). Disruption of the sleep-wake cycle and diurnal fluctuation of β-amyloid in mice with Alzheimer’s disease pathology. Science Translational Medicine, 4(150), 150ra122 10.1126/scitranslmed.3004291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh JH, Jiang H, Finn MB, Stewart FR, Mahan TE, Cirrito JR, … Holtzman DM (2014). Potential role of orexin and sleep modulation in the pathogenesis of Alzheimer’s disease. The Journal of Experimental Medicine, 211(13), 2487–2496. 10.1084/jem.20141788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman V, Van der Borght K, Leemburg SA, Van der Zee EA, & Meerlo P (2005). Sleep restriction by forced activity reduces hippocampal cell proliferation. Brain Research, 1065(1–2), 53–59. 10.1016/J.BRAINRES.2005.10.020 [DOI] [PubMed] [Google Scholar]
- Rothman SM, Herdener N, Frankola KA, Mughal MR, & Mattson MP (2013). Chronic mild sleep restriction accentuates contextual memory impairments, and accumulations of cortical Aβ and pTau in a mouse model of Alzheimer’s disease. Brain Research, 1529, 200–208. 10.1016/j.brainres.2013.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu S, Kauser H, Ray K, Kishore K, Kumar S, & Panjwani U (2013). Caffeine and modafinil promote adult neuronal cell proliferation during 48 h of total sleep deprivation in rat dentate gyrus. Experimental Neurology, 248, 470–481. 10.1016/J.EXPNEUROL.2013.07.021 [DOI] [PubMed] [Google Scholar]
- Scammell TE, Matheson JK, Honda M, Thannickal TC, & Siegel JM (2012). Coexistence of narcolepsy and Alzheimer’s disease. Neurobiology of Aging, 33(7), 1318–1319. 10.1016/j.neurobiolaging.2010.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider F, Baldauf K, Wetzel W, & Reymann KG (2014). Behavioral and EEG changes in male 5xFAD mice. Physiology & Behavior, 135, 25–33. 10.1016/j.physbeh.2014.05.041 [DOI] [PubMed] [Google Scholar]
- Singh A, Das G, Kaur M, & Mallick BN (2019). Noradrenaline acting on alpha1 adrenoceptor as well as by chelating iron reduces oxidative burden on the brain: Implications with rapid eye movement sleep. Frontiers in Molecular Neuroscience, 12 10.3389/fnmol.2019.00007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somarajan BI, Khanday MA, & Mallick BN (2016). Rapid Eye Movement Sleep Deprivation Induces Neuronal Apoptosis by Noradrenaline Acting on Alpha1 Adrenoceptor and by Triggering Mitochondrial Intrinsic Pathway. Frontiers in Neurology, 7, 25 10.3389/fneur.2016.00025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto-Rodriguez S, Lopez-Armas G, Luquin S, Ramos-Zuñiga R, Jauregui-Huerta F, Gonzalez-Perez O, & Gonzalez-Castañeda RE (2016). Rapid Eye Movement Sleep Deprivation Produces Long-Term Detrimental Effects in Spatial Memory and Modifies the Cellular Composition of the Subgranular Zone. Frontiers in Cellular Neuroscience, 10(May), 1–13. 10.3389/fncel.2016.00132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spano GM, Banningh SW, Marshall W, de Vivo L, Bellesi M, Loschky SS, … Cirelli C (2019). Sleep Deprivation by Exposure to Novel Objects Increases Synapse Density and Axon-Spine Interface in the Hippocampal CA1 Region of Adolescent Mice. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 39(34), 6613–6625. 10.1523/JNEUROSCI.0380-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Hilaire MA, Rüger M, Fratelli F, Hull JT, Phillips AJK, & Lockley SW (2017). Modeling neurocognitive decline and recovery during repeated cycles of extended sleep and chronic sleep deficiency. Sleep, 40(1). 10.1093/sleep/zsw009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner B, Kronenberg G, Jessberger S, Brandt MD, Reuter K, & Kempermann G (2004). Differential regulation of gliogenesis in the context of adult hippocampal neurogenesis in mice. Glia, 46(1), 41–52. 10.1002/glia.10337 [DOI] [PubMed] [Google Scholar]
- Suchecki D, Lobo L, Hipolide D, & Tufik S (1998). Increased ACTH and corticosterone secretion induced by different methods of paradoxical sleep deprivation. Journal of Sleep Research, 7(4), 276–281. 10.1046/j.1365-2869.1998.00122.x [DOI] [PubMed] [Google Scholar]
- Tabuchi M, Lone SR, Liu S, Liu Q, Zhang J, Spira AP, & Wu MN (2015). Sleep interacts with Aβ to modulate intrinsic neuronal excitability. Current Biology, 25(6), 702–712. 10.1016/j.cub.2015.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke E. a., Grønborg M, Riedel D, … Jahn R (2006). Molecular Anatomy of a Trafficking Organelle. Cell (Vol. 127). 10.1016/j.cell.2006.10.030 [DOI] [PubMed] [Google Scholar]
- Toossi H, del Cid-Pellitero E, & Jones BE (2017). Homeostatic changes in GABA and acetylcholine muscarinic receptors on GABAergic neurons in the mesencephalic reticular formation following sleep deprivation. ENeuro, 4(6). 10.1523/ENEURO.0269-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung A, Takase L, Fornal C, & Jacobs B (2005). Effects of sleep deprivation and recovery sleep upon cell proliferation in adult rat dentate gyrus. Neuroscience, 134(3), 721–723. 10.1016/J.NEUROSCIENCE.2005.06.008 [DOI] [PubMed] [Google Scholar]
- van der Borght K, Ferrari F, Klauke K, Roman V, Havekes R, Sgoifo A, … Meerlo P (2006). Hippocampal cell proliferation across the day: Increase by running wheel activity, but no effect of sleep and wakefulness. Behavioural Brain Research, 167(1), 36–41. 10.1016/J.BBR.2005.08.012 [DOI] [PubMed] [Google Scholar]
- Van Dongen HPA, Maislin G, Mullington JM, & Dinges DF (2003). The Cumulative Cost of Additional Wakefulness: Dose-Response Effects on Neurobehavioral Functions and Sleep Physiology From Chronic Sleep Restriction and Total Sleep Deprivation. Sleep, 26(2), 117–126. 10.1093/sleep/26.2.117 [DOI] [PubMed] [Google Scholar]
- Wadhwa M, Kumari P, Chauhan G, Roy K, Alam S, Kishore K, … Panjwani U (2017). Sleep deprivation induces spatial memory impairment by altered hippocampus neuroinflammatory responses and glial cells activation in rats. Journal of Neuroimmunology, 312, 38–48. 10.1016/J.JNEUROIM.2017.09.003 [DOI] [PubMed] [Google Scholar]
- Wadhwa M, Prabhakar A, Anand JP, Ray K, Prasad D, Kumar B, & Panjwani U (2019). Complement activation sustains neuroinflammation and deteriorates adult neurogenesis and spatial memory impairment in rat hippocampus following sleep deprivation. Brain, Behavior, and Immunity, 82, 129–144. 10.1016/j.bbi.2019.08.004 [DOI] [PubMed] [Google Scholar]
- Wadhwa M, Prabhakar A, Ray K, Roy K, Kumari P, Jha PK, … Panjwani U (2017). Inhibiting the microglia activation improves the spatial memory and adult neurogenesis in rat hippocampus during 48 h of sleep deprivation. Journal of Neuroinflammation, 14(1), 222 10.1186/s12974-017-0998-z [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wang C, & Holtzman DM (2019). Bidirectional relationship between sleep and Alzheimer’s disease: role of amyloid, tau, and other factors. Neuropsychopharmacology, 1–17. 10.1038/s41386-019-0478-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Balaji V, Kaniyappan S, Krüger L, Irsen S, Tepper K, … Mandelkow EM (2017). The release and trans-synaptic transmission of Tau via exosomes. Molecular Neurodegeneration, 12(1). 10.1186/s13024-016-0143-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, & Gundersen HJG (1990). Unbiased Stereological Estimation of the Number of Neurons i n the Human Hippocampus. The Journal of Comparative Neurology, 296, 1–22. [DOI] [PubMed] [Google Scholar]
- Winston CN, Aulston B, Rockenstein EM, Adame A, Prikhodko O, Dave KN, … Yuan SH (2019). Neuronal Exosome-Derived Human Tau is Toxic to Recipient Mouse Neurons in vivo. Journal of Alzheimer’s Disease : JAD, 67(2), 541–553. 10.3233/JAD-180776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisor JP, Pasumarthi RK, Gerashchenko D, Thompson CL, Pathak S, Sancar A, … Kilduff TS (2008). Sleep deprivation effects on circadian clock gene expression in the cerebral cortex parallel electroencephalographic differences among mouse strains. Journal of Neuroscience, 28(28), 7193–7201. 10.1523/JNEUROSCI.1150-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, … Duff KE (2016). Neuronal activity enhances tau propagation and tau pathology in vivo. Nature Neuroscience, 19(8), 1085–1092. 10.1038/nn.4328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Xie L, Kang H, Xu Q, Chen MJ, Liao Y, … Nedergaard M (2013). Sleep Drives Metabolite Clearance from the Adult Brain. Science, 342(October), 373–378. 10.1126/science.1241224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Holth JK, Liao F, Stewart FR, Mahan TE, Jiang H, … Holtzman DM (2014). Neuronal activity regulates extracellular tau in vivo. Journal of Experimental Medicine, 211(3), 387–393. 10.1084/jem.20131685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin M, Chen Y, Zheng H, Pu T, Marshall C, Wu T, & Xiao M (2017). Assessment of mouse cognitive and anxiety-like behaviors and hippocampal inflammation following a repeated and intermittent paradoxical sleep deprivation procedure. Behavioural Brain Research, 321, 69–78. 10.1016/J.BBR.2016.12.034 [DOI] [PubMed] [Google Scholar]
- Zhang B, Veasey SC, Wood MA, Leng LZ, Kaminski C, Leight S, … Trojanowski JQ (2005). Impaired rapid eye movement sleep in the Tg2576 APP murine model of Alzheimer’s disease with injury to pedunculopontine cholinergic neurons. The American Journal of Pathology, 167(5), 1361–1369. 10.1016/S0002-9440(10)61223-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhu Y, Zhan G, Fenik P, Panossian L, Wang MM, … Veasey S (2014). Extended wakefulness: Compromised metabolics in and degeneration of locus ceruleus neurons. Journal of Neuroscience, 34(12), 4418–4431. 10.1523/JNEUROSCI.5025-12.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhang HQ, Liang XY, Zhang HF, Zhang T, & Liu FE (2013). Melatonin ameliorates cognitive impairment induced by sleep deprivation in rats: Role of oxidative stress, BDNF and CaMKII. Behavioural Brain Research, 256, 72–81. 10.1016/j.bbr.2013.07.051 [DOI] [PubMed] [Google Scholar]
- Zhao H-Y, Wu H-J, He J-L, Zhuang J-H, Liu Z-Y, Huang L-Q, & Zhao Z-X (2017). Chronic Sleep Restriction Induces Cognitive Deficits and Cortical Beta-Amyloid Deposition in Mice via BACE1-Antisense Activation. CNS Neuroscience & Therapeutics, 23(3), 233–240. 10.1111/cns.12667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Fenik P, Zhan G, Somach R, Xin R, & Veasey S (2016). Intermittent short sleep results in lasting sleep wake disturbances and degeneration of locus coeruleus and orexinergic neurons. Sleep, 39(8), 1601–1611. 10.5665/sleep.6030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Zhan G, Fenik P, Brandes M, Bell P, Francois N, … Veasey S (2018). Chronic Sleep Disruption Advances the Temporal Progression of Tauopathy in P301S Mutant Mice. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 38(48), 10255–10270. 10.1523/JNEUROSCI.0275-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]