

Abstract
Anticancer drug-loaded nanoparticles have been explored extensively to decrease side effects while improving their therapeutic efficacy. However, due to the low drug loading content, premature drug release, non-standardized carrier structure, and difficulty in predicting the fate of the carrier, only a few nanomedicines have been approved by the FDA. Hereby, we developed a carrier-free nanoparticle based on the self-assembly of the curcumin-erlotinib conjugate (EPC), which has a size of about 105 nm. The EPC nano-assembly exhibited more potent cell killing, better anti-migration and anti-invasion effects for BxPC-3 pancreatic cancer cells than the combination of free curcumin and erlotinib. Furthermore, benefited from both passive and active tumor targeting effect, EPC nano-assembly could effectively accumulate in the tumor tissue in a xenograft pancreatic tumor mouse model. Consequently, EPC effectively reduced the growth of pancreatic tumors and extended the median survival time of the tumor-bearing mice from 22 days to 68 days. In addition, no systemic toxicity was detected in the major organs from the mice receiving EPC treatment. Attributed to the uniformity of the curcumin-erlotinib conjugate and easiness of scaling up, we expect the EPC could be translated into a powerful tool in fighting against pancreatic cancer and other EGFR positive cancers.
Keywords: Carrier-free, curcumin, erlotinib, nano-assembly, pancreatic cancer
Graphical Abstract
Carrier-free nano-assembly, EPC, is fabricated from a curcumin-erlotinib conjugate, which has the merit of uniform structure and easy scaling-up. Due to the existence of erlotinib outlayer, EPC effectively accumulates in the tumor and enters EGFR positive cancer cells. Benefit from the synergetic effect of curcumin and erlotinib, EPC reduces the growth of tumors and extends the survival time of the animal with pancreatic cancer.
1. Introduction
To date, cancer is still one of the most lethal causes of death around the world.[1] Pancreatic cancer, amongst all the types of cancer, is one of the extremely malignant ones. The 5-year survival rate for pancreatic cancer has remained lower than 10% for over four decades.[1–2] Given the stubborn and metastatic properties of pancreatic cancer, chemotherapy is the most common and reliable therapeutic method for the treatment. However, gemcitabine, which has been the standard care of chemotherapy for pancreatic cancer for two decades since it was approved by the FDA in 1996, can only achieve a median survival rate of about six months.[3] As a consequence, the development of more effective chemotherapeutic agents for pancreatic cancer is urgently needed.
In 2005, erlotinib (ELT) was approved by the FDA for the treatment of advanced and metastatic pancreatic cancer in combination with gemcitabine. ELT is an epidermal growth factor receptor (EGFR) inhibitor, which has been utilized to treat non-small cell lung cancer, pancreatic cancer, and other types of cancer.[4] The combination of erlotinib with gemcitabine prolonged the median survival rate of gemcitabine monotherapy.[4a] Curcumin (CCM), a natural yellow powder derived from the rhizome of a turmeric plant that is broadly grown in Southeast Asia,[5] possesses a variety of biological and pharmacological activities, including analgesic, antiseptic, anti-oxidant, anti-inflammatory, and anticancer activities.[6] However, the biological applications of CCM are severely limited by some undesired properties such as poor water solubility, short serum half-life, and low bioavailability.[7] Many researchers found that the combination of CCM and other anticancer drugs exhibit boosted therapeutic effect in treating breast cancer,[8] prostate cancer,[9] lung cancer,[10] and ovarian cancer.[11]
Recently, a synergistic effect was found between ELT and CCM, and the combination therapy with ELT and CCM has been used to treat many types of cancer.[12] It was reported that the combined usage of ELT and CCM could affect the drug resistance signaling pathways by decreasing the αvβ3 integrin and increasing PDK4 gene expression, resulting in higher anticancer efficacy.[12b] Despite the better performance of the combination therapy compared to individual drugs, the aforementioned limitations of CCM are still unresolved, which restricts its applications in the biomedical field. Herein, we developed an ELT and CCM conjugated carrier-free nano-assembly (EPC) to circumvent the limitations of CCM and enhance the synergistic anticancer effect of the two drugs.
2. Results and Discussion
2.1. EPC nano-assembly preparation and characterization
Two anticancer drugs erlotinib (ELT) and curcumin (CCM) were linked together by a bifunctional polyethylene glycol (PEG) chain, which contains both azide and carboxylic acid groups, to produce our desired compound erlotinib-PEG-curcumin (EPC) in two steps (Figure 1A). ELT was first conjugated to PEG via click chemistry, followed by the attachment of CCM through an esterification reaction. The chemical structures of ELT-PEG and EPC were confirmed by mass spectroscopy (MS) and nuclear magnetic resonance (NMR) analysis (Figure S1–5).
Figure 1.
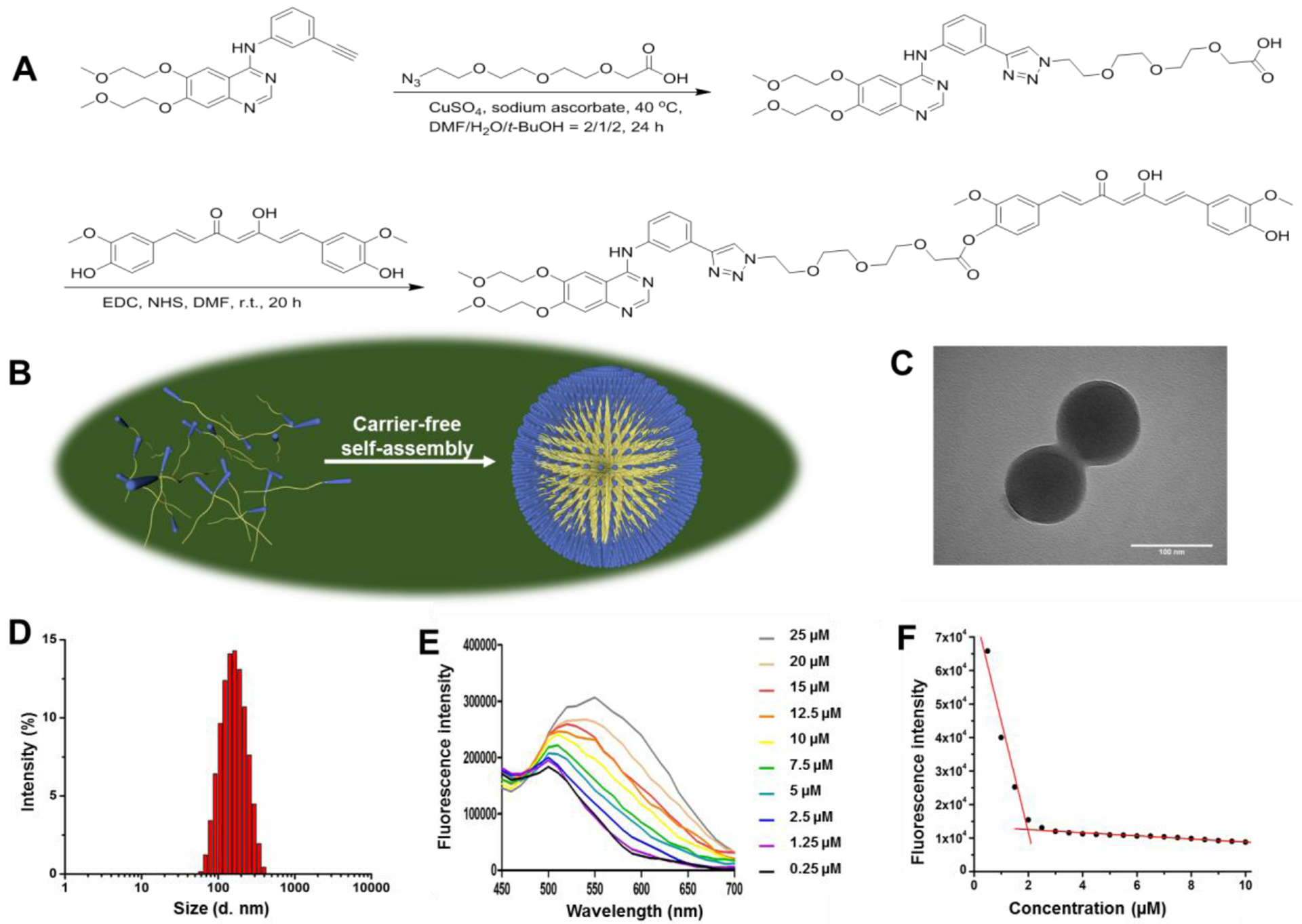
The development of EPC nano-assembly and its characterization. (A) The synthesis scheme of EPC molecule. (B) The self-assembly of EPC nano-assembly. The TEM image (C), size distribution (D), fluorescence spectra (E), and CMC determination (F) of EPC nano-assembly.
A nano-assembly of EPC was fabricated through a one-step nano-precipitation process. When it was dispersed into water, EPC self-assembled into uniform nano-assemblies because of its amphiphilic property (Figure 1B). Transmission electron microscopy (TEM) revealed that EPC had a spherical-shaped morphology and a size of about 105 nm (Figure 1C). Dynamic light scattering (DLS) showed that the EPC nano-assembly had a hydrodynamic size of 146.3 nm with a PDI of 0.157 (Figure 1D) and a zeta potential of −7.3 mV. The critical micellar concentration (CMC) of EPC was determined to be 1.95 μM by measuring the fluorescence of EPC at varied concentrations in water (Figure 1E and 1F).
2.2. Cellular uptake of EPC nano-assembly
Confocal laser scanning microscopy (CLSM) and flow cytometry were adopted to evaluate the cellular uptake of EPC in human pancreatic cancer cells BxPC-3. As shown in Figure 2A, the fluorescence emitted from cells indicated that both CCM and EPC could be taken up by cells. Cells incubated with CCM emitted stronger fluorescence than those with EPC because the fluorescence intensity of CCM is much higher than that of EPC at the same concentration (Figure S6). To compare the absolute amount of CCM and EPC entered BxPC-3 cells, cells were harvested and lysed. The fluorescence intensities of the cell lysates were compared with the corresponding calibration curve of CCM and EPC to calculate their intracellular concentrations. It was found that the intracellular CCM and EPC were 0.023 and 0.292 μM, respectively, suggesting that EPC entered BxPC-3 cells much more effectively than free CCM. Flow cytometry analysis found that CCM treated cells exhibited stronger fluorescence signal than EPC, which is in good agreement with the outcomes achieved from CLSM (Figure 2B and 2C). Furthermore, the addition of free ELT inhibited the cellular uptake of EPC, which validates that EPC enters BxPC-3 cells through EGFR receptor-mediated endocytosis, which is the main cause for the boosted EPC uptake.
Figure 2.
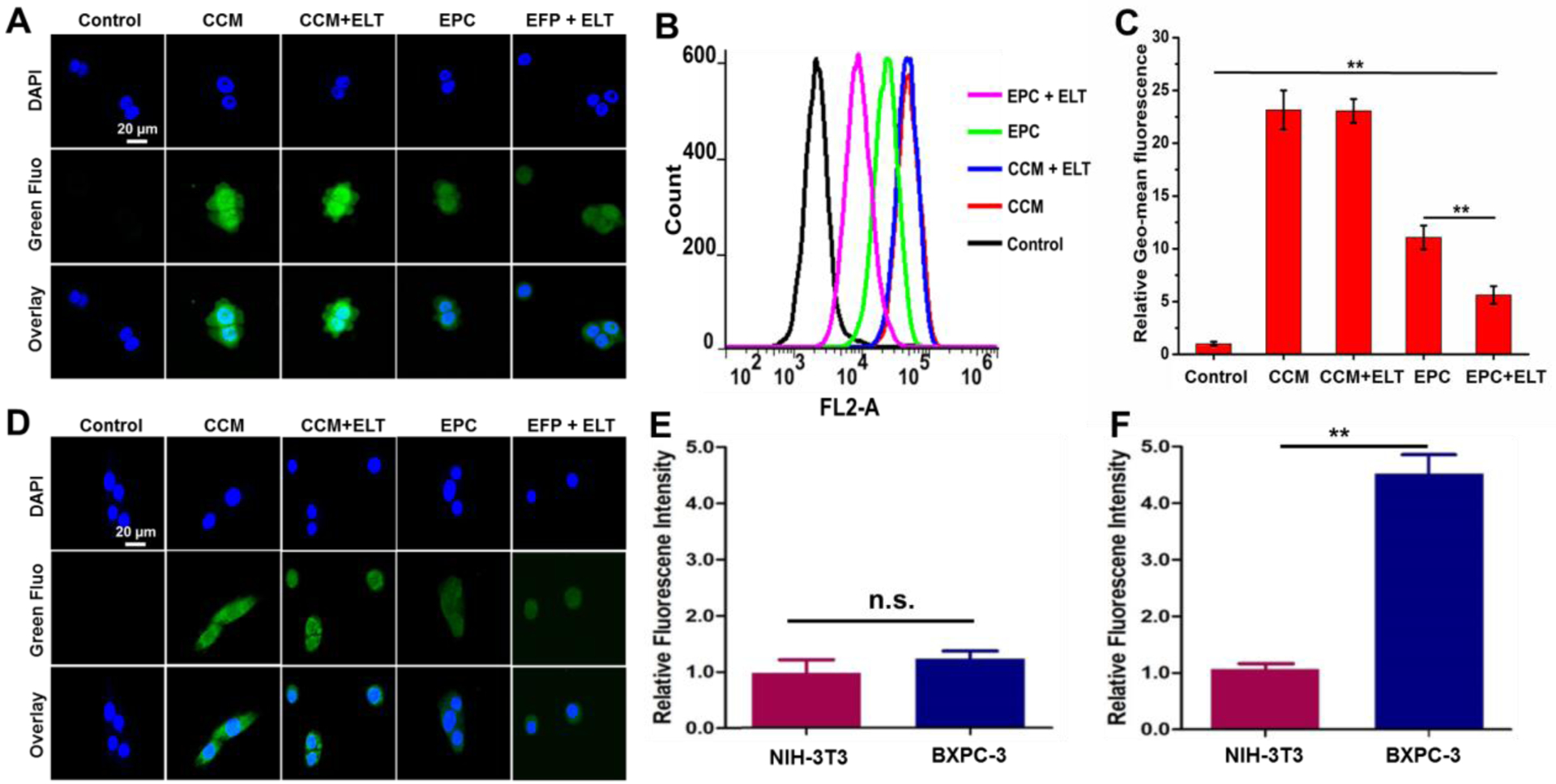
Cellular uptake of CCM, CCM + ELT, EPC, and EPC + ELT. Representative CLSM images of BxPC-3 (A) and NIH-3T3 (D) cells after various treatments for 3 h. Cell nuclei were stained with Hoechst 33342. The green fluorescence (Green Fluo channel) is from CCM/EPC and the blue fluorescence (DAPI channel) is from Hoechst 33342. The scale bar is 20 μm. Flow cytometry spectra (B) and mean fluorescence value (C) of BxPC-3 cells after various treatments for 3h. Relative fluorescence intensity of CCM (E) and EPC (F) treated NIH-3T3 and BxPC-3 cells. n = 3, **P < 0.01.
To investigate the cancer cell selectivity of EPC, cellular uptake of CCM and EPC was also studied in NIH-3T3 cells and analyzed by confocal microscopy and protein extraction assay. The fluorescence intensity of CCM in both BXPC-3 and NIH-3T3 cells are similar as observed with confocal microscopy (Figure 2A and 2D) and quantified by protein extraction method (Figure 2E). Since EPC in serum containing environment (Figure S6), such as cytoplasm, exhibits relatively dim fluorescence signal, it is hard to tell EPC intensity difference in BXPC-3 and NIH-3T3 cells with confocal microscopy (Figure 2A and 2D). However, protein extraction method revealed that EPC exhibited a much stronger capacity in entering BxPC-3 cells than NIH-3T3 cells (Figure 2F).
2.3. Cytotoxicity of EPC nano-assembly
The cytotoxicity of EPC was evaluated in both BxPC-3 cells and NIH-3T3 cells by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay at varying concentrations. As expected, the combination of CCM and ELT killed more cancer cells than CCM and ELT alone (Figure 3A). EPC exhibited an inhibitory effect on cell proliferation in a dose-dependent manner. More importantly, EPC was more potent than the combination of CCM and free ELT in killing cancer cells. Furthermore, compared with the free drug counterpart, EPC was much less toxic to NIH-3T3 cells, suggesting that EPC can selectively kill cancer cells ((Figure 3B).
Figure 3.
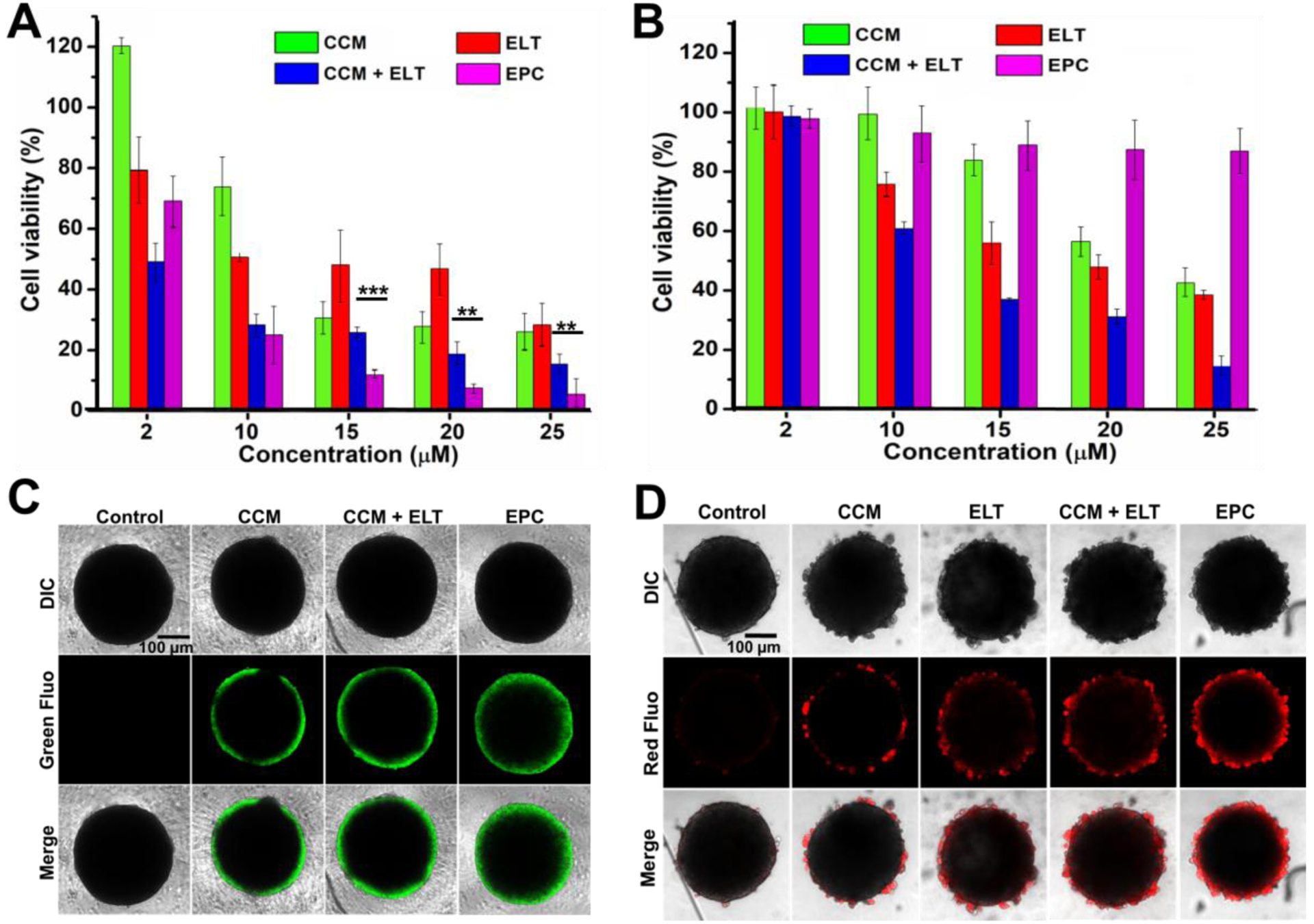
Cytotoxicity of CCM and EPC in 2D and 3D culture. Cell viability of BxPC-3 cells (A) and NIH-3T3 cells (B) after receiving various treatments in 96 well plate 2D culture for 24 h. n = 3, **P < 0.01 and ***P < 0.001. Fluorescence images of drug penetration (C) and cell killing effect (D) in BxPC-3 tumor spheroids. Green fluorescence signal of CCM and EPC in (C), and red fluorescence signal propidium iodide staining for dead cell in (D). Scale bars in (C) and (D) are 100 μm.
To some extent, 2-D cell culture has some limitations in predicting anticancer efficacy of drugs in vivo. The anticancer efficacy of nanomedicines is affected not only by cellular uptake but also by the depth that a particle can reach in a solid tumor mass. A multicellular tumor spheroid model reflects the anticancer efficacy of nanoparticles in vivo more accurately because it resembles many features of solid tumor in vivo, such as poor vascularization, high interstitial pressure, and oxygen/nutrient gradients.[13] Therefore, we evaluated the tumor inhibition effect of EPC in tumor spheroid prior to the in vivo investigation. Upon the formation of BxPC-3 tumor spheroid in a 96-well ultra-low attachment plate, the penetration ability of EPC was investigated. As shown in Figure 3C, tumor spheroids incubated with EPC exhibited thicker fluorescent shell compared to those with CCM, which indicates that EPC could penetrate deeper into the tumor spheroid. Moreover, the co-incubation of CCM and ELT did not affect the penetration of CCM in the tumor spheroid.
Thanks to its better tumor-penetrating ability, EPC is more efficient in destroying tumor spheroid than CCM, ELT, or the combination of CCM and ELT. As displayed in Figure 3D, the tumor spheroid in the control group shows a clear smooth boundary under a microscope, whereas an irregular layer of buddings was observed on the tumor spheroids treated with CCM, ELT, and their combination, suggesting that these tumor spheroids were partially damaged. More importantly, the totally broken edges of tumor spheroids incubated with EPC indicated that they were severely damaged by EPC. In order to visually evaluate the antitumor effect of those treatments, dead cells in the tumor spheroid were stained with propidium iodide (PI). As shown in Figure 3D, a thicker reddish layer on the tumor spheroid treated with EPC than other treatment groups was observed, suggesting much more dead cells were found in EPC treated tumor spheroids than other treatment groups, which confirmed the potent antitumor efficacy of EPC.
2.4. Anti-migration and anti-invasion effect of EPC nano-assembly
In addition to cell viability test, wound healing assay, cell adhesion assay, and Transwell invasion assay were carried out to investigate the effectiveness of CCM, ELT, CCM+ELT, and EPC in inhibiting the migration and invasion of cancer cells. As displayed in Figure 4A and 4B, apparent healing progress was observed for the wound of cells in the control group after 24 h. However, the treatment of CCM, ELT, and CCM+ELT effectively delayed the healing process of the wound to varying degrees. Surprisingly, almost no visible healing effect was detected in the cells treated with EPC, which suggested that EPC effectively prevented the wound from getting healed. Cell adhesion assay revealed that all the treatments significantly hindered the adhesion of cancer cells to Matrigel (Figure 4C and 4D). The results of Transwell invasion assay further demonstrated that the treatments of CCM, ELT, CCM+ELT, and EPC inhibited the invasion of cancer cells through the Matrigel-coated membrane, indicating that they all have an anti-invasive function on the cancer cells (Figure 4E and 4F). Amongst them, EPC exhibited the highest anti-invasive efficacy.
Figure 4.
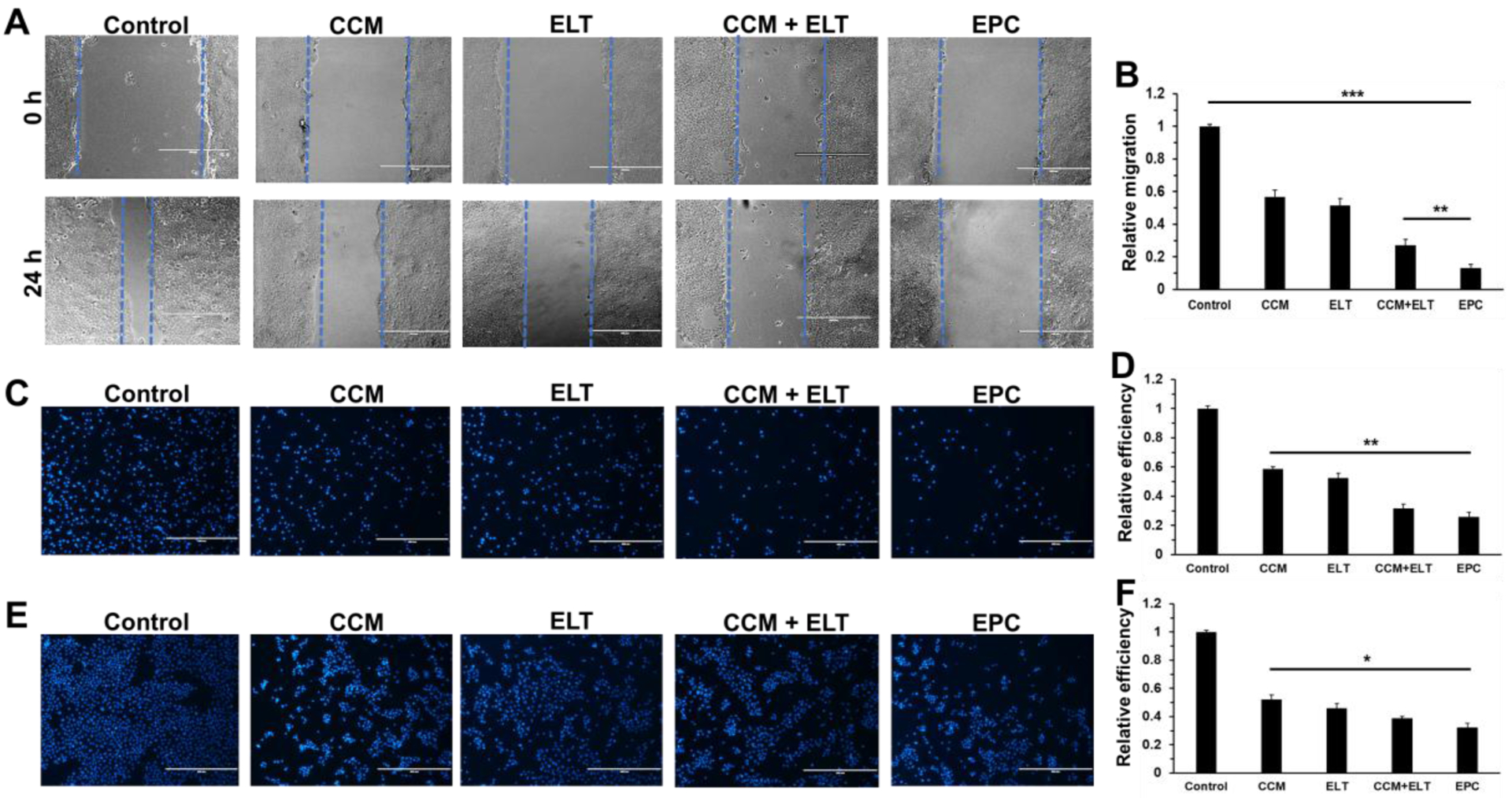
Wound healing, adhesion, and invasion assays of BxPC-3 cells after being treated with CCM, ELT, CCM+ELT, and EPC for 24 h. Images of wound at 0 h and 24 h (A) after receiving different treatments and relative cell migration during 24 h of treatment (B). Images of cells after 24 h of adhesion assay (C) and the relative adhesion efficiency of different treatments (D). Images of cells after 24 h of Transwell invasion assay (E) and the relative invasion efficiency of different treatments (F). Cells were stained with DAPI in (C) and (E). Scale bars in A, C, and E are 400 μm. n = 3, *P < 0.05; **P < 0.01; and ***P < 0.001.
2.5. Biodistribution of EPC nano-assembly
Based on the above promising in vitro results, we further investigated the antitumor efficacy of EPC in vivo. BxPC-3 cells were injected subcutaneously to female nude mice to establish a tumor model. The biodistribution of EPC nano-assembly was examined in BxPC-3 tumor-bearing mice. Figure 5A and 5B revealed that there were much stronger fluorescence signals in tumors of mice administrated with EPC nano-assemblies compared to those of mice treated with CCM, suggesting a higher tumor accumulation and retention of EPC nano-assemblies in tumor tissues than free CCM.
Figure 5.
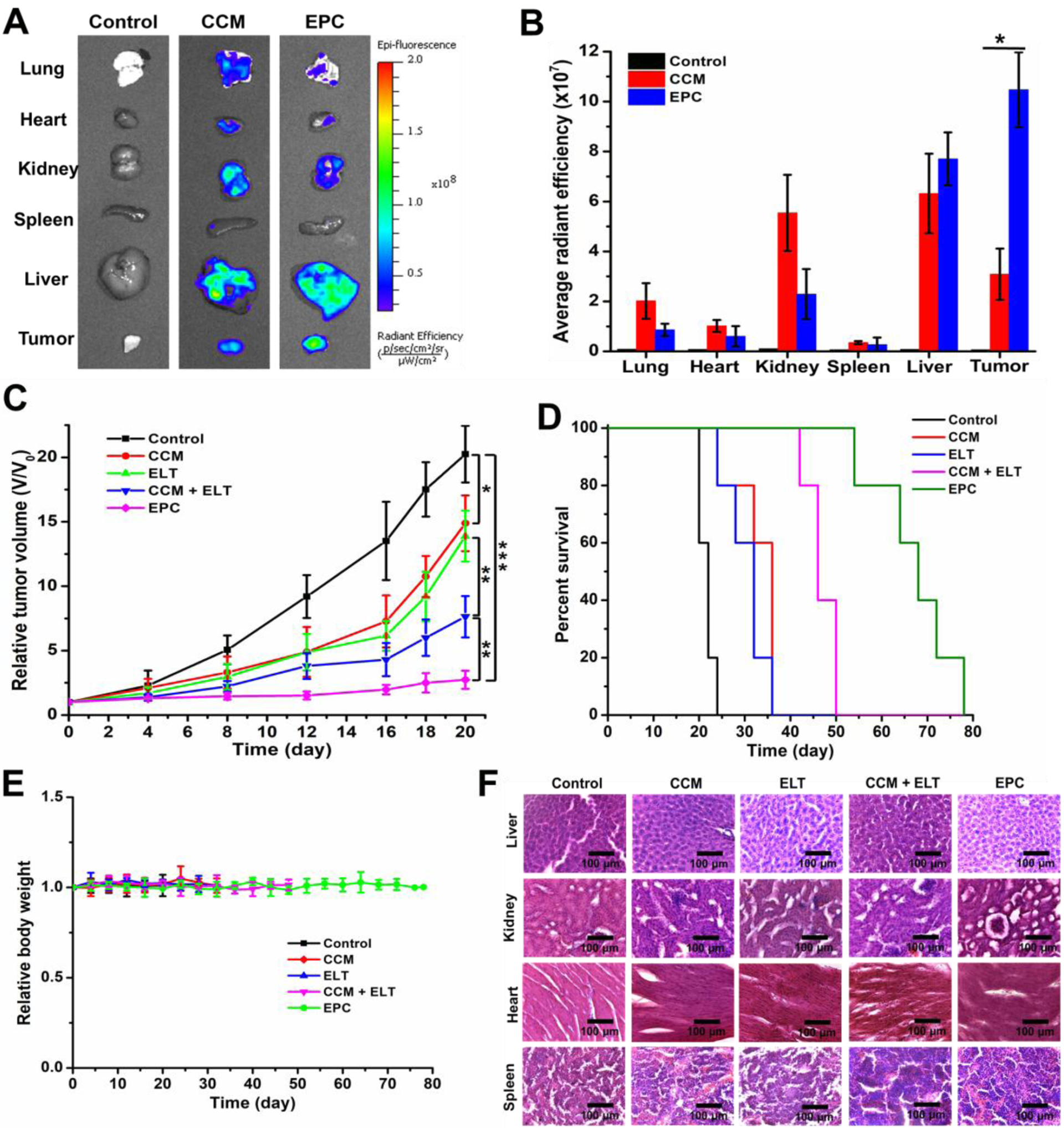
In vivo study of the EPC nano-assembly in a pancreatic xenograft mouse tumor model. Ex vivo biodistribution (A) and quantitative fluorescence intensity (B) of CCM and EPC in different organs. Tumor growth profiles (C) and survival curve (D) for mice treated with different formulations. n=5, *P < 0.05; **P < 0.01; and ***P < 0.001. Body weight changes of mice receiving different treatments (E). Images of H&E stained major organs tissue sections of different treatment groups (F). Scale bars in (F) are 100 μm.
2.6. Tumor growth inhibitory effect of EPC nano-assembly
To evaluate the in vivo anticancer efficacy of EPC, tumor-bearing mice were intravenously administrated with PBS, CCM, ELT, CCM+ELT, or EPC nano-assemblies twice a week. On day 20 post-administration, the tumor volumes of mice in the control group increased by about 20-fold (Figure 5C), while the tumor volumes of CCM, ELT, and CCM+ELT treated mice increased by 15-fold, 14-fold, and 7.6-fold, respectively. In contrast, only 2.7-fold enlargement was found in tumor volumes of mice administrated with EPC during the same period, which confirmed the excellent inhibitory effect of EPC towards tumor growth. Kaplan–Meier survival analysis was carried out for 11 weeks. The control group showed 100% lethal by 24 days (Figure 5D). Free CCM and ELT alone treatments slightly extended the 100% lethal time to 36 days, while the combination of CCM and ELT, as well as EPC postponed the total lethal time to 50 and 78 days, respectively. Correspondingly, the median survival time for control, CCM, ELT, combination of CCM and ELT, and EPC were 22, 36, 32, 46, and 68 days, respectively.
2.7. Systemic toxicity analysis
There is no significant change was observed in the body weight of all mice during the entire process of treatment (Figure 5E), which suggested that EPC did not induce systemic toxicity to the mice. Furthermore, histological analysis did not detect any obvious pathological change in the liver, kidney, heart, and spleen tissues among all treatment groups (Figure 5F).
3. Discussion
As a deadly threat to human health, pancreatic cancer has become more and more prominent while significant progress has been in the treatment of other types of cancer, which is partially attributed to the lack of efficient chemotherapeutic agents. Erlotinib and curcumin both have demonstrated anticancer effectiveness against pancreatic cancer,[14] and a synergistic effect has been discovered between them when they are given in combination for cancer treatment.[12] The poor water solubility and low bioavailability of CCM severely limit the biomedical applications of this combination chemotherapy. Some research attempted to solve the problem by employing drug carriers such as micelles to deliver CCM and positive results were achieved.[15] The strategy we provide here does not need any delivery vehicles, named carrier-free drug delivery.
There is a triple bond in the chemical structure of ELT, which allows for introducing modifications to ELT through click chemistry.[16] We utilized a hydrophilic bifunctional PEG, which bears an azido group and a carboxylic acid group on either end, to link ELT and CCM via click chemistry and esterification reaction, respectively. The resulted compound EPC is composed of both hydrophilic and hydrophobic moieties, which renders it capable of self-assembling into nano-assemblies through a one-step nano-precipitation process. The spherical shape and a uniform size of 105 nm of the EPC nano-assembly, as observed by TEM, makes it a qualified candidate for biological applications. Moreover, DLS determination showed that it has high colloidal stability in aqueous media, suggesting that the EPC nano-assembly will be stable in blood circulation.
The EPC molecule can emit fluorescence, resulting from the fluorescent CCM moiety. Such a preferred property makes it convenient to measure the critical micellar concentration (CMC) of EPC and to investigate the cellular uptake of the EPC nano-assemblies. The low CMC value of EPC, 1.95 μM, ensures that EPC is stable after being injected into the blood stream. Due to the overexpressing of EGFR receptor in BxPC-3 cells, EPC nano-assembly could efficiently enter cancer cells via receptor-mediated endocytosis as evidenced by flow cytometry analysis. The in vitro cytotoxicity test showed that the combination usage of ELT and CCM is more effective in suppressing the growth of cancer cells than used alone. Moreover, EPC nano-assemblies exhibit higher anti-proliferation efficacy for cancer cells in comparison with its free drug counterpart. This result should be ascribed to the boosted uptake of EPC than the combined use of free ELT and CCM. The wound healing assay, cell adhesion assay, and Transwell invasion assay further confirmed the effectiveness of the combination therapy of ELT and CCM, and the higher anticancer efficacy of EPC nano-assembly.
Multicellular tumor spheroid is more reliable in evaluating the antitumor efficiency of drugs in vitro than 2-D cell culture, since it can mimic the in vivo tumor condition. First, the tumor spheroid model can test the tumor-penetrating ability of anticancer agents. As a form of nanoparticle, EPC nano-assembly penetrates deeper into the tumor spheroid than free drug CCM and the combination therapy, which is evidenced by the thickness of the fluorescent shell in the fluorescence images of tumor spheroids after incubation with different drugs. Next, the tumor spheroid model can also be utilized to assess the cancer cell-killing efficiency of drugs. The more damaged morphology and more dead cells observed in the tumor spheroids treated with EPC nano-assemblies indicate that EPC is more effective in inhibiting tumor growth than the other treatments.
The in vivo investigation of the antitumor efficacy of EPC nano-assembly was performed in a pancreatic tumor model using BxPC-3 tumor-bearing nude mice. The ex vivo biodistribution study demonstrates that more EPC nano-assemblies accumulate in tumor tissues than free CCM, especially considering that the fluorescence intensity of EPC is lower than that of CCM in a protein containing environment. This result should be attributed to the enhanced permeability and retention (EPR) effect of EPC nano-assemblies and the active targeting effect of the EGFR receptor-mediated endocytosis.[17] Furthermore, the enhanced tumor accumulation of EPC nano-assembly leads to more effective inhibition against tumor growth, as evidenced by the smaller tumor volumes and prolonged survival of mice administrated with EPC nano-assemblies. In addition, no treatment associated side effects or body weight loss were found in mice during the whole process of experiment, suggesting that the EPC nano-assembly is a safe chemotherapeutic agent.
4. Conclusion
In summary, we developed a carrier-free nano-assembly based on the conjugate of ELT and CCM with the help of a bifunctional PEG molecule. Benefit from the ELT outlayer, EPC nano-assembly enters BxPC-3 cells via EGFR receptor-mediated endocytosis and selectively kills cancer cells. EPC shows stronger cell killing, better anti-migration and anti-invasion effects for BxPC-3 pancreatic cancer cells than free CCM, ELT, and the combination of free CCM and ELT. Furthermore, owing to both passive and active tumor targeting effects, EPC nano-assembly can effectively accumulate in the tumor tissue in a xenograft pancreatic tumor mouse model. Consequently, EPC reduced the growth of pancreatic tumor and extended the median survival time of the tumor-bearing mice from 22 days to 68 days. In addition, no systemic toxicity was detected in the major organs from the mice receiving EPC treatment. Attributed to the uniformity of the curcumin-erlotinib conjugate and easiness of scaling up, we expect the EPC could be quickly translated into a powerful tool in fighting against pancreatic cancer and other EGFR positive cancers.
5. Experimental Section
Materials:
N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), Curcumin (CCM), silica gel (spherical, 100 μm), and (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Tokyo Chemical Industry (TCI) Co., Ltd (Portland, OR, USA). Sodium ascorbate, N-hydroxysuccinimide (NHS), copper sulfate pentahydrate, TWEEN®80, and phosphate buffered saline (PBS) were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Erlotinib (ELT) was purchased from Cayman Chemical Co. (Ann Arbor, MI, USA). Azido-PEG3-acid (C₉H₁₇N₃O₅, CAS Number:1056024-94-2) was purchased from BroadPharm Inc. (San Diego, CA, USA). Invitrogen™ Hoechst 33342, Gibco™ Dulbecco’s modified Eagle’s medium (DMEM), trypsin-EDTA, penicillin-streptomycin (PS), and fetal bovine serum (FBS) were purchased from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Deuterated solvents were acquired from Cambridge Isotope Laboratories, Inc. (Andover, MA, USA). All the solvents used in this study were bought from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA) and used directly without further purification unless specified.
Synthesis of PEG modified erlotinib (ELT-PEG):
In a 25 mL round-bottom flask, ELT (944 mg, 2.4 mmol), azido-PEG3-acid (466 mg, 2.0 mmol), CuSO4·5H2O (100 mg, 0.4 mmol), and sodium ascorbate (158 mg, 0.8 mmol) was dissolved in 10 mL mixed solvents DMF/H2O/t-BuOH = 2/1/2 under nitrogen atmosphere with stirring. The reaction mixture was heated to 40 ◦C and kept stirring for 24 h. Then it was condensed and extracted with dichloromethane (DCM). The organic phase was dried with Na2SO4 and condensed under vacuum. The raw product was purified by flash column chromatography with silica gel (100 μm) using the gradient elution solvents of methanol and DCM (10/90) to afford ELT-PEG as a light-yellow oil (921 mg, 73.5%).
Synthesis of PEG linked erlotinib-curcumin conjugate (EPC):
NHS (115 mg, 1.0 mmol) was added to a 10 mL DMF solution of mixed ELT-PEG (627 mg, 1.0 mmol) and EDC (230 mg, 1.5 mmol) stirred under nitrogen atmosphere for 90 min in a 25 mL round-bottom flask. CCM (1.105 g, 3.0 mmol) was added into the above reaction mixture and kept stirring at room temperature overnight. The resulting mixture was condensed under vacuum with a rotary evaporator, and then extracted with DCM/H2O and washed three times with brine. The organic phase was dried over anhydrous Na2SO4, and then condensed under vaccum. The crude product was separated by flash column chromatography with silica gel (100 μm) using the gradient elution solvents of methanol and DCM (5/95) to afford EPC as an orange solid (520 mg, 53.2%). The chemical structure of EPC was confirmed by MS and NMR results.
Preparation of EPC nano-assembly:
EPC nano-assembly was prepared by a one-step nano-precipitation method. The compound EPC was first dissolved in acetone and then added dropwise into deionized water with a syringe pump at 20 mL/h rate under vigorous stirring. Self-assembly of the NP occurred spontaneously. Acetone in the nano-formulation was removed at room temperature under vacuum. The particle size, polydispersity index (PDI), and zeta potential of the NPs were measured by dynamic light scattering (DLS, Zetasizer Nano ZS, Malvern Instruments Ltd, Malvern, UK). The morphology of the NP was observed using Hitachi HT7800 transmission electron microscopy (TEM, Hitachi High-Technologies Corporation, Tokyo, Japan).
Critical micellar concentration:
Critical micellar concentration (CMC) of EPC was determined by measuring the fluorescence emission of EPC with various concentrations in water. The fluorescence intensity of EPC NP dispersion in deionized water with a series of concentrations (from 0.5 μM to 10 μM) was measured (λex = 420 nm, λem = 530 nm). The CMC value of EPC was calculated according to a plot of relative fluorescence intensity versus concentration.
Cell culture:
BxPC-3pancreatic cancer cells were cultured in Gibco™ DMEM supplemented with 10% FBS, 100 units/mL of penicillin, and 100 μg/mL of streptomycin at 37 °C with 5% CO2 under a humidified atmosphere. Cells were sub-cultured when the cell confluency reached ~80%.
Cellular uptake:
The cellular uptake of EPC by BxPC-3 cells was qualitatively evaluated by confocal laser scanning microscopy (CLSM). Cells were seeded in 35 mm2 glass bottom dish for 24 h. After being washed with PBS (pH 7.4), cells were incubated with CCM, ELT, or EPC at a concentration of 10 μM for 3h. Cells without receiving any treatment were utilized as control. Cells were processed following our reported method and imaged under a confocal microscope (LSM 700, Carl-Zeiss Inc.).[18] Flow cytometry: The uptake of EPC by BxPC-3 cells was further quantitatively evaluated by flow cytometry. Cells were seeded in 6-well plates (300,000 cells/well) and incubated for 24 h. After being washed with PBS (pH 7.4), cells were incubated with 10 μM CCM, ELT, or EPC for 3h and then harvested following our published protocol.[18] The cellular fluorescence intensities of different treatment groups were quantified by flow cytometer (BD Accuri C6, BD Biosciences).
Cellular uptake quantitative determination:
The intracellular levels of CCM and EPC were determined by fluorescence spectrometer. Cells were seeded in 6-well plates at a density of 700,000 cells/well for 24 h at 37 °C with 5% CO2. Cells were washed with PBS and then incubated with 20 μM CCM or EPC for 3h. Cells with no treatment were utilized as control. After being washed with PBS, cells were trypsinized with trypsin-EDTA, and harvested in 100 μL ice-cold deionized water and ultrasonicated for 20 min at 4 °C. 100 μL DMSO was added and the resulted mixture was centrifuged at 10,000 rpm for 10 min. The fluorescence emission of the supernatant was measured (λex = 420 nm, λem = 530 nm).
Cytotoxicity assay:
The anticancer activity of the EPC NP against BxPC-3 cells was determined by MTT assay. Cells were seeded in 96-well plates (5,000 cells/well) for 24 h prior to the assay with 5% CO2 at 37 °C. Cells were treated with different concentrations of CCM, ELT, CCM+ELT, or EPC in fresh medium and further incubated for another 20 h. After that, the viability of the cells were determined following our published protocol with a microplate reader (ELX808, Bio-Tech Instrument, Inc.).[19]
Tumor spheroid assay:
BxPC-3 cells were seeded in Corning® Ultra-Low Attachment 96-well plate at a density of 50,000 cells/well. Cells were incubated for 5 days to form tumor spheroid and then incubated with CCM, ELT, CCM+ELT, or EPC at a concentration of 20 μM for 6 h. Tumor spheroids without any treatment were utilized as control. Then the tumor spheroids were washed with PBS and imaged with the confocal microscope. To investigate the cytotoxicity that the EPC nanoparticles exerted to the tumor spheroids, the spheroids were incubated with 20 μM CCM, ELT, CCM+ELT, or EPC for 24 h. Tumor spheroids treated with PBS were utilized as control. The morphology change of tumor spheroids after receiving different treatments was observed by light microscopy. To visually evaluate the cytotoxicity effect of EPC, red-emissive propidium iodide was used to stain dead cells. The tumor spheroids were stained with propidium iodide (5 μM) for 2 h and then were washed with PBS and imaged with the confocal microscope.
Wound healing assay:
BxPC-3 cells were seeded in a 6-well plate at the concentration of 2,000,000 cells/well. When the cell confluency reached almost 100%, the supernatant was aspirated and then the cells were scratched with a yellow pipette tip to generate the wound. After being washed with PBS, the cells were incubated with medium containing 10 μM CCM, ELT, CCM+ELT, or EPC for 24 h. The scratched areas were monitored and photographed with light microscopy.
Cell adhesion assay:
Cellular adhesion test was performed in a 24-well plate coated with 0.1 mg/mL Matrigel. In brief, BxPC-3 cells treated with 10 μM CCM, ELT, CCM+ELT, or EPC in 0.5 mL serum-free medium were transferred into each well at the density of 100,000 cells/well. The plate was incubated at 37˚C for 1 h, after which it was washed with PBS to remove unattached cells. Cells attached to Matrigel were fixed with methanol for 15 min and then counted in five random optical fields as determined by light microscopy.
Transwell invasion assay:
Cell invasion assay was carried out using a 24-well plate coupled with 8 μm pore size Transwell inserts (Costar Corp., Cambridge, MA). Briefly, 50 μL Matrigel was added into each insert and solidified at 37 °C for 30 minutes to generate a thin gel layer. BxPC-3 cells treated with 10 μM CCM, ELT, CCM+ELT, or EPC in 200 μL serum-free medium were transferred into upper chambers at the concentration of 100,000 cells/well. The bottom chambers contained 600 μL complete medium with the same concentration of respective drugs. After incubation for 24 h at 37 °C, cells retained in the upper chamber were removed, while the invaded cells attached to the underside surface of the membrane were fixed with 4% paraformaldehyde for 30 min and then counted under a light microscope.
Animal model:
All animal experiments were conducted in accordance with NIH regulations and approved by the Institutional Animal Care and Use Committee of the University of South Carolina. In brief, 2,000,000 BxPC-3 cells suspended in 100 μL DMEM culture medium were inoculated subcutaneously to a female nude mouse (8–10 weeks old, ~20 g, Jackson Laboratories). The size of the tumor was measured by a digital caliper and calculated according to the following formula: Tumor volume = 1/2 × (tumor length) × (tumor width)2. Tumor volumes were monitored every other day. The body weight and signs of pain of the animals were observed throughout the duration of experiments.
In vivo biodistribution of EPC:
Three weeks after the inoculation of BxPC-3 cells, the tumor-bearing mice were administered with CCM and EPC by intravenous injection at a dose of 10 mg/kg equivalent to CCM. PBS (pH 7.4) was used as control. Animals were sacrificed after 6 h post-injection, and major organs and tumors were harvested and imaged ex vivo with an IVIS Lumina III whole body imaging system.
Anti-tumor efficacy:
When the tumor volume of BxPC-3 tumor-bearing mice reached 100 mm3, the mice were randomly assigned into five groups (n = 5) and were intravenously administrated with PBS, CCM, ELT, CCM+ELT, or EPC at a dose of 10 mg kg−1 equivalent to CCM (10.7 mg/kg equivalent to ELT) twice a week. Tumor volumes (V) and body weight of the mice were recorded every other day. Tumor size change during the treatment process was denoted as V/V0 (V0 is the tumor volume when the treatment was initiated). Mice were sacrificed when the tumor volume reached 2,000 mm3 or tumor ulceration observed, and the organs and tumors were harvested for further analysis.
Histological analysis:
The collected major organs (liver, spleen, heart, and kidney) and tumors were precessed following our published H&E protocol and analyzed under a light microscope.[18] The histology assay was performed by professional personnel at the University of South Carolina in a blinded fashion.
Statistical analysis:
All data were processed and expressed as means with standard deviations (mean ± SD). No outliers were identified and removed prior to the test. Student’s t-test was utilized to analyze the statistical difference between parallel groups. P < 0.05 from a two-tailed test was considered statistically significant.
Supplementary Material
Acknowledgments
The authors want to thank the National Institutes of Health (1R01AG054839-01A1 and 1R21CA252360-01) for financial support of the research.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
References
- [1].Siegel RL, Miller KD, Jemal A, CA: A Cancer Journal for Clinicians 2019, 69, 7–34. [DOI] [PubMed] [Google Scholar]
- [2].Wolfgang CL, Herman JM, Laheru DA, Klein AP, Erdek MA, Fishman EK, Hruban RH, CA: A Cancer Journal for Clinicians 2013, 63, 318–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Chugh R, Sangwan V, Patil SP, Dudeja V, Dawra RK, Banerjee S, Schumacher RJ, Blazar BR, Georg GI, Vickers SM, Saluja AK, Science translational medicine 2012, 4, 156ra139–156ra139; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabernero J, Hidalgo M, Goldstein D, Van Cutsem E, Wei X, Iglesias J, Renschler MF, The New England journal of medicine 2013, 369, 1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M, Parulekar W, Journal of Clinical Oncology 2007, 25, 1960–1966; [DOI] [PubMed] [Google Scholar]; b) Chen J, Li D, Huo B, Zhang F, Zhao X, Yuan G, Chen D, Song M, Xue J, Molecular Pharmaceutics 2019 [DOI] [PubMed] [Google Scholar]; c) Pennell NA, Neal JW, Chaft JE, Azzoli CG, Jänne PA, Govindan R, Evans TL, Costa DB, Wakelee HA, Heist RS, Shapiro MA, Muzikansky A, Murthy S, Lanuti M, Rusch VW, Kris MG, Sequist LV, Journal of Clinical Oncology 2018, 37, 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Willenbacher E, Khan ZS, Mujica CS, Trapani D, Hussain S, Wolf D, Willenbacher W, Spizzo G, Seeber A, International Journal of Molecular Sciences 2019, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Naeini MB, Momtazi AA, Jaafari MR, Johnston TP, Barreto G, Banach M, Sahebkar A, Journal of Cellular Physiology 2019, 234, 14743–14758; [DOI] [PubMed] [Google Scholar]; b) Rodrigues FC, Anil Kumar NV, Thakur G, European Journal of Medicinal Chemistry 2019, 177, 76–104; [DOI] [PubMed] [Google Scholar]; c) Wilken R, Veena MS, Wang MB, Srivatsan ES, Molecular cancer 2011, 10, 12–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Feng T, Wei Y, Lee RJ, Zhao L, International journal of nanomedicine 2017, 12, 6027–6044; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Saw PE, Lee S, Jon S, Advanced Therapeutics 2019, 2, 1800146. [Google Scholar]
- [8].a) Chen S, Liang Q, Liu E, Yu Z, Sun L, Ye J, Shin MC, Wang J, He H, J Mater Chem B 2017, 5, 4060–4072; [DOI] [PubMed] [Google Scholar]; b) Falah RR, Talib WH, Shbailat SJ, Ther Adv Med Oncol 2017, 9, 235–252; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Saghatelyan T, Tananyan A, Janoyan N, Tadevosyan A, Petrosyan H, Hovhannisyan A, Hayrapetyan L, Arustamyan M, Arnhold J, Rotmann AR, Hovhannisyan A, Panossian A, Phytomedicine 2020, 70, 153218. [DOI] [PubMed] [Google Scholar]
- [9].a) Cabrespine-Faugeras A, Bayet-Robert M, Bay JO, Chollet P, Barthomeuf C, Nutr Cancer 2010, 62, 148–153; [DOI] [PubMed] [Google Scholar]; b) Li J, Xiang S, Zhang Q, Wu J, Tang Q, Zhou J, Yang L, Chen Z, Hann SS, J Exp Clin Cancer Res 2015, 34, 46; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Eslami SS, Jafari D, Montazeri H, Sadeghizadeh M, Tarighi P, Nutr Cancer 2020, 1–14. [DOI] [PubMed] [Google Scholar]
- [10].a) Hong Y, Che S, Hui B, Yang Y, Wang X, Zhang X, Qiang Y, Ma H, Biomed Pharmacother 2019, 112, 108614; [DOI] [PubMed] [Google Scholar]; b) Hong Y, Che S, Hui B, Wang X, Zhang X, Ma H, Drug Des Devel Ther 2020, 14, 2263–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Liu Z, Zhu YY, Li ZY, Ning SQ, Oncol Lett 2016, 12, 3944–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Chen S, Liang Q, Xie S, Liu E, Yu Z, Sun L, Shin MC, Lee SJ, He H, Yang VC, Frontiers of Chemical Science and Engineering 2016, 10, 383–388; [Google Scholar]; b) Javadi S, Rostamizadeh K, Hejazi J, Parsa M, Fathi M, Phytotherapy Research 2018, 32, 355–364; [DOI] [PubMed] [Google Scholar]; c) Yamauchi Y, Izumi Y, Yamamoto J, Nomori H, Phytotherapy Research 2014, 28, 728–735. [DOI] [PubMed] [Google Scholar]
- [13].Zanoni M, Piccinini F, Arienti C, Zamagni A, Santi S, Polico R, Bevilacqua A, Tesei A, Scientific Reports 2016, 6, 19103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Bimonte S, Barbieri A, Leongito M, Piccirillo M, Giudice A, Pivonello C, de Angelis C, Granata V, Palaia R, Izzo F, Nutrients 2016, 8, 433; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Díaz Osterman CJ, Wall NR, Journal of Nature and Science 2015, 1, e124; [Google Scholar]; c) Kanai M, World journal of gastroenterology 2014, 20, 9384–9391; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kelley RK, Ko AH, Biologics: targets and therapy 2008, 2, 83–95; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Su J, Zhou X, Wang L, Yin X, Wang Z, American journal of cancer research 2016, 6, 1949–1962; [PMC free article] [PubMed] [Google Scholar]; f) Wang JP, Wu C-Y, Yeh Y-C, Shyr Y-M, Wu Y-Y, Kuo C-Y, Hung Y-P, Chen M-H, Lee W-P, Luo J-C, Chao Y, Li C-P, Oncotarget 2015, 6, 18162–18173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Gong F, Chen D, Teng X, Ge J, Ning X, Shen Y.-l., Li J, Wang S, Molecular Pharmaceutics 2017, 14, 2585–2594; [DOI] [PubMed] [Google Scholar]; b) Yang X, Li Z, Wang N, Li L, Song L, He T, Sun L, Wang Z, Wu Q, Luo N, Yi C, Gong C, Scientific reports 2015, 5, 10322–10322; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhao S, Ma L, Cao C, Yu Q, Chen L, Liu J, International journal of nanomedicine 2017, 12, 2489–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Gao Y, Zhang H, Zhang Y, Lv T, Zhang L, Li Z, Xie X, Li F, Chen H, Jia L, Molecular Pharmaceutics 2018, 15, 5146–5161; [DOI] [PubMed] [Google Scholar]; b) Yu W, Jiang L, Shen C, Zhang P, Drug Development Research 2016, 77, 319–325. [DOI] [PubMed] [Google Scholar]
- [17].a) Maeda H, Journal of Controlled Release 2012, 164, 138–144; [DOI] [PubMed] [Google Scholar]; b) Overchuk M, Zheng G, Biomaterials 2018, 156, 217–237. [DOI] [PubMed] [Google Scholar]
- [18].Sui BL, Cheng C, Wang MM, Hopkins E, Xu PS, Advanced Functional Materials 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bahadur KCR, Thapa B, Xu P, Macromolecular Bioscience 2012, 12, 637–646. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.