

 Potent α-N-heterocyclic carboxaldehyde thiosemicarbazone (HCT) antiproliferatives were synthesized through iterative rounds of methylation and fluorination modifications, with anticancer activities being potentiated by physiological levels of copper.
Potent α-N-heterocyclic carboxaldehyde thiosemicarbazone (HCT) antiproliferatives were synthesized through iterative rounds of methylation and fluorination modifications, with anticancer activities being potentiated by physiological levels of copper.
Abstract
A potent class of isoquinoline-based α-N-heterocyclic carboxaldehyde thiosemicarbazone (HCT) compounds has been rediscovered; based upon this scaffold, three series of antiproliferative agents were synthesized through iterative rounds of methylation and fluorination modifications, with anticancer activities being potentiated by physiologically relevant levels of copper. The lead compound, HCT-13, was highly potent against a panel of pancreatic, small cell lung carcinoma, prostate cancer, and leukemia models, with IC50 values in the low-to-mid nanomolar range. Density functional theory (DFT) calculations showed that fluorination at the 6-position of HCT-13 was beneficial for ligand-copper complex formation, stability, and ease of metal-center reduction. Through a chemical genomics screen, we identify DNA damage response/replication stress response (DDR/RSR) pathways, specifically those mediated by ataxia-telangiectasia and Rad3-related protein kinase (ATR), as potential compensatory mechanism(s) of action following HCT-13 treatment. We further show that the cytotoxicity of HCT-13 is copper-dependent, that it promotes mitochondrial electron transport chain (mtETC) dysfunction, induces production of reactive oxygen species (ROS), and selectively depletes guanosine nucleotide pools. Lastly, we identify metabolic hallmarks for therapeutic target stratification and demonstrate the in vivo efficacy of HCT-13 against aggressive models of acute leukemias in mice.
Introduction
The diverse therapeutic potential of α-N-heterocyclic carboxaldehyde thiosemicarbazones (HCTs) has been investigated since the 1940s, with tuberculostatic activity being first observed in vivo as early as 1946.1 This class of compounds was subsequently shown to possess antitumor and antimicrobial activities, prompting decades of research and development.2–7 In particular, isoquinoline-based HCTs such as 2-(isoquinolin-1-ylmethylene)hydrazine-1-carbothioamide (IQ-1, HCT-1)3 were the subject of early interest due to evidence of preclinical anti-tumor efficacy (Fig. 1).5 While the mechanisms of action of HCTs are multi-modal and have not yet been fully defined,8–15 their biological activities generally stem from the ability to chelate transition metals through their heterocyclic nitrogen, Schiff base nitrogen, and thiosemicarbazone sulfur. The resulting HCT-metal complexes undergo redox cycles and generate cytotoxic reactive oxygen species (ROS) through Fenton and/or Haber–Weiss processes.16 HCTs are particularly adept at binding copper,17 which can be either beneficial or detrimental to the compound's biological activity. For instance, physiological concentrations of copper in human plasma (11–18 μM)18,19 interfere with the ribonucleotide reductase-inhibitory activity of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP),17 while the cytotoxicities of Dp44mT20,21 and NSC-31972622 are potentiated by copper. Binding of this transition metal is intriguing from an anticancer therapy standpoint, as cancers rely upon higher intracellular levels of copper, relative to healthy cells, to promote angiogenesis, tumor growth, and metastasis.23,24 Indeed, several therapeutic strategies have employed small molecules to disrupt copper homeostasis in cancers, either through chelation-mediated copper sequestration, or by increasing intracellular copper to cytotoxic levels through ionophoric modalities.22,25
Fig. 1. Cu(ii) supplementation increases the potency of a subset of HCT compounds. MIAPACA2 cells were treated with HCT compound with 20 μM CuCl2 supplementation or with HCT compound alone (compound-only IC90 value shown in parentheses) for 72 h. Cell viability was then measured with CellTiter-Glo and IC90 values determined. Lead compound HCT-13 is highlighted.

The cytotoxic effects of HCTs coupled with their ability to bind copper make them a compelling scaffold from which to develop copper-mediated therapeutics. Our group has identified isoquinoline-based HCTs as a viable scaffold for this purpose, as previously reported analogs demonstrated both in vitro and in vivo efficacy and the chemical space and mechanism(s) of action remain insufficiently explored. Confounding factors have included lack of cancer tissue-specificity, difficulty stratifying susceptible cancer subtypes, poorly defined mechanisms of action, and potency and toxicity issues arising in part from aberrant metal chelation.12 Herein, the design and synthesis of a series of novel isoquinoline-based HCTs is reported, with identification of a promising lead compound – HCT-13 – preceding mechanistic and pre-clinical studies. The lead compound potently inhibited proliferation of a panel of cancer models at nanomolar concentrations in the presence of physiologically relevant levels of copper, while non-cancerous cells were significantly less affected. A mechanism of action was delineated, with HCT-13 being found to induce mitochondrial electron transport chain (mtETC) dysfunction and production of reactive oxygen species (ROS), deplete guanosine nucleotide pools, and engage DNA damage response/replication stress response pathways. The activity of HCT-13 was potentiated by co-administration of the glycolysis inhibitor 2-deoxy-d-glucose (2-DG), indicating that cancers which rely more heavily upon oxidative phosphorylation, such as certain leukemias, will be more susceptible to HCT-13 therapy. Lastly, we demonstrate the in vivo efficacy of a copper:HCT-13 (Cu[HCT-13]) complex in murine models of aggressive acute leukemias.
Results
Synthesis of isoquinoline-based HCT compounds
Among the previously reported isoquinoline HCTs, the 5-, 7-, and 8-fluoro analogs of HCT-1 were of particular interest to our group. These compounds varied in terms of potency and toxicity,6 demonstrating that the effects of fluorination were dependent upon the position of isoquinoline substitution. The benefits that fluorination endows upon small-molecule drugs are well documented, and are also reflected in the marketplace, as approximately 20% of all pharmaceuticals are fluorinated.26–28 Importantly, fluorination is associated with increased lipophilicity and may modulate redox activity upon metal complexation by virtue of its high electronegativity, a property which we wished to explore given our desire to leverage the copper-chelating potential of the scaffold.29 With this in mind, we were curious as to how the previously unreported 4- and 6-fluoro analogs of HCT-1 would perform in antiproliferative assays against cancer models, both with and without copper supplementation. Additionally, we were intrigued as to whether fluorination at these positions would complement 4′ amine alkylation, as Kowol et al. discovered that 4′ amine alkylation potentiated the cytotoxicity of 3-AP analogs.30
A total of 11 novel isoquinoline-based HCTs were synthesized, as well as four previously reported compounds (HCT-1, HCT-4, HCT-5, HCT-6) for comparison purposes. Our synthetic approach began with methylation of the appropriate isoquinoline 1 to generate 2 (Scheme 1). Depending upon the desired 5-position substituent, 2 was then subjected to either Route A (5-hydrido), Route B (5-amino), or Route C (5-methylamino). Syntheses of HCTs 1–3, HCTs 6–8, and HCTs 11–13 were carried out by route A, wherein the methyl substituent of 2 was oxidized using selenium dioxide (SeO2) to furnish the carboxaldehyde 3. Condensation with the appropriate thiosemicarbazide under acidic conditions yielded the desired HCT. HCT-5 and HCT-10 were synthesized via route B, which began with nitration of 2 followed by an iron-mediated reduction to the amine, which was subsequently Boc-protected and oxidized to produce carboxaldehyde 4. This intermediate was then simultaneously Boc-deprotected and condensed with the appropriate thiosemicarbazide under acidic conditions to furnish the target HCT. Syntheses of HCT-4, HCT-9, HCT-14, and HCT-15via route C proceeded from 2 with installation of a nitro group, subsequent conversion to the mono-Boc-methylamine, and SeO2-mediated oxidation to furnish 5. Concurrent Boc-deprotection and thiosemicarbazide condensation were again achieved under acidic conditions to provide the desired HCT compound. While characterizing the HCTs, we occasionally observed the presence of a minor Z-isomeric product, particularly for HCTs 11–15, which was inseparable by HPLC from the E-isomer. When encountered, the isomeric mixtures were used for in vitro investigations, as previous studies have reported no significant difference in potency for these geometric isomers.31
Scheme 1. Synthesis of HCT compounds 1–15 from simple isoquinolines. (a) Allyl chloroformate, MeMgBr, THF; (b) Pd(PPh3)4, morpholine; DDQ, CH2Cl2; (c) SeO2, 1,4-dioxane, 60 °C; (d) appropriate thiosemicarbazide, HCl, EtOH, reflux or microwave 50 °C; (e) KNO3, H2SO4; (f) Fe, HCl, MeOH, reflux; (g) Boc2O, DMAP, TEA, THF; (h) Boc2O, DMAP, TEA, THF; NaHCO3, MeOH, reflux or K2CO3, MeOH, reflux; (i) NaH, THF; MeI. HCTs 1–3, 6–8, and 11–13 synthesized through route A; HCTs 5 and 10 synthesized through route B; HCTs 4, 9, 14, and 15 synthesized through route C. See Experimental section for full synthetic details.

Fluorination and dimethylation display synergistic effects further potentiated by copper(ii) supplementation
IC90 values against MIAPACA2 cells were first determined using conventional cell culture conditions (Fig. 1, compound-only IC90 values reported in parentheses). Compounds were separated into three series – 4′ primary amines, 4′ secondary amines, and 4′ tertiary amines – to reflect the relative degrees of 4′ amine methylation. We first synthesized a series of non-methylated 4′ primary amine compounds, with known compounds HCT-1, HCT-4, and HCT-5 included to gauge whether fluorination of the isoquinoline proved beneficial for biological activity.32,33 Within the 4′ primary amine series, fluorination at the isoquinoline 4-position (HCT-2) did not show an increase in potency relative to unsubstituted analog HCT-1. However, fluorination at the 6-position (HCT-3) showed a 3-fold increase, demonstrating that the fluorine position impacts the activity of these compounds.
In the 4′ secondary amine series, we observed a synergistic effect when combining isoquinoline fluorination with 4′ amine methylation. The 4-fluorine substituted HCT-7 and the 6-fluorine substituted HCT-8 were each significantly more potent than their corresponding non-fluorinated analog HCT-6, as well as more potent than their 4′ primary amine counterparts (HCT-2 and HCT-3, respectively). The trend of isoquinoline substitution and 4′ secondary amine combining to enhance the potency of the drug held well for the 5-methylamino substituted HCT-9 and 5-amino substituted HCT-10 analogs. Together, these results suggested that combining isoquinoline substitution, particularly 4- or 6-fluorination, with 4′ amine methylation produced greater-than-additive antiproliferative effects when compared with either modification alone.
The effects of fluorine substitution became significantly more pronounced for the dimethylated 4′-tertiary amine compounds HCT-12 and HCT-13, whose IC90 values were in the nM range and were roughly 110- and 270-fold more potent, respectively, when compared to their non-fluorinated analog HCT-11. Fluorination at the 6-position (HCT-13) was found to be superior to fluorination at the 4-position (HCT-12), a trend which also held for the fluorine-substituted 5-methylamino compounds HCT-14 and HCT-15.
HCT compounds are known to be copper chelators,34–36 and a recent publication by Stockwell and coworkers demonstrated that the activity of a known 4′ tertiary amine HCT (NSC-319726) was significantly potentiated by the addition of copper.22 To test whether our compounds were similarly potentiated, we determined IC90 values against MIAPACA2 cells in media supplemented with physiologically relevant levels of copper (Fig. 1, ESI‡ Fig. S1a). While the activity of HCT-5 was attenuated, all other compounds displayed significant increases in potency under copper-supplemented conditions, with +Cu IC90 values improving as the degree of 4′ amine methylation increased. Fluorine substitution led to greater potency when compared with corresponding non-fluorinated analogs, and compounds bearing 4′ tertiary amines were the most active, achieving +Cu IC90 values as low as 21.6 nM (HCT-13). Our results demonstrated that physiologically relevant levels of copper potentiated the activity of isoquinoline HCTs, and that 4′ amine methylation worked cooperatively with fluorine substitution. Due to its potency and straightforward synthesis, we chose HCT-13 as our lead compound for in-depth mechanism of action studies.
Computational analysis shows fluorination increases the stability of copper:HCT complex
To elucidate potential contributing factors to HCT-13's potency, density functional theory (DFT) calculations were performed and the relative energies of formation and Cu(ii) reduction were determined for selected ligand–Cu(ii) complexes (Table 1). All calculations were performed with Gaussian 1637 using the B3LYP functional and the 6-31G(d) basis set. A correlation between IC90 value and energy of complex formation was found, with the latter consisting of ligand deprotonation and copper binding energies. HCT-5, which possesses an electron-donating substituent at the isoquinoline 6-position and was the least potent compound tested, bonded most weakly to Cu(ii). Conversely, HCT-12 and HCT-13, which bear electron-withdrawing fluorine substituents at the 4- and 6-positions, respectively, bonded more strongly. HCT-11, which possessed no isoquinoline substituents, had an energy of coordination to Cu(ii) that was 4.0 kcal mol–1 lower than that of HCT-5. These studies revealed that the ease of deprotonation in the thiosemicarbazone chain most strongly contributed to the trends observed in the energies of complex formation, where ligands with electron-withdrawing substituents had lower energies of deprotonation. It was also found that the reduction of Cu(ii) to Cu(i) was most exergonic in the Cu:HCT-13 complex, implying that this compound is most readily capable of participating in redox processes.
Table 1. DFT calculations of relative deprotonation, binding, and complexation energies of selected Cu(ii):HCT-13 complexes and Cu(ii) to Cu(i) reduction energies.
| ||||
| Compound | Rel. deprotonation energy (kcal mol–1) | Rel. binding energy (kcal mol–1) | Rel. complexation energy (kcal mol–1) | Rel. energy of reduction (kcal mol–1) |
| HCT-5 | 0 | 0 | 0 | –0.9 |
| HCT-11 | –4.6 | 0.6 | –4 | 0 |
| HCT-12 | –9.6 | 4.3 | –5.5 | –1.5 |
| HCT-13 | –10.5 | 4.4 | –6.1 | –1.5 |
HCT-13 induces copper-dependent toxicity and is highly potent against a panel of cancer models
To begin investigating the mechanism of action of HCT-13, its ionophoric capabilities were evaluated using inductively coupled plasma mass spectrometry (ICP-MS). Intracellular levels of copper increased when cells were treated with HCT-13 in Cu(ii)-supplemented media, but not with HCT-13 or Cu(ii) supplementation alone (Fig. 2a). To measure the effect of copper upon the lead compound's antiproliferative activity, the viability of MIAPACA2 cells was assessed following treatment with HCT-13 in Cu(ii)-supplemented media with and without bathocuproine disulfonate (BCPS), a membrane-impermeable copper chelator. The addition of BCPS completely abrogated the cytotoxicity of HCT-13, suggesting that the growth inhibitory effect of our lead compound is dependent upon copper availability (Fig. 2b). We next determined the effects of supplementation with selected metals, namely iron and zinc, upon the antiproliferative activity of HCT-13 and found its potency was highest in the presence of Cu(ii), diminished but still active in the presence of Fe(ii), and largely inactive in the presence of Zn(ii) (Fig. 2c, 20 μM supplementation). Collectively, these data suggest that HCT-13 is a Cu(ii) ionophore which increases intracellular copper concentration and whose cytotoxicity is copper-dependent.
Fig. 2. Copper is required for HCT-13 activity. (a) Intracellular concentrations of copper measured by inductively coupled plasma mass spectrometry (ICP-MS) in MIAPACA2 PDAC cells treated with HCT-13 (25 nM) for 24 h ± Cu(ii) (20 μM); *** P < 0.001; V: vehicle. (b) Viability of MIAPACA2 cells treated with HCT-13 (25 nM) + Cu(ii) (20 μM) for 24 h ± bathocuproine disulfonate (BCPS, 300 μM) measured by trypan blue exclusion; * P < 0.05; ** P < 0.01; V: vehicle, NS: no supplement. (c) Proliferation rate of MIAPACA2 PDAC cells measured by CellTiterGlo following HCT-13 treatment for 72 h ± Cu(ii), Fe(ii) or Zn(ii) (20 μM). (mean ± SD, n = 2, one-way ANOVA corrected for multiple comparisons by Bonferroni adjustment).

To determine whether HCT-13 displayed heterogeneous effects against different cell types, it was screened against a panel containing aggressive cancer models and a non-cancerous cell line. Pancreatic ductal adenocarcinoma (PDAC), small cell lung carcinoma (SCLC), and prostate cancer (PC) models were evaluated as these cancers rely upon elevated copper levels to sustain growth, and serum copper levels are elevated (>20 μM) in individuals with these types of tumors.24,38–43 Aggressive leukemia models were also evaluated, as these may be more directly targeted relative to solid tumors and treatments for hematological malignancies such as acute myeloid leukemia (AML) remain an unmet need.44 PDAC, SCLC, and PC cancer models were cultured in media supplemented with physiologically relevant levels of copper (20 μM CuCl2) and treated with HCT-13, while the leukemia models were treated with pre-formed copper:HCT-13 (Cu[HCT-13]) complexes, as free copper is known to be toxic towards these cells (antiproliferative activity of Cu[HCT-13] was consistent with that of HCT-13 + Cu(ii)) (ESI‡ Fig. S1b). In this assay, HCT-13 was shown to be a highly potent cancer cell growth inhibitor, with IC50 values ranging from 1 nM to 200 nM (Fig. 3). Importantly, the non-cancerous human epithelial cell line HPDE was markedly more resistant to treatment than the cancer models evaluated, while the cancer models displayed differential susceptibility. Together, these results indicate that HCT-13 possesses a high degree of cancer-specific cytotoxicity, while its differential effects against the cancer models suggest its mechanism(s) of action is dependent upon specific cancer-subtypes/characteristics.
Fig. 3. Activity of HCT-13 in a panel of cancer cell lines and non-transformed cells. IC50 values in a panel of human and mouse prostate cancer (PC), small cell lung carcinoma (SCLC), pancreatic ductal adenocarcinoma (PDAC), leukemia, and non-transformed epithelial models treated with HCT-13 + Cu(ii) (20 μM) for 72 h measured by CellTiterGlo.

Chemical genomics screen identifies DNA damage response and replication stress response as potential mechanisms of action
To begin identifying the mechanisms of action of HCT-13, we performed an unbiased pharmacological inhibition screen using a chemical genomics platform consisting of 430 kinase inhibitors. MIAPACA2 cells were treated with the 430-member library, covering a 7-point concentration range spanning between 6.5 nM and 5 μM, with and without 25 nM HCT-13 in presence of 20 μM CuCl2. After 72 h, ATP content was measured using CellTiter-Glo (Fig. 4a, ESI‡ Fig. S2a). A composite synergy score was calculated for each combination, defined as the sum of the Bliss additivity score (% proliferation inhibition observed – % proliferation inhibition expected). A positive synergy score indicates greater-than-additive interaction, and a negative score indicates less-than-additive interaction i.e. antagonism.
Fig. 4. A chemical genomics screen identifies the replication stress response pathway as an actionable co-dependency of HCT-13-treated cells. (a) Experimental design of a synthetic lethality screen using a library of protein kinase inhibitors against HCT-13-treated cells in the presence of Cu(ii) (20 μM). (b) Radar plot of screen results. (c) Z-score values for kinase inhibitors within the DNA damage response/replication stress response (DDR/RSR) pathway module. (d) Annexin V/PI staining in MIAPACA2 cells to validate the synergistic interaction of HCT-13 (25 nM) + Cu(ii) (20 μM) with ATRi (250 nM VE-822) treated for 72 h; **** P < 0.0001. (e) Representative immunoblot of DDR pathway and cell death biomarkers in MIAPACA2 PDAC cells treated with HCT-13 (10 nM) + Cu(ii) (20 μM); V: vehicle. (f) Nucleotide pool measurements by LC-MS/MS-MRM in MIAPACA-2 cells treated with 25 nM HCT-13 + Cu(ii) (20 μM) for 48 h (mean ± SD, n = 3, comparison made between glucose-labeled pools using two-way ANOVA corrected for multiple comparisons by Bonferroni adjustment, * P < 0.05; *** P < 0.001); V: vehicle.

The ten highest scoring compounds were kinase inhibitors contained in the DNA damage response/replication stress response (DDR/RSR) module, with the ataxia telangiectasia mutated serine/threonine kinase (ATM)/checkpoint kinase 2 (CHK2) and Rad3-related serine/threonine kinase (ATR)/CHK1 pathways featuring as the most prominent codependencies (Fig. 4b and c).45–47 All eight ATR and CHEK1 inhibitors included in the library scored positively in the screen, implying that the DDR/RSR pathways are activated by HCT-13. The synergistic interaction of HCT-13 with ATR inhibition was further validated by annexin V/PI (apoptosis) and trypan blue viability staining in PDAC (MIAPACA2, CFPAC-1) and PC (C4-2) cell lines (Fig. 4d, ESI‡ Fig. S2b and c). Furthermore, phosphorylation of CHEK1 and CHEK2 kinases was consistently observed upon HCT-13 + Cu(ii) treatment, as well as the induction of DNA damage marker pH2A.X and cleavage of the apoptotic marker caspase 3 (Fig. 4e). These observations are indicative of DDR/RSR pathway activation, which was hypothesized to arise from nucleotide insufficiency and/or, given the ability of copper to generate ROS, from ROS-mediated DNA damage.48–50
To test this hypothesis, nucleotide levels were measured in treated and non-treated cells using liquid chromatography-tandem mass spectrometry with multiple reaction monitoring (LC-MS/MS-MRM) as previously described.51 Both dGTP and rGTP pools were decreased in HCT-13-treated cells, while levels of the other nucleotides either increased or the change was statistically insignificant (Fig. 4f). These findings indicate that HCT-13 preferentially decreases the dGTP and rGTP pools, which is potentially mediated by oxidative processes as guanine is the most readily oxidized nucleobase.52 Together with the immunoblot results (Fig. 4e), these data suggest that dGTP pool insufficiency in HCT-13-treated cells triggers the activation of the intra-S checkpoint, as measured by increased pChk1 levels, which in turn renders these cells dependent upon the activity of the replication stress response pathway.
HCT-13 induces ROS production, displays mitochondrial-dependent toxicity and targets OXPHOS
Given that our dGTP and rGTP pool measurements pointed towards a guanosine-depleting mechanism of action, we set out to determine whether our lead compound was giving rise to ROS. We found that HCT-13 treatment induced ROS generation detectable by CM-H2DCFDA staining in MIAPACA2 cells (Fig. 5a).53 ROS generation was also observed in the mitochondria, as measured by mitochondria-specific dye MitoSOX (Fig. 5b).53 To further probe the ramifications of the HCT-13-generated ROS, we inquired as to whether oxidative phosphorylation (OXPHOS) in MIAPACA2 cells was altered following treatment. A Seahorse assay was performed to measure the alteration in overall OXPHOS, and an electron flow assay was performed in isolated mitochondria to assess which electron transport chain (ETC) complex is impacted (Fig. 5c and d, ESI‡ Fig. S3a and b).54–56 These assays showed that cell respiratory capacity decreased significantly upon HCT-13 treatment as measured by reduced oxygen consumption rate (OCR), that OXPHOS capacity was impaired, and that ETC complex 1 was most dramatically affected.
Fig. 5. HCT-13 + Cu(ii) induces oxidative stress and has selective mitochondrial toxicity. (a) Reactive oxygen species (ROS) measurements using CM-H2DCFDA after HCT-13 (25 nM) ± Cu(ii) (20 μM) treatment for 24 h; mean fluorescence intensity (MFI); mean ± SD, n = 2, Student t-test, ** P < 0.01. (b) Mitochondrial ROS detection using MitoSOX in MIAPACA2 PDAC cells treated with HCT-13 (25 nM) + Cu(ii) (20 μM) for 24 h. mean ± SD, n = 2, Student t-test, *** P < 0.001. (c) Oxygen consumption rate (OCR) of MIAPACA2 PDAC cells treated with HCT-13 (25 nM) + Cu(ii) (20 μM) for 24 h; ** P < 0.01; *** P < 0.001. (d) OCR of isolated mitochondria measured with or without HCT-13 (25 nM) + Cu(ii) (20 μM); * P < 0.05; ** P < 0.01. (e) Viability of 143 BTK parental (wild type, WT) and ρ0 cells after 48 h of the indicated HCT-13 concentration + Cu(ii) (20 μM) treatment, assessed with trypan blue staining; mean ± SD, n = 2, Student t-test, * P < 0.05; *** P < 0.001; V: vehicle. (f) Cell viability by trypan blue staining in MIAPACA2 PDAC cells to interrogate the interaction of HCT-13 (25 nM) + Cu(ii) (20 μM) with 2-DG (2 mM) for 48 h; mean ± SD, n = 2, Student t-test, ** P < 0.01; V: vehicle.

These findings suggested that HCT-13 inhibited mtETC activity but did not indicate whether our lead compound's cytotoxicity stemmed from effects independent of mitochondrial function. Another HCT compound, Dp44mT, has been reported to induce ROS, though its cytotoxicity was not attributed to the functionality of the mitochondria.57 To determine whether the cytotoxicity of HCT-13 was ETC-dependent, we examined its effects upon 143 BTK ρ0, a mitochondrial DNA (mtDNA)-deficient osteosarcoma cell line. Both 143 BTK ρ0 and parental (wild type, WT) cells were treated with HCT-13 + 20 μM Cu(ii) for 48 h, after which cell viability was determined (Fig. 5e). Compared to WT, the ρ0 cells were significantly less sensitive to the treatment, with a concurrent decrease in levels of S-phase arrest (ESI‡ Fig. S3c and d). Together, these results indicate that the cytotoxic effects of HCT-13 are mitochondria-dependent and suggest that HCT-13 may preferentially target tumor cells which rely more heavily upon OXPHOS than on glycolysis. To further explore this potentially stratifying metabolic hallmark, cells were treated with HCT-13 with and without 2-deoxyglucose (2-DG), a molecule that competitively inhibits glycolysis, thereby forcing cells to rely upon OXPHOS for energy production (Fig. 5f).58 Co-administration of 2-DG significantly potentiated the activity of HCT-13, conceivably by forcing the cancer cells to rely more heavily upon OXPHOS which was in turn impaired by HCT-13.
HCT-13 pre-complexed with copper is effective against aggressive B-ALL and AML models in vivo
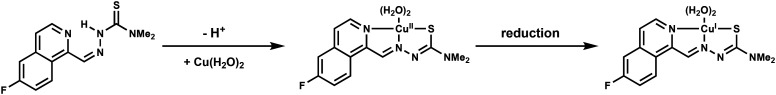
With a mechanism of action outlined and a potentially stratifying metabolic hallmark identified, we next investigated the in vivo efficacy and tolerability of HCT-13. Two leukemia models were chosen – a primary murine BCR-ABL-expressing Arf-null pre-B (p185BCR-ABLArf–/–) ALL model (p185) and a human systemic acute myeloid leukemia (AML) model (MV4-11) – as these leukemias possess aggressive phenotypes, have high intrinsic levels of OXPHOS, and there remains a persistent unmet need for effective therapeutic options, particularly in the case of AML. Both p18559–61 and MV4-1162,63 cell lines were engineered to express luciferase to monitor the systemic leukemic burden by bioluminescence imaging (BLI). To bypass the need for systemic copper supplementation, a one-to-one complex of copper and HCT-13 (Cu[HCT-13]) was prepared for in vivo administration according to reported procedures for similar compounds64 and characterized by UV-HPLC and HR-MS (ESI‡ Fig. S4). The in vitro antiproliferative activity of Cu[HCT-13] was consistent with that of HCT-13 + Cu(ii) in all cell lines tested.
Treatment in both p185 and MV4-11 murine models was initiated on day six post-inoculation of cells, when all mice showed evidence of systemic disease. Mice in treatment groups of the pre-B ALL and AML arms of the study were administered 1 mg kg–1Cu[HCT-13] q.d. for 8 and 13 days, respectively (Fig. 6a and d). The treatment was well tolerated as indicated by body weight measurements (ESI‡ Fig. S5). 14 days after treatment initiation in the pre-B ALL arm, HCT-13-treated mice displayed significantly lower systemic disease burden than mice in the control group (Fig. 6b and c). Similarly, treatment group mice in the MV4-11 portion of the study had significantly lower disease burden on day 19 compared to the control group mice (Fig. 6e and f).
Fig. 6. Cu[HCT-13] is effective in aggressive models of systemic B-ALL and AML. (a) Dose and schedule for the efficacy study in p185 pre-B ALL bearing mice. (b and c) Bioluminescence images and quantification of whole body radiance of mice treated with Cu[HCT-13] (n = 8) or vehicle control (n = 5). (d) Dose and schedule for the efficacy study in MV4-11 AML bearing mice. (e and f) Bioluminescence images and quantification of whole body radiance of mice treated with Cu[HCT-13] (n = 5) or vehicle control (n = 5) (mean ± SD, n = 2, Student's paired t-test, * P < 0.05; ** P < 0.01; *** P < 0.001). q.d. once per day.

Discussion
This study aimed to leverage the general ability of HCTs to bind copper and the anticancer activity of isoquinoline-based HCTs towards developing novel copper-binding analogs with a defined mechanism of action and in vivo efficacy against cancer models. Two potential modifications of the previously reported HCT-1 (IQ-1) scaffold drew particular interest during our synthetic planning: fluorination of the isoquinoline ring, and sequential methylation of the 4′ amine. With the goal of leveraging the copper chelating ability of the isoquinoline HCT scaffold against cancer cells, we sought to synthesize the novel 4- and 6-fluoro analogs of HCT-1 and investigate what effects sequential 4′ amine methylation would have upon antiproliferative activity. Following our synthetic campaign, analysis of the antiproliferative data revealed several trends. In conditions without copper supplementation, fluorination at either the 4- or 6-position of the isoquinoline ring led to an increase in potency for five out of six compounds, when compared with their corresponding non-fluorinated analogs (Fig. 1). In some cases, the change was dramatic – the IC90 of HCT-13 was nearly 270-fold lower than its non-fluorinated analog HCT-11. Secondly, 4′ amine methylation in the absence of isoquinoline substitution or copper supplementation was detrimental to activity, as demonstrated by the decrease in potencies for HCTs 6 and 11 when compared with 4′ primary amine HCT-1. However, combining 4′ amine methylation and isoquinoline substitution in a single compound, as in HCTs 7–10, 12 and 13, produced greater-than-additive antiproliferative effects when compared with either their 4′ primary amine or unsubstituted isoquinoline analogs; HCT-13 again exemplified this trend. Interestingly, the fluorinated compounds HCT-12 and HCT-13 were qualitatively more stable than non-substituted HCT-11, the latter of which quickly began to decompose when solubilized in DMSO, whereas the former were stable for months when stored as a DMSO solution at room temperature, as monitored by 1H NMR. This serendipitous discovery factored heavily into our decision to continue generating fluorinated analogs. The underlying mechanisms responsible for the observed synergy and stability remain under study by our group. Finally, the antiproliferative activities of all but one of our isoquinoline HCTs were potentiated by supplementation with physiologically relevant levels of copper, with HCT-5 being the exception. DFT calculations were employed to gain further insight into the observed trends, revealing that presence of an electron withdrawing fluorine at the 4- and particularly the 6-position led to lower barriers to ligand deprotonation and greater complex stability. Additionally, the most exergonic reduction of Cu(ii) to Cu(i) was observed in the Cu:HCT-13 complex, implying that this compound is most readily able to participate in redox processes. The observed trends culminated with identification of our lead compound as HCT-13, which was uncomplicated in its synthesis and highly potent.
The use of copper-chelating small molecules in anticancer therapy is an established strategy which is executed either through sequestration of copper from tumor tissue, or through increasing intracellular copper to cytotoxic levels.65HCT-13 behaves as an ionophore and increases intracellular levels of copper, a property that is essential for its cytotoxicity as sequestration of copper via BCPS-chelation negated our lead compound's growth inhibitory effects (Fig. 2b). The potency of HCT-13 is highlighted by its nanomolar IC50 values against a panel of PDAC, SCLC, PC, and leukemia cancer models (Fig. 3). Our chemical genomics screen pointed towards activation of DDR/RSR pathways following HCT-13 treatment. Upon further investigation, it was found that HCT-13 reduced dGTP and rGTP pools in treated cells, an effect consistent with oxidative damage. These results implicate a mechanism by which HCT-13 activates DDR/RSR pathways following oxidative depletion of guanosine nucleotide pools (Fig. 4f). Incorporation of oxidized nucleotides into newly synthesized DNA leads to DNA damage, while their removal from nucleotide pools may engender replication stress response due to insufficient nucleotide availability. Further probing of this mechanism led to confirmation of ROS presence in HCT-13-treated cells and the discovery of mitochondrial ROS and suppression of mitochondrial OXPHOS (Fig. 5a–d). Strikingly, the cytotoxicity of HCT-13 was greatly attenuated in cells lacking the mitochondrial ETC (Fig. 5e). Conversely, 2-DG-mediated suppression of glycolysis sensitized MIAPACA2 cells to treatment (Fig. 5f). Cumulatively, these data outline a mechanism in which in the presence of copper, HCT-13 targets the mtETC and interferes with its function, generating ROS which in turn deplete GTP nucleotide pools and damage DNA either through nucleotide oxidation or through ROS-mediated ssDNA or dsDNA breaks.
The delineated mechanism of action illustrates a means for stratifying potential oncogenic targets – cancers which are more dependent upon OXPHOS and/or which possess defects in DDR are more likely to respond to HCT-13 therapy than those without. Leukemias are reported to rely heavily upon OXPHOS in order to meet their energetic demands,66–72 and its inhibition has been efficacious against preclinical models and is currently the subject of clinical investigation.73 Additionally, preleukemic syndromes and myeloid malignancies rely heavily upon error-prone non-homologous end joining for the repair of double-strand breaks, a type of DNA damage commonly caused by ROS.74–76 Coupled with preexisting oxidative stress, this reliance can lead to irreparable damage when challenged by treatment-induced ROS, a process which preferentially affects these malignant cells.77 These characteristics informed our choice of pre-B ALL and AML models for preclinical studies, as leukemias fulfill the identified criteria for potential responders to HCT-13 treatment. Gratifyingly, in vivo efficacy was realized, with a one-to-one copper:HCT-13 complex (Cu[HCT-13]) being administered to avoid potential complications of systemic copper supplementation. Leukemic burden in both preclinical models as measured by BLI was significantly reduced in treatment group mice relative to control group mice, the latter of which had all succumbed to disease by the day of measurement (Fig. 6). In general, cancer cells exhibit higher levels of ROS and higher baseline oxidative stress than healthy cells, which may imbue our lead compound with selectivity towards cancerous tissue and explain the observed resistance of non-cancerous HPDE cells (Fig. 3).
While the data supporting our proposed mechanism of action is compelling, we cannot rule out additional mechanisms through which HCT-13 may be effecting its anticancer activity. A recent report on the disulfiram metabolite diethyldithiocarbamate (DTC) details a mechanism by which DTC chelates copper in vivo and preferentially accumulates in tumor tissue before binding and aggregating NPL4, a protein which enables p97-mediated protein degradation.78 Given the fact that NPL4 possesses two zinc finger domains which bind metal complexes, it is possible that HCT-13 may derive some of its activity through mechanisms similar to the DTC–copper complex.79,80 Additionally, the observed mechanisms of action suggest that HCT-13 may synergize with radiotherapy, as therapeutic ionizing radiation increases ROS, thereby increasing oxidative stress and DNA damage in the targeted area(s). Therefore, HCT-13 could also function as a radiosensitizer by further increasing the load of ROS, oxidative stress, and DNA damage when administered in combination with radiation therapy. Further studies regarding mechanisms of action and potentials for combination therapy are ongoing in our group. Also of current interest is the use of alternative delivery strategies for administration of pre-complexed Cu[HCT-13]. Pre-complexation may prevent toxicity resulting from aberrant metal chelation following systemic administration when compared with HCT-13 alone, and could promote preferential accumulation in cancer tissue, as has been previously observed.78 Utilizing albumin-based formulations for delivery of this complex may bring further benefits for in vivo therapy and is currently under investigation.81 Collectively, we think that identifying viable combination therapies and optimizing drug delivery will enhance the clinical efficacy of this already promising drug when combined with our elucidation of HCT-13's mechanism(s) of action and identification of target-stratifying hallmarks.
Conclusion
We have expanded upon a class of isoquinoline-based HCTs to produce a set of novel antiproliferative compounds. We demonstrated the synergistic effects of combining 4′ amine methylation with isoquinoline substitution and identified HCT-13 as a highly potent antiproliferative agent which is active against a panel of PDAC, SCLC, PC, and leukemia cancer models. We showed that presence of physiologically-relevant levels of Cu(ii) greatly potentiated our lead compound's activity, and subsequent investigation into HCT-13's mechanism of action revealed that it acts as a copper ionophore and requires copper to effect its cytotoxicity. Furthermore, HCT-13 induces ROS production and mitochondrial dysfunction, decreases guanosine nucleotide pools, engages DDR/RSR pathways and synergizes with ATR inhibition, possesses mitochondrial-dependent cytotoxicity, and targets high-OXPHOS cells. Lastly, a one-to-one copper:HCT-13 (Cu[HCT-13]) complex was demonstrated to be efficacious in preclinical models of aggressive leukemias. Taken together, the delineated mechanism of action of HCT-13 and its demonstrated potency as a single agent therapeutic against aggressive tumor models in vitro and in vivo signal its promise as a therapeutic agent and warrant further investigations.
Experimental section
Chemistry
All chemicals, reagents and solvents were obtained from commercial sources and were used without further purification. Unless otherwise noted, reactions were carried out in oven-dried glassware under an atmosphere of argon using commercially available anhydrous solvents. Tetrahydrofuran (THF) was distilled from sodium under an argon atmosphere. Dichloromethane was distilled from calcium hydride. Solvents used for extractions and chromatography were not anhydrous. Analytical TLC was carried out on precoated silica gel (Merck silica gel 60, F254) and visualized with UV light. Column chromatography was performed with silica (Fisher, 230–400 mesh). 1H NMR, 13C NMR, and 19F NMR spectra were measured in CDCl3 or DMSO-d6 on Bruker AV spectrometers at 400 or 500 MHz. Chemical shifts were reported in parts per million (δ) relative to residual solvent signals. The signals observed were described as follows: s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), dt (doublet of triplets), ddd (doublet of doublet of doublets), tt (triplet of triplets), tdd (triplet of doublet of doublets), m (multiplet), br s (broad singlet). Mass spectra were obtained on a Waters LCT Premier with ACQUITY UPLC mass spectrometer under electrospray ionization (ESI) or Thermo Fisher Scientific Exactive Plus with direct analysis in real time (DART) ionization. Purity of all compounds used in biological assays was determined to be >95% pure on a Hewlett Packard 1090 HPLC system using an Aquasil C18 column (250 mm × 2 mm, 5 μm, Keystone Scientific) with an acetonitrile/water solvent system containing 0.1% TFA with detection performed at 254 nm (minute/% acetonitrile: 0/0, 8/0, 35/95, 43/95, 45/0, 55/0). All microwave-assisted reactions were carried out in a CEM Discover 908005 Microwave synthesizer system.
All final compounds (HCT 1–15) were synthesized according to the following synthetic procedure for HCT13, when incorporating the appropriate intermediates. The synthetic details and characterization information for all intermediates are shown in the ESI.‡
(E)-2-((6-Fluoroisoquinolin-1-yl)methylene)-N,N-dimethylhydrazine-1-carbothioamide and (Z)-2-((6-fluoroisoquinolin-1-yl)methylene)-N,N-dimethylhydrazine-1-carbothioamide (HCT13)
To a solution of S12 (8.6 mg, 0.0491 mmol) in EtOH (0.5 mL) was added 4,4-dimethyl-3-thiosemicarbazide (5.9 mg, 0.0491 mmol) and HCl (49 μL, 0.294 mmol, 6 M in H2O). The mixture was stirred and refluxed for 1.5 hours and then cooled to room temperature (22 °C). The hydrochloride salt that formed was then neutralized with saturated aqueous NaHCO3 solution (0.5 mL). The precipitate of the desired compound was collected by filtration, the filter cake sequentially washed with water and EtOH, and then dried to yield the isoquinoline HCT13 as a pale-yellow solid containing a mixture of E- and Z-isomers (7.4 mg, 55%). 1H NMR (500 MHz, DMSO-d6) δ 15.90 (s, 0.21H), 11.30 (s, 1H), 9.87 (dd, J = 9.5, 5.9 Hz, 1H), 8.91 (dd, J = 9.4, 5.4 Hz, 0.21H), 8.66 (m, 1.21H), 8.59 (s, 0.21H), 8.55 (d, J = 5.6 Hz, 1H), 7.97 (d, J = 5.6 Hz, 0.21H), 7.91 (dd, J = 9.6, 2.7 Hz, 0.21H), 7.79–7.82 (m, 2H), 7.73 (td, J = 9.1, 2.7 Hz, 0.21H), 7.62 (ddd, J = 9.6, 8.6, 2.8 Hz, 1H), 3.40 (s, 1.26H), 3.33 (s, 6H). 13C NMR (125 MHz, DMSO-d6) δ 180.78 (2C), 163.19 (d, 1JC–F = 251.7 Hz), 162.66 (d, 1JC–F = 250.6 Hz), 151.99 (d, 5JC–F = 1.2 Hz), 150.63 (d, 5JC–F = 0.9 Hz), 147.57, 143.42, 141.42, 138.83 (d, 3JC–F = 15.5 Hz), 138.76 (d, 4JC–F = 10.7 Hz), 132.08 (d, 3JC–F = 9.3 Hz), 131.62, 128.64 (d, 3JC–F = 9.9 Hz), 124.14, 123.10, 122.12 (d, 4JC–F = 5.2 Hz), 121.56 (d, 4JC–F = 5.1 Hz), 119.54 (d, 2JC–F = 25.6 Hz), 119.14 (d, 2JC–F = 24.4 Hz), 111.48 (d, 2JC–F = 20.8 Hz), 110.90 (d, 2JC–F = 20.7 Hz), 42.04 (4C). 19F NMR (376 MHz, DMSO-d6) δ –106.34, –107.95. DART-MS: m/z calcd. for C13H14FN4S (M + H)+ 277.09232, found 277.0905.
(E)-2-(Isoquinolin-1-ylmethylene)hydrazine-1-carbothioamide (HCT1)
1H NMR (500 MHz, DMSO-d6) δ 11.74 (s, 1H), 9.19 (d, J = 8.5 Hz, 1H), 8.60–8.54 (m, 2H) 8.49 (br s, 1H), 8.02 (d, J = 8.1 Hz, 1H), 7.86 (d, J = 5.6 Hz, 1H), 7.84–7.78 (m, 2H), 7.75 (ddd, J = 8.3, 6.8, 1.4 Hz, 1H). 13C NMR (125 MHz, DMSO-d6) δ 178.41, 150.78, 145.99, 142.13, 136.24, 130.47, 129.08, 127.22, 126.94, 125.58, 121.77. DART-MS: m/z calcd. for C11H10N4S (M + H)+ 231.06989, found 231.06938.
(E)-2-((4-Fluoroisoquinolin-1-yl)methylene)hydrazine-1-carbothioamide (HCT2)
1H NMR (500 MHz, DMSO-d6) δ 11.70 (s, 1H), 9.28 (d, J = 8.5 Hz, 1H), 8.56 (d, J = 1.5 Hz, 1H), 8.53 (s, 1H), 8.48 (s, 1H), 8.13 (d, J = 8.2 Hz, 1H), 7.94 (ddd, J = 8.2, 7.0, 0.9 Hz, 1H), 7.85 (m, 2H). 13C NMR (125 MHz, DMSO-d6) δ 178.84, 154.73 (d, 1JC–F = 262.2 Hz), 148.03 (d, 3JC–F = 5.2 Hz), 145.84, 131.75, 130.69, 128.10 (d, 2JC–F = 23.3 Hz), 127.75, 127.35 (d, 4JC–F = 3.0 Hz), 126.51 (d, 2JC–F = 14.9 Hz), 119.79 (d, 3JC–F = 4.6 Hz). 19F NMR (376 MHz, DMSO-d6) δ –137.31. DART-MS: m/z calcd. for C11H9FN4S (M + H)+ 249.06047, found 249.05042.
(E)-2-((6-Fluoroisoquinolin-1-yl)methylene)hydrazine-1-carbothioamide (HCT3)
1H NMR (500 MHz, DMSO-d6) δ 11.74 (s, 1H), 9.30 (dd, J = 9.4, 5.8 Hz, 1H), 8.55 (d, J = 5.6 Hz, 1H), 8.51 (s, 2H), 7.80–7.85 (m, 3H), 7.57 (td, J = 9.0, 2.8 Hz, 1H). 13C NMR (125 MHz, DMSO-d6) δ 178.88, 162.70 (d, 1JC–F = 250.4 Hz), 151.35, 146.33, 143.50, 138.62 (d, 3JC–F = 10.7 Hz), 131.52 (d, 3JC–F = 9.5 Hz), 123.24, 121.79 (d, 4JC–F = 5.0 Hz), 119.36 (d, 2JC–F = 24.5 Hz), 110.86 (d, 2JC–F = 20.7 Hz). 19F NMR (376 MHz, DMSO-d6) δ –107.79, extraneous peak found at –106.49. DART-MS: m/z calcd. for C11H10FN4S (M + H)+ 249.06047, found 249.05984.
(E)-2-((5-(Methylamino)isoquinolin-1-yl)methylene)hydrazine-1-carbothioamide (HCT4)
1H NMR (500 MHz, DMSO-d6) δ 12.32 (s, 1H), 9.07 (br s, 1H), 8.92 (s, 1H), 8.90 (s, 1H), 8.57 (d, J = 6.6 Hz, 1H), 8.52 (d, J = 6.7 Hz, 1H), 7.85 (t, J = 8.2 Hz, 1H), 7.56 (d, J = 8.3 Hz, 1H), 7.30 (br s, 1H), 7.01 (d, J = 8.0 Hz, 1H), 2.91 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 179.40, 146.25, 146.00, 133.09, 130.09, 128.72, 126.75 (2C), 119.38, 111.17, 110.66, 30.39. DART-MS: m/z calcd. for C12H14N5S (M + H)+ 260.09644, found 260.09501.
(E)-2-((5-Aminoisoquinolin-1-yl)methylene)hydrazine-1-carbothioamide (HCT5)
1H NMR (500 MHz, DMSO-d6) δ 11.66 (br s, 1H), 8.57 (s, 1H), 8.42 (d, J = 5.8 Hz, 1H), 8.31 (br s, 1H), 8.25 (d, J = 8.5 Hz, 1H), 7.98 (d, J = 5.8 Hz, 1H), 7.60 (br s, 1H), 7.42 (t, J = 8.1 Hz, 1H), 6.89 (d, J = 7.1 Hz, 1H), 6.02 (s, 2H). 13C NMR (125 MHz, DMSO-d6) δ 178.46, 150.36, 145.86, 144.62, 140.01, 129.74, 126.78. 125.83. 116.50, 113.12, 110.74. DART-MS: m/z calcd. for C11H11N5S (M + H)+ 246.08079, found 246.08020.
(E)-2-(Isoquinolin-1-ylmethylene)-N-methylhydrazine-1-carbothioamide (HCT6)
1H NMR (500 MHz, DMSO-d6) δ 11.78 (br s, 1H), 9.11 (br s, 1H), 8.61 (s, 1H), 8.56 (d, J = 5.6 Hz, 1H), 8.31 (br s, 1H), 8.02 (d, J = 8.1 Hz, 1H), 7.89–7.79 (m, 2H), 7.76 (t, J = 7.7 Hz, 1H), 3.07 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 178.36, 151.06, 144.76, 142.15, 136.25, 130.42, 128.86, 127.21, 126.82, 125.64, 121.48, 31.34. DART-MS: m/z calcd. for C12H13N4S (M + H)+ 245.08554, found 245.08505.
(E)-2-((4-Fluoroisoquinolin-1-yl)methylene)-N-methylhydrazine-1-carbothioamide (HCT7)
1H NMR (500 MHz, DMSO-d6) δ 11.76 (s, 1H), 9.19 (d, J = 8.6 Hz, 1H), 8.56 (d, J = 1.4 Hz, 1H), 8.56 (s, 1H), 8.34 (d, J = 4.4 Hz, 1H), 8.14 (d, J = 8.3 Hz, 1H), 7.94–7.97 (m, 1H), 7.85–7.89 (m, 1H), 3.06 (d, J = 4.6 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 178.84, 154.73 (d, 1JC–F = 262.2 Hz), 148.03 (d, 3JC–F = 5.2 Hz), 145.84, 131.75, 130.69, 128.10 (d, 2JC–F = 23.3 Hz), 127.75, 127.35 (d, 4JC–F = 3.0 Hz), 126.51 (d, 2JC–F = 14.9 Hz), 119.79 (d, 3JC–F = 4.6 Hz), 31.86. 19F NMR (376 MHz, DMSO-d6) δ –137.53, extraneous peak found at –134.32. DART-MS: m/z calcd. for C12H12FN4S (M + H)+ 263.07612, found 263.07520.
(E)-2-((6-Fluoroisoquinolin-1-yl)methylene)-N-methylhydrazine-1-carbothioamide (HCT8)
1H NMR (500 MHz, DMSO-d6) δ 11.80 (s, 1H), 9.20 (dd, J = 9.4, 5.7 Hz, 1H), 8.55 (d, J = 5.6 Hz, 1H), 8.54 (s, 1H), 8.35 (d, J = 4.7 Hz, 1H), 7.83 (dd, J = 9.2, 3.9 Hz, 2H) 7.60 (td, J = 9.0, 2.7 Hz, 1H), 3.06 (d, J = 4.5 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 178.56, 162.71 (d, 1JC–F = 250.4 Hz), 151.55, 145.22, 143.53, 138.62 (d, 3JC–F = 10.6 Hz), 131.28 (d, 3JC–F = 9.5 Hz), 123.29, 121.67 (d, 4JC–F = 5.1 Hz), 119.23 (d, 2JC–F = 24.8 Hz), 110.89 (d, 2JC–F = 20.8 Hz), 31.85. 19F NMR (376 MHz, DMSO-d6) δ –106.55, extraneous peak found at –107.74. DART-MS: m/z calcd. for C12H12FN4S (M + H)+ 263.07612, found 263.07538.
(E)-N-Methyl-2-((5-(methylamino)isoquinolin-1-yl)methylene)hydrazine-1-carbothioamide and (Z)-N-methyl-2-((5-(methylamino)isoquinolin-1-yl)methylene)hydrazine-1-carbothioamide (HCT9)
1H NMR (500 MHz, DMSO-d6) δ 14.74 (s, 0.15H), 12.22 (s, 1H), 9.39 (br s, 1H), 8.93 (q, J = 4.7 Hz, 0.15H), 8.78 (s, 1H), 8.54 (d, J = 5.9 Hz, 0.15H), 8.50 (d, J = 6.5 Hz, 1H), 8.38 (s, 1H), 8.17 (s, 0.15H), 8.12 (d, J = 5.9 Hz, 0.15H), 7.82 (d, J = 8.6 Hz, 0.15H), 7.76 (t, J = 8.1 Hz, 1H), 7.69 (s, 1H), 7.56 (t, J = 8.1 Hz, 0.15H), 7.08 (br s, 1H), 6.92 (d, J = 7.7 Hz, 1.15H), 6.72 (d, J = 7.8 Hz, 0.15H), 3.07 (d, J = 4.6 Hz, 3H), 3.02 (d, J = 4.6 Hz, 0.45 H), 2.88 (s, 3H), 2.86 (s, 0.45H). 13C NMR (125 MHz, DMSO-d6) δ 178.84, 178.38, 150.11, 147.37, 145.83 (2C), 145.56, 138.94, 132.45, 130.58, 129.19, 128.23, 128.22, 126.87 (2C), 126.80, 118.47, 117.04, 111.57, 110.13, 109.63, 106.57, 31.59, 31.42, 30.42 (2C). DART- MS: m/z calcd. for C13H16N5S (M + H)+ 274.11209, found 274.11104.
(E)-2-((5-Aminoisoquinolin-1-yl)methylene)-N-methylhydrazine-1-carbothioamide and (Z)-2-((5-aminoisoquinolin-1-yl)methylene)-N-methylhydrazine-1-carbothioamide (HCT10)
1H NMR (500 MHz, DMSO-d6) δ 14.80 (s, 0.08H), 11.66 (br s, 1H), 8.95 (d, J = 4.9 Hz, 0.08H), 8.62 (s, 1H), 8.52 (d, J = 5.9 Hz, 0.8H), 8.42 (d, J = 5.8 Hz, 1H), 8.25 (d, J = 3.3 Hz, 1H), 8.18 (s, 0.08H), 8.11–8.15 (m, 1.08H), 7.99 (d, J = 5.9 Hz, 1H), 7.83 (d, J = 8.4 Hz, 0.08H), 7.48 (t, J = 7.9 Hz, 0.08H), 7.43 (t, J = 8.0 Hz, 1H), 6.97 (d, J = 7.6 Hz, 0.08H), 6.91 (dd, J = 7.6, 0.9 Hz, 1H), 6.21 (s, 0.16H), 6.04 (s, 2H), 3.05–3.07 (m, 3.24H). 13C NMR (125 MHz, DMSO-d6) δ 178.85, 178.52, 150.83, 150.19, 145.53, 145.20, 145.10, 140.49, 138.40, 130.28, 130.10, 129.16, 128.41, 127.36, 126.27, 126.16, 117.81, 116.96, 113.24, 111.66, 111.18, 110.61, 31.74, 31.59. DART-MS: m/z calcd. for C12H14N5S (M + H)+ 260.09644, found 260.09563.
(E)-2-(Isoquinolin-1-ylmethylene)-N,N-dimethylhydrazine-1-carbothioamide and (Z)-2-(isoquinolin-1-ylmethylene)-N,N-dimethylhydrazine-1-carbothioamide (HCT11)
1H NMR (500 MHz, DMSO-d6) δ 15.99 (s, 0.33H), 11.26 (br s, 1H), 9.77 (dd, J = 8.8, 5.1 Hz, 1H), 8.81 (d, J = 8.6 Hz, 0.33H), 8.70 (d, J = 1.7 Hz, 1H), 8.69 (d, J = 1.2 Hz, 0.33H), 8.63 (s, 0.33H), 8.55 (d, J = 5.5 Hz, 1H), 8.12 (d, J = 8.2 Hz, 0.33H), 8.01–7.96 (m, 1.33H), 7.92 (ddd, J = 8.1, 7.0, 1.1 Hz, 0.33H), 7.88–7.76 (m, 2.33H), 7.72 (ddd, J = 8.4, 6.8, 1.4 Hz, 1H), 3.43 (s, 1.98H), 3.35 (s, 6H). 13C NMR (125 MHz, DMSO-d6) δ 180.88, 180.81, 151.94, 150.58, 147.81, 142.50, 140.45, 136.86, 136.82, 131.80, 130.77, 129.48, 129.16, 128.21, 128.17 (2C), 127.68, 126.83, 125.87, 124.60, 122.53, 121.93, 42.08 (4C). DART-MS: m/z calcd. for C13H15N4S (M + H)+ 259.10119, found 259.10080.
(E)-2-((4-Fluoroisoquinolin-1-yl)methylene)-N,N-dimethylhydrazine-1-carbothioamide and (Z)-2-((4-fluoroisoquinolin-1-yl)methylene)-N,N-dimethylhydrazine-1-carbothioamide (HCT12)
1H NMR (500 MHz, DMSO-d6) δ 15.52 (s, 0.15H), 11.28 (s, 1H), 9.87 (d, J = 8.7 Hz, 1H), 8.87 (d, J = 9.0 Hz, 0.15H), 8.77 (d, J = 1.9 Hz, 0.15H), 8.69 (s, 1H), 8.57 (d, J = 1.6 Hz, 1.15H), 8.23 (d, J = 8.2 Hz, 0.15H), 8.15 (d, J = 8.2 Hz, 1H), 8.02–8.05 (m, 0.15H), 7.92–7.97 (m, 1.15H), 7.86 (ddd, J = 8.3, 6.9, 1.3 Hz, 1H), 3.42 (s, 0.90H), 3.36 (s, 6H). 13C NMR (125 MHz, DMSO-d6) δ 180.79, 180.73, 154.60 (d, 1JC–F = 261.7 Hz), 154.13 (d, 1JC–F = 261.8 Hz), 148.75 (d, 3JC–F = 5.1 Hz), 147.76 (d, 3JC–F = 5.7 Hz), 147.05, 132.52, 131.59 (d, 4JC–F = 5.1 Hz), 131.10, 130.49, 130.30, 128.60 (d, 4JC–F = 3.3 Hz), 128.41 (d, 4JC–F = 1.0 Hz), 127.90 (d, 2JC–F = 23.3 Hz), 127.19 (d, 4JC–F = 2.6 Hz), 126.88 (d, 2JC–F = 14.8 Hz), 126.71 (d, 2JC–F = 14.7 Hz), 126.35 (d, 2JC–F = 25.2 Hz),124.98, 120.23 (d, 3JC–F = 4.3 Hz), 119.79 (d, 3JC–F = 4.7 Hz), 42.04 (4C). 19F NMR (376 MHz, DMSO-d6) δ –134.93, –138.02. DART-MS: m/z calcd. for C13H14FN4S (M + H)+ 277.09177, found 277.09096.
(E)-2-((6-Fluoro-5-(methylamino)isoquinolin-1-yl)methylene)-N,N-dimethylhydrazine-1-carbothioamide and (Z)-2-((6-fluoro-5-(methylamino)isoquinolin-1-yl)methylene)-N,N-dimethylhydrazine-1-carbothioamide (HCT14)
1H NMR (500 MHz, DMSO-d6) δ 15.96 (s, 0.17H), 11.22 (Br s, 1H), 9.20 (s, 1H), 8.62–8.54 (m, 1.17H), 8.52 (s, 0.17H), 8.34 (d, J = 5.5 Hz, 1H), 8.20 (d, J = 6.2 Hz, 0.17H), 8.07 (dd, J = 9.3, 4.2 Hz, 0.17H), 7.87 (br s, 1H), 7.56 (dd, J = 13.6, 9.2 Hz, 0.17H), 7.33 (dd, J = 13.4, 9.5 Hz, 1H), 6.10 (br s, 0.17H), 5.69 (br s, 1H), 3.41 (s, 1.02H), 3.27 (s, 6H), 3.10 (t, J = 5.5 Hz, 0.51H), 3.05 (t, J = 5.2 Hz, 3H). A 13C NMR was not obtained. 19F NMR (376 MHz, CDCl3) δ –129.05, –129.53. DART-MS: m/z calcd. for C14H17FN5S (M + H)+ 306.11832, found 306.11719.
(E)-2-((4-Fluoro-5-(methylamino)isoquinolin-1-yl)methylene)-N,N-dimethylhydrazine-1-carbothioamide and (Z)-2-((4-fluoro-5-(methylamino)isoquinolin-1-yl)methylene)-N,N-dimethylhydrazine-1-carbothioamide (HCT15)
1H NMR (500 MHz, DMSO-d6) δ 15.46 (s, 0.33H), 11.13 (br s, 1H), 8.91 (dd, J = 8.4, 2.9 Hz, 1H), 8.62 (s, 1H), 8.50 (d, J = 5.1 Hz, 0.33H), 8.39 (s, 0.33H), 8.32 (d, J = 4.8 Hz, 1H), 7.89 (dd, J = 8.5, 2.9 Hz, 0.33H), 7.65 (t, J = 8.2 Hz, 0.33H), 7.57 (t, J = 8.2 Hz, 1H), 6.82 (d, J = 8.0 Hz, 0.33H), 6.73 (d, J = 7.9 Hz, 1H), 6.55 (dd, J = 11.9, 5.2 Hz, 0.33H), 6.39 (dd, J = 12.4, 5.0 Hz, 1H), 3.37 (s, 1.98H), 3.31 (s, 6H), 2.86–2.84 (m, 3.99H). 13C NMR (125 MHz, DMSO-d6) δ 180.95, 180.72, 156.41 (d, J = 260.4 Hz), 147.99 (d, 4JC–F = 4.3 Hz), 147.41, 147.16, 144.92, 144.61 (d, 4JC–F = 3.7 Hz), 131.90, 131.69, 130.83, 130.78, 129.29 (d, 4JC–F = 2.4 Hz), 127.41 (d, 2JC–F = 28.8 Hz), 125.43 (d, 2JC–F = 30.5 Hz), 115.98 (d, 2JC–F = 7.6 Hz), 113.89, 113.84, 110.26, 108.50, 107.78, 42.15 (4C), 30.95 (2C), one low-field carbon were either not observed or is overlapping with another low-field carbon. 19F NMR (376 MHz, DMSO-d6) δ –125.86, –129.02. DART-MS: m/z calcd. for C14H17FN5S (M + H)+ 306.11832, found 306.11716.
Cu[HCT13]
HR-MS (ESI+) data: m/z calculated for [C13H12CuFN4S]+ = 338.0057; found 338.0038; m/z calculated for [C13H12CuFN4S + MeCN]+ = 379.0323; found 379.0297 (Thermo LTQ-Orbitrap XL). Refer to Supplementary Information for HPLC trace and ESI data.
Cell culture and culture conditions
Pancreatic adenocarcinoma cell lines: PATU8988T, MIAPACA2, SU8686, PSN1, HPAC, BXPC3, DANG, SUIT2, A13A, CAPAN2, T3M4, A2.1, HUPT4, XWR200, L36PL, YAPC, PANC0327, PANC1, PATU8902, HPAF11, ASPC1, PANC0813, PANC0203, HS766T, SW1990, and CFPAC1; prostate cancer cell lines: 22Rv1, LNCaP, RM1 and C4-2; and small cell lung carcinoma cell lines: NCI-H526, NCI-H146, and NCI-H1963 were obtained from American Type Culture Collection (ATCC). 143 BTK WT and 143 BTK ρ0, BJ WT and BJ ρ0 cells were gifts from Prof. Michael Teitell in UCLA. Murine Prostate cancer cell lines MyC CaP was a kind gift from Prof. DLJ Thorek at WUSTL. Murine Pancreatic cancer cells KP4662 was kind gift from Prof. Robert Vonderheide at UPenn. Immortalized human pancreatic ductal epithelial (HPDE) cells were a kind gift from Dr. Ming-Sound Tsao at Ontario Cancer Institute (Invitrogen Cat#20). With a few exceptions, cell lines were cultured in DMEM (Corning) or RPMI (Corning) containing 10% fetal bovine serum (FBS, Omega Scientific) and were grown at 37 °C, 20% O2 and 5% CO2. All cultured cells were incubated in antibiotic free media and were regularly tested for mycoplasma contamination using MycoAlert kit (Lonza) following the manufacturer's instructions, except that the reagents were diluted 1 : 4 from their recommended amount.
HCT-13 stock solution
HCT-13 was solubilized up to a concentration of 20 mg mL–1 in an aqueous solution of 40% Captisol with 1% DMSO with the aid of heating at 50 °C and sonication for 15 minutes.
Proliferation assay
Cells were plated in 384-well plates (500 cells per well for adherent cell lines in 30 μl volume). Drugs were serially diluted to the desired concentrations and an equivalent volume of DMSO was added to vehicle control. Following 72 h incubation, ATP content was measured using CellTiter-Glo reagent according to manufacturer's instructions (Promega, CellTiter-Glo Luminescent Cell Viability Assay), and analyzed by SpectraMax luminometer (Molecular Devices). IC50 and IC90 values, concentrations required to inhibit proliferation by 50% and 90% respectively compared to DMSO treated cells, were calculated using Prism 6.0 h (Graphpad Software). The 430-member protein kinase inhibitor library used for the chemical genomics screen was purchased from Selleckchem, catalog no. L1200.
Western blot
Cells were lysed using RIPA buffer supplemented with protease (ThermoFisher, 78430) and phosphatase (ThermoFisher, 78420) inhibitors, scraped, sonicated, and centrifuged (20 000g at 4 °C). Protein concentrations in the supernatant were determined using the Micro BCA Protein Assay kit (Thermo), and equal amounts of protein were resolved on pre-made bis-tris polyacrylamide gels (Life Technologies). Primary antibodies: pAMPKT172 (cell signaling, #2535, 1 : 1000), HO-1 (cell signaling, #5061S, 1 : 1000), pS345 CHEK1(cell signaling, #2348L, 1 : 1000), pT68 CHEK2 (cell signaling, #2197S, 1 : 1000), pS139 H2A.X (Millipore, 05-636, 1 : 1000), clvd. Casp3 (cell signaling, #9662, 1 : 1000), and anti-actin (Cell Signaling Technology, 9470, 1 : 10 000). Primary antibodies were stored in 5% BSA (Sigma-Aldrich) and 0.1% NaN3 in TBST solution. Anti-rabbit IgG HRP-linked (Cell Signaling Technology, 7074, 1 : 2500) and anti-mouse IgG HRP-linked (Cell Signaling Technology, 7076, 1 : 2500) were used as secondary antibodies. Chemiluminescent substrates (ThermoFisher Scientific, 34077 and 34095) and autoradiography film (Denville) were used for detection.
Viability/apoptosis assay
Viable cells were measured by trypan blue staining using a Vi-Cell counter (Beckman Coulter, CA, USA). Apoptosis and cell death were assayed using annexin V-FITC and PI according to manufacturer's instructions (FITC annexin V apoptosis detection kit, BD Sciences, #556570).
Cell cycle
Cell cycle was assessed using propidium iodide staining at indicated timepoints. Cells were pulsed with EdU 1 h before collection at different time points. Cells were fixed 4% paraformaldehyde, permeabilized with perm/wash reagent (Invitrogen), stained with Azide-AF647 (using click-chemistry, Invitrogen; Click-iT EdU Flow cytometry kit, #C10634) and FxCycle-Violet (Invitrogen), and then analyzed by flow cytometry.
ROS measurements
Cellular ROS measurement was assayed with CM-H2DCFDA staining after treatment according to manufacturer's instructions (reactive oxygen species (ROS) detection reagents, Invitrogen, #D399). The cells were then incubated with 5 μM of CM-H2DCFDA for 30 min, spun down at 450g for 4 min, and the supernatant was replaced with fresh media containing lethal compounds and/or Cu(ii). Then, the cells were incubated for 30 min, spun down, and the supernatant was replaced with PBS. The samples were analyzed using flow cytometry.
Mitochondrial ROS was measured using MitoSOX staining according to manufacturer's instructions (MitoSOX, Invitrogen, #M36008). Cells were treated with HCT-13, washed and treated with MitoSOX. Cells were then incubated for 30 minutes at 37 °C. After incubation, media is aspirated and cells are washed with PBS and analyzed by flow cytometry.
Mito stress test and electron flow assay
All oxygen consumption rate (OCR) was measured using a XF24 Analyzer (Agilent) and normalized per μg protein. For cellular OCR, cells were incubated in unbuffered DMEM containing 25 mM glucose, 1 mM pyruvate and 2 mM glutamine. OCR was measured before (total respiration) and after the sequential injection of 1 μM oligomycin (complex V inhibitor), 0.75 μM FCCP (uncoupler), and 1 μM of rotenone and myxothiazol (complex I and III inhibitors, respectively), as described previously.54 Mitochondrial respiration was calculated by subtracting the non-mitochondrial respiration left after rotenone and myxothiazol injection. Oligomycin-sensitive respiration represents ATP-linked respiration (coupled respiration).
To measure electron transport chain complex activity from cells, cells were incubated in MAS buffer with 10 mM pyruvate (complex I substrate), 2 mM malate, 4 μM FCCP, 4 mM ADP, and 1 nM of XF plasma membrane permeabilizer (PMP) reagent (Agilent). OCR was measured before and after the sequential injection of 2 μM rotenone, 10 mM succinate (complex II substrate), 4 μM antimycin A (complex III inhibitor), and a mix of 10 mM ascorbate and 100 μM TMPD (complex IV substrates), as described previously.55 Antimycin A-sensitive respiration represents the complex III respiration.
To measure OCR directly from mitochondria, mitochondria were isolated from fresh mouse liver by dual centrifugation at 800g and 8000g and seeded by centrifugation.55 Mitochondria were incubated with 1 mM pyruvate (complex I), 2 mM malate, 4 μM FCCP in MAS buffer, as well as the “corresponding drugs” for 30 min at 37 °C. OCR was measured before and after the sequential injections described in the previous paragraph.
Intracellular Cu(ii) measurement
Cells were plated in 6-well plates and cultured for one day. Vehicle of HCT-13 were added to the cells the following day and incubated for 24 hours. The plates were then washed 2 times with PBS containing 1 mM EDTA and 2 times with PBS alone. The concentration of Cu(ii) was measured using inductive coupled plasma mass spectrometry (ICP-MS) using standard procedure.
FACS analyses
All flow cytometry data were acquired on a five-laser LSRII cytometer (BD), and analyzed using the FlowJo software (Tree Star).
Isotopic labeling in cell culture
Cells were transferred into DMEM without glucose and supplemented with 10% dialyzed FBS (Gibco) containing [U-13C6]glucose (Sigma-Aldrich, 389374) at 11 mM. The cells were incubated for 48 h before sample collection and processing as previously described.51
Animal studies
Mice were housed under specific pathogen-free conditions and were treated in accordance with UCLA Animal Research Committee protocol guidelines. All C57BL/6 female mice were purchased from the UCLA Radiation Oncology breeding colony. All NCG female mice were purchased from the Jackson Labs (JAX).
In vivo leukemia models and treatment regimens
All animal studies were approved by the UCLA Animal Research Committee (ARC). For development of systemic murine B ALL model, C57Bl/6 female mice were injected intravenously with 200 000 firefly luciferase expressing p185BCR-ABL Arf–/– pre-B-ALL cells (kindly gifted by Dr. Nidal Boulos and the CERN Foundation).59,82 For development of the systemic human AML model, NCG female mice (from Jackson Labs) were injected intravenously with 5 × 106 firefly luciferase expressing MV4-11 cells.83 The leukemic burden was monitored using bioluminescence imaging. All Cu[HCT-13] treatments were performed using a formulation consisting of 40% Captisol and 1% DMSO. The treatments were performed by intra-peritoneal (i.p.) injections using 100 μL volume daily.
Abbreviations used
- HCTs
α-N-Heterocyclic carboxaldehyde thiosemicarbazones
- 3-AP
3-Aminopyridine-2-carboxaldehyde thiosemicarbazone
- RNR
Ribonucleotide reductase
- ROS
Reactive oxygen species
- PDAC
Pancreatic ductal adenocarcinoma
- SCLC
Small cell lung carcinoma
- PC
Prostate cancer
- BCPS
Bathocuproine disulfonate
- mtETC
Mitochondrial electron transport chain
- DDR/RSR
DNA damage response/replication stress response
- ATR
Ataxia-telangiectasia mutated (ATM) and Rad3-related protein kinase
- dGTP
Deoxyguanosine triphosphate
- OXPHOS
Oxidative phosphorylation
- 2-DG
2-Deoxy-d-glucose
- BLI
Bioluminescence imaging
- OCR
Oxygen consumption rate
Conflicts of interest
The authors declare the following competing financial interest(s): C. G. R., J. C., and M. E. J. are co-founders of Trethera Corporation. They and the University of California hold Equity in Trethera Corporation. The University of California has patented additional intellectual property for thiosemicarbazone-based antiproliferative compounds.
Supplementary Material
Acknowledgments
We thank Dr. Nagichettiar Satyamurthy for his expert chemistry advice, and Dr. Kym Faull at the Pasarow Mass Spectrometry Laboratory for continued tutelage regarding mass spectrometry. We additionally thank Joseph R. Capri, Anthony E. Cabebe, and Wesley R. Armstrong for their excellent technical assistance. This work was supported by the National Cancer Institute [Grant R01CA187678-01] administered by the National Institutes of Health, Trethera Corporation [Contract Number 20162965]. This material is additionally based upon work supported by the National Science Foundation Graduate Research Fellowship Program [Grant No. DGE-1650504 to E. W. R.].
Footnotes
†The authors declare the following competing financial interest(s): C. G. R., J. C., and M. E. J. are co-founders of Trethera Corporation. They and the University of California hold equity in Trethera Corporation. The University of California has patented additional intellectual property for thiosemicarbazone-based antiproliferative compounds.
‡Electronic supplementary information (ESI) available. See DOI: 10.1039/c9md00594c
References
- Brockman R. W., Thomson J. R., Bell M. J., Skipper H. E. Cancer Res. 1956;16(2):167–170. [PubMed] [Google Scholar]
- Beraldo H., Gambino D. Mini-Rev. Med. Chem. 2004;4(1):31–39. doi: 10.2174/1389557043487484. [DOI] [PubMed] [Google Scholar]
- Sartorelli A. C. Biochem. Biophys. Res. Commun. 1967;27(1):26–32. doi: 10.1016/s0006-291x(67)80034-2. [DOI] [PubMed] [Google Scholar]
- Liu M. C., Lin T. C., Sartorelli A. C. J. Med. Chem. 1992;35(20):3672–3677. doi: 10.1021/jm00098a012. [DOI] [PubMed] [Google Scholar]
- Agrawal K. C., Mooney P. D., Sartorelli A. C. J. Med. Chem. 1976;19(7):970–972. doi: 10.1021/jm00229a023. [DOI] [PubMed] [Google Scholar]
- French F. A., Blanz E. J., DoAmaral J. R., French D. A. J. Med. Chem. 1970;13(6):1117–1124. doi: 10.1021/jm00300a024. [DOI] [PubMed] [Google Scholar]
- French F. A., Blanz, Jr E. J. Cancer Res. 1965;29(9):1454–1458. [PubMed] [Google Scholar]
- Finch R. A., Liu M. C., Cory A. H., Cory J. G., Sartorelli A. C. Adv. Enzyme Regul. 1999;39:3–12. doi: 10.1016/s0065-2571(98)00017-x. [DOI] [PubMed] [Google Scholar]
- Yu Y., Wong J., Lovejoy D. B., Kalinowski D. S., Richardson D. R. Clin. Cancer Res. 2006;12(23):6876–6883. doi: 10.1158/1078-0432.CCR-06-1954. [DOI] [PubMed] [Google Scholar]
- Alvero A. B., Chen W., Sartorelli A. C., Schwartz P., Rutherford T., Mor G. J. Soc. Gynecol. Invest. 2006;13(2):145–152. doi: 10.1016/j.jsgi.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Yu Y., Gutierrez E., Kovacevic Z., Saletta F., Obeidy P., Suryo Rahmanto Y., Richardson D. R. Curr. Med. Chem. 2012;19(17):2689–2702. doi: 10.2174/092986712800609706. [DOI] [PubMed] [Google Scholar]
- Merlot A. M., Kalinowski D. S., Richardson D. R. Antioxid. Redox Signaling. 2013;18(8):973–1006. doi: 10.1089/ars.2012.4540. [DOI] [PubMed] [Google Scholar]
- Chaston T. B., Richardson D. R., Lovejoy D. B., Watts R. N. Clin. Cancer Res. 2003;9:402–414. [PubMed] [Google Scholar]
- Cory J. G., Cory A. H., Rappa G., Lorico A., Mao-Chin L., Tai-Shun L., Sartorelli A. C. Biochem. Pharmacol. 1994;48(2):335–344. doi: 10.1016/0006-2952(94)90105-8. [DOI] [PubMed] [Google Scholar]
- Richardson D. R., Kalinowski D. S., Richardson V., Sharpe P. C., Lovejoy D. B., Islam M., Bernhardt P. V. J. Med. Chem. 2009;52(5):1459–1470. doi: 10.1021/jm801585u. [DOI] [PubMed] [Google Scholar]
- Popović-Bijelić A., Kowol C. R., Lind M. E. S., Luo J., Himo F., Enyedy É. A., Arion V. B., Gräslund A. J. Inorg. Biochem. 2011;105(11):1422–1431. doi: 10.1016/j.jinorgbio.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro K., Lin Z. P., Penketh P. G., Shyam K., Zhu R., Baumann R. P., Zhu Y.-L., Sartorelli A. C., Rutherford T. J., Ratner E. S. Biochem. Pharmacol. 2014;91(3):312–322. doi: 10.1016/j.bcp.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denoyer D., Clatworthy S. A. S., Masaldan S., Meggyesy P. M., Cater M. A. Prostate. 2015;75(14):1510–1517. doi: 10.1002/pros.23022. [DOI] [PubMed] [Google Scholar]
- Madsen E., Gitlin J. D. Annu. Rev. Neurosci. 2007;30(1):317–337. doi: 10.1146/annurev.neuro.30.051606.094232. [DOI] [PubMed] [Google Scholar]
- Jansson P. J., Yamagishi T., Arvind A., Seebacher N., Gutierrez E., Stacy A., Maleki S., Sharp D., Sahni S., Richardson D. R. J. Biol. Chem. 2015;290(15):9588–9603. doi: 10.1074/jbc.M114.631283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitnall M., Howard J., Ponka P., Richardson D. R. Proc. Natl. Acad. Sci. U. S. A. 2006;103(40):14901–14906. doi: 10.1073/pnas.0604979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K., Reznik E., Stokes M. E., Krishnamoorthy L., Bos P. H., Song Y., Quartararo C. E., Pagano N. C., Carpizo D. R., deCarvalho A. C., Lo D. C., Stockwell B. R. Cell Chem. Biol. 2018;25(5):585–594. doi: 10.1016/j.chembiol.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D., Cui Q. C., Yang H., Dou Q. P. Cancer Res. 2006;66(21):10425–10433. doi: 10.1158/0008-5472.CAN-06-2126. [DOI] [PubMed] [Google Scholar]
- Brewer G. J., Dick R. D., Grover D. K., LeClaire V., Tseng M., Wicha M., Pienta K., Redman B. G., Jahan T., Sondak V. K., Strawderman M., LeCarpentier G., Merajver S. D. Clin. Cancer Res. 2000;6(1):1–10. [PubMed] [Google Scholar]
- Cen D., Gonzalez R. I., Buckmeier J. A., Kahlon R. S., Tohidian N. B., Meyskens F. L. Mol. Cancer Ther. 2002;1(3):197–204. [PubMed] [Google Scholar]
- Müller K., Faeh C., Diederich F. Science. 2007;317(5846):1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- Purser S., Moore P. R., Swallow S., Gouverneur V. Chem. Soc. Rev. 2008;37(2):320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- Gillis E. P., Eastman K. J., Hill M. D., Donnelly D. J., Meanwell N. A. J. Med. Chem. 2015;58(21):8315–8359. doi: 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- Belle C., Beguin C., Gautier-Luneau I., Hamman S., Philouze C., Pierre J. L., Thomas F., Torelli S., Saint-Aman E., Bonin M. Inorg. Chem. 2002;41(3):479–491. doi: 10.1021/ic010534g. [DOI] [PubMed] [Google Scholar]
- Kowol C. R., Trondl R., Heffeter P., Arion V. B., Jakupec M. A., Roller A., Galanski M., Berger W., Keppler B. K. J. Med. Chem. 2009;52(16):5032–5043. doi: 10.1021/jm900528d. [DOI] [PubMed] [Google Scholar]
- Kowol C. R., Miklos W., Pfaff S., Hager S., Kallus S., Pelivan K., Kubanik M., Enyedy É. A., Berger W., Heffeter P., Keppler B. K. J. Med. Chem. 2016;59(14):6739–6752. doi: 10.1021/acs.jmedchem.6b00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal K. C., Booth B. A., Sartorelli A. C. J. Med. Chem. 1968;11(4):700–703. doi: 10.1021/jm00310a014. [DOI] [PubMed] [Google Scholar]
- Mooney P. D., Booth B. A., Moore E. C., Agrawal K. C., Sartorelli A. C. J. Med. Chem. 1974;17(11):1145–1150. doi: 10.1021/jm00257a004. [DOI] [PubMed] [Google Scholar]
- Zaltariov M. F., Hammerstad M., Arabshahi H. J., Jovanović K., Richter K. W., Cazacu M., Shova S., Balan M., Andersen N. H., Radulović S., Reynisson J., Andersson K. K., Arion V. B. Inorg. Chem. 2017;56(6):3532–3549. doi: 10.1021/acs.inorgchem.6b03178. [DOI] [PubMed] [Google Scholar]
- Malarz K., Mrozek-Wilczkiewicz A., Serda M., Rejmund M., Polanski J., Musiol R. Oncotarget. 2018;9(25):17689–17710. doi: 10.18632/oncotarget.24844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K. C., Fouani L., Jansson P. J., Wooi D., Sahni S., Lane D. J. R., Palanimuthu D., Lok H. C., Kovacevic Z., Huang M. L. H., Kalinowski D. S., Richardson D. R. Metallomics. 2016;8(9):874–886. doi: 10.1039/c6mt00105j. [DOI] [PubMed] [Google Scholar]
- Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Petersson G. A., Nakatsuji H., Li X., Caricato M., Marenich A. V., Bloino J., Janesko B. G., Gomperts R., Mennucci B., Hratchian H. P., Ortiz J. V., Izmaylov A. F., Sonnenberg J. L., Williams-Young D., Ding F., Lipparini F., Egidi F., Goings J., Peng B., Petrone A., Henderson T., Ranasinghe D., Zakrzewski V. G., Gao J., Rega N., Zheng G., Liang W., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Throssell K., Montgomery, Jr. J. A., Peralta J. E., Ogliaro F., Bearpark M. J., Heyd J. J., Brothers E. N., Kudin K. N., Staroverov V. N., Keith T. A., Kobayashi R., Normand J., Raghavachari K., Rendell A. P., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Millam J. M., Klene M., Adamo C., Cammi R., Ochterski J. W., Martin R. L., Morokuma K., Farkas O., Foresman J. B. and Fox D. J., Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016.
- Denoyer D., Pearson H. B., Clatworthy S. A. S., Smith Z. M., Francis P. S., Llanos R. M., Volitakis I., Phillips W. A., Meggyesy P. M., Masaldan S., Cater M. A. Oncotarget. 2016;7(24):37064–37080. doi: 10.18632/oncotarget.9245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cater M. A., Pearson H. B., Wolyniec K., Klaver P., Bilandzic M., Paterson B. M., Bush A. I., Humbert P. O., La Fontaine S., Donnelly P. S., Haupt Y. ACS Chem. Biol. 2013;8(7):1621–1631. doi: 10.1021/cb400198p. [DOI] [PubMed] [Google Scholar]
- Safi R., Nelson E. R., Chitneni S. K., Franz K. J., George D. J., Zalutsky M. R., McDonnell D. P. Cancer Res. 2014;74(20):5819–5831. doi: 10.1158/0008-5472.CAN-13-3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodydkova J., Vavrova L., Stankova B., Macasek J., Krechler T., Zak A. Pancreas. 2013;42(4):614–621. doi: 10.1097/MPA.0b013e318288360a. [DOI] [PubMed] [Google Scholar]
- Lener M. R., Scott R. J., Wiechowska-Kozłowska A., Serrano-Fernández P., Baszuk P., Jaworska-Bieniek K., Sukiennicki G., Marciniak W., Muszyńska M., Kładny J., Gromowski T., Kaczmarek K., Jakubowska A., Lubiński J. Cancer Res. Treat. 2016;48(3):1056–1064. doi: 10.4143/crt.2015.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Yang Q. J. Int. Med. Res. 2018;46(12):4863–4873. doi: 10.1177/0300060518798507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiernik P. H. Med. Oncol. 2014;31(8):136. doi: 10.1007/s12032-014-0136-z. [DOI] [PubMed] [Google Scholar]
- Maréchal A., Zou L. Cold Spring Harbor Perspect. Biol. 2013;5(9):a012716. doi: 10.1101/cshperspect.a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A. M., Ryan A. J. Pharmacol. Ther. 2015;149:124–138. doi: 10.1016/j.pharmthera.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Yang J., Yu Y., Hamrick H. E., Duerksen-Hughes J. Carcinogenesis. 2003;24(10):1571–1580. doi: 10.1093/carcin/bgg137. [DOI] [PubMed] [Google Scholar]
- Halliwell B., Gutteridge J. M. Methods Enzymol. 1990;186:1–85. doi: 10.1016/0076-6879(90)86093-b. [DOI] [PubMed] [Google Scholar]
- Duncan C., White A. R. Metallomics. 2012;4(2):127–138. doi: 10.1039/c2mt00174h. [DOI] [PubMed] [Google Scholar]
- Willis J., Patel Y., Lentz B. L., Yan S. Proc. Natl. Acad. Sci. U. S. A. 2013;110(26):10592–10597. doi: 10.1073/pnas.1301445110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le T. M., Poddar S., Capri J. R., Abt E. R., Kim W., Wei L., Uong N. T., Cheng C. M., Braas D., Nikanjam M., Rix P., Merkurjev D., Zaretsky J., Kornblum H. I., Ribas A., Herschman H. R., Whitelegge J., Faull K. F., Donahue T. R., Czernin J., Radu C. G. Nat. Commun. 2017;8(1):1–14. doi: 10.1038/s41467-017-00221-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kino K., Hirao-Suzuki M., Morikawa M., Sakaga A., Miyazawa H. Genes Environ. 2017;39:21. doi: 10.1186/s41021-017-0081-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalov S. I., Harrison D. G. Antioxid. Redox Signaling. 2014;20(2):372–382. doi: 10.1089/ars.2012.4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M., Neilson A., Swift A. L., Moran R., Tamagnine J., Parslow D., Armistead S., Lemire K., Orrell J., Teich J., Chomicz S., Ferrick D. A. Am. J. Physiol. 2007;292(1):C125–C136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- Vergnes L., Davies G. R., Lin J. Y., Yeh M. W., Livhits M. J., Harari A., Symonds M. E., Sacks H. S., Reue K. J. Clin. Endocrinol. Metab. 2016;101(11):4440–4448. doi: 10.1210/jc.2016-2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand M. D., Nicholls D. G. Biochem. J. 2011;435(2):297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishan S., Richardson D. R., Sahni S. Biochim. Biophys. Acta, Mol. Cell Res. 2016;1863(12):2916–2933. doi: 10.1016/j.bbamcr.2016.09.011. [DOI] [PubMed] [Google Scholar]
- Dar S., Chhina J., Mert I., Chitale D., Buekers T., Kaur H., Giri S., Munkarah A., Rattan R. Sci. Rep. 2017;7(1):8760. doi: 10.1038/s41598-017-09206-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulos N., Mulder H. L., Calabrese C. R., Morrison J. B., Rehg J. E., Relling M. V., Sherr C. J., Williams R. T. Blood. 2011;117(13):3585–3595. doi: 10.1182/blood-2010-08-301267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes T. P., Saglio G., Quintás-Cardama A., Mauro M. J., Kim D. W., Lipton J. H., Bradley-Garelik M. B., Ukropec J., Hochhaus A. Leukemia. 2015;29(9):1832–1838. doi: 10.1038/leu.2015.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R. T., Roussel M. F., Sherr C. J. Proc. Natl. Acad. Sci. U. S. A. 2006;103(17):6688–6693. doi: 10.1073/pnas.0602030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmani M., Nkwocha J., Hawkins E., Pei X., Parker R. E., Kmieciak M., Leverson J. D., Sampath D., Ferreira-Gonzalez A., Grant S. Cancer Res. 2018;78(11):3075–3086. doi: 10.1158/0008-5472.CAN-17-3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaura T., Nakatani T., Uda K., Ogura H., Shin W., Kurokawa N., Saito K., Fujikawa N., Date T., Takasaki M., Terada D., Hirai A., Akashi A., Chen F., Adachi Y., Ishikawa Y., Hayakawa F., Hagiwara S., Naoe T., Kiyoi H. Blood. 2018;131(4):426–438. doi: 10.1182/blood-2017-05-786657. [DOI] [PubMed] [Google Scholar]
- Jansson P. J., Sharpe P. C., Bernhardt P. V., Richardson D. R. J. Med. Chem. 2010;53(15):5759–5769. doi: 10.1021/jm100561b. [DOI] [PubMed] [Google Scholar]
- Englinger B., Pirker C., Heffeter P., Terenzi A., Kowol C. R., Keppler B. K., Berger W. Chem. Rev. 2018;119(2):1519–1624. doi: 10.1021/acs.chemrev.8b00396. [DOI] [PubMed] [Google Scholar]
- Goto M., Miwa H., Suganuma K., Tsunekawa-Imai N., Shikami M., Mizutani M., Mizuno S., Hanamura I., Nitta M. BMC Cancer. 2014;14(1):76–79. doi: 10.1186/1471-2407-14-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto M., Miwa H., Shikami M., Tsunekawa-Imai N., Suganuma K., Mizuno S., Takahashi M., Mizutani M., Hanamura I., Nitta M. Cancer Invest. 2014;32(6):241–247. doi: 10.3109/07357907.2014.907419. [DOI] [PubMed] [Google Scholar]
- Simsek T., Kocabas F., Zheng J., DeBerardinis R. J., Mahmoud A. I., Olson E. N., Schneider J. W., Zhang C. C., Sadek H. A. Cell Stem Cell. 2010;7(3):380–390. doi: 10.1016/j.stem.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Škrtić M., Sriskanthadevan S., Jhas B., Gebbia M., Wang X., Wang Z., Hurren R., Jitkova Y., Gronda M., Maclean N., Lai C. K., Eberhard Y., Bartoszko J., Spagnuolo P., Rutledge A. C., Datti A., Ketela T., Moffat J., Robinson B. H., Cameron J. H., Wrana J., Eaves C. J., Minden M. D., Wang J. C. Y., Dick J. E., Humphries K., Nislow C., Giaever G., Schimmer A. D. Cancer Cell. 2011;20(5):674–688. doi: 10.1016/j.ccr.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriskanthadevan S., Jeyaraju D. V., Chung T. E., Prabha S., Xu W., Škrtić M., Jhas B., Hurren R., Gronda M., Wang X., Jitkova Y., Sukhai M. A., Lin F.-H., Maclean N., Laister R., Goard C. A., Mullen P. J., Xie S., Penn L. Z., Rogers I. M., Dick J. E., Minden M. D., Schimmer A. D. Blood. 2015;125(13):2120–2130. doi: 10.1182/blood-2014-08-594408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagadinou E. D., Sach A., Callahan K., Rossi R. M., Neering S. J., Minhajuddin M., Ashton J. M., Pei S., Grose V., O'Dwyer K. M., Liesveld J. L., Brookes P. S., Becker M. W., Jordan C. T. Cell Stem Cell. 2013;12(3):329–341. doi: 10.1016/j.stem.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samudio I., Harmancey R., Fiegl M., Kantarjian H., Konopleva M., Korchin B., Kaluarachchi K., Bornmann W., Duvvuri S., Taegtmeyer H., Andreeff M. J. Clin. Invest. 2010;120(1):142–156. doi: 10.1172/JCI38942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina J. R., Sun Y., Protopopova M., Gera S., Bandi M., Bristow C., McAfoos T., Morlacchi P., Ackroyd J., Agip A.-N. A., Al-Atrash G., Asara J., Bardenhagen J., Carrillo C. C., Carroll C., Chang E., Ciurea S., Cross J. B., Czako B., Deem A., Daver N., de Groot J. F., Dong J.-W., Feng N., Gao G., Gay J., Do M. G., Greer J., Giuliani V., Han J., Han L., Henry V. K., Hirst J., Huang S., Jiang Y., Kang Z., Khor T., Konoplev S., Lin Y.-H., Liu G., Lodi A., Lofton T., Ma H., Mahendra M., Matre P., Mullinax R., Peoples M., Petrocchi A., Rodriguez-Canale J., Serreli R., Shi T., Smith M., Tabe Y., Theroff J., Tiziani S., Xu Q., Zhang Q., Muller F., DePinho R. A., Toniatti C., Draetta G. F., Heffernan T. P., Konopleva M., Jones P., Di Francesco M. E., Marszalek J. R. Nat. Med. 2018;24(7):1036–1046. doi: 10.1038/s41591-018-0052-4. [DOI] [PubMed] [Google Scholar]
- Rassool F. V., Gaymes T. J., Omidvar N., Brady N., Beurlet S., Pla M., Reboul M., Lea N., Chomienne C., Thomas N. S. B., Mufti G. J., Padua R. A. Cancer Res. 2007;67(18):8762–8771. doi: 10.1158/0008-5472.CAN-06-4807. [DOI] [PubMed] [Google Scholar]
- Mills K. D., Ferguson D. O., Alt F. W. Immunol. Rev. 2003;194(1):77–95. doi: 10.1034/j.1600-065x.2003.00060.x. [DOI] [PubMed] [Google Scholar]
- Fan J., Li L., Small D., Rassool F. Blood. 2010;116(24):5298–5305. doi: 10.1182/blood-2010-03-272591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hole P. S., Darley R. L., Tonks A. Blood. 2011;117(22):5816–5826. doi: 10.1182/blood-2011-01-326025. [DOI] [PubMed] [Google Scholar]
- Skrott Z., Mistrik M., Andersen K. K., Friis S., Majera D., Gursky J., Ozdian T., Bartkova J., Turi Z., Moudry P., Kraus M., Michalova M., Vaclavkova J., Dzubak P., Vrobel I., Pouckova P., Sedlacek J., Miklovicova A., Kutt A., Li J., Mattova J., Driessen C., Dou Q. P., Olsen J., Hajduch M., Cvek B., Deshaies R. J., Bartek J. Nature. 2017;552(7684):194–199. doi: 10.1038/nature25016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lass A., McConnell E., Fleck K., Palamarchuk A., Wójcik C. Exp. Cell Res. 2008;314(14):2715–2723. doi: 10.1016/j.yexcr.2008.06.008. [DOI] [PubMed] [Google Scholar]
- Voráčková I., Suchanová Š., Ulbrich P., Diehl W. E., Ruml T. Protein Expression Purif. 2011;79(1):88–95. doi: 10.1016/j.pep.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M. T., Kuhlmann M., Hvam M. L., Howard K. A. Mol. Cell. Ther. 2016;4(1):3. doi: 10.1186/s40591-016-0048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanson D. A., Armijo A. L., Tom M., Li Z., Dimitrova E., Austin W. R., Nomme J., Campbell D. O., Ta L., Le T. M., Lee J. T., Darvish R., Gordin A., Wei L., Liao H.-I., Wilks M., Martin C., Sadeghi S., Murphy J. M., Boulos N., Phelps M. E., Faull K. F., Herschman H. R., Jung M. E., Czernin J., Lavie A., Radu C. G. J. Exp. Med. 2014;211(3):473–486. doi: 10.1084/jem.20131738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacque N., Ronchetti A. M., Larrue C., Meunier G., Birsen R., Willems L., Saland E., Decroocq J., Maciel T. T., Lambert M., Poulain L., Hospital M. A., Sujobert P., Joseph L., Chapuis N., Lacombe C., Moura I. C., Demo S., Sarry J. E., Recher C., Mayeux P., Tamburini J., Bouscary D. Blood. 2015;126(11):1346–1356. doi: 10.1182/blood-2015-01-621870. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.