

 Identified as an HIV-1 inhibitor in cellulo, this pyrazole does not belong to the three main classes of anti HIV-drugs, a feature of prime interest in the context of viral resistance.
Identified as an HIV-1 inhibitor in cellulo, this pyrazole does not belong to the three main classes of anti HIV-drugs, a feature of prime interest in the context of viral resistance.
Abstract
Inspired by the antiviral activity of known pyrazole-based HIV inhibitors, we screened our in-house library of pyrazole-based compounds to evaluate their in cellulo activity against HIV-1 replication. Two hits with very similar structures appeared from single and multiple-round infection assays to be non-toxic and active in a dose-dependent manner. Chemical expansion of their series allowed an in-depth and consistent structure–activity-relationship study (SAR) to be built. Further ADME evaluation led to the selection of 4-amino-3-cyano-1-(2-benzyloxyphenyl)-1H-pyrazole-5-carboxylate with an advantageous pharmacokinetic profile. Finally, examination of its mode of action revealed that this compound does not belong to the three main classes of anti-HIV drugs, a feature of prime interest in the context of viral resistance.
Introduction
Human immunodeficiency virus (HIV) and the resulting acquired immunodeficiency syndrome (AIDS) still represent a major global public health threat. According to the Joint United Nations Program on HIV and AIDS (UNAIDS), since the beginning of the epidemic, more than three decades ago, 77.3 million people have become infected with HIV and 35.4 million of them have died from AIDS-related illnesses.1 Highly active anti-retroviral therapy (HAART), introduced in 1996, led to a significant decrease of AIDS-related deaths by 38% and helped to save 11.4 million lives.2 HAART has also improved the life expectancy of HIV/AIDS patients which nowadays is approaching that of the general population.3
However, HAART is still not curative,4 and this life-long treatment suffers from several pitfalls such as inherent drug toxicity,5 emergence or transmission of drug-resistant HIV-1 strains,6–8 and the decline in patient adherence.9 In response to these concerns, a global effort is required to pursue the development of novel and potent anti-HIV-1 drugs, which represents a continuing challenge.10 In this context, prompted by the promising antiretroviral activities of several pyrazole-based inhibitors such as PNU-32945 I,11 Lersivirine II12 and benzylamino derivatives III13 (Fig. 1), we decided to screen a pyrazole-based chemical library, which was previously developed in our laboratory.14,15
Fig. 1. Structures of known pyrazole-based HIV inhibitors.

We report herein the discovery of new pyrazolic HIV-1 inhibitors, their synthesis, biological activities and ADMET properties. Interestingly, methyl 4-amino-3-cyano-1-(2-benzyloxyphenyl)-1H-pyrazole-5-carboxylate that was selected as a lead compound does not inhibit any of the viral enzymes that are classically targeted in antiviral therapy.
Results and discussion
Discovery of the hit compounds through a double screening
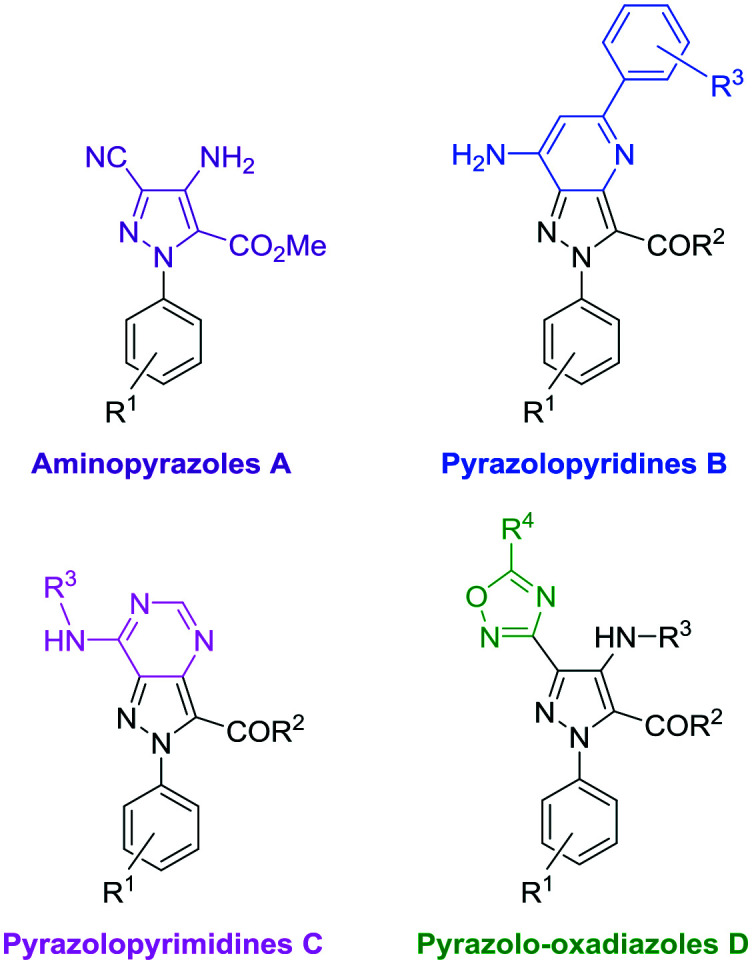
Our in-house chemical library, which was originally designed to target a kinase domain, is composed of 110 pyrazole-based molecules that are well balanced between four heteroaromatic collections: aminopyrazoles A, pyrazolopyridines B, pyrazolopyrimidines C and pyrazolo-oxadiazoles D (Fig. 2). Interestingly, while compounds from families C and D behave like kinase inhibitors, those from series A and B are not active against this kind of target.16
Fig. 2. Molecular scaffolds included in the screened chemical library.
These compounds were tested for their potential inhibitory activity against HIV-1 replication in cell cultures. This primary screening was carried out using HeLa P4 cells containing a stably integrated LTR linked to a Lac Z reporter gene. These cells were infected with HIV-1 Laï viruses, and were incubated for 48 h with 50 μM of each compound, using zidovudine as a positive control (see the ESI†). This kind of phenotypic assay was chosen because it allows for several steps of the HIV replication cycle to be evaluated at once. In parallel, the cytotoxicity of each compound was evaluated under the same culture conditions according to the MTS standard protocol (see the ESI†). This test is also necessary to assess the relevancy of inhibition values.
After this first screening at 50 μM, none of the 24 pyrazolopyrimidines C nor the 27 pyrazolo-oxadiazoles D showed enough activity to be selected (the full table of results can be found in the ESI†). However, three compounds displayed an inhibition activity higher than 50%, and a toxicity value lower than 15%: aminopyrazoles A.20 and A.21, as well as pyrazolopyridine B.19 (Fig. 3). Interestingly, the benzyloxy group present in both A.20 and A.21 is likely to be part of the pharmacophoric requirements.
Fig. 3. Structures of the three selected hits and biological results @50 μM.

Dose-dependent assay
To further validate these hits, and to rule out the possibility of false positives, the inhibition assay was repeated in a dose-dependent manner using a 1.0 to 50 μM concentration range of the inhibitors (see the ESI†).
The resulting dose–response curves allowed for the concentration at which 50% of the replication of HIV is inhibited to be determined (EC50 value). These experiments revealed that A.20 and A.21 are able to inhibit the HIV replication in a dose-dependent manner. In contrast, it was found that B.19 is likely to be a false positive because its apparent inhibition potency is related to its toxicity and not to its activity, and therefore it was discarded. Encouragingly, the EC50 values of A.20 and A.21 were found to be 19.4 μM and 15.2 μM, respectively, which are values that are comparable to the 13.3 μM EC50 of tenofovir (TDF, clinically used inhibitor) which we determined under the same conditions.17 Hence, A.20 and A.21 were validated as hits and selected for the next step of biological evaluation.
Multiple-round infection assay
With the aim to ascertain their antiviral potency, the validated hits were then evaluated in different cell-types, with another source of the virus, and also incubated for a longer amount of time. For this multiple-round infection assay, human lymphoblastoid MT2 cells were incubated for 3 days with Laï virus at MOI 3 and the antiviral effect of A.20 and A.21 was monitored in a concentration range from 0 to 50 μM using zidovudine as a positive control (see the ESI†). According to these results, A.20 and A.21 are undoubtedly efficient in counteracting HIV-1 replication, even at this high concentration of virus. To further develop this series of aminopyrazoles, the synthesis of analogous compounds was therefore undertaken.
Chemical expansion of the A series
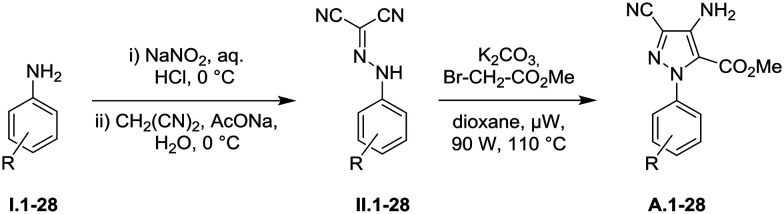
To study and verify the impact of substituents on the phenyl ring of pyrazoles A, new analogues were synthesized according to the two-step strategy which we previously developed (Table 1).14,15 Starting from commercially available anilines I.1–28, firstly a diazotization reaction with sodium nitrite in aqueous hydrochloric acid was performed. This was immediately followed by condensation with malononitrile in basic medium to afford the corresponding dicyanohydrazones II.1–28 in good to excellent yields. The second step is a Thorpe–Ziegler reaction which was performed with methyl bromoacetate and potassium carbonate in dioxane under microwave irradiation. Desired aminopyrazoles A.1–28 were obtained in moderate to good yields ranging from 16 to 83% over two steps. Thanks to this synthetic effort, we obtained a homogeneous series of aminopyrazoles with comparable substitution patterns which are suitable for structure–activity relationship studies.
Table 1. Two-step synthesis of aminopyrazoles A.1–28 a .
| ||
| Compound | –R | Overall yield (%) |
| A.1 | H | 69 |
| A.2 | 2-NO2 | 38 |
| A.3 | 3-NO2 | 48 |
| A.4 | 4-NO2 | 16 |
| A.5 b | 2-Br | 35 |
| A.6 | 3-Br | 56 |
| A.7 | 4-Br | 44 |
| A.8 b | 2-Cl | 35 |
| A.9 | 3-Cl | 51 |
| A.10 | 4-Cl | 38 |
| A.11 b | 2-F | 48 |
| A.12 | 3-F | 32 |
| A.13 | 4-F | 38 |
| A.14 b | 2-I | 44 |
| A.15 b | 3-CF3 | 82 |
| A.16 b | 4-CF3 | 83 |
| A.17 b | 2-CO2tBu | 35 |
| A.18 | 3-CO2tBu | 68 |
| A.19 | 4-CO2tBu | 42 |
| A.20 | 2-OBn | 42 |
| A.21 | 3-OBn | 61 |
| A.22 b | 4-OBn | 18 |
| A.23 b | 2-OMe | 50 |
| A.24 | 3-OMe | 73 |
| A.25 b | 4-OMe | 22 |
| A.26 b | 2-C CH | 31 |
| A.27 | 3-C CH | 77 |
| A.28 | 4-C CH | 44 |
aYields are calculated over two steps.
bHydrazones II.x and pyrazoles A.x are described in the ESI.†
Structure–activity relationship (SAR)
To draw out the SAR pattern, the inhibition and toxicity values of the previously and newly synthesized pyrazoles are gathered in the following table (Table 2). For the sake of clarity, we used a minimum threshold of 30% for inhibition, and a maximum one of 15% for toxicity. Indeed, beyond this value, the cell mortality distorts the value of inhibition significantly. The application of this double filter revealed 11 interesting compounds (Table 2).
Table 2. In vitro evaluation of the inhibitory activity and toxicity of aminopyrazoles A.1–28 a .
| Compound | –R | Inhibition (%) | Toxicity (%) |
| A.1 | H | <30 | 7.0 |
| A.2 | 2-NO2 | 42 | 0 |
| A.3 | 3-NO2 | 47 | 0 |
| A.4 | 4-NO2 | 43 | 0 |
| A.5 | 2-Br | 32 | 4.8 |
| A.6 | 3-Br | 42 | 9.0 |
| A.7 | 4-Br | 67 | >15 |
| A.8 | 2-Cl | 30 | 2.4 |
| A.9 | 3-Cl | <30 | 11.0 |
| A.10 | 4-Cl | 56 | >15 |
| A.11 | 2-F | <30 | 3.6 |
| A.12 | 3-F | 48 | 0.0 |
| A.13 | 4-F | <30 | 6.0 |
| A.14 | 2-I | <30 | >15 |
| A.15 | 3-CF3 | <30 | >15 |
| A.16 | 4-CF3 | <30 | 14 |
| A.17 | 2-CO2tBu | <30 | 2.8 |
| A.18 | 3-CO2tBu | 36 | 0 |
| A.19 | 4-CO2tBu | 47 | 14.0 |
| A.20 | 2-OBn | 63 | 9.0 |
| A.21 | 3-OBn | 65 | 11.0 |
| A.22 | 4-OBn | <30 | 3.8 |
| A.23 | 2-OMe | <30 | 9.2 |
| A.24 | 3-OMe | <30 | 6.0 |
| A.25 | 4-OMe | <30 | 14.6 |
| A.26 | 2-C CH | <30 | 7.0 |
| A.27 | 3-C CH | <30 | 14.0 |
| A.28 | 4-C CH | Nd | Nd |
aData represent mean values of two experiments in duplicate.
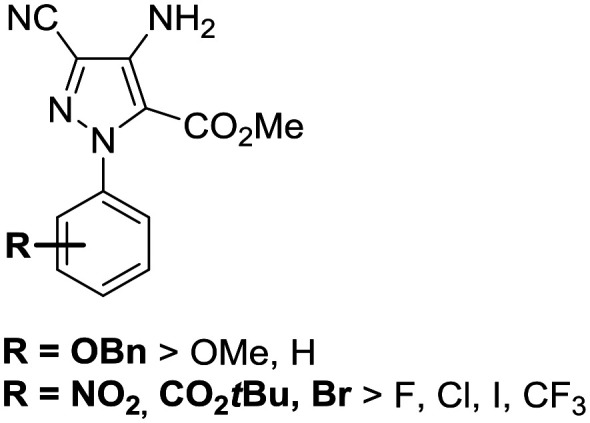
First of all, the activity of N-phenyl-pyrazole A.1 (without any substituents) remains below the critical threshold of 30% inhibition. When compared to the activity of hits A.20 and A.21 (63% and 65% respectively), this low value supports our hypothesis regarding the key role that might be played by the benzyloxy group. Then, nine compounds which show good inhibition all share the common feature of bearing electron withdrawing groups such as NO2 (A.2–4), Br (A.5–6), Cl (A.8), F (A.12), or CO2tBu (A.18–19). Besides the pure electronic effect on the phenyl ring, these groups could also be involved in non-covalent interactions such as hydrogen or halogen-bonding. Finally, none of the newly synthesized pyrazoles were found to be more efficient; with A.20 and A.21, bearing a benzyloxy substituent, being the best inhibitors within this series. When compared to their methyloxy counterparts (A.23–24), these hits are at least twice as active, suggesting that the benzyl moiety is likely to interact with a hydrophobic pocket. These observations are summarized in the following figure (Fig. 4). As no other compound appeared to be as potent as A.20 and A.21, these two hits were further evaluated in order to assess their drugability.
Fig. 4. General trends resulting from SAR analysis.
ADME properties
A full pharmacokinetic profile has been established for A.20 and A.21. The overall results are gathered in Table 3.
Table 3. ADME properties of hits A.20 and A.21.
| ADME properties | A.20 | A.21 |
| PBS solubility | 9.6 ± 0.5 μM | 1.03 ± 0.06 μM |
| Octanol/water partition coefficient | log D7.4 = 3.69 ± 0.06 | log D7.4 = 3.84 ± 0.05 |
| Plasma protein binding | 95 ± 1% | 100 ± 0% |
| Permeability (PAMPA) | log Pe = –5.41 ± 0.04 | log Pe = –5.97 ± 0.04 |
| Metabolic stability – half life | t 1/2 = 3.0 ± 0.5 min | t 1/2 = 4.6 ± 0.5 min |
| In vitro stability | 100% | 100% |
| Maximum plasma concentration after intravenous administration in mice | 238 ± 29 ng mL–1 at 10 min | 241 ± 26 ng mL–1 at 10 min |
| Maximum plasma concentration after oral administration in mice | 18 ± 4 ng mL–1 at 10 min | Nd |
Although the structures of A.20 and A.21 are very similar, A.20 shows a better overall profile than A.21. Being slightly less lipophilic, A.20 is ten times more soluble; it displays better membrane permeability by passive diffusion. As the compound doesn't contain any hydrolysable group, its plasma stability is excellent. Its low metabolic stability is offset by a high level of protein binding, which should ensure good availability over time. According to these results, A.20 has the best drugability potential, therefore we decided to pursue our study with this compound.
Identification of the potential target of hit A.20
At present, 30 drugs have been officially approved for the treatment of HIV/AIDS.18 These molecules target essential steps or viral enzymes that are involved in the HIV replication cycle: CCR5 co-receptor,19 viral fusion,20 reverse transcriptase (RT),21 integrase (IN)22,23 and protease (PR).24 It is noteworthy to say that the current WHO preferred first-line regimens predominantly consist of RT inhibitors (RTI), in some instances associated with PR or IN strand-transfer inhibitors (PRI or INSTI).25,26 However, despite tremendous efforts to develop new therapeutic agents, no novel target exploitation is to be expected from the few inhibitors in clinical trials at the moment.27
In this context, to explore the mechanism of action of A.20, we tried to discover its biological target. Since A.20 was selected through a screening assay that cannot detect PR inhibitors (one single cycle of HIV replication does not allow detection of post-integrative step inhibitors), no further investigation was performed on this target. We therefore focused our study on RT and IN strand transfer (INST) activities.
To evaluate the inhibitory activity of compound A.20 on the RT of HIV-1, its relative efficiency of incorporation was measured using subtype B WT HIV-1 RT in an in vitro susceptibility assay, using nevirapine and AZT as positive controls (see the ESI†). Under these conditions, A.20 appeared to be completely ineffective toward RT (IC50 > 1000).
To check the propensity of A.20 to inhibit the reaction mediated by IN, strand transfer assays were carried out with increasing concentrations of inhibitors, from 100 nM to 333 μM, using raltegravir as a positive control (see the ESI†). Regardless of the concentration used, no effect on the strand transfer reaction efficiency was observed, thereby demonstrating that A.20 does not impact the catalytic properties of INST at all.
From these results, we can conclude that our lead compound, A.20, does not belong to the three main classes of anti-HIV drugs (RTI, PI and INSTI), and thus is likely to inhibit an underexploited or unknown target.
Conclusion
It is still of key importance to develop new inhibitors of HIV replication in order to counteract the rapid and unavoidable virus mutations, with a preference for those which are orthogonal to the current drugs. In this study, we identified two promising hits A.20 and A.21 with interesting EC50 values, similar to that of tenofovir. Further multiple round infection assays confirmed their inhibitory activity on long term incubation. Based on the same aminopyrazole scaffold, a series of derivatives were synthesized to draw an SAR study that ascertained A.20 and A.21 as hits, and the OBn substituent as an essential group for activity. ADME evaluations allowed us to select A.20 as the better lead compound. The final part of our study revealed that A.20 does not behave as usual anti-HIV drugs (RTI, PI and INSTI) and thus belongs to the sought-out category of inhibitors which could advantageously slow down the development of resistance.
This study provided us with A.20 as a validated lead compound, and a solid SAR basis to design and develop more potent inhibitors. Moreover, fostered by the complete lack of activity on the classical targets, further studies will be dedicated to uncover its mechanism of action.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The authors thank the “Agence Nationale de Recherches sur le Sida et les hépatites virales” for support (ANRS, ref ECTZ20422), the “Ministère de l'Enseignement supérieur, de la Recherche et de l'Innovation” for J. F. PhD grant and the UMR 8601 CNRS, Université Paris Descartes for HRMS and NMR facilities.
Footnotes
†Electronic supplementary information (ESI) available: Biology: experimental procedures, full results of the first screening, dose–response curves; chemistry: general procedures, compound characterization, 1H and 13C NMR spectra. See DOI: 10.1039/d0md00025f
References
- UNAIDS - Global HIV & AIDS statistics http://www.unaids.org/en/resources/fact-sheet.
- World Health Organization- Key facts http://www.who.int/mediacentre/factsheets/fs360/en/.
- Marcus J. L., Chao C. R., Leyden W. A., Xu L., Quesenberry C. P., Klein D. B., Towner W. J., Horberg M. A., Silverberg M. J. J. Acquired Immune Defic. Syndr. 2016;73:39. doi: 10.1097/QAI.0000000000001014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks S. G., Lewin S. R., Ross A. L., Ananworanich J., Benkirane M., Cannon P., Chomont N., Douek D., Lifson J. D., Lo Y.-R., Kuritzkes D., Margolis D., Mellors J., Persaud D., Tucker J. D., Barre-Sinoussi F., International AIDS Society Towards a Cure Working Group, Alter G., Auerbach J., Autran B., Barouch D. H., Behrens G., Cavazzana M., Chen Z., Cohen É. A., Corbelli G. M., Eholié S., Eyal N., Fidler S., Garcia L., Grossman C., Henderson G., Henrich T. J., Jefferys R., Kiem H.-P., McCune J., Moodley K., Newman P. A., Nijhuis M., Nsubuga M. S., Ott M., Palmer S., Richman D., Saez-Cirion A., Sharp M., Siliciano J., Silvestri G., Singh J., Spire B., Taylor J., Tolstrup M., Valente S., van Lunzen J., Walensky R., Wilson I., Zack J. Nat. Med. 2016;22:839. doi: 10.1038/nm.4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr A. Nat. Rev. Drug Discovery. 2003;2:624. doi: 10.1038/nrd1151. [DOI] [PubMed] [Google Scholar]
- Perno C.-F., Moyle G., Tsoukas C., Ratanasuwan W., Gatell J., Schechter M. J. Med. Virol. 2008;80:565. doi: 10.1002/jmv.21034. [DOI] [PubMed] [Google Scholar]
- Hué S., Gifford R. J., Dunn D., Fernhill E., Pillay D. J. Virol. 2009;83:2645. doi: 10.1128/JVI.01556-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menéndez-Arias L. Antiviral Res. 2013;98:93. doi: 10.1016/j.antiviral.2013.01.007. [DOI] [PubMed] [Google Scholar]
- Mathes T., Pieper D., Antoine S.-L., Eikermann M. HIV Med. 2013;14:583. doi: 10.1111/hiv.12051. [DOI] [PubMed] [Google Scholar]
- Dabis F., Bekker L.-G. Science. 2017;357:335. doi: 10.1126/science.aao4197. [DOI] [PubMed] [Google Scholar]
- Genin M. J., Biles C., Keiser B. J., Poppe S. M., Swaney S. M., Tarpley W. G., Yagi Y., Romero D. L. J. Med. Chem. 2000;43:1034–1040. doi: 10.1021/jm990383f. [DOI] [PubMed] [Google Scholar]
- Platten M., Fätkenheuer G. Expert Opin. Invest. Drugs. 2013;22:1687. doi: 10.1517/13543784.2013.846325. [DOI] [PubMed] [Google Scholar]
- Mizuhara T., Kato T., Hirai A., Kurihara H., Shimada Y., Taniguchi M., Maeta H., Togami H., Shimura K., Matsuoka M., Okazaki S., Takeuchi T., Ohno H., Oishi S., Fujii N. Bioorg. Med. Chem. Lett. 2013;23:4557–4561. doi: 10.1016/j.bmcl.2013.06.026. [DOI] [PubMed] [Google Scholar]
- Corre L. L., Tak-Tak L., Guillard A., Prestat G., Gravier-Pelletier C., Busca P. Org. Biomol. Chem. 2015;13:409. doi: 10.1039/c4ob01951b. [DOI] [PubMed] [Google Scholar]
- Corre L. L., Lang M. C. D., Garbay C., Gravier-Pelletier C., Busca P., Ethève-Quelquejeu M., Braud E. Synthesis. 2016;48:4569. [Google Scholar]
- Unpublished results
- Unpublished results
- Mehellou Y., De Clercq E. J. Med. Chem. 2010;53:521. doi: 10.1021/jm900492g. [DOI] [PubMed] [Google Scholar]
- Shah H. R., Savjani J. K. Eur. J. Med. Chem. 2018;147:115. doi: 10.1016/j.ejmech.2018.01.085. [DOI] [PubMed] [Google Scholar]
- Eggink D., Berkhout B., Sanders R. W. Curr. Pharm. Des. 2010;16:3716. doi: 10.2174/138161210794079218. [DOI] [PubMed] [Google Scholar]
- Holec A. D., Mandal S., Prathipati P. K., Destache C. J. Curr. HIV Res. 2017;15:411. doi: 10.2174/1570162X15666171120110145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y., Johnson A. A., Marchand C. Nat. Rev. Drug Discovery. 2005;4:236. doi: 10.1038/nrd1660. [DOI] [PubMed] [Google Scholar]
- Nair V., Chi G. Rev. Med. Virol. 2007;17:277. doi: 10.1002/rmv.539. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K., Osswald H. L., Prato G. J. Med. Chem. 2016;59:5172. doi: 10.1021/acs.jmedchem.5b01697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Department of Health and Human Services - Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents Living with HIV, https://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf.
- World Health Organization – The Public Health Response to Pretreatment HIV drug resistance, WHO Guide, 2017, https://www.who.int/hiv/pub/guidelines/hivdr-guidelines-2017/en/.
- Tieu H.-V., Taylor B. S., Jones J., Wilkin T. J. Top. Antivir. Med. 2018;26:40. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.