

 A quantum chemistry guided optimisation (leading to piceatannol analogue 3t) with a good understanding of the catechol binding mode to the bimanganese cluster of arginase.
A quantum chemistry guided optimisation (leading to piceatannol analogue 3t) with a good understanding of the catechol binding mode to the bimanganese cluster of arginase.
Abstract
Arginase is involved in a wide range of pathologies including cardiovascular diseases and infectious diseases whilst it is also a promising target to improve cancer immunotherapy. To date, only a limited number of inhibitors of arginase have been reported. Natural polyphenols, among them piceatannol, are moderate inhibitors of arginase. Herein, we report our efforts to investigate catechol binding by quantum chemistry and generate analogues of piceatannol. In this work, we synthesized a novel series of amino-polyphenols which were then evaluated as arginase inhibitors. Their structure–activity relationships were elucidated by deep quantum chemistry modelling. 4-((3,4-Dihydroxybenzyl)amino)benzene-1,2-diol 3t displays a mixed inhibition activity on bovine and human arginase I with IC50 (Ki) values of 76 (82) μM and 89 μM, respectively.
Introduction
Arginase (EC 3.5.3.1) is a binuclear manganese transaminidase expressed as two isoforms whose active sites exhibit complete homology.1 Arginase catalyses the hydrolysis of l-arginine into urea and l-ornithine. l-Ornithine is then converted into l-proline, the main component of collagen, and polyamines involved in cell proliferation. Since it is well known that arginase contributes to a wide range of pathologies, interest in this enzyme as a target for drug discovery is rising. For instance, arginase is tightly involved in nitric oxide (NO) homeostasis since both nitric-oxide synthase (NOS) and arginase share arginine as a substrate. Therefore, up regulation of arginase activity contributes to cardiovascular diseases and endothelial dysfunction2 that is involved in a high degree of cardiovascular risk and morbidity due to cardiovascular and non-cardiovascular diseases (rheumatoid arthritis and diabetes).3,4 In infectiology, arginase is a key target for leishmaniasis since parasitic arginase produces the trypanothione defensive molecule and host arginase decreases cytotoxic NO secreted by macrophages.5 Particular interest is focused on immuno-oncology because the depletion of arginine by myeloid-derived suppressor cells contributes to an immunosuppressive tumour microenvironment that inhibits the proliferation of effector lymphocytes.6 In murine models no apparent toxicity was observed after long-term administration of arginase inhibitors.7,8 Arginase dysregulation in patients has been corroborated with benefits from local administration of arginase inhibitors.6,9–11 The above confirms the wide therapeutic interest in arginase and the subsequent need to find effective inhibitors.
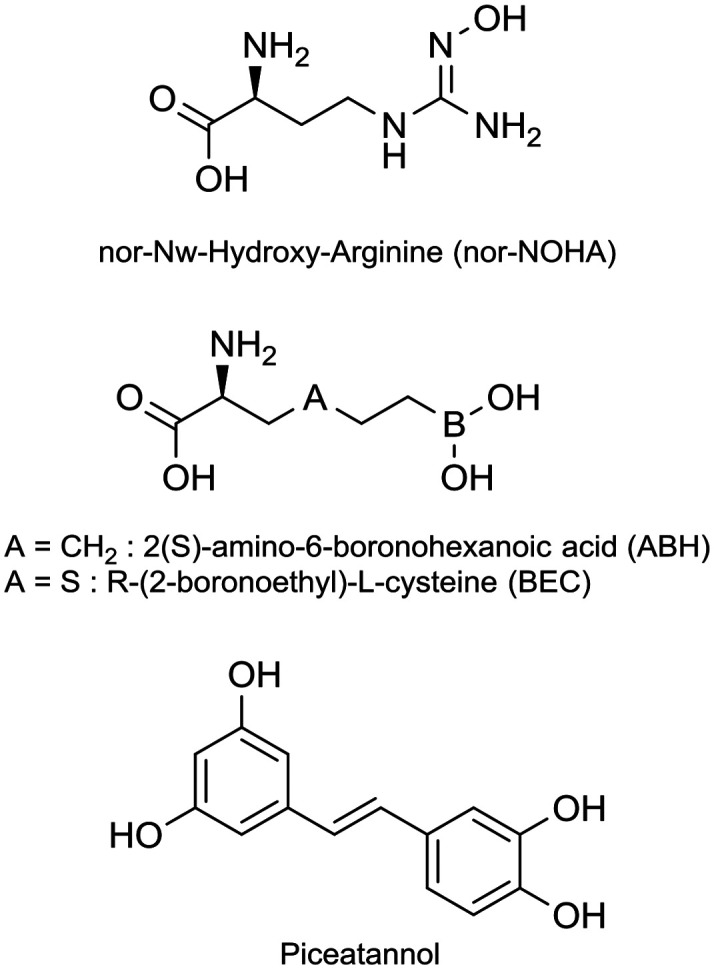
While there is a need, only a limited number of arginase inhibitors have been reported.12 Reference inhibitors (Fig. 1) are modified α-amino acids: nor-Nω-hydroxy-arginine (nor-NOHA),13,14 and two boronic acids, 2(S)-amino-6-boronohexanoic acid (ABH)15 and R-(2-boronoethyl)-l-cysteine (BEC).16 α,α-Disubstituted amino acid-based arginase inhibitors17–20 have been directly derived from ABH, and among them, one is in a phase I/II clinical trial in solid tumour patients (CX-1158-101). Very recently Guo et al.21 described suicide inhibitors based on 1,6-elimination that revealed a para-azaquinone methide as a strong electrophile. Nor-NOHA has a short half-life22,23 and boronic acids suffer from poor oral bioavailability and/or fast clearance17,19 and nothing is mentioned about the pharmacokinetics parameters of the latest irreversible inhibitors. Therefore, these compounds are not suitable for clinical use and drugs complying with systemic administration are now clearly warranted. In parallel, some arginase inhibitors of natural origin have been discovered. They belong for the most part to the secondary metabolite class of polyphenols.24 The catechol core is the recurrent structural feature of these natural arginase inhibitors.25–27 Among them piceatannol, a stilbene derivative, is one of the most efficient.26,28
Fig. 1. Reference inhibitors of arginase.
We are engaged in the development of synthetic analogues29 of natural products and in this study we have developed a series of inhibitors of arginase derived from piceatannol with an amine insert as a linker between the resorcinol and catechol parts (Fig. 2). A rational investigation was used for the development. Firstly, the binding modes of catechol to the active site of arginase were unravelled using computational simulations, corroborated by in vitro evaluation. In practice, computational simulations were carried out through a 21-amino acid/241 atom model of the enzymatic pocket (see Fig. S3 in the ESI†), fully treated with quantum chemistry methods (DFT).
Fig. 2. Rational approach used in this work for designing piceatannol inspired arginase inhibitors.

This level of theory was required by the different types of non-covalent interactions within the active sites (metal–ligand, pi-stacking, different types of hydrogen bonds, etc.), which cannot be accurately described without the use of quantum chemistry. In vitro inhibition was evaluated by the percentage of inhibition and IC50 determination on bovine arginase (b-ARG). The prediction of the complexation energies of some relevant catechol derivatives leads us to the pharmacomodulation of the resorcinol part and a structure–activity relationship (SAR) study was conducted. The most active compounds were evaluated with respect to human arginase (h-ARG). Finally, the mechanism of inhibition and the Ki of the most active compounds and piceatannol, which was used as a reference compound, have also been determined. The sampling of the possible docking conformations of the inhibitors was performed with quantum chemistry methods only, since molecular mechanics docking methods proved unable to treat the manganese-inhibitor interactions accurately. For each inhibitor considered, 5 to 10 different starting geometries were built manually and fully optimized. Only the lowest potential energy per inhibitor is reported in this paper.
Results and discussion
Catechol binding modes
The aim of the first computational investigation was to determine the most favourable binding mode of the catechol moiety to the antiferromagnetically coupled bi-manganese cluster. The incomplete coordination sphere of both ions suggests that they can bind potent inhibitor molecules. We obtained two main binding possibilities, named “double” and “bridge” respectively hereafter. In the double configuration, the distance separating the two oxygen atoms of catechol (about 2.68 Å) allows them to bind respectively to the manganese ions that are separated by a distance of 3.27 Å. In this situation, the bonding with the two manganese ions is reinforced by two strong hydrogen bonds between the catechol phenol groups and the residues Glu277 on one side, and Thr246 (Fig. 3A) or Asp234 (see Fig. S1 in the ESI†) on the other side. A slight pi-stacking effect also seems to take place between the aromatic rings of catechol and the two neighbouring histidine residues (His126, shown in Fig. 3, and His141), although their planes are not strictly parallel. The binding modes of powerful arginase inhibitors nor-NOHA,13 BE and ABH30 suggest that catechol could also bind in a ‘bridging’ fashion, with only one phenol group bound to the manganese ions, and the other forming a strong hydrogen bond with Glu277 (Fig. 3B). The structures of these two binding modes were carefully optimised and unambiguously proved to be the most stable positions of catechol inside the arginase pocket. Energetically, the ‘double’ binding mode is determined to be slightly more stable than the ‘bridging’ one, by up to 4.9 kcal mol–1 depending on the inhibitor considered. This potential energy difference is not significant enough to exclude one of the binding modes, although the ‘double’ binding mode is likely to be preferred in most cases.
Fig. 3. Probable binding modes of the catechol moiety to the manganese cluster of arginase: A) ‘double’ binding mode; B) ‘bridging’ binding mode. Distances are shown in angstroms. Metal–ligand bonds are drawn in purple dotted lines and hydrogen bonds in pale blue dotted lines.

In principle, the deprotonated form of catechol can also bind to the manganese ions in this bridging fashion (see Fig. S2 in the ESI†), as does the hydroxide31 and nor-NOHA,13 for example. This deprotonated form of the unsubstituted catechol is clearly in the minority but chemical groups added to the ring can significantly lower the pKa of at least one phenol group, down to 7.17 for 4-nitrocatechol or 7.46 in the case of 2,3-dihydroxybenzaldehyde.32 However, the inhibition of these two compounds is weak, which suggests that the contribution of the deprotonated binding mode is very limited. This can possibly be explained by the electrostatic interactions between the negatively charged inhibitors and the (negatively charged) enzyme pocket, and/or by modified pKa values in the less solvated protein environment, at the bottom of the pocket. As a consequence, this binding mode was no longer considered.
Catechol derivative evaluation
Arginase inhibition was evaluated using a previously described colorimetric assay33,34 with slight modifications. Preliminary SAR was investigated by utilising the inhibition percentage at 500 μM (final concentration of catechol derivatives).
First, a total loss of inhibition was observed by locking the catechol function with methylidene which confirmed the role of the catechol function (Fig. 4A). Resorcinol displayed almost no activity whereas naphthalene-1,8-diol was still potent, which showed that the distance between the two oxygen atoms is obviously crucial (Fig. 4B). Indeed, the distance between the two oxygens of catechol, resorcinol and naphthalene-1,8-diol is respectively 2.68 Å, 4.71 Å and 2.62 Å (see Fig. S4 in the ESI†). These observations are completely in line with the above results obtained previously by quantum chemistry calculations for the ‘double’ configuration. The replacement of one phenolic group of the catechol moiety by aliphatic alcohol, carboxylic acid and other derivatives greatly decreased the inhibition (Fig. 4C).
Fig. 4. Inhibition of b-ARG1 by catechol derivatives; A) locking of the catechol function; B) distance between metal binding atoms; C) salicyl derivatives; D) soft ‘metal coordinating’ atoms; E) substituted catechol derivatives.

When the oxygen of the catechol was replaced by nitrogen or sulphur, considered as softer metal binding elements, a decrease in activity was observed. The only exception to this trend was 1,2-dithiophenol, which displayed unexpected activity, but this derivative was not viewed as an attractive lead due to its sensitivity towards oxidation. (Fig. 4D).
Electronic effects have been extensively studied from the catechol substituted library (see Table S1 in the ESI†). As evaluation shows, high and also moderate differences were observed, therefore the analysis was deepened by compiling dose–response curves and the IC50 values were determined (Fig. 4E). The results suggest that four-substituted compounds with an electron-donating group tend to do better. Our quantum chemistry calculations on an extended library confirm that there is no significant improvement of the enzyme inhibitor complexation energy, with respect to that of unsubstituted catechol. We also found that the best compounds usually feature an electron-donating group in position 4, but the complexation energy is only improved by 4.5 kcal mol–1 in the best case (left part of Fig. 5, blue bars). This result led us to explore compounds that take better advantage of the whole size of the enzymatic pocket, and could form hydrogen bonds with the outer asparagine, serine, aspartate and glutamate residues.
Fig. 5. Calculated complexation energies of some inhibitors with arginase, given in kcal mol–1, with respect to that of unsubstituted catechol.

Pharmacomodulation
Catechol–aniline derivatives are a promising group of possible inhibitors: their size matches that of the pocket (about 1.2 nm), their flexibility is greater than that of piceatannol, and the amino group can form indirect hydrogen bonds with carboxyl or carboxylate groups located on the walls of the pocket. For these reasons, we carried out complexation energy calculations on ten of these derivatives (right part of Fig. 5, orange bars), and our calculations show unambiguously that compounds featuring hydrogen bond donor or acceptor groups are excellent inhibitor candidates, with complexation energies improved by between 8.1 to 13.4 kcal mol–1 with respect to that of unsubstituted catechol (see bright orange bars in Fig. 5). Those compounds unable to form hydrogen bonds with the residues of the mouth of the cavity display low complexation energies (from 1.9 to –0.4 kcal mol–1, pale orange bars in Fig. 5). The fine analysis of the structures suggests that hydrogen bonding to the carboxylate groups of glutamate and aspartate are significantly stabilising factors, which invites us to consider the hydrogen bond donor groups on the aniline aromatic ring.
The synthesis of such derivatives using the procedure shown in Fig. 6 was thus undertaken. Methoxymethyl- (MOM), tert-butyldimethylsilyl (TBDMS) and diphenylmethylene ketal (C(Ph)2) protected 3,4-dihydroxybenzaldehyde 1a, 1b and 1c were respectively prepared from MOM bromide, TBDMS chloride and 1,1-dichloro-1,1-diphenylmethane. Amino derivatives were efficiently prepared from the reductive amination with sodium cyanoborohydride to give compounds from 2a to 2t. Deprotection under acidic conditions lead to catechol ammonium salts, 3a to 3t, which were assessed in the arginase inhibition assay.
Fig. 6. Synthesis of compounds 1a–c, 2a–t and 3a–t. Reagents and conditions: A: (1a) MOMBr (4.2 equiv.), DIPEA (5.2 equiv.), DCM, room temp., 4 h, 92%; (1b) TBDMSCl (2.5 equiv.), DBU (3 equiv.), Et2O, room temp., 16 h, 76%; (1c) (Ph)2CCl2 (1.05 equiv.), K2CO3 (2.2 equiv.), ACN, room temp., 16 h, 55%; B: i) R'NH2 (1.1 to 3 equiv.), AcOH (1.5 equiv.), MgSO4 (3 equiv.), MeOH, room temp., 1 h, ii) NaBH3CN (1.5 equiv.), MeOH, room temp., 16 h, 40–70% over two steps; C: HCl in MeOH (5 to 10 equiv.), 0 °C to room temp., 15 min, 20–90%; H2O (20 equiv.), TFA, room temp., 15 min, 40–50%.

Arginase inhibition was investigated with catechol compounds 3a–t and the corresponding results are shown in Fig. 7. Phenyl and benzyl moieties (respectively 3l and 3f) were well tolerated although they did not significantly improve the activity. However, aliphatic, hetero-aromatic and phenylethyl moieties (respectively 3a–c, 3d and 3e) clearly decreased the activity (IC50 over 200 μM). From this observation, substitutions over phenyl or benzyl derivatives were explored. It appeared that substitutions of benzyl derivatives (3g–k) significantly altered the inhibition (IC50 going over 200 μM) while substitutions of the phenyl moiety (3m–t) were tolerated. Moreover, functionalisation of the phenyl ring moiety in some cases provided added potency. Unexpectedly, compound 3s was not potent (IC50 = 141 ± 15 μM) although it is a pure analogue of piceatannol, which was the starting point of our investigation. Compounds 3q and 3r, which bear salicylic moieties, were slightly potent (IC50 = 108 ± 9 μM and 187 ± 18 μM respectively). Interestingly, compound 3p, bearing a hydroxyl at position three, displayed good activity with an IC50 of 83 ± 4 μM that is slightly superior to that of piceatannol (IC50 = 47 ± 7 μM). The 3,4-dihydroxyl compound 3t was also a good inhibitor with an IC50 of 76 ± 6 μM whereas the 4-hydroxyl compound 3o was less potent with an IC50 of 238 ± 32 μM. These results revealed that having the hydroxyl group at position three is critical with respect to the potency of the molecule while having the hydroxyl group at position four is optional. Finally, switching from 3-hydroxy (3p) to 3-methoxy (3n), avoiding the H-bond donor character, resulted in a drastic decrease of inhibition which suggests that an H-donor group is mandatory. In this case, quantum chemistry calculations will confirm these observations.
Fig. 7. b-ARG1 inhibition by compounds 3a to 3t. IC50 represented as the mean ± SD of 3 independent assays. Test conditions: b-ARG1 (5.89 μg mL–1), arginine (14.3 mM), magnesium dichloride (4.3 mM), 37 °C, 60 min. Revealed using o-phthalaldehyde (2.3 mM), primaquine (900 μM), boric acid (30 mM), sulphuric acid (15%), room temp., 15 min.

Mechanistic and binding assessment of compound 3t
The most active compound (3t) was studied in detail through the measurement of the inhibition constant (Ki) over bovine arginase I (b-ARG1). Dose–response experiments were carried out with several concentrations of arginine in the absence or presence of various concentrations of inhibitors. As shown in Fig. 8-I, the Dixon plot linear transformation and the Cornish–Bowden plot linear transformation indicated that the straight lines intersected in the second quadrant (counter clockwise).35
Fig. 8. Dose–response curves, IC50, Ki and mechanism on b-ARG1; IC50, enzymatic and cell based assay on h-ARG1. I & II. Dose response curves of 3t & piceatannol: A) Dixon (reciprocal enzyme reaction velocity vs. inhibitor concentrations), B) Cornish–Bowden (arginine concentration multiplied by the reciprocal enzyme reaction velocity vs. inhibitor concentrations), slopes are significantly different (p-value < 0.05), C) Lineweaver–Burk (reciprocal velocities vs. reciprocal of substrate concentrations) D) (slopes of Lineweaver–Burk plot vs. inhibitor concentration) plots to allow the determination of the inhibition type and the Ki. III. IC50, Ki, mechanism on b-ARG1; enzymatic and cell based assay on h-ARG1. Values are means ± SD from 3 separate experiments.

The IC50 values were measured over human arginase I (h-ARG1). While piceatannol displays an IC50 of 69 ± 7 μM, 3t shows an IC50 of 89 ± 8 μM which could be considered in the same range (nor-NOHA used as a control displayed an IC50 of 6.0 ± 0.4 μM under the same conditions). However, on CHO-K1 cells transiently transfected with h-ARG1 cDNA, piceatannol displayed poor inhibition reflecting poor cell uptake and 3t showed no significant improvement (Fig. 8-III).
The primary Lineweaver–Burk plot, the Dixon plot and the Cornish–Bowden plot (see Fig. 8-I-A–C) show that the straight lines intersected at a common point in the second quadrant (counterclockwise). Taken together, these results strongly suggest the mixed inhibition of 3t on arginase. The nonlinear regression model with the Michaelis–Menten model yields an α value of 8.0 which confirms mixed inhibition (while Asano et al. reported α values of 0.53 and 24.5 respectively for a pure non-competitive inhibitor and the reference competitive inhibitor, BEC).36 In the same way, piceatannol displays mixed inhibition on bovine arginase (see Fig. 8-II-A–C). The Ki values of 3t and piceatannol, obtained from the secondary Lineweaver–Burk plot (see Fig. 8-I-D and II-D), are 82 μM and 136 μM, respectively.
As shown in the docking pose on h-Arg1 in Fig. 9, the primary catechol group in 3t engages in two strong binding interactions with the two manganese nuclei as the ‘double’ binding mode, strengthened by a strong hydrogen bond with Glu277 and a weaker one with Thr246 (Fig. 9A). The secondary catechol moiety of compound 3t interacts with Asp183 by providing two strong hydrogen bonds, with Asn130 by accepting a weaker one, and with Ser137 (H-bond between its alcohol group and the aromatic ring). It is worth noting that residues Asn130, Asp183, Glu277, Ser137, and Thr246 to a minor extent are also strongly involved in the binding of ABH derivatives.37 More significantly, the phenol group at position three of 3p is also predicted to interact with the same residue (see Fig. S5 in the ESI†), corroborating previous SAR results where an increase in activity was observed for compounds 3p and 3t and the phenol group in position four could be considered as optional. Interestingly the space left by 3t in the active site could be explored through substitutions on the linker to reach new interactions with Asp128 or Glu186 for instance.
Fig. 9. Docking of 3t in the active site of h-Arg1. A) Interactions with residues Asp183, Asn130, Asp232 and Thr246; B) volume of the active site.

Woo et al. were the first to show arginase inhibition by piceatannol 3′-O-glucoside from murine liver and kidney lysates.28 Further investigations link arginase inhibition by piceatannol 3′-O-glucoside with regulation of NO production and endothelial dysfunction improvement in isolated mouse aorta, human endothelial cells28 and atherogenic model mice.38 The effect of piceatannol aglycone in restoring the endothelial function was demonstrated by Frombaum et al.39 They failed to observe arginase inhibition in vitro probably due to the use of a low concentration (1 μM). Polyphenols inhibit arginase from tens of micromolar to a few hundred micromolars.
Due to technical variations in enzymatic assays, inhibition by piceatannol on b-ARG1 and murine tissue lysate shifts from an IC50 of 12.1 μM (ref. 26) to 30% at 10 μM (ref. 40) respectively. We found piceatannol inhibition on b-ARG1 and h-ARG1 respectively at 47 μM and 69 μM consistent with preceding results. Compound 3t displays similar inhibition values respectively of 76 μM and 89 μM on b-ARG1 and h-ARG1.
The clinical use of piceatannol is restricted by low solubility and poor bioavailability rather than moderate absorption suggesting first-pass metabolism.25,41 The only series of derivatives previously evaluated40 suggest that ethylene linkage could be reduced without hindering arginase inhibition. In the present work, amine linkage was used due to their putative solubility.
However, we failed to obtain active compounds without the phenolic function instead of the resorcinol part and consequently didn't succeed in improving piceatannol activity in cell based assays.
Catechol is the common feature of effective polyphenols.12,24 In vitro evaluation of catechol derivatives and especially blockade by methylendioxybenzene reinforces the catechol determinant hypothesis. The electronic effect of catechol substitution on inhibition imply charge transfers between the hydroxyl groups and the manganese cluster and first simulations of the catechol binding mode suggest simultaneous different non-covalent interactions requiring a high level of theory approached by quantum chemistry methods. Experimental results are correlated with computational simulations and putative binding modes of piceatannol and 3t clearly indicate hydroxyl as the most stabilizing chemical group. Currently there are no crystallographic data for a polyphenol–arginase complex but our results are in line with the crystallographic data for amino acid based inhibitors13,30 showing a H-bonding network.
Conclusions
Arginase is a fundamental enzyme and its overexpression is associated with cardiovascular and non-cardiovascular diseases. Despite being recognised as an attractive target, only a few inhibitors of arginase have been found hitherto and among them piceatannol is one of the most potent natural polyphenols. In this report, we prepared and evaluated a series of amino-analogues of piceatannol that yielded compounds 3p and 3t with IC50 on b-ARG1 of respectively 83 ± 4 μM and 76 ± 6 μM which is slightly superior to that of piceatannol (IC50 = 47 ± 7 μM). On h-ARG1, the IC50 values of 3t and piceatannol are in the same range (respectively 89 ± 8 μM and 69 ± 7 μM). Catechol binding has been deeply studied through quantum chemistry modelling, corroborated by in vitro results, which suggest two modes of binding. These experiments allowed the rigorous determination of the most probable interactions of 3p and 3t which suggests that aminopolyphenols are a promising avenue for the development of new arginase inhibitors.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
J. M. thanks the Conseil Régional de Bourgogne Franche-Comté for grants 2017Y-07543 and 2017Y-06422. The authors would like to acknowledge the computing facilities of the Mésocentre de Franche-Comté and their staff for the allocation of computing time, and the analysis platform of UTINAM UMR CNRS 6213 for NMR.
Footnotes
†Electronic supplementary information (ESI) available: Table S1, Fig. S1 to S5, protocols for quantum chemistry modelling, enzymatic assays and functional cell assays, preparation, HRMS, and 1H and 13C NMR spectra of all new compounds. See DOI:10.1039/d0md00011f
References
- Vockley J. G., Jenkinson C. P., Shukla H., Kern R. M., Grody W. W., Cederbaum S. D. Genes Genomics. 1996;38(2):118–123. doi: 10.1006/geno.1996.0606. [DOI] [PubMed] [Google Scholar]
- Steppan J., Nyhan D. and Berkowitz D. E., Development of Novel Arginase Inhibitors for Therapy of Endothelial Dysfunction. Front Immunol. 2013, [cité 29 oct 2019];4. Disponible sur: http://journal.frontiersin.org/article/10.3389/fimmu.2013.00278/abstract. [DOI] [PMC free article] [PubMed]
- Pernow J., Jung C. Curr. Vasc. Pharmacol. 2016;14(2):155–162. doi: 10.2174/1570161114666151202205617. [DOI] [PubMed] [Google Scholar]
- Caldwell R. B., Toque H. A., Narayanan S. P., Caldwell R. W. Trends Pharmacol. Sci. 2015;36(6):395–405. doi: 10.1016/j.tips.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acuña S. M., Aoki J. I., Laranjeira-Silva M. F., Zampieri R. A., Fernandes J. C. R., Muxel S. M., Floeter-Winter L. M. PLoS One. 2017;12(11):e0187186. doi: 10.1371/journal.pone.0187186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steggerda S. M., Bennett M. K., Chen J., Emberley E., Huang T., Janes J. R. J. Immunother. Cancer. 2017;5(1):101. doi: 10.1186/s40425-017-0308-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnost T., Ma L., da Silva R. F., Rezakhaniha R., Houdayer C., Stergiopulos N. Cardiovasc. Res. 2010;87(3):569–577. doi: 10.1093/cvr/cvq081. [DOI] [PubMed] [Google Scholar]
- Olivon V. C., Fraga-Silva R. A., Segers D., Demougeot C., de Oliveira A. M., Savergnini S. S. Atherosclerosis. 2013;227(2):236–243. doi: 10.1016/j.atherosclerosis.2012.12.014. [DOI] [PubMed] [Google Scholar]
- Holowatz L. A., Kenney W. L. J. Physiol. 2007;581(2):863–872. doi: 10.1113/jphysiol.2007.128959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovamees O., Shemyakin A., Eriksson M., Angelin B., Pernow J. J. Intern. Med. 2016;279(5):477–484. doi: 10.1111/joim.12461. [DOI] [PubMed] [Google Scholar]
- Kövamees O., Shemyakin A., Checa A., Wheelock C. E., Lundberg J. O., Östenson C.-G. J. Clin. Endocrinol. Metab. 2016;101(11):3952–3958. doi: 10.1210/jc.2016-2007. [DOI] [PubMed] [Google Scholar]
- Pudlo M., Demougeot C., Girard-Thernier C. Med. Res. Rev. 2017;37(3):475–513. doi: 10.1002/med.21419. [DOI] [PubMed] [Google Scholar]
- Di Costanzo L., Ilies M., Thorn K. J., Christianson D. W. Arch. Biochem. Biophys. 2010;496(2):101–108. doi: 10.1016/j.abb.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colleluori D. M., Ash D. E. Biochemistry. 2001;40(31):9356–9362. doi: 10.1021/bi010783g. [DOI] [PubMed] [Google Scholar]
- Baggio R., Elbaum D., Kanyo Z. F., Carroll P. J., Cavalli R. C., Ash D. E. J. Am. Chem. Soc. 1997;119(34):8107–8108. [Google Scholar]
- Kim N. N., Cox J. D., Baggio R. F., Emig F. A., Mistry S. K., Harper S. L. Biochemistry. 2001;40(9):2678–2688. doi: 10.1021/bi002317h. [DOI] [PubMed] [Google Scholar]
- Golebiowski A., Paul Beckett R., Van Zandt M., Ji M. K., Whitehouse D., Ryder T. R. Bioorg. Med. Chem. Lett. 2013;23(7):2027–2030. doi: 10.1016/j.bmcl.2013.02.024. [DOI] [PubMed] [Google Scholar]
- Golebiowski A., Whitehouse D., Beckett R. P., Van Zandt M., Ji M. K., Ryder T. R. Bioorg. Med. Chem. Lett. 2013;23(17):4837–4841. doi: 10.1016/j.bmcl.2013.06.092. [DOI] [PubMed] [Google Scholar]
- Van Zandt M. C., Whitehouse D. L., Golebiowski A., Ji M. K., Zhang M., Beckett R. P. J. Med. Chem. 2013;56(6):2568–2580. doi: 10.1021/jm400014c. [DOI] [PubMed] [Google Scholar]
- Van Zandt M. C., Jagdmann G. E., Whitehouse D. L., Ji M., Savoy J., Potapova O. J. Med. Chem. 2019;62(17):8164–8177. doi: 10.1021/acs.jmedchem.9b00931. [DOI] [PubMed] [Google Scholar]
- Guo X., Chen Y., Seto C. T. Bioorg. Med. Chem. 2018;26(14):3939–3946. doi: 10.1016/j.bmc.2018.06.015. [DOI] [PubMed] [Google Scholar]
- Havlinova Z., Babicova A., Hroch M., Chladek J. Xenobiotica. 2013;43(10):886–894. doi: 10.3109/00498254.2013.780672. [DOI] [PubMed] [Google Scholar]
- Havlínová Z., Hroch M., Nagy A., Šišpera L., Holeček M., Chládek J. Gen. Physiol. Biophys. 2014;33(02):189–198. doi: 10.4149/gpb_2013078. [DOI] [PubMed] [Google Scholar]
- Girard-Thernier C., Pham T.-N., Demougeot C. Mini-Rev. Med. Chem. 2015;15(10):798–808. doi: 10.2174/1389557515666150511153852. [DOI] [PubMed] [Google Scholar]
- Minozzo B., Fernandes D., Beltrame F. Planta Med. 2018;84(05):277–295. doi: 10.1055/s-0044-100398. [DOI] [PubMed] [Google Scholar]
- Bordage S., Pham T.-N., Zedet A., Gugglielmetti A.-S., Nappey M., Demougeot C. Planta Med. 2016;83(07):647–653. doi: 10.1055/s-0042-118711. [DOI] [PubMed] [Google Scholar]
- Manjolin L. C., dos Reis M. B. G., Maquiaveli C. do C., Santos-Filho O. A., da Silva E. R. Food Chem. 2013;141(3):2253–2262. doi: 10.1016/j.foodchem.2013.05.025. [DOI] [PubMed] [Google Scholar]
- Woo A., Min B., Ryoo S. Exp. Mol. Med. 2010;42(7):524. doi: 10.3858/emm.2010.42.7.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham T.-N., Bordage S., Pudlo M., Demougeot C., Thai K.-M., Girard-Thernier C. Int. J. Mol. Sci. 2016;17(10):1656. doi: 10.3390/ijms17101656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Costanzo L., Sabio G., Mora A., Rodriguez P. C., Ochoa A. C., Centeno F. Proc. Natl. Acad. Sci. U. S. A. 2005;102(37):13058–13063. doi: 10.1073/pnas.0504027102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Costanzo L., Pique M. E., Christianson D. W. J. Am. Chem. Soc. 2007;129(20):6388–6389. doi: 10.1021/ja071567j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The predicted pKa calculations were provided by the website www.chemicalize.com [Internet]. [cité 26 nov 2019]. Disponible sur: https://chemaxon.com/products.
- Jung D., Biggs H., Erikson J., Ledyard P. U. Clin. Chem. 1975;21(8):1136–1140. [PubMed] [Google Scholar]
- Zawada R. J. X., Kwan P., Olszewski K. L., Llinas M., Huang S.-G. Biochem. Cell Biol. 2009;87(3):541–544. doi: 10.1139/o09-011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish-Bowden A. Biochem. J. 1974;137(1):143–144. doi: 10.1042/bj1370143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano W., Takahashi Y., Kawano M., Hantani Y. SLAS Discovery. 2019;24(4):457–465. doi: 10.1177/2472555218812663. [DOI] [PubMed] [Google Scholar]
- Velázquez-Libera J. L., Navarro-Retamal C., Caballero J. Int. J. Mol. Sci. 2018;19(10):2956. doi: 10.3390/ijms19102956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo A., Shin W., Cuong T. D., Min B., Lee J. H., Jeon B. H. Int. J. Mol. Med. 2013;31(4):803–810. doi: 10.3892/ijmm.2013.1261. [DOI] [PubMed] [Google Scholar]
- Frombaum M., Therond P., Djelidi R., Beaudeux J.-L., Bonnefont-Rousselot D., Borderie D. Free Radical Res. 2011;45(3):293–302. doi: 10.3109/10715762.2010.527337. [DOI] [PubMed] [Google Scholar]
- Moon I., Damodar K., Kim J.-K., Ryoo S., Jun J.-G. Bull. Korean Chem. Soc. 2017;38(3):342–349. [Google Scholar]
- Abdelkawy K. S., Lack K., Elbarbry F. Eur. J. Drug Metab. Pharmacokinet. 2017;42(3):355–370. doi: 10.1007/s13318-016-0381-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.