

Glucose catabolism is the backbone of metabolism in most organisms. In spite of numerous studies and extensive knowledge, major controls on glycolysis and its connections to the other metabolic pathways remain to be discovered. A striking example is provided by the extreme glucose sensitivity of the yeast tps1Δ mutant, which undergoes apoptosis in the presence of just a few millimolar glucose. Previous work has shown that the conspicuous glucose-induced hyperaccumulation of the glycolytic metabolite fructose-1,6-bisphosphate (Fru1,6bisP) in tps1Δ cells triggers apoptosis through activation of the Ras-cAMP-protein kinase A (PKA) signaling pathway. However, the molecular cause of this Fru1,6bisP hyperaccumulation has remained unclear. We now provide evidence that the persistent drop in intracellular pH upon glucose addition to tps1Δ cells likely compromises the activity of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a major glycolytic enzyme downstream of Fru1,6bisP, due to its unusually high pH optimum. Our work highlights the potential importance of intracellular pH fluctuations for control of major metabolic pathways.
KEYWORDS: glycolysis, intracellular pH, glyceraldehyde-3-phosphate dehydrogenase, trehalose-6-phosphate synthase, Saccharomyces cerevisiae, glucose metabolism, TPS1
ABSTRACT
Whereas the yeast Saccharomyces cerevisiae shows great preference for glucose as a carbon source, a deletion mutant in trehalose-6-phosphate synthase, tps1Δ, is highly sensitive to even a few millimolar glucose, which triggers apoptosis and cell death. Glucose addition to tps1Δ cells causes deregulation of glycolysis with hyperaccumulation of metabolites upstream and depletion downstream of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The apparent metabolic barrier at the level of GAPDH has been difficult to explain. We show that GAPDH isozyme deletion, especially Tdh3, further aggravates glucose sensitivity and metabolic deregulation of tps1Δ cells, but overexpression does not rescue glucose sensitivity. GAPDH has an unusually high pH optimum of 8.0 to 8.5, which is not altered by tps1Δ. Whereas glucose causes short, transient intracellular acidification in wild-type cells, in tps1Δ cells, it causes permanent intracellular acidification. The hxk2Δ and snf1Δ suppressors of tps1Δ restore the transient acidification. These results suggest that GAPDH activity in the tps1Δ mutant may be compromised by the persistently low intracellular pH. Addition of NH4Cl together with glucose at high extracellular pH to tps1Δ cells abolishes the pH drop and reduces glucose-6-phosphate (Glu6P) and fructose-1,6-bisphosphate (Fru1,6bisP) hyperaccumulation. It also reduces the glucose uptake rate, but a similar reduction in glucose uptake rate in a tps1Δ hxt2,4,5,6,7Δ strain does not prevent glucose sensitivity and Fru1,6bisP hyperaccumulation. Hence, our results suggest that the glucose-induced intracellular acidification in tps1Δ cells may explain, at least in part, the apparent glycolytic bottleneck at GAPDH but does not appear to fully explain the extreme glucose sensitivity of the tps1Δ mutant.
INTRODUCTION
Identification of the mutant allele in several glucose-negative strains of the yeast Saccharomyces cerevisiae and cloning of the trehalose-6-phosphate (Tre6P) synthase gene unexpectedly converged on the same gene, TPS1 (1–8). Trehalose-6-phosphate synthase uses glucose-6-phosphate (Glu6P) and UDP-glucose (UDPG) as the substrates to catalyze the first step in trehalose biosynthesis, followed by TPS2-encoded trehalose-6-phosphate phosphatase, which dephosphorylates Tre6P into trehalose (9). The tps1 mutants, but not the tps2 mutants, are unable to grow on glucose or other rapidly fermentable sugars. The addition of glucose to tps1 cells causes hyperaccumulation of glycolytic metabolites upstream of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and depletion of metabolites downstream of GAPDH (1, 5, 10–16). Glu6P, fructose-6-phosphate, and especially fructose-1,6-bisphosphate (Fru1,6bisP) hyperaccumulate, while dihydroxyacetone phosphate and glyceraldehyde-3-phosphate (GA3P) increase to a lesser extent, likely because the equilibrium of the aldolase reaction is located toward Fru1,6bisP. Hence, the extent of Fru1,6bisP hyperaccumulation is a good indicator of the apparent glycolytic bottleneck at the level of GAPDH in tps1Δ cells. This is consistent with data on the control of the overactive glycolytic flux in cancer cells, where GAPDH was also identified as the rate-limiting step in the pathway and the level of Fru1,6bisP found to be predictive for the rate and control of glycolytic flux at GAPDH (17).
Inactivation of hexokinase 2 (Hxk2) restores normal growth of tps1 mutants on glucose (13), and tps1 mutants also show normal growth on galactose, for which the catabolism bypasses the hexokinase step in glycolysis (1–6, 8). These observations suggested that unregulated hexokinase activity was responsible for the growth defect of tps1 mutants on glucose. This was confirmed by the discovery that Tre6P is a potent competitive inhibitor of hexokinase (18). On the other hand, the extreme sensitivity of tps1 mutants to just a few millimolar glucose in the presence of 100 mM galactose (14), the partial complementation by the Escherichia coli TPS-encoding otsA (19), the discrepancies between Tre6P levels and the glycolytic deregulation (20), and the absence of a glucose growth defect in yeast cells expressing Tre6P-insensitive hexokinase from Schizosaccharomyces pombe (21) suggest that the absence of Tre6P inhibition of hexokinase is not the sole cause of the glucose growth defect and deregulation of glycolysis. Whereas the suppression of the tps1Δ growth defect on glucose by reduction of hexokinase activity suggests a plausible underlying mechanism, several other suppressors of tps1Δ have been identified, for which the action mechanism remains unclear: the overexpression of Mig1 (2), enhanced glycerol production (22, 23), inactivation of specific components of the electron transport chain (24), inhibition of respiration by antimycin (2, 24), and inactivation of Snf1 (25). In general, however, many of these suppressors seem to act by deviating the high levels of sugar phosphates into glycerol production, thus providing ample NAD+ and Pi for GAPDH activity but, at the same time, also preventing the induction of apoptosis by hyperaccumulation of Fru1,6bisP.
GAPDH catalyzes the first step in lower glycolysis by converting glyceraldehyde-3-phosphate (GA3P) to 1,3-bisphosphoglycerate (1,3bisPG) at the expense of Pi and reduction of NAD+ (26). It has remained unclear why glycolysis stalls at the level of GAPDH in tps1 mutants. One possible explanation is that GAPDH uses free phosphate (Pi) as a substrate and that the hyperaccumulation of sugar phosphates depletes the Pi level to such an extent that it compromises in vivo GAPDH activity (1, 15). Suppression of the tps1Δ growth defect on glucose by stimulation of glycerol biosynthesis is consistent with this explanation (27), since it recovers free phosphate but, on the other hand, also reduces sugar phosphate hyperaccumulation. The recent discovery that Fru1,6bisP is a potent stimulator of reactive oxygen species (ROS) formation and apoptosis in the tps1Δ strain in the presence of glucose through activation of the Ras-cAMP-protein kinase A (PKA) pathway therefore provides an alternative explanation for the suppression of tps1Δ by stimulation of glycerol production (12). This mechanism is supported by a recent report that the antioxidant N-acetylcysteine restores growth of the tps1Δ strain on low glucose (28). In addition, increasing the Pi level by itself does not revert the inefficiency of permeabilized tps1Δ spheroplasts to ferment glucose into ethanol (29), and extracellular addition of up to 50 mM Pi and/or the overexpression of Pi transporters failed to rescue the tps1Δ strain on glucose medium (22). This suggests that other mechanisms are involved that constrict glycolytic flux through GAPDH. Experimental observations also point toward GAPDH as an important bottleneck in the overactive glycolytic flux or Warburg effect in cancer cells (17, 30). It has been reported that the catalytic cysteine of GAPDH in mammalian cells is sensitive to oxidation by reactive oxygen species (ROS) and possibly helps to rewire flux toward the oxidative pentose phosphate pathway to generate more reducing power in the form of NADPH (31, 32).
In S. cerevisiae, GAPDH is a homotetramer composed of three isoforms, encoded by TDH1, TDH2, and TDH3 (33). These three genes have high sequence similarity (>90%) but are differently regulated at the transcriptional level (34). During the exponential phase, TDH3 is by far the most abundantly expressed, while TDH2 transcription is low and TDH1 barely expressed. As cells approach the stationary phase, expression levels of TDH3 decline by 50%, while TDH1 levels progressively increase and TDH2 levels peak at the end of the exponential phase. In the stationary phase, expression levels of TDH1 and TDH3 are similar, while the expression of TDH2 is low (34). The expression of TDH1 is rapidly enhanced under various stress conditions (35–38). The catalytic activities of the three isoenzymes are different, with Tdh1 (Km = 0.86 mM; kcat = 29.04 s−1) having the highest, Tdh2 intermediate (Km = 0.42 mM; kcat = 16.22 s−1), and Tdh3 the lowest activity (Km = 0.25 mM; kcat = 9.12 s−1), while the affinity ranks in the opposite order (39). The combined deletion of TDH2 and TDH3 is synthetically lethal, likely due to the low expression of TDH1, whereas the other double deletion strains are viable (34).
The addition of glucose to wild-type cells causes a rapid drop in the intracellular pH and ATP level, apparently due to rapid phosphorylation of the incoming glucose by hexokinase (1, 15, 40–42). However, in wild-type cells, this drop in intracellular pH and ATP level is only transient, and its recovery is faster under aerobic conditions than anaerobic conditions (41). On the other hand, in tps1Δ cells, the drop in the ATP level and the intracellular acidification are permanent (1, 23, 42). The consumption of ATP in the initiation of glycolysis and the generation of extra ATP downstream in glycolysis are no longer balanced, and the overactive sugar phosphorylation results in an ATP trap (43).
In the present study, we aim to understand the mechanism behind the glycolytic bottleneck at the level of GAPDH upon the addition of glucose to tps1Δ cells. We have explored the possibility that the persistent intracellular acidification observed after the addition of glucose to tps1Δ cells might be limiting glycolytic flux at the level of GAPDH because of its unusually high pH optimum. We provide evidence that the prevention of the glucose-induced pH drop partially suppresses sugar phosphate hyperaccumulation and metabolic defects in tps1Δ cells. Our results highlight the importance of intracellular pH in the control of glycolytic flux at the level of GAPDH and reveal novel roles for Tps1 in the control of glycolysis beyond its role in limiting hexokinase activity.
RESULTS
Influence of single GAPDH isoforms on the tps1Δ glucose growth defect.
To investigate whether a specific GAPDH isoform played a significant role in the glucose sensitivity of the tps1Δ strain, single TDH deletions were introduced in wild-type and tps1Δ strains. Subsequently a spot test was performed to test growth of these strains on different respiratory carbon sources (ethanol, glycerol, and galactose) as well as different glucose concentrations (Fig. 1). Consistent with the literature, the deletion of single GAPDH isoforms did not affect growth in the W303 wild-type background on any of the carbon sources tested (Fig. 1a and b). In the tps1Δ strain, on the other hand, the additional deletion of TDH1 or TDH2 worsened the tps1Δ growth defect at low glucose concentrations (Fig. 1b), while growth on the respiratory carbon sources was unaffected (Fig. 1a). The deletion of TDH3 in the tps1Δ strain, on the other hand, completely abolished growth at low glucose concentrations, down to 0.5 mM, with which the tps1Δ strain still grew to some extent. Since Tdh3 is responsible for most of the GAPDH activity when yeast is growing with glucose (39), this aggravated glucose sensitivity suggests that the deletion of TDH3 increases the metabolic bottleneck at the level of GAPDH. Unexpectedly, however, the additional deletion of TDH3 in the tps1Δ strain also completely abolished growth on galactose and allowed only residual growth on glycerol. In the wild-type strain, the deletion of TDH3 did not cause any effect for growth on galactose or glycerol. Growth on ethanol, for which TDH3 is not required, was not affected in any strain. These results indicate a novel interaction between Tps1 and the metabolism of galactose and glycerol, since hexokinase, the only known target of Tre6P, the product of Tps1, is not involved in the metabolism of galactose and glycerol.
FIG 1.

Single TDH isoform deletions increase the glucose sensitivity of the tps1Δ strain but do not affect growth on glucose of the wild-type strain. Wild-type and tps1Δ cells and their respective TDH single-deletion derivatives were spotted on YP agar in serial 5-fold dilutions after pregrowth on 3% glycerol. Cells were spotted onto plates containing 220 mM ethanol, 325 mM glycerol, or 100 mM galactose (a) or increasing concentrations of glucose: 0.5, 1, 2.5, 5 or 10 mM (b). Pictures were taken after 3 days.
To know if the reduced growth capacity of the tps1Δ tdh3Δ strain on galactose medium involves a similar bottleneck in the glycolytic pathway as that observed on glucose (12), we measured intracellular Fru1,6bisP levels after addition of 100 mM galactose or 100 mM glucose to glycerol-grown tps1Δ tdh3Δ cells (Fig. 2). Clearly, addition of galactose did not cause Fru1,6bisP hyperaccumulation in the tps1Δ tdh3Δ strain compared to that in the tps1Δ strain. In contrast, the deletion of TDH3 in the tps1Δ strain caused a further increase in Fru1,6bisP hyperaccumulation compared to that in the tps1Δ strain. This result indicates that the extent of Fru1,6bisP hyperaccumulation is inversely correlated with the residual GAPDH activity in the tps1Δ strain and that GAPDH acts as a bottleneck for glycolytic flux in the tps1Δ strain. These results also indicate that the growth defects of the tps1Δ tdh3Δ strain on glucose and galactose are apparently caused by different mechanisms and that the growth defect on galactose is not due to insufficient GAPDH activity.
FIG 2.
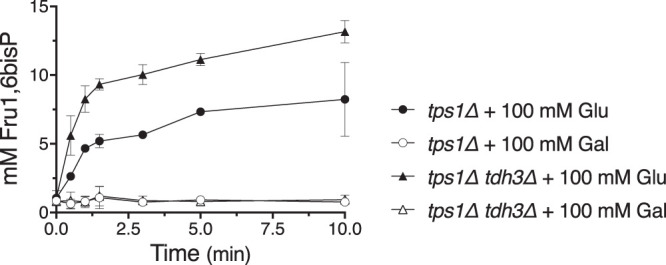
The deletion of TDH3 enhances Fru1,6bisP accumulation in the tps1Δ strain after the addition of glucose but not after addition of galactose. Fru1,6bisP accumulation was measured after addition of 100 mM glucose (closed symbols) or galactose (open symbols) to cells of the tps1Δ strain (circles) or tps1Δ tdh3Δ strain (triangles). Cells were pregrown on complete synthetic medium with 3% glycerol as carbon source.
To evaluate whether the apparent bottleneck of glycolytic flux in the tps1Δ strain at the level of GAPDH is merely due to insufficient catalytic activity, we next investigated whether the overexpression of GAPDH isoforms could overcome the tps1Δ growth defect on glucose. For this purpose, the multicopy p426 plasmid was used for the overexpression of the GAPDH isogenes TDH2 and TDH3 using the constitutive TEF1 promoter. Since TDH1 is mostly expressed in stationary-phase cells (35), we did not select this isogene for overexpression. Glycerol-grown precultures were spotted onto rich solid nutrient medium containing different carbon sources (glycerol, galactose, or glucose) (Fig. 3a) or a series of low glucose concentrations ranging between 0.5 and 10 mM (Fig. 3b). In the wild-type strain, the overexpression of TDH2 or TDH3 had no effect for growth on any carbon source or any concentration of glucose tested. The glucose growth defect of the tps1Δ strain was also not overcome by the overexpression of TDH2 at any glucose concentration tested, while the overexpression of TDH3 even aggravated the glucose-sensitive phenotype.
FIG 3.

The overexpression of TDH2 or TDH3 does not rescue the glucose growth defect of the tps1Δ strain. Plasmid-transformed cells of wild-type and tps1Δ strains were spotted in serial 5-fold dilutions on YP agar supplemented with 325 mM glycerol, 100 mM galactose, or 100 mM glucose (a) or different concentrations of glucose: 0.5, 1, 2.5, 5 or 10 mM (b). TDH2 and TDH3 were overexpressed from the p426 multicopy plasmid behind the strong constitutive TEF1 promoter. Strains containing the p426 plasmid without an insert served as controls. Pictures were taken after 3 days.
GAPDH activity is highly sensitive to changes in cytosolic pH.
Since the glycolytic deregulation seems to hint at compromised GAPDH activity in the tps1Δ strain, we first measured the pH optimum of GAPDH in permeabilized spheroplasts of wild-type cells (Fig. 4a). This in situ technique allows us to manipulate experimental conditions while preserving the native protein environment in the cells. While specific GAPDH activity was highest around pH 8.0, almost no activity was measured below pH 6.0. From these results, a near linear relationship of GAPDH activity with respect to pH within the physiological range between pH 6 and 7.5 becomes apparent. We also compared the pH dependency and maximal activity of GAPDH activity in situ between wild-type and tps1Δ cells (Fig. 4b). However, both strains displayed similar pH profiles and maximal activities, making it unlikely that the absence of Tps1 compromises glycolytic flux by directly influencing GAPDH activity. We also studied the influence of the two most relevant metabolites, Tre6P and Fru1,6bisP, on GAPDH activity at the physiologically relevant pH of 6.8. However, addition of neither Tre6P, nor Fru1,6bisP significantly influenced GAPDH activity in permeabilized wild-type cells, suggesting that these metabolites do not play a significant role in the deregulation of glycolysis through stimulation or inhibition of GAPDH (Fig. 4c and d).
FIG 4.
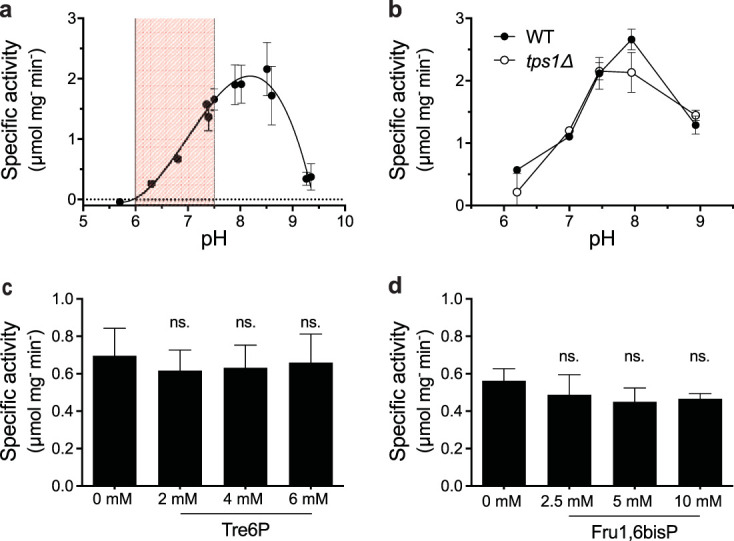
The in situ GAPDH activity is strongly compromised at low pH and not affected by Tre6P or Fru1,6bisP. GAPDH activity was measured in permeabilized spheroplasts of wild-type cells studied over a wide pH range (highlighted: physiological pH range from 6 to 7.5) (a) and wild-type compared to tps1Δ cells (b). GAPDH activity was measured in permeabilized wild-type cells at pH 6.8 in the absence or presence of increasing concentrations of Tre6P (2, 4, or 6 mM) (c) and Fru1,6bisP (2.5, 5, or 10 mM) (d). Statistical analysis was performed by one-way analysis of variance (ANOVA) with Dunnett’s multiple-comparison test. No significant difference was observed between the samples with and without Tre6P or Fru1,6bisP. ns, not significant, P > 0.05.
Given the strong pH dependency and high pH optimum of GAPDH activity and the fact that glucose addition to tps1Δ cells is known to trigger intracellular acidification (1, 23, 42), we set out to investigate whether low cytosolic pH could be a cause of the apparent metabolic bottleneck in the glycolytic flux at the level of GAPDH after the addition of glucose to tps1Δ cells. To measure the intracellular pH (pHi) more precisely, we applied a fluorescence-based technique by expressing the cytosolic pH-sensitive green fluorescent protein (GFP) variant, pHluorin, in the cells of interest (44). We have compared the pHi profiles following glucose addition to pHluorin-expressing cells of wild-type, tps1Δ, and the tps1Δ suppressor strains tps1Δ hxk2Δ and tps1Δ snf1Δ. As previously reported (1, 15, 40–42), the pHi of wild-type cells transiently dropped and rapidly recovered to approximately neutral pHi in response to glucose addition, while tps1Δ cells were unable to recover from the initial pH drop and showed persistent intracellular acidification (Fig. 5a). The transient or persistent drop in pHi coincided with a transient or persistent increase in the Glu6P level, respectively. Interestingly, the initial Glu6P peak (45 s after glucose addition) in wild-type cells corresponded to a pHi of approximately 6.0, at which specific GAPDH activity measured in vitro is minimal. Moreover, the tps1Δ suppressor strains, tps1Δ hxk2Δ and tps1Δ snf1Δ, clearly regained the ability to rapidly recover from the initial intracellular acidification after glucose addition, although recovery was somewhat less robust (Fig. 5b). Taken together, we observed a clear correlation between the maintenance of a proper pHi and the ability to grow on glucose.
FIG 5.
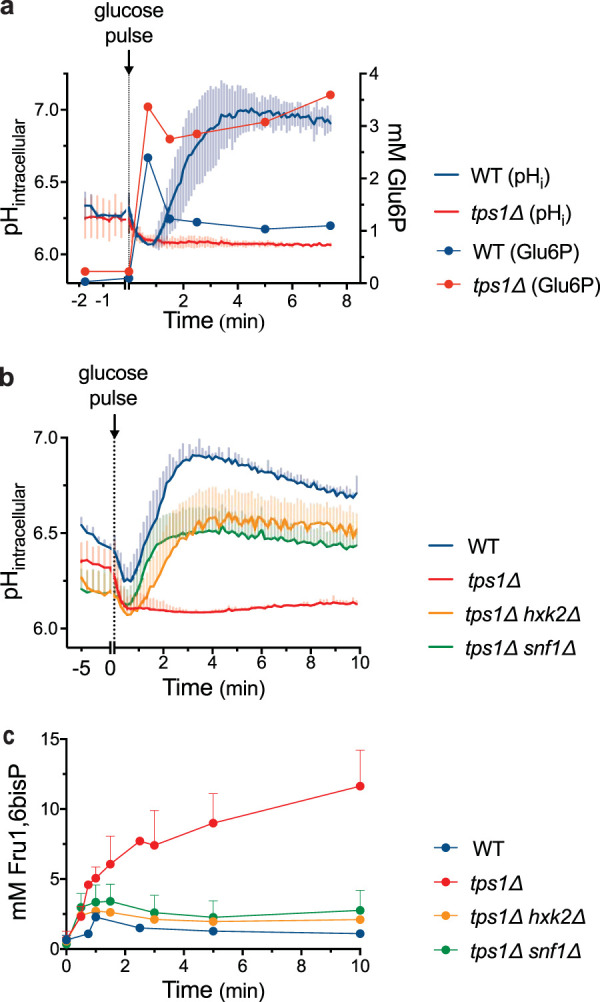
Intracellular acidification correlates with Fru1,6bisP accumulation. (a) Intracellular pH (full lines without symbols) and Glu6P levels (full lines with circles) after addition of 100 mM glucose in wild-type (blue) and tps1Δ (red) strains. (b) Intracellular pH profile of wild-type (blue), tps1Δ (red), tps1Δ hxk2Δ (yellow), and tps1Δ snf1Δ (green) strains after addition of 100 mM glucose. Standard deviations were calculated from at least 9 technical repeats per strain. (c) Fru1,6bisP levels after addition of 100 mM glucose to wild-type (blue), tps1Δ (red), tps1Δ hxk2Δ (yellow), and tps1Δ snf1Δ (green) strains. Cells were always pregrown on complete synthetic medium supplemented with 2% galactose.
In the next step, we examined if the pHi profile correlated with the apparent bottleneck in glycolytic flux at the level of GAPDH by measuring intracellular Fru1,6bisP accumulation (Fig. 5c). Indeed, among the four strains, wild type, tps1Δ, tps1Δ hxk2Δ, and tps1Δ snf1Δ, the capacity to recover pHi after glucose addition was inversely correlated with the extent of intracellular Fru1,6bisP accumulation and thus with the apparent bottleneck at the level of GAPDH. Moreover, the transient increase in Fru1,6bisP concentration observed in wild-type cells, with a maximum at approximately 1 min after glucose addition, coincided approximately with the minimum pHi value after glucose addition. Hence, our results are consistent with low pHi being a limiting factor for glycolytic flux at the level of GAPDH, as inferred from Fru1,6bisP accumulation, in wild-type cells and especially in cells of the tps1Δ strain.
Preventing intracellular acidification partially counteracts glycolytic deregulation.
The previous experiments suggested that the pHi dependency of GAPDH might, at least in part, constitute a causal factor for Fru1,6bisP hyperaccumulation in tps1Δ cells after glucose addition. Hence, we have applied experimental conditions to prevent the cytosolic acidification in the tps1Δ strain by supplementing NH4Cl at high extracellular pH (41). At an extracellular pH closer to the protonation equilibrium (pKa,NH4+ = 9.24) (45), ammonium (NH4+) shifts toward its un-ionized form, ammonia (NH3), which can diffuse through the plasma membrane. Once in the cell, where the pHi is lower, NH3 converts to NH4+ taking up one proton and raising the pHi. By using this approach, we were able to prevent intracellular acidification in both wild-type and tps1Δ cells after glucose addition together with 200 mM NH4Cl in extracellular medium buffered with 200 mM Tris-HCl at pH 8, 8.5, or 9 (see Fig. S1a and b in the supplemental material). At an extracellular pH of 9, we were able to maintain pHi between approximately 7.4 and 6.8 for 60 min in both wild-type and tps1Δ cells (Fig. 6a). However, at an extracellular pH of 7.9, growth was already completely prevented both in wild-type and tps1Δ cells (see Fig. S2a and b). This made it impossible to evaluate the rescue of growth in the tps1Δ strain by the prevention of intracellular acidification using this approach. On the other hand, we were able to determine the effect of the maintenance of proper pHi on the short-term deregulation of glycolysis in tps1Δ cells.
FIG 6.
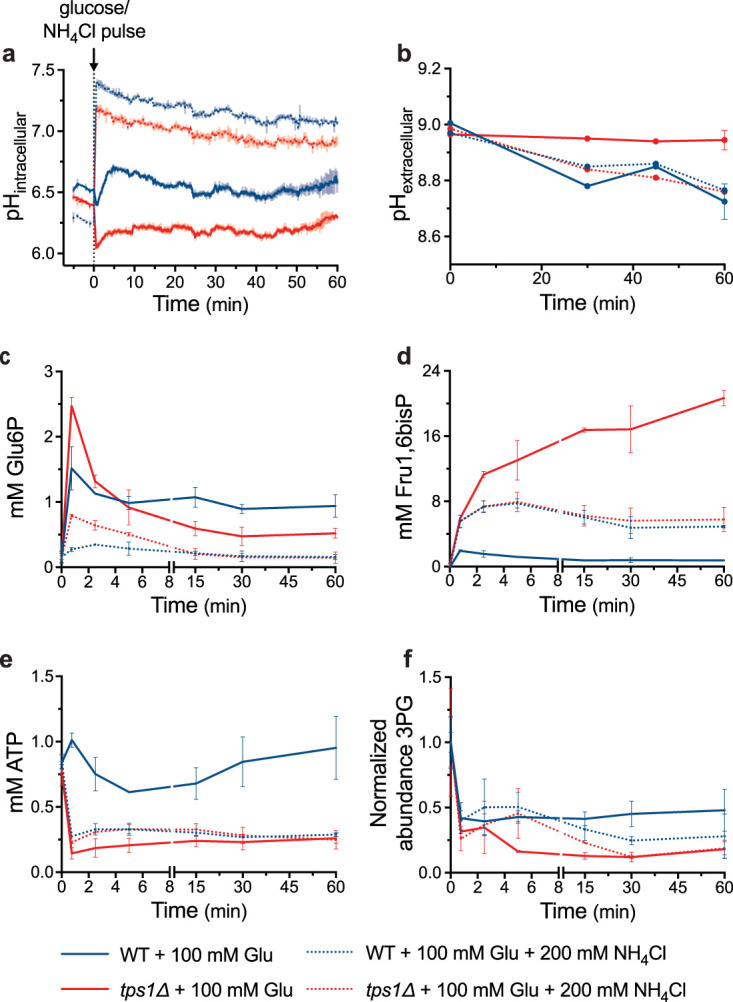
The prevention of intracellular acidification reduces Glu6P and Fru1,6bisP hyperaccumulation and partially counteracts metabolic deregulation in tps1Δ cells. Wild-type (blue) and tps1Δ (red) galactose-grown cells were resuspended in 200 mM Tris-HCl, pH 9. At time point zero, cells were provided with 100 mM glucose (full lines) or with 100 mM glucose and 200 mM NH4Cl (dotted lines). Graphs represent the time course profiles of intracellular pH (a), extracellular pH (b), Glu6P level (c), Fru1,6bisP level (d), ATP level (e), and normalized level of 3PG (f).
Intracellular acidification after the addition of glucose is prevented in 200 mM Tris-HCl medium at high pH in the presence of 200 mM NH4Cl. Intracellular pH was monitored after addition at time point zero of 100 mM glucose with (full lines) or without (dotted lines) 200 mM NH4Cl. Wild-type (a) and tps1Δ (b) cells in 200 mM Tris-HCl medium at pH 8 (light blue), pH 8.5 (dark blue), and pH 9 (purple). Cells were grown in uracil-deficient medium with 2% galactose to retain the pHluorin plasmid. Download FIG S1, EPS file, 2.5 MB (2.4MB, eps) .
Copyright © 2020 Van Leemputte et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
High extracellular pH inhibits growth. Growth in complete synthetic medium supplemented with either 100 mM glucose (full lines) or 100 mM galactose (dotted lines) was followed over 48 h by measuring OD595 for wild-type (blue) and tps1Δ (red) cells in regular liquid medium at pH 5.5 (a) or 50 mM Tris-HCl medium at pH 8 (b). Cells were pregrown on galactose medium. Download FIG S2, EPS file, 2.7 MB (3.2MB, eps) .
Copyright © 2020 Van Leemputte et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Addition of 200 mM NH4Cl buffered at pH 9 together with glucose effectively abolished intracellular acidification in both wild-type and tps1Δ cells (Fig. 6a). We next measured the extracellular pH over the course of the experiment. In spite of the high alkaline pH and the strong buffering of the medium, wild-type cells were still able to lower the pH of the medium from 9 to approximately 8.8 in 60 min after the addition of glucose, both in the presence and the absence of NH4Cl, which is indicative of active fermentation and proton pumping by the plasma membrane H+-ATPase (Fig. 6b). On the other hand, the tps1Δ strain was only able to lower the extracellular pH to the same extent as wild-type cells when NH4Cl was added together with glucose. In the absence of NH4Cl, there was no drop in medium pH after the addition of glucose to tps1Δ cells (Fig. 6b), consistent with previous reports on the absence of glucose-induced activation of plasma membrane H+-ATPase and a precipitous glucose-induced drop in ATP in the tps1Δ mutant (1, 42). This result suggests that the prevention of intracellular acidification in tps1Δ cells was able to restore glycolytic flux to such an extent that enough ATP could be produced for the restoration of proton pumping activity and extracellular medium acidification.
We next determined the major glycolytic metabolites under the same conditions of glucose addition with and without NH4Cl at extracellular pH 9 (Fig. 6c to f). The transient Glu6P accumulation was strongly reduced in the presence of NH4Cl (Fig. 6c). However, this was also the case in the wild-type strain, which could be attributed to decreased glucose transport because of the high extracellular pH and/or stimulation of phosphofructokinase by NH4+ (46). The latter mechanism might provide at least a partial explanation, since the glucose-induced increase in Fru1,6bisP was strongly enhanced in wild-type cells (Fig. 6d). On the other hand, the dramatic glucose-induced hyperaccumulation of Fru1,6bisP in tps1Δ cells was strongly reduced in the presence of NH4Cl and now actually overlapped with the enhanced increase of Fru1,6bisP in wild-type cells, consistent with improved glycolytic flux at the level of GAPDH (Fig. 6d). The latter was also supported by the slightly lower drop in the ATP level in tps1Δ cells (Fig. 6e) and higher concentration of 3-phosphoglycerate (3PG), a metabolite downstream of GAPDH in glycolysis, as measured by mass spectrometry (Fig. 6f). Glucose addition caused a much stronger drop in 3PG in tps1Δ cells than in wild-type cells, consistent with the bottleneck at the level of GAPDH in tps1Δ cells (Fig. 6f). In wild-type cells, the presence of NH4Cl caused a precipitous drop in the ATP level (Fig. 6e), consistent with the dramatic increase in the Fru1,6bisP level (Fig. 6d). The presence of NH4Cl had little effect on the 3PG level in wild-type cells in the short term but caused a drop in the longer term (Fig. 6f). These results are consistent with stimulation of glycolytic flux at the level of GAPDH in tps1Δ cells by counteracting the dramatic glucose-induced drop in pHi and thus suggest that the persistent intracellular acidification is, at least in part, responsible for the apparent bottleneck in glycolytic flux in tps1Δ cells.
We also evaluated the possible alternative interpretation that the addition of 200 mM NH4Cl at high extracellular pH of 9 in 200 mM Tris-HCl buffer reduced glucose uptake to such an extent that the glucose-induced hyperaccumulation of sugar phosphates in tps1Δ cells was counteracted because of reduced glucose influx. Hence, we first measured glucose uptake rate in wild-type and tps1Δ cells in the absence and presence of NH4Cl at pH 9. The uptake of 100 mM glucose was reduced 30% and 35% in wild-type and tps1Δ strains, respectively (Fig. 7a). To determine whether such a reduction in glucose uptake rate could counteract the glucose-induced hyperaccumulation of Glu6P and Fru1,6bisP in tps1Δ cells to the same extent as we observed in the presence of NH4Cl at pH 9, we deleted five HXT glucose transporter genes, HXT6, -7, -2, -4, and -5, in the tps1Δ strain. This did not restore the capacity to grow on 100 mM glucose (see Fig. S3).
FIG 7.
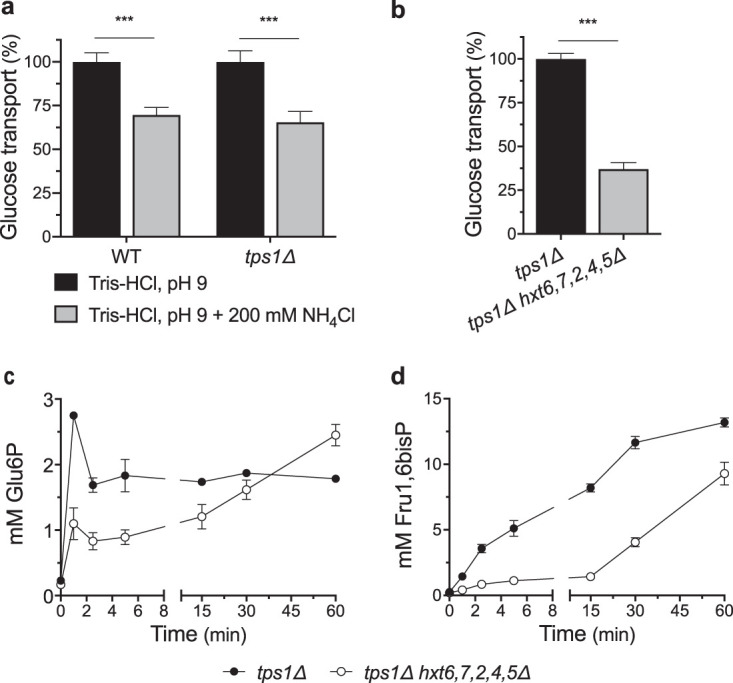
Reduced glucose transport delays but does not prevent deregulation of glycolytic flux in tps1Δ cells. Relative uptake rate of 100 mM glucose in wild-type and tps1Δ cells in 200 mM Tris-HCl, pH 9, in the absence (black bars) or presence (gray bars) of 200 mM NH4Cl (a) and tps1Δ (black bar) and tps1Δ hxt6,7,2,4,5Δ (gray bar) cells in complete synthetic medium (b). Metabolite profiles for Glu6P (c) and Fru1,6bisP (d) after addition of 100 mM glucose to galactose-grown tps1Δ (closed symbols) or tps1Δ hxt7,6,2,4,5Δ (open symbols) cells. Statistical analysis was performed by two-way ANOVA with Bonferroni’s correction (a) and an unpaired Student's t test (b). ***, P < 0.001.
Absence of growth on 100 mM glucose in the tps1Δ hxt6,7,2,4,5Δ strain. Cultures of the wild-type, tps1Δ, hxt6,7,2,4,5Δ, and tps1Δ hxt6,7,2,4,5Δ strains were spotted in 5-fold dilutions on YP agar with 100 mM galactose or 100 mM glucose as carbon source. Pictures were taken after three days. Download FIG S3, TIF file, 2.5 MB (2.5MB, tif) .
Copyright © 2020 Van Leemputte et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The tps1Δ hxt6,7,2,4,5Δ cells were pregrown on glycerol to avoid the expression of the high-affinity galactose (and glucose) transporter GAL2. The tps1Δ hxt6,7,2,4,5Δ strain displayed a reduction in the glucose (100 mM) transport rate of 65% (Fig. 7b). This is approximately double the transport reduction caused by NH4Cl addition. The initial transient Glu6P increase was considerably reduced in the tps1Δ hxt6,7,2,4,5Δ strain compared to that in the tps1Δ strain, but Glu6P kept accumulating until it reached levels as high as in the tps1Δ strain (Fig. 7c). Similarly, Fru1,6bisP accumulation in the tps1Δ hxt6,7,2,4,5Δ strain was only delayed and not prevented (Fig. 7d). Hence, the reduction of glucose uptake of 65% in the tps1Δ hxt6,7,2,4,5Δ strain was not enough to prevent the deregulation of glycolysis, which is consistent with the inability of that strain to grow on 100 mM glucose. Since glucose uptake was reduced twice as much in the tps1Δ hxt6,7,2,4,5Δ strain compared to that under the condition with NH4Cl, these results make it unlikely that the reduction of glucose uptake was responsible for the diminished Glu6P and Fru1,6bisP hyperaccumulation in tps1Δ cells after the addition of glucose plus NH4Cl at high pH (Fig. 6).
DISCUSSION
The addition of glucose to tps1Δ cells of the yeast S. cerevisiae causes hyperaccumulation of all glycolytic metabolites upstream and depletion of all metabolites downstream of GAPDH, suggesting that the deletion of Tps1 in some way creates a bottleneck in glycolysis at the level of GAPDH (27). Measurements of the specific activity of the glycolytic enzymes in cell extracts as well as determination of initial glucose uptake rates did not reveal significant differences between the wild-type and tps1 strains that could explain the glycolytic bottleneck in tps1Δ cells (1, 11). More detailed measurements of the glucose uptake rate and the pH dependency of GAPDH in the present work have underscored the conclusion that there is no difference in the inherent activity of these two crucial components in the tps1Δ strain. Hence, the bottleneck appears to be due to a metabolic or regulatory problem at the level of GAPDH that is not maintained in cell extracts and is not apparent from the Vmax or Km of initial glucose uptake rates in intact cells.
A possible reason for reduced GAPDH activity in vivo in tps1Δ cells might have been a direct regulatory action of Tre6P on GAPDH. This would have been in accordance with previous observations that Tre6P stimulates ethanolic fermentation in permeabilized spheroplasts of tps1Δ cells (29). However, we did not detect any significant influence of Tre6P on GAPDH activity in permeabilized spheroplasts (Fig. 4c), and GAPDH activity also did not show any significant deviation as a function of pH in extracts of tps1Δ versus that of wild-type cells (Fig. 4b). In the absence of glucose, GAPDH activity itself does not seem to be affected, since the tps1Δ strain displays normal growth on galactose and on nonfermentable carbon sources, for which GAPDH activity is also essential.
Since Pi and NAD+ are substrates of GAPDH, their depletion could in principle compromise in vivo GAPDH activity. NAD+ levels, however, appear to be unaffected (16), and conditions that enhance the NADH availability strongly counteract the glucose growth defect of the tps1Δ strain (2, 15, 24). NAD+ is also unlikely to be limiting, since no NAD+-consuming steps are present in the first part of glycolysis. Although the Pi level drops somewhat more upon the addition of glucose to tps1Δ cells compared to that for wild-type cells, the difference is small (1). The suppression of the tps1Δ growth defect on glucose by stimulation of glycerol biosynthesis, which causes recovery of free phosphate, initially appeared to be consistent with restriction of GAPDH activity by Pi limitation. However, supply of high external Pi levels or stimulation of Pi uptake by the overexpression of the Pho84 phosphate carrier does not suppress the tps1Δ growth defect on glucose (22, 27). In addition, the depletion of all other phosphate-containing metabolites in tps1Δ cells to specifically sustain the hyperaccumulation of the upstream sugar phosphates in glycolysis (16, 23) apparently provides sufficient free Pi for this continued buildup, which requires the maintenance of ATP production from ADP and free Pi. This makes it unlikely that Pi limitation is the main cause of the apparent bottleneck at GAPDH, although it cannot be excluded that it reinforces the bottleneck.
We also explored the possible contribution of the three Tdh isoforms in the apparent GAPDH bottleneck. Although the deletion of any one of the Tdh isoforms did not affect the growth of wild-type cells, the deletion of a single isoform, and especially of the most active isoform, Tdh3, exacerbated the glucose sensitivity of the tps1Δ strain (Fig. 1). Moreover, the absence of Tdh3 resulted in much higher levels of Fru1,6bisP after glucose addition (Fig. 2). This reinforces the conclusion that GAPDH activity is limiting in tps1Δ cells and that this is a major factor for their high glucose sensitivity. On the other hand, the overexpression of Tdh isoforms did not rescue the growth defect of the tps1Δ strain to any extent, not even at very low glucose concentrations (Fig. 3). This indicates that the factor limiting GAPDH activity in tps1Δ cells overrides any increase in intrinsic GAPDH specific activity that could be established in vivo. An inappropriate intracellular pH could constitute such a factor, since it would severely limit the actual in vivo GAPDH activity, whatever the potential activity under optimal conditions.
In this work, we have explored an alternative explanation as a major cause for the apparent glycolytic bottleneck at the level of GAPDH by concentrating on the unusually high pH optimum of GAPDH and the persistent glucose-induced drop in intracellular pH in tps1Δ cells. When glucose is added to respiring wild-type yeast cells, there is a short transient drop in the intracellular pH, which is likely caused by the very rapid proton-producing phosphorylation of glucose (7, 41). The plasma membrane H+-ATPase Pma1, which is posttranslationally activated by glucose (47, 48), supports proton extrusion to the medium, while the vacuolar V-ATPase pumps protons into the vacuole, which together cause rapid recovery of the cytosolic pHi to approximately neutral pH (49–51). The tps1Δ strain is deficient in glucose-induced activation of plasma membrane H+-ATPase (1, 42), and the rapid depletion of the ATP level likely compromises proton pumping activity by both Pma1 and V-ATPase (1, 42) (Fig. 6e). This explains the persistent intracellular acidification in tps1Δ cells (Fig. 6a) (23, 42). The limitation of proton export by the plasma membrane H+-ATPase likely explains the absence of glucose-induced medium acidification in tps1Δ cells (Fig. 6b).
Our data show that GAPDH activity is particularly sensitive to pHi, with a very atypical optimum at pH 8 and thus reduced activity in the physiological pH range of 6.5 to 7.5 (Fig. 4a). Between pH 6.5 and 6, the activity drops further to less than 10% of the optimal activity at pH 8. Moreover, our results indicate that, coinciding with the sugar phosphate hyperaccumulation in the tps1Δ strain after the addition of glucose, the intracellular pH drops to approximately 6.1 to 6.2 and does not recover, as opposed to the rapid recovery in the wild-type and tps1Δ suppressor strains (Fig. 5a and b). This suggests that the persistent intracellular acidification might be responsible for the apparent block of glycolysis at the level of GAPDH, causing the continuous increase in Fru1,6bisP levels in tps1Δ cells. In wild-type cells, the drop in intracellular pH after the addition of glucose is very transient, recovering within 1 to 2 min, and it may be supporting the transient overshoot in Glu6P and Fru1,6bisP levels that happens at that time (Fig. 5). As opposed to that in tps1Δ cells, the ATP level remains high in wild-type cells after the addition of glucose (Fig. 6e), consistent with rapid initiation of proper flux downstream in glycolysis and proper support of H+-pumping activity to establish a regular intracellular pH.
To evaluate the possible involvement of persistent intracellular acidification in tps1Δ cells after the addition of glucose in causing the apparent block at GAPDH and the resulting hyperaccumulation of Fru1,6bisP and other metabolic defects, we suppressed intracellular acidification by adding 200 mM NH4Cl at pH 9 together with glucose. This was very effective and maintained the intracellular pH at or above pH 7 for at least 60 min. The maintenance of a proper intracellular pH strongly reduced the hyperaccumulation of Fru1,6bisP, as well as the overshoot of Glu6P, and also restored the extracellular medium acidification for which proper ATP provision and proton pumping are required (Fig. 6). Hence, these results support that the maintenance of a proper intracellular pH caused the restoration of downstream glycolytic flux, at least to some extent, in the tps1Δ cells. This was further supported by a slightly smaller drop in the ATP level and a slight increase in 3PG levels (Fig. 6). A recent report that protonophores, such as dinitrophenol and carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), stimulate glucose consumption and support growth on low glucose of tps1Δ cells is also consistent with this explanation (28). Interestingly, in wild-type cells, the overshoot in Glu6P was also greatly suppressed when glucose was added with 200 mM NH4Cl at pH 9, while the increase in Fru1,6bisP was strongly elevated (Fig. 6d). The strong elevation of Fru1,6bisP might be due to the well-known stimulating effect of NH4+ on phosphofructokinase activity (46), which is consistent with the significant simultaneous drop in ATP (Fig. 6e). Since this stimulation likely happened also in tps1Δ cells and since the Fru1,6bisP levels in wild-type and tps1Δ cells coincided when glucose was added with 200 mM NH4Cl at pH 9, the maintenance of a proper intracellular pH appears to have largely normalized the Fru1,6bisP profile in the tps1Δ cells. Hence, these results further support that the persistently low pHi acts as a major driver at least for the initial glycolytic bottleneck in tps1Δ cells, which might be aggravated further by other factors such as the stronger reduction in intracellular Pi than that in wild-type cells. Since both wild-type and tps1Δ cells were unable to grow at the high extracellular pH of 9 and since the addition of 200 mM NH4Cl at pH 9 also caused deregulation of glycolytic metabolite levels in wild-type cells, it was not possible to assess the effect of the restored intracellular pH on long-term recovery of growth on glucose in tps1Δ cells under these experimental conditions.
A possible alternative explanation for the partial restoration of glycolytic metabolite levels when glucose was added together with 200 mM NH4Cl at pH 9, as opposed to those at pH 9 without NH4Cl, is that the high NH4Cl concentration reduced glucose uptake to such an extent that glucose influx in glycolysis became limiting and the absence of the hexokinase Tre6P control therefore was no longer relevant. It is well established that a reduction of glucose transport can effectively lower glycolytic flux in S. cerevisiae (52). This is likely also the reason why tps1Δ cells can still grow at low glucose concentrations of 0.5 to 2 mM (Fig. 1b). This hypothesis could explain the strong reduction in sugar phosphate accumulation in tps1Δ cells (Fig. 6c and d), but it is more difficult to reconcile with the strong elevation of Fru1,6bisP levels in wild-type cells when glucose was added together with 200 mM NH4Cl at pH 9 (Fig. 6d). To evaluate this alternative explanation more rigorously, we first measured the glucose uptake rate at pH 9 in the absence and presence of 200 mM NH4Cl. This revealed a rather modest drop in glucose uptake activity of 30% to 35% (Fig. 7a). To assess the relevance of such a drop in the glucose uptake rate for the deregulation of glycolytic metabolite levels in tps1Δ cells, we constructed a tps1Δ strain with the additional deletion of five HXT glucose carrier genes, resulting in a strain with only low-affinity glucose uptake. This strain displayed a much higher reduction (65%) in glucose uptake but still was not able to grow on 100 mM glucose. Consistently, glucose-induced hyperaccumulation of sugar phosphates in that strain was only delayed and not prevented (Fig. 7c and d). These results contradict the alternative explanation that the experimental conditions used to prevent the persistent intracellular pH drop in tps1Δ cells after glucose addition caused a reduction in glucose influx in glycolysis high enough to weaken the deregulation of glycolysis as significantly as we observed. Hence, we are bound to conclude that the aberrant pHi profile in tps1Δ cells after glucose addition is likely a major contributor to the deregulation of glycolysis, reducing GAPDH activity to such an extent that Fru1,6bisP starts to accumulate to very high levels, triggering ROS formation and apoptosis by hyperactivation of the Ras-cAMP-PKA pathway (12).
Our results support the importance of cytosolic pH for metabolic regulation. Under normal physiological conditions, the intracellular pH acidifies rather gradually toward the end of fermentation or drops suddenly in response to acute glucose starvation (53). A study on single-cell glycolytic oscillations showed that fluctuations in cytosolic pH as small as 0.1 to 0.3 can affect NADH concentrations (54). It might also affect GAPDH activity given its high sensitivity to changes in pH. A strong reduction in GAPDH activity has been described in the case of acute oxidative stress. The catalytic site of GAPDH is known to be very sensitive to oxidation, causing glycolytic flux to be rerouted to the pentose phosphate pathway for increased production of NADPH to combat ROS toxicity (31, 32). Interestingly, several conditions that are associated with increased ROS production, such as carbon starvation and the stationary phase (55), are associated with lowered cytosolic pH (53, 56).
Our work has also revealed novel regulatory effects caused by the deletion of TPS1. Although the allosteric inhibition of hexokinase activity by Tre6P provided a seemingly logical explanation for the overactive and persistent sugar phosphate accumulation and resulting glycolytic deregulation in tps1Δ cells (18), multiple experimental results have been obtained indicating that Tps1 must exert additional controls on yeast metabolism (19, 21, 27, 29). For instance, the expression of Tre6P-insensitive hexokinase from Schizosaccharomyces pombe in S. cerevisiae does not cause glycolytic deregulation (21). It should also be pointed out that the deletion of HXK2 has important consequences besides lowering hexokinase activity, which might contribute to the restoration of growth on glucose of the tps1Δ strain, such as the loss of the glucose-repressed state (57) and the mislocalization of Ras to the mitochondria (58). In addition, tps1 mutants also show defects on media with respiratory carbon sources, i.e., in the absence of glucose, such as the change in regulatory properties of glycogen synthase (11) and the sporulation defect (1). By examining the contribution of the different Tdh isoenzymes of GAPDH in the tps1Δ growth defect, we unexpectedly discovered that the deletion of TDH3 in a tps1Δ strain severely compromises growth on galactose, glycerol, and ethanol, whereas it produces no noticeable defect in a wild-type strain. This points to the existence of an unknown regulatory connection between Tps1 and GAPDH, since hexokinase activity is not required for growth on galactose, glycerol, and ethanol and the absence of its Tre6P control therefore would not be expected to have any relevance. On the other hand, GAPDH activity is required for growth on galactose, glycerol, and ethanol, and since only the deletion of TDH3 caused this growth problem, the results indicate that Tps1 is required in some way for the maintenance of proper Tdh1 and Tdh2 activity in the absence of Tdh3. Future work will have to reveal the precise underlying mechanism of this novel connection between Tps1 and GAPDH.
Conclusions.
Our work reveals that the apparent bottleneck in glycolysis at the level of GAPDH after the addition of glucose to cells of the tps1Δ mutant is, at least in part, due to the combination of the unusually high pH optimum of GAPDH and the persistent intracellular acidification caused by the deregulation of glycolysis. Counteraction of the intracellular acidification strongly reduced Fru1,6bisP hyperaccumulation and restored extracellular medium acidification, consistent with partial restoration of glycolytic flux. Our work provides further evidence for the importance of intracellular pH regulation in the control of metabolism. In addition, we provide evidence for novel regulatory defects caused by tps1Δ in combination with tdh3Δ, which are independent of the Tps1 control on hexokinase activity.
MATERIALS AND METHODS
Yeast strain and plasmid overview.
All yeast strains used in this work share the same W303 genetic background. Strains and plasmids used and generated in this study are listed in Tables 1 and 2, respectively. For gene deletions, coding sequences of antibiotic resistance genes or auxotrophic markers were PCR amplified using primers with tails designed to be homologous to the 50 bp directly upstream or downstream of the gene of interest. Genes were deleted by means of homologous recombination. Both linear and plasmid DNA were introduced through the lithium acetate heat shock protocol at 42°C reported by Gietz and Schiestl (59). For transformations with antibiotic selection, cells were recovered in liquid medium without selection for 4 h prior to plating on medium with the antibiotic. For transformations with auxotrophic selection, cells were immediately plated after heat shock.
TABLE 1.
List of the strains used and generated in this work
| Strain/main genotype | Complete genotype | Source or reference |
|---|---|---|
| W303-1A (JT 9019) | MATa leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 GAL SUC mal | 63 |
| tps1Δ (JT 9020) | MATa leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 GAL SUC mal tps1::TRP1 | 13 |
| tps1Δ tdh1Δ | MATa leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 GAL SUC mal tps1::TRP1 tdh1::KanMX | This study |
| tps1Δ tdh2Δ | MATa leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 GAL SUC mal tps1::TRP1 tdh2::KanMX | This study |
| tps1Δ tdh3Δ | MATa leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 GAL SUC mal tps1::TRP1 tdh3::KanMX | This study |
| tps1Δ hxk2Δ | MATa leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 GAL SUC mal tps1::TRP1 hxk2::LEU2 | This study |
| tps1Δ snf1Δ | MATa leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 GAL SUC mal tps1::TRP1 snf1::HIS3 | 25 |
| hxt6,7,2,4,5Δ | MATa leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 GAL SUC mal HXT6,7::looped HXT2::looped HXT4::NatMX HXT5::KanMX | This study |
| tps1Δ hxt6,7,2,4,5Δ | MATa leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 GAL SUC mal tps1::HphMX HXT6,7::looped HXT2::looped HXT4::NatMX HXT5::KanMX | This study |
TABLE 2.
List of the plasmids used and generated in this work
| Plasmid backbone | Vector element | Selection marker | Insert | Source or reference |
|---|---|---|---|---|
| p426 | Multicopy 2 μ, TEF1 promotor | KanMX | None | A. Claes (MCB lab) |
| TDH2 | T. Nicolaï (MCB lab) | |||
| TDH3 | T. Nicolaï (MCB lab) | |||
| pYES2 | Multicopy 2 μ, ACT1 promotor | URA3 | pHluorin | 53 |
General growth conditions.
Yeast cells were grown in either rich or minimal medium. For rich medium, cells were grown in YP (1% [wt/vol] yeast extract, 2% [wt/vol] bacteriological peptone) supplemented with 100 mg/liter adenine. In case of minimal medium, cells were grown in complete synthetic medium (MP biomedicals) containing 0.17% (wt/vol) yeast nitrogen base without amino acids and ammonium sulfate (Oxoid), supplemented with 0.5% (wt/vol) ammonium sulfate (Sigma-Aldrich) and 100 mg/liter adenine. The same medium with the appropriate amount of essential amino acid(s) was used for growth of auxotrophic strains. As a carbon source, either 100 mM glucose, 100 mM galactose, 220 mM ethanol, or 325 mM glycerol was added to the growth media. The pH was adjusted to 5.5 for liquid medium and 6.5 for solid medium (2% agar) with KOH. Strains were grown at 30°C under continuous shaking for liquid cultures (180 rpm). For assay purposes, cells were typically pregrown overnight and inoculated the next day in fresh medium. Cells were harvested in the exponential phase unless mentioned otherwise.
Assessment of cell proliferation.
(i) Spot tests. For spot tests of growth capacity, cells were pregrown overnight on 3% glycerol in rich medium until the late-exponential early stationary phase. For cells harboring the p426 overexpression plasmid, 100 mg/liter Geneticin was supplemented in both liquid and solid media to retain the plasmid. Next, cells were washed and spotted at a 5-fold serial dilution (starting from optical density at 600 nm [OD600] = 0.5) on freshly poured solid media with the carbon source of interest. Pictures were taken after 2 to 3 days of incubation at 30°C.
(ii) Growth curves. To determine growth curves, cells from an overnight preculture on galactose were harvested, washed, and resuspended to an OD600 of 0.1 in a 96-well plate. OD595 was measured every 30 min for 48 h using the Thermo Scientific Multiskan FC microplate photometer. Plates were shaken every 10 min for 1 min at medium speed to keep cells in suspension.
Metabolite measurements.
Metabolites were extracted and measured as described previously Peeters et al. (12). General procedures will be mentioned in short. All auxiliary enzymes were purchased from Sigma-Aldrich.
(i) Sample collection. Cells for metabolite measurements were cultured in complete synthetic medium with 2% galactose (unless stated otherwise) to the exponential phase, harvested by centrifugation, and washed twice with 25 mM morpholineethanesulfonic acid (MES), pH 6. Cells were resuspended at a concentration of 75 mg (wet weight)/ml in complete synthetic medium with no added sugar and incubated at 30°C for 30 min of acclimatization. For metabolite determination after glucose or galactose addition, the sugars were added to a final concentration of 100 mM. Samples were taken by quenching cell suspensions in 60% methanol at −40°C (60). Quenched cells were centrifuged to remove the supernatant, after which, the pellet was resuspended in 0.5 ml 1 M HClO4 and transferred to a screw-cap tube. After mechanical lysis, an additional 0.5 ml of 1 M HClO4 was added, after which, the samples were stored at −20°C.
(ii) Sample processing. Thawed cell lysates were spun down at high speed and supernatants were collected. From this point on, cell lysates needed to undergo a process of neutralization. First, 50 μl of 5 M K2CO3 was added to 250 μl cell lysate together with 10 μl thymol blue (0.025%) to monitor the pH visually. Samples were allowed to degas on ice for 15 min. Next, samples were spun down at high speed, and 200 μl supernatant was added to 100 μl 1 M HCl and 10 μl Tris-HCl (pH 7.5). Samples were stored at −20°C.
(iii) Metabolite measurements. Metabolite concentrations were determined by endpoint measurement of the absorption of NADH or NADPH at 340 nm through coupled enzymatic reactions. The increase or decrease of absorption correlates linearly with the metabolite’s concentration and was calculated by applying Lambert’s law. The general reaction buffer was composed of 50 μl sample incubated with 150 μl 100 mM Tris-HCl, pH 7.5. Other components were added to the reaction buffer depending on the metabolite to be measured. For measurement of Glu6P, basal absorption was measured at 340 nm from samples added to reaction buffer supplemented with 0.8 mg/ml NADP+ and 10 mM MgCl2. After addition of Glu6P dehydrogenase at a final concentration of 50 μg/ml, the difference in OD340 values was used to calculate the absolute concentration of Glu6P. For measurement of ATP, the stabilized absorption spectrum of the Glu6P samples was taken as the new basal level. Next, hexokinase was added to a final concentration of 100 μg/ml together with 0.5 mM glucose. Stabilized spectra were used for calculating the corresponding ATP concentration.
For measurement of Fru1,6bisP, the general reaction buffer was supplemented with 0.8 μg/ml NADH, 25 μg/ml triosephosphate isomerase, and 25 μg/ml glycerol-3-phosphate dehydrogenase. After measuring basal absorption, 200 μg/ml aldolase was added, and the difference in OD340 values was used to calculate Fru1,6bisP levels. To express metabolite levels in terms of cytosolic concentration, an intracellular volume of 12 μl/mg protein was assumed.
Determination of in situ GAPDH activity.
(i) Preparation of permeabilized spheroplasts. The protocol for preparation of spheroplasts was adapted from previous reports (29). Strains were grown on 2% galactose in rich medium to the exponential phase. Cells were harvested, washed with 25 mM MES (pH 6), and resuspended at a concentration of 200 mg (wet weight)/ml in digestion buffer containing 1.2 M sorbitol, 60 mM potassium phosphate buffer (pH 7.5) (K2HPO4 plus KH2PO4), 1 mM EDTA (pH 8), 10 μl/ml β-mercaptoethanol, and 100 U/ml lyticase. Cell wall digestion at 30°C was followed by sampling the cell suspension for decrease in OD600 when treated with 5% SDS. When the OD600 dropped to around 80%, spheroplasts were considered appropriate for use, and lyticase digestion was stopped by addition of four volumes ice-cold 1.2 M sorbitol buffer. Spheroplasts were washed twice with ice-cold 1.2 M sorbitol and resuspended in spheroplast buffer (1.2 M sorbitol, 0.75 mM EDTA, 2 mM MgSO4, 1.8 mM NaCl, and 10 mM potassium phosphate buffer, pH 6.8). Protein concentration was measured in order to dilute the spheroplast suspension to a final protein concentration of 2 mg/ml. To permeabilize the spheroplasts, they were incubated for 10 min at 30°C with 20 μl/ml nystatin prior to measuring enzyme activity.
(ii) Determination of specific GAPDH activity. To measure GAPDH activity using permeabilized spheroplasts, the reaction buffer was composed of spheroplast buffer, 2 mM GA3P, 4 mM NAD+, and 20 mM Tris-HCl. The pH of the reaction buffer was adjusted with HCl/KOH to obtain the desired pH values. The influence of Tre6P and Fru1,6bisP on GAPDH activity was examined at pH 6.8. The reaction was initiated by adding 5 μl spheroplast suspension (2 mg/ml) to 145 μl reaction buffer. The initial reaction velocity was calculated from the measured linear increase of absorbance at 340 nm using the Synergy H1 Hybrid reader. Specific GAPDH activity was expressed as nanomoles per minute per milligram protein.
Determination of cytosolic pH.
For determination of intracellular pH, the pHluorin plasmid was transformed into strains of interest. The cells were grown in uracil dropout medium to retain the plasmid but with LoFlo formulation (ForMedium) to minimize background fluorescence. After reaching the exponential phase on 2% galactose, cells were harvested, washed, and resuspended in LoFlo medium at pH 5.5. Emission spectra were monitored at 510 nm after excitation at the wavelengths of 485 nm (510Ex: 485 nm) and 390 nm (510Ex: 390 nm). The ratio (510Ex: 485 nm/510Ex: 390 nm) was used to extrapolate the corresponding pH value using a calibration curve obtained by incubation of cell suspensions in calibration buffer at various pHs (25 mM MES, 25 mM HEPES, 25 mM KCl, 25 mM NaCl, 0.1 M ammonium acetate, 5 mM NaN3, and 0.18 mM 2-deoxyglucose). For experiments involving addition of NH4Cl, LoFlo medium was buffered with 200 mM Tris-HCl at the desired pH. Prior to treatment, baseline fluorescence was measured for 5 min. Using an integrated dispensing system that allows for quasisimultaneous pipetting and measurement (Thermo Scientific Fluoroskan Ascent FL with 390/510 and 485/510 filter sets), glucose and NH4Cl at final concentrations of 100 mM and 200 mM, respectively, were added at time point zero, after which, fluorescence was measured for the desired time periods. Per strain, emission spectra were measured for at least 9 technical repeats to consider the variability between experiments.
Determination of glucose uptake.
Transport of [U-14C]glucose was measured in accordance with previous studies by Reifenberger et al. (61) and Özcan et al. (62). In short, cells were grown in complete synthetic medium with 2% galactose. After reaching the exponential phase, cells were washed once with ice-cold 25 mM MES (pH 6), weighed, and resuspended in complete synthetic medium to a final concentration of 45 mg (wet weight)/ml. The cell suspension was dispensed into test tubes on ice and through a rotating schedule, each sample was temperature acclimated for 10 min at 30°C prior to the measurement of glucose uptake. For uptake of 100 mM glucose, an adequate amount of [U-14C]glucose was added to obtain after 10 s a response of at least 1,000 counts per min to create a sufficient signal-to-noise ratio. At time point zero, labeled glucose was added to the cells, and the suspension was quickly vortexed and quenched after 10 s with 5 ml ice-cold H2O, after which, the cell suspension was immediately filtered over a glass microfiber filter (Whatman GF/C). Cells were additionally washed on the filter two more times with water, after which, the loaded filter was transferred to a scintillation vial containing 3 ml liquid scintillation cocktail (Ultima-Flo M; PerkinElmer). To account for background signal originating from filter-attached radiolabeled glucose, three blank measurements per strain were included, for which cells where quenched prior to adding the labeled glucose. In experiments in which the effect of NH4Cl was assessed (Fig. 7a), Tris-HCl (pH 9) was added to the cells 5 s prior to glucose and NH4Cl addition. The final concentration of Tris-HCl and NH4Cl was 200 mM. Scintillation counting was performed using the Hidex 300 SL.
ACKNOWLEDGMENTS
We thank Filip Rolland (KU Leuven) for provision of the tps1Δ snf1Δ strain and Thomas Nicolaï and Arne Claes (MCB lab) for provision of plasmids. We also thank Nico Vangoethem for expertise and help with preparation of the figures.
This work was supported by PhD fellowships from the Agency for Innovation by Science and Technology (IWT-Flanders) to F.V.L. and W.V., the Fund for Scientific Research—Flanders, Interuniversity Attraction Poles Network P7/40, and the Research Fund of the KU Leuven (Concerted Research Actions) to J.M.T.
F.V.L. and J.M.T. conceived the project, F.V.L., W.V., and J.M.T. conceived the experimental approaches, F.V.L., W.V., and S.W. performed the experiments, F.V.L., W.V., G.V.Z., and J.M.T. analyzed the data, and F.V.L., W.V., G.V.Z., and J.M.T. wrote the manuscript.
Footnotes
Citation Van Leemputte F, Vanthienen W, Wijnants S, Van Zeebroeck G, Thevelein JM. 2020. Aberrant intracellular pH regulation limiting glyceraldehyde-3-phosphate dehydrogenase activity in the glucose-sensitive yeast tps1Δ mutant. mBio 11:e02199-20. https://doi.org/10.1128/mBio.02199-20.
REFERENCES
- 1.Van Aelst L, Hohmann S, Bulaya B, de Koning W, Sierkstra L, Neves MJ, Luyten K, Alijo R, Ramos J, Coccetti P, Martegani E, de Magalhães-Rocha NM, Brandão RL, Van Dijck P, Vanhalewyn M, Durnez P, Jans AWH, Thevelein JM. 1993. Molecular cloning of a gene involved in glucose sensing in the yeast Saccharomyces cerevisiae. Mol Microbiol 8:927–943. doi: 10.1111/j.1365-2958.1993.tb01638.x. [DOI] [PubMed] [Google Scholar]
- 2.Hohmann S, Huse K, Valentin E, Mbonyi K, Thevelein JM, Zimmermann FK. 1992. Glucose-induced regulatory defects in the Saccharomyces cerevisiae byp1 growth initiation mutant and identification of MIG1 as a partial suppressor. J Bacteriol 174:4183–4188. doi: 10.1128/jb.174.12.4183-4188.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cannon JF, Pringle JR, Fiechter A, Khalil M. 1994. Characterization of glycogen-deficient glc mutants of Saccharomyces cerevisiae. Genetics 136:485–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bell W, Klaassen P, Ohnacker M, Boller T, Herweijer M, Schoppink P, Van der Zee P, Wiemken A. 1992. Characterization of the 56-kDa subunit of yeast trehalose-6-phosphate synthase and cloning of its gene reveal its identity with the product of CIF1, a regulator of carbon catabolite inactivation. Eur J Biochem 209:951–959. doi: 10.1111/j.1432-1033.1992.tb17368.x. [DOI] [PubMed] [Google Scholar]
- 5.van de Poll KW, Kerkenaar A, Schamhart DH. 1974. Isolation of a regulatory mutant of fructose-1,6-diphosphatase in Saccharomyces carlsbergensis. J Bacteriol 117:965–970. doi: 10.1128/JB.117.3.965-970.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gonzalez MI, Stucka R, Blazquez MA, Feldmann H, Gancedo C. 1992. Molecular cloning of CIF1, a yeast gene necessary for growth on glucose. Yeast 8:183–192. doi: 10.1002/yea.320080304. [DOI] [PubMed] [Google Scholar]
- 7.Navon G, Shulman RG, Yamane T, Eccleshall TR, Lam KB, Baronofsky JJ, Marmur J. 1979. Phosphorus-31 nuclear magnetic resonance studies of wild-type and glycolytic pathway mutants of Saccharomyces cerevisiae. Biochemistry 18:4487–4499. doi: 10.1021/bi00588a006. [DOI] [PubMed] [Google Scholar]
- 8.Vuorio OE, Kalkkinen N, Londesborough J. 1993. Cloning of two related genes encoding the 56-kDa and 123-kDa subunits of trehalose synthase from the yeast Saccharomyces cerevisiae. Eur J Biochem 216:849–861. doi: 10.1111/j.1432-1033.1993.tb18207.x. [DOI] [PubMed] [Google Scholar]
- 9.De Virgilio C, Burckert N, Bell W, Jeno P, Boller T, Wiemken A. 1993. Disruption of TPS2, the gene encoding the 100-kDa subunit of the trehalose-6-phosphate synthase/phosphatase complex in Saccharomyces cerevisiae, causes accumulation of trehalose-6-phosphate and loss of trehalose-6-phosphate phosphatase activity. Eur J Biochem 212:315–323. doi: 10.1111/j.1432-1033.1993.tb17664.x. [DOI] [PubMed] [Google Scholar]
- 10.Alonso A, Pascual C, Herrera L, Gancedo JM, Gancedo C. 1984. Metabolic imbalance in a Saccharomyces cerevisiae mutant unable to grow on fermentable hexoses. Eur J Biochem 138:407–411. doi: 10.1111/j.1432-1033.1984.tb07930.x. [DOI] [PubMed] [Google Scholar]
- 11.van de Poll KW, Schamhart DHJ. 1977. Characterization of a regulatory mutant of fructose 1,6-bisphosphatase in Saccharomyces carlsbergensis. Mol Gen Genet 154:61–66. doi: 10.1007/BF00265577. [DOI] [PubMed] [Google Scholar]
- 12.Peeters K, Van Leemputte F, Fischer B, Bonini BM, Quezada H, Tsytlonok M, Haesen D, Vanthienen W, Bernardes N, Gonzalez-Blas CB, Janssens V, Tompa P, Versees W, Thevelein JM. 2017. Fructose-1,6-bisphosphate couples glycolytic flux to activation of Ras. Nat Commun 8:922. doi: 10.1038/s41467-017-01019-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hohmann S, Neves MJ, de Koning W, Alijo R, Ramos J, Thevelein JM. 1993. The growth and signalling defects of the ggs1 (fdp1/byp1) deletion mutant on glucose are suppressed by a deletion of the gene encoding hexokinase PII. Curr Genet 23:281–289. doi: 10.1007/BF00310888. [DOI] [PubMed] [Google Scholar]
- 14.Neves MJ, Hohmann S, Bell W, Dumortier F, Luyten K, Ramos J, Cobbaert P, de Koning W, Kaneva Z, Thevelein JM. 1995. Control of glucose influx into glycolysis and pleiotropic effects studied in different isogenic sets of Saccharomyces cerevisiae mutants in trehalose biosynthesis. Curr Genet 27:110–122. doi: 10.1007/BF00313424. [DOI] [PubMed] [Google Scholar]
- 15.van Heerden JH, Wortel MT, Bruggeman FJ, Heijnen JJ, Bollen YJ, Planque R, Hulshof J, O'Toole TG, Wahl SA, Teusink B. 2014. Lost in transition: start-up of glycolysis yields subpopulations of nongrowing cells. Science 343:1245114. doi: 10.1126/science.1245114. [DOI] [PubMed] [Google Scholar]
- 16.Gibney PA, Chen A, Schieler A, Chen JC, Xu Y, Hendrickson DG, McIsaac RS, Rabinowitz JD, Botstein D. 2020. A tps1Δ persister-like state in Saccharomyces cerevisiae is regulated by MKT1. PLoS One 15:e0233779. doi: 10.1371/journal.pone.0233779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shestov AA, Liu X, Ser Z, Cluntun AA, Hung YP, Huang L, Kim D, Le A, Yellen G, Albeck JG, Locasale JW. 2014. Quantitative determinants of aerobic glycolysis identify flux through the enzyme GAPDH as a limiting step. Elife 3:e03342. doi: 10.7554/eLife.03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blázquez MA, Lagunas R, Gancedo C, Gancedo JM. 1993. Trehalose-6-phosphate, a new regulator of yeast glycolysis that inhibits hexokinases. FEBS Lett 329:51–54. doi: 10.1016/0014-5793(93)80191-v. [DOI] [PubMed] [Google Scholar]
- 19.Bonini BM, Van Vaeck C, Larsson C, Gustafsson L, Ma P, Winderickx J, Van Dijck P, Thevelein JM. 2000. Expression of Escherichia coli otsA in a Saccharomyces cerevisiae tps1 mutant restores trehalose 6-phosphate levels and partly restores growth and fermentation with glucose and control of glucose influx into glycolysis. Biochem J 350:261–268. doi: 10.1042/bj3500261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hohmann S, Bell W, Neves MJ, Valckx D, Thevelein JM. 1996. Evidence for trehalose-6-phosphate-dependent and -independent mechanisms in the control of sugar influx into yeast glycolysis. Mol Microbiol 20:981–991. doi: 10.1111/j.1365-2958.1996.tb02539.x. [DOI] [PubMed] [Google Scholar]
- 21.Bonini BM, Van Dijck P, Thevelein JM. 2003. Uncoupling of the glucose growth defect and the deregulation of glycolysis in Saccharomyces cerevisiae tps1 mutants expressing trehalose-6-phosphate-insensitive hexokinase from Schizosaccharomyces pombe. Biochim Biophys Acta 1606:83–93. doi: 10.1016/s0005-2728(03)00086-0. [DOI] [PubMed] [Google Scholar]
- 22.Luyten K, Albertyn J, Skibbe WF, Prior BA, Ramos J, Thevelein JM, Hohmann S. 1995. Fps1, a yeast member of the MIP family of channel proteins, is a facilitator for glycerol uptake and efflux and is inactive under osmotic stress. EMBO J 14:1360–1371. doi: 10.1002/j.1460-2075.1995.tb07122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Aelst L, Hohmann S, Zimmermann FK, Jans AW, Thevelein JM. 1991. A yeast homologue of the bovine lens fibre MIP gene family complements the growth defect of a Saccharomyces cerevisiae mutant on fermentable sugars but not its defect in glucose-induced RAS-mediated cAMP signalling. EMBO J 10:2095–2104. doi: 10.1002/j.1460-2075.1991.tb07742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blázquez MA, Gancedo C. 1995. Mode of action of the gcr9 and cat3 mutations in restoring the ability of Saccharomyces cerevisiae tps1 mutants to grow on glucose. Mol Gen Genet 249:655–664. doi: 10.1007/BF00418035. [DOI] [PubMed] [Google Scholar]
- 25.Deroover S, Ghillebert R, Broeckx T, Winderickx J, Rolland F. 2016. Trehalose-6-phosphate synthesis controls yeast gluconeogenesis downstream and independent of SNF1. FEMS Yeast Res 16:fow036. doi: 10.1093/femsyr/fow036. [DOI] [PubMed] [Google Scholar]
- 26.Dashko S, Compagno C, Piskur J. 2014. Introduction to carbon metabolism in yeast, p 1–21, In Piskur J, Compagno C. (ed), Molecular mechanisms in yeast carbon metabolism. Springer-Verlag, Berlin, Germany. doi: 10.1007/978-3-642-55013-3_1. [DOI] [Google Scholar]
- 27.Thevelein JM, Hohmann S. 1995. Trehalose synthase: guard to the gate of glycolysis in yeast? Trends Biochem Sci 20:3–10. doi: 10.1016/s0968-0004(00)88938-0. [DOI] [PubMed] [Google Scholar]
- 28.Sokolov SS, Smirnova EA, Markova OV, Kireeva NA, Kirsanov RS, Khailova LS, Knorre DA, Severin FF. 2020. Lipophilic cations rescue the growth of yeast under the conditions of glycolysis overflow. Biomolecules 10:1345. doi: 10.3390/biom10091345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noubhani A, Bunoust O, Rigoulet M, Thevelein JM. 2000. Reconstitution of ethanolic fermentation in permeabilized spheroplasts of wild-type and trehalose-6-phosphate synthase mutants of the yeast Saccharomyces cerevisiae. Eur J Biochem 267:4566–4576. doi: 10.1046/j.1432-1327.2000.01511.x. [DOI] [PubMed] [Google Scholar]
- 30.Liberti MV, Dai Z, Wardell SE, Baccile JA, Liu X, Gao X, Baldi R, Mehrmohamadi M, Johnson MO, Madhukar NS, Shestov AA, Chio IIC, Elemento O, Rathmell JC, Schroeder FC, McDonnell DP, Locasale JW. 2017. A predictive model for selective targeting of the Warburg effect through GAPDH inhibition with a natural product. Cell Metab 26:648.e8–659.e8. doi: 10.1016/j.cmet.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ralser M, Wamelink MM, Kowald A, Gerisch B, Heeren G, Struys EA, Klipp E, Jakobs C, Breitenbach M, Lehrach H, Krobitsch S. 2007. Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress. J Biol 6:10–10. doi: 10.1186/jbiol61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reisz JA, Wither MJ, Dzieciatkowska M, Nemkov T, Issaian A, Yoshida T, Dunham AJ, Hill RC, Hansen KC, D'Alessandro A. 2016. Oxidative modifications of glyceraldehyde 3-phosphate dehydrogenase regulate metabolic reprogramming of stored red blood cells. Blood 128:e32–e42. doi: 10.1182/blood-2016-05-714816. [DOI] [PubMed] [Google Scholar]
- 33.McAlister L, Holland MJ. 1985. Isolation and characterization of yeast strains carrying mutations in the glyceraldehyde-3-phosphate dehydrogenase genes. J Biol Chem 260:15013–15018. [PubMed] [Google Scholar]
- 34.McAlister L, Holland MJ. 1985. Differential expression of the three yeast glyceraldehyde-3-phosphate dehydrogenase genes. J Biol Chem 260:15019–15027. [PubMed] [Google Scholar]
- 35.Boucherie H, Bataille N, Fitch IT, Perrot M, Tuite MF. 1995. Differential synthesis of glyceraldehyde-3-phosphate dehydrogenase polypeptides in stressed yeast cells. FEMS Microbiol Lett 125:127–133. doi: 10.1111/j.1574-6968.1995.tb07348.x. [DOI] [PubMed] [Google Scholar]
- 36.Stanley D, Chambers PJ, Stanley GA, Borneman A, Fraser S. 2010. Transcriptional changes associated with ethanol tolerance in Saccharomyces cerevisiae. Appl Microbiol Biotechnol 88:231–239. doi: 10.1007/s00253-010-2760-7. [DOI] [PubMed] [Google Scholar]
- 37.Valadi H, Valadi A, Ansell R, Gustafsson L, Adler L, Norbeck J, Blomberg A. 2004. NADH-reductive stress in Saccharomyces cerevisiae induces the expression of the minor isoform of glyceraldehyde-3-phosphate dehydrogenase (TDH1). Curr Genet 45:90–95. doi: 10.1007/s00294-003-0469-1. [DOI] [PubMed] [Google Scholar]
- 38.Norbeck J, Blomberg A. 2000. The level of cAMP-dependent protein kinase A activity strongly affects osmotolerance and osmo-instigated gene expression changes in Saccharomyces cerevisiae. Yeast 16:121–137. doi:. [DOI] [PubMed] [Google Scholar]
- 39.Linck A, Vu XK, Essl C, Hiesl C, Boles E, Oreb M. 2014. On the role of GAPDH isoenzymes during pentose fermentation in engineered Saccharomyces cerevisiae. FEMS Yeast Res 14:389–398. doi: 10.1111/1567-1364.12137. [DOI] [PubMed] [Google Scholar]
- 40.den Hollander JA, Ugurbil K, Brown TR, Shulman RG. 1981. Phosphorus-31 nuclear magnetic resonance studies of the effect of oxygen upon glycolysis in yeast. Biochemistry 20:5871–5880. doi: 10.1021/bi00523a034. [DOI] [PubMed] [Google Scholar]
- 41.Thevelein JM, Beullens M, Honshoven F, Hoebeeck G, Detremerie K, Griewel B, den Hollander JA, Jans AW. 1987. Regulation of the cAMP level in the yeast Saccharomyces cerevisiae: the glucose-induced cAMP signal is not mediated by a transient drop in the intracellular pH. J Gen Microbiol 133:2197–2205. doi: 10.1099/00221287-133-8-2197. [DOI] [PubMed] [Google Scholar]
- 42.Walther T, Mtimet N, Alkim C, Vax A, Loret MO, Ullah A, Gancedo C, Smits GJ, Francois JM. 2013. Metabolic phenotypes of Saccharomyces cerevisiae mutants with altered trehalose 6-phosphate dynamics. Biochem J 454:227–237. doi: 10.1042/BJ20130587. [DOI] [PubMed] [Google Scholar]
- 43.Teusink B, Walsh MC, van Dam K, Westerhoff HV. 1998. The danger of metabolic pathways with turbo design. Trends Biochem Sci 23:162–169. doi: 10.1016/s0968-0004(98)01205-5. [DOI] [PubMed] [Google Scholar]
- 44.Marešová L, Hošková B, Urbánková E, Chaloupka R, Sychrová H. 2010. New applications of pHluorin—measuring intracellular pH of prototrophic yeasts and determining changes in the buffering capacity of strains with affected potassium homeostasis. Yeast 27:317–325. doi: 10.1002/yea.1755. [DOI] [PubMed] [Google Scholar]
- 45.Kadam PC, Boone DR. 1996. Influence of pH on ammonia accumulation and toxicity in halophilic, methylotrophic methanogens. Appl Environ Microbiol 62:4486–4492. doi: 10.1128/AEM.62.12.4486-4492.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoshino M, Murakami K. 1985. AMP deaminase reaction as a control system of glycolysis in yeast. Role of ammonium ion in the interaction of phosphofructokinase and pyruvate kinase activity with the adenylate energy charge. J Biol Chem 260:4729–4732. [PubMed] [Google Scholar]
- 47.dos Passos JB, Vanhalewyn M, Brandao RL, Castro IM, Nicoli JR, Thevelein JM. 1992. Glucose-induced activation of plasma membrane H+-ATPase in mutants of the yeast Saccharomyces cerevisiae affected in cAMP metabolism, cAMP-dependent protein phosphorylation and the initiation of glycolysis. Biochim Biophys Acta 1136:57–67. doi: 10.1016/0167-4889(92)90085-p. [DOI] [PubMed] [Google Scholar]
- 48.Serrano R. 1983. In vivo glucose activation of the yeast plasma membrane ATPase. FEBS Lett 156:11–14. doi: 10.1016/0014-5793(83)80237-3. [DOI] [PubMed] [Google Scholar]
- 49.Kane PM. 2016. Proton transport and pH control in fungi. Adv Exp Med Biol 892:33–68. doi: 10.1007/978-3-319-25304-6_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carmelo V, Santos H, Sa-Correia I. 1997. Effect of extracellular acidification on the activity of plasma membrane ATPase and on the cytosolic and vacuolar pH of Saccharomyces cerevisiae. Biochim Biophys Acta 1325:63–70. doi: 10.1016/s0005-2736(96)00245-3. [DOI] [PubMed] [Google Scholar]
- 51.Orij R, Brul S, Smits GJ. 2011. Intracellular pH is a tightly controlled signal in yeast. Biochim Biophys Acta 1810:933–944. doi: 10.1016/j.bbagen.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 52.Elbing K, Larsson C, Bill RM, Albers E, Snoep JL, Boles E, Hohmann S, Gustafsson L. 2004. Role of hexose transport in control of glycolytic flux in Saccharomyces cerevisiae. Appl Environ Microbiol 70:5323–5330. doi: 10.1128/AEM.70.9.5323-5330.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Orij R, Postmus J, Ter Beek A, Brul S, Smits GJ. 2009. In vivo measurement of cytosolic and mitochondrial pH using a pH-sensitive GFP derivative in Saccharomyces cerevisiae reveals a relation between intracellular pH and growth. Microbiology (Reading) 155:268–278. doi: 10.1099/mic.0.022038-0. [DOI] [PubMed] [Google Scholar]
- 54.Dodd BJT, Kralj JM. 2017. Live cell imaging reveals pH oscillations in Saccharomyces cerevisiae during metabolic transitions. Sci Rep 7:13922–13922. doi: 10.1038/s41598-017-14382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trancíková A, Weisová P, Kiššová I, Zeman I, Kolarov J. 2004. Production of reactive oxygen species and loss of viability in yeast mitochondrial mutants: protective effect of Bcl-xL. FEMS Yeast Res 5:149–156. doi: 10.1016/j.femsyr.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 56.Dolz-Edo L, van der Deen M, Brul S, Smits GJ. 2019. Caloric restriction controls stationary phase survival through protein kinase A (PKA) and cytosolic pH. Aging Cell 18:e12921. doi: 10.1111/acel.12921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vega M, Riera A, Fernández-Cid A, Herrero P, Moreno F. 2016. Hexokinase 2 is an intracellular glucose sensor of yeast cells that maintains the structure and activity of Mig1 protein repressor complex. J Biol Chem 291:7267–7285. doi: 10.1074/jbc.M115.711408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Amigoni L, Martegani E, Colombo S. 2013. Lack of HXK2 induces localization of active Ras in mitochondria and triggers apoptosis in the yeast Saccharomyces cerevisiae. Oxid Med Cell Longev 2013:678473–678473. doi: 10.1155/2013/678473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gietz RD, Schiestl RH. 2007. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2:31–34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
- 60.de Koning W, van Dam K. 1992. A method for the determination of changes of glycolytic metabolites in yeast on a subsecond time scale using extraction at neutral pH. Anal Biochem 204:118–123. doi: 10.1016/0003-2697(92)90149-2. [DOI] [PubMed] [Google Scholar]
- 61.Reifenberger E, Boles E, Ciriacy M. 1997. Kinetic characterization of individual hexose transporters of Saccharomyces cerevisiae and their relation to the triggering mechanisms of glucose repression. Eur J Biochem 245:324–333. doi: 10.1111/j.1432-1033.1997.00324.x. [DOI] [PubMed] [Google Scholar]
- 62.Özcan S, Freidel K, Leuker A, Ciriacy M. 1993. Glucose uptake and catabolite repression in dominant HTR1 mutants of Saccharomyces cerevisiae. J Bacteriol 175:5520–5528. doi: 10.1128/jb.175.17.5520-5528.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thomas BJ, Rothstein R. 1989. Elevated recombination rates in transcriptionally active DNA. Cell 56:619–630. doi: 10.1016/0092-8674(89)90584-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Intracellular acidification after the addition of glucose is prevented in 200 mM Tris-HCl medium at high pH in the presence of 200 mM NH4Cl. Intracellular pH was monitored after addition at time point zero of 100 mM glucose with (full lines) or without (dotted lines) 200 mM NH4Cl. Wild-type (a) and tps1Δ (b) cells in 200 mM Tris-HCl medium at pH 8 (light blue), pH 8.5 (dark blue), and pH 9 (purple). Cells were grown in uracil-deficient medium with 2% galactose to retain the pHluorin plasmid. Download FIG S1, EPS file, 2.5 MB (2.4MB, eps) .
Copyright © 2020 Van Leemputte et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
High extracellular pH inhibits growth. Growth in complete synthetic medium supplemented with either 100 mM glucose (full lines) or 100 mM galactose (dotted lines) was followed over 48 h by measuring OD595 for wild-type (blue) and tps1Δ (red) cells in regular liquid medium at pH 5.5 (a) or 50 mM Tris-HCl medium at pH 8 (b). Cells were pregrown on galactose medium. Download FIG S2, EPS file, 2.7 MB (3.2MB, eps) .
Copyright © 2020 Van Leemputte et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Absence of growth on 100 mM glucose in the tps1Δ hxt6,7,2,4,5Δ strain. Cultures of the wild-type, tps1Δ, hxt6,7,2,4,5Δ, and tps1Δ hxt6,7,2,4,5Δ strains were spotted in 5-fold dilutions on YP agar with 100 mM galactose or 100 mM glucose as carbon source. Pictures were taken after three days. Download FIG S3, TIF file, 2.5 MB (2.5MB, tif) .
Copyright © 2020 Van Leemputte et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.